

Abstract
CTL-associated antigen 4 (CTLA-4) blockade can induce tumor regression and improved survival in cancer patients. This treatment can enhance adaptive immune responses without an exogenous vaccine, but the immunologic biomarkers associated with improved clinical outcome in cancer patients are not fully established. A phase Ib trial in patients with metastatic, castration resistant prostate cancer (mCRPC) was performed combining ipilimumab with sargramostim (GM-CSF). In addition to evaluating ipilimumab dose, patients were followed clinically for response and overall survival, and for immunomodulation of circulating T cells. PSA declines of ≥50% and radiographic responses were observed at doses of ≥3 mg/kg/dose. Timing of clinical responses could be either immediate or delayed. Durable responses were also observed off treatment. A subset of patients experienced long-term survival with or without objective clinical responses. The relationship between T-cell phenotype in peripheral blood and overall survival were examined retrospectively. We found that the treatment induced an increase in the levels of CD4+ effector T (Teff) cells, regulatory T (Treg) cells, PD-1+ CD4 Teff cells, and PD-1+ CD8 T cells. However, these increased levels were not associated with overall survival. Instead, low pre-treatment baseline levels of PD-1+ CD4 Teff cells were found to correlate with longer overall survival. Furthermore, baseline levels of PD-1+ CD4 Teff cells from patients with shorter overall survival were higher than from cancer-free male controls. These results suggest that pre-existing expression of immunologic checkpoint marker PD-1 on CD4 Teff cells may help identify patients that may benefit from ipilimumab treatment.
Keywords: anti-CTLA-4, prostate cancer, PD-1, CTLA-4, PBMC, survival
Introduction
Cytotoxic T-lymphocyte antigen-4 (CTLA-4) is an immune checkpoint receptor expressed on T cells that provides inhibitory signaling following activation of naïve and memory T cells to maintain immune homeostasis (1, 2). Blocking CTLA-4 may serve to remove this inhibition of T-cell responses in the setting of an immunosuppressive tumor environment thereby leading to immune responses against the tumor. In animal models, CTLA-4 blockade with monoclonal antibodies can enhance T-cell responses and may also deplete intratumoral regulatory T cells (Treg) enabling tumor regression (3, 4).
Ipilimumab is a fully humanized monoclonal antibody targeting CTLA-4 that is FDA approved for the treatment of unresectable or metastatic melanoma at 3 mg/kg/dose (5). In two phase III studies in advanced melanoma, ipilimumab was shown to significantly prolong overall survival (OS) (6, 7). In the pivotal clinical trial, melanoma patients were treated with ipilimumab plus gp100 (a melanoma peptide vaccine), ipilimumab alone or gp100 alone (6). The median OS were 10.0, 10.1, and 6.4 months, respectively. Although improvement in median OS was modest, a subset of patients was observed in these and other melanoma clinical trials to have durable long-term survival benefit (8, 9). Notably, long-term survival can occur without accompanying objective tumor response. Improved OS was also observed with ipilimumab in combination with dacarbazine versus dacarbazine plus placebo in a phase III clinical trial of patients with metastatic melanoma who received no prior treatment (11.2 months versus 9.1 months) (7). Additionally, treatment with ipilimumab plus sargramostim (GM-CSF) resulted in improved median OS and lower toxicity compared to ipilimumab alone (17.5 months versus 12.7 months) in a phase II clinical trial with unresectable melanoma (10).
In a phase III clinical trial for patients with metastatic castration-resistant prostate cancer (mCRPC) who had received prior chemotherapy, the results demonstrated no significant difference in OS between treatments with 10 mg/kg of ipilimumab versus placebo following local radiotherapy to a metastatic site (11). The median OS was 11.2 months for the ipilimumab-treated group and 10.0 months for the placebo group. However, it was observed that the hazard ratio (HR) decreased over time favoring the ipilimumab arm, suggesting that ipilimumab treatment is associated with better survival at later time points. HR was 1.46 (95% CI 1.10 – 1.95) for 0 – 5 months and 0.6 (95% CI 0.43 – 0.86) for beyond 12 months.
Here we present survival outcome along with updated ipilimumab dose evaluation of 42 mCRPC patients treated with a combination of ipilimumab and sargramostim in a phase Ib trial (12). As of censor date of the trial on October 21st 2014, all except two patients have died. Clinical responses, designated as ≥ 50% PSA declines from the level at start of treatment or objective tumor responses, were not observed at dose levels less than 3 mg/kg of ipilimumab. A subset of patients experienced long-term survival with and without clinical responses. The relationship between survival and immune subsets was evaluated in an exploratory level with patients from the 3 mg/kg and above dose groups. We found that improved overall survival was correlated with baseline expression levels of programmed death-1 (PD-1) on CD4 effector T (Teff) cells.
Materials and Methods
Clinical trial
Results for the lower-dose levels up to 3 mg/kg/dose for this phase 1b trial have been described (12). Inclusion criteria for patients were histologically proven metastatic castration-resistant adenocarcinoma of the prostate with progression as defined by the PSA Working Group Consensus Criteria (13), and no prior treatment with steroids, chemotherapy or immunotherapy. For patients with measurable disease, progressive CRPC was defined as at least a 20% increase in the sum of the longest diameter of target lesions or the appearance of one or more new lesions, as per Response Evaluation Criteria in Solid Tumors (RECIST) criteria (14); for patients with no measurable disease, a positive bone scan and a PSA level of at least 5 ng/ml which had risen on at least 2 successive occasions, at least 2 weeks apart were required. Patients received escalating doses of ipilimumab (Bristol-Myers Squibb) with a fixed dose of sargramostim (Sanofi). The initial design included dose escalation of ipilimumab from 0.5 mg/kg to 3 mg/kg (0.5, 1.5, and 3) every 4 weeks for 4 doses (12). The study was subsequently modified to include 5 and 10 mg/kg dose levels, as well as an expansion cohort of 6 patients at 3 mg/kg/dose (cohort 5A) (Table 1). Sargramostim at 250 μg/m2/dose on days 1–14 of 28 days cycles was administered subcutaneously and continued until disease progression or grade 3 or 4 treatment-related toxicity.
Table 1.
Clinical responses per cohort
| Dose Levela | ≥50% PSA Response (Best decline %) | Objective Tumor Responseb | TTP c (months) | Median Overall Survival (months) |
|---|---|---|---|---|
| 1 (0.5 mg/kg x 4) | 0/3 | 0/3 | 25 | |
| 2 (0.5 mg/kg x 3, 1.5 mg/kg x 1) | 0/7 | 0/7 | 26 | |
| 3 (1.5 mg/kg x 4) | 0/5 | 0/5 | 28 | |
| 4 (1.5 mg/kg x 3, 3 mg/kg x 1) | 0/3 | 0/3 | 12 | |
| 5 (3 mg/kg x 4) | 3/6 (79, 95, 97) | 2/6 | 20, 25.75, 89.25 | 56 |
| 6 (5 mg/kg x 4) | 0/6 | 0/6 | 13 | |
| 7 (10 mg/kg x 4) | 2/6 (50, 80) | 0/6 | 9.75, 18 | 19 |
| 5A (3 mg/kg x 4) | 0/6 | 0/6 | 20 | |
| Cumulative | 5/42 | 2/42 | 20 (median) | 23.6 |
Dosage of ipilimumab and the number of doses are given in brackets ();
Objective tumor response defined by RECIST;
TTP is time to progression calculated from the time of initial response.
The primary endpoint of safety was graded according to National Cancer Institute (NCI) Common Terminology Criteria for Adverse Events version 3.0. Dose-limiting toxicity (DLT) included grade 3 or 4 treatment-related toxicity but excluded grade 3 immune-related adverse events (with the exception of ocular events) that did not require the use of steroids. Exploratory endpoints included T-cell activation, objective tumor responses (decrease in tumor size and/or lesions) as defined by RECIST (14), and PSA declines of ≥ 50% in PSA levels confirmed 4 weeks later as defined by the PSA Working Group Consensus Criteria.
Progression is defined as a 50% rise in PSA above the nadir or back to baseline, whichever is lower, on at least two consecutive measurements at least two weeks apart; or the appearance of one or more new lesions occurring more than one month after the initiation of therapy. Bone scans (and CT scans if abnormal) were repeated every 12 weeks and at the time of PSA progression. Best PSA decline was the maximum percentage (%) decline from initial PSA levels before treatment. OS was calculated from date of first treatment to date of death (n = 40) or censor date of trial on October 21st 2014 (n = 2).
Flow cytometry
Staining for flow cytometry was carried out on cryopreserved peripheral blood mononuclear cells (PBMC). In addition to study participants, PBMCs were also obtained from men undergoing prostate cancer screening without a subsequent diagnosis of cancer (cancer-free male controls). Cells were incubated with DNAse I (15 U/ml, Roche Diagnostics) for 30 min at 30°C and washed twice with FACs buffer (PBS with 2% FBS and 2 mM EDTA). Cell surface staining was performed in FACS buffer for 30 min at 4°C. Intracellular FoxP3 was performed using the FoxP3 fix/perm buffer set (Biolegend, Inc.) according to the manufacturer’s protocol. The following anti-human antibodies were used: (Alexa Fluor 700)-CD3 (clone HIT3a), (Brilliant violet 570)-CD4 (clone RPA-T4), (Brilliant violet 650)-CD25 (clone BC96), (Alexa Fluor 647)-CD127 (clone A019D5), (Alexa Fluor 488)-FoxP3 (clone 206D), and (Brilliant violet 421)-PD-1 (clone EH12.2H7). Stained cells were fixed with Fluorofix buffer (Biolegend, Inc.) according to manufacturer’s instructions and analyzed with an LSRII (BD Biosciences) flow cytometer. Data analysis was performed with Flowjo software (Treestar). Percentage (%) of positive cells was gated based on appropriate isotype control. Absolute count (per μl of blood) for each immune subset is calculated by multiplying the percentage of each subset with the preceding parent subset and with the absolute lymphocyte count quantified on the day of blood drawn.
Statistical Analysis
Distributions of percentage of paired immune subsets at week 0 (pre-treatment) were compared with that at week 4 (cycle 1) or at week 8 (cycle 2) using Wilcoxon matched-pairs signed rank test using Prism (GraphPad) software. The number of patients with PBMCs at the various time points differed based on availability.
Distributions of categorical patient characteristics such as Eastern Cooperative Oncology group (ECOG) status, Gleason score, prior radical prostatectomy, prior radiation, subsequent therapies, and clinical responses between long-term survivors (LTS, OS range: 25.4 months – 99.7 months) (n=11) and short-term survivors (STS, OS range: 1.9 months – 22.4 months) (n=12), were compared using Fisher’s exact test with Prism (Graphpad) software.
Distributions of continuous patient characteristics such as age, baseline PSA levels, lactate dehydrogenase (LDH) levels, months on study, and percentage of immune subsets between long-term survivors and short-term survivors as described above were compared using Mann-Whitney U-test with Prism (GraphPad) software. Distributions of percentage of immune subsets between cancer-free male controls (n=7) and LTS or STS were similarly compared using Mann-Whitney U-test.
Statistical significance was declared based on alpha level of 0.05 with Bonferroni correction to adjust for multiple testing as needed. Due to the small sample size, all significant outcomes should be considered as hypothesis generating and confirmation with a larger sample size is needed.
Results
Patient Characteristics
A total of 42 patients underwent treatment. Patient characteristics for each cohort (media and range) are presented in Supplementary Table S1. For all 42 patients, the median age was 70.5 years (range 47 – 82). Eight patients had a Gleason score of ≤ 6, 12 patients had a Gleason score of 7, and 20 patients had a Gleason score of 8 – 10. Gleason scores were not available for two patients. 31 and 11 patients had an ECOG performance status of 0 and 1, respectively. At pre-treatment, the median LDH was 172 U/L (range 136 – 557), and the median alkaline phosphatase was 92 U/L (range 28 – 1725). The median PSA at entry was 37.45 ng/mL (range 6.72 – 435.10). 25 patients had bone only disease, 5 patients had soft tissue only disease, and 12 patients had both bone and soft tissue disease.
Toxicity
Consistent with other studies of ipilimumab, toxicity was primarily immune in nature with the most common adverse events being diarrhea and rash. All adverse events are delineated by cohort in Supplementary Table S2. Seven patients experienced diarrhea, with three of these being grade 1 and four being grade 3. One of these events required steroids, which is defined as a DLT. Eight patients experienced a rash, one was grade 1, three were grade 2, and four were grade 3. Two of the patients required steroids, making them DLTs. Other immune-related adverse events included adrenal insufficiency (grade 2), panhypopituitarism (grade 3), pneumonitis (grade 2), and temporal arteritis (grade 3). Aside from the expected immune-related adverse events, cardiovascular events were also observed with 2 occurrences of atrial fibrillation (both grade 3), two cerebrovascular incidences (CVA, one grade 3 and one grade 4), and one grade 3 deep venous thrombosis (DVT). One patient died from pulmonary embolism (PE) and not from disease progression. The maximum tolerated dose was not established for this trial as the 2 DLTs were not observed in the highest dose level (10mg/kg/dose).
Clinical Outcomes
A waterfall plot of nadir PSA values (Fig. 1A) demonstrates that 23 of 42 patients (54%) had some decline in PSA. Five of 42 patients (11.9%) experienced a 50% or greater decline in PSA (Table 1). The median time to PSA nadir was 5.9 weeks (range 1.9 – 39.1 weeks) for patients with any PSA decline and 15.9 weeks (range 11.9 – 39.1 weeks) for patients with ≥ 50% PSA decline (Fig. 1B). Objective tumor response and ≥ 50% PSA decline was not observed in cohorts treated at < 3 mg/kg/dose level. Three of 12 patients treated at 3 mg/kg/dose experienced ≥ 50% PSA decline, two of these three patients had objective responses with regression of liver metastasis in one patient and of bone metastasis in the other (cohort 5). One patient in the expansion cohort at 3mg/kg experienced a 49% decline (cohort 5A). In the cohort treated at 5 mg/kg/dose, none of 6 patients demonstrated ≥ 50% PSA decline or objective tumor response. Of the 6 patients treated at the 10 mg/kg, two had ≥ 50% PSA decline. There was no accompanying objective tumor response.
Figure 1.

Clinical outcomes of 42 mCRPC patients in a Phase Ib ipilimumab (anti-CTLA-4) and sargramostim (GM-CSF) clinical trial. A, Waterfall plot of the maximum percentage change in PSA from baseline of each patient until nadir or off study. Dashed line shows 50% decline in PSA. B, Spider plot shows change in PSA with time from baseline of each patient until nadir or off study. Dashed line shows 50% decline in PSA. C, Graph showing the duration of study treatment, duration of response, time to disease progression, and time to ≥50% decline in PSA for each patient. D, Overall survival curves for all patients based on the analysis on the censor date. Dotted lines below and above the survival curve (solid line) show lower and upper 95% confidence intervals, respectively. Vertical tick marks indicate OS of patients who were still alive as of the censor date.
As of censor date of the trial, all patients had come off study. One patient came off treatment by patient’s choice. 13 patients came off treatment for PSA progression prior to the first set of scans, 16 patients came off treatment with PSA progression following the first set of scans, 6 patients came off treatment for tumor progression by scans, and 6 patients came off treatment for immune-related adverse toxicities. However, two patients from the 3 mg/kg/dose group that came off treatment due to immune-related toxicities demonstrated durable responses with their PSA levels remaining less than 50% of their pre-treatment levels for 19 and 85 months after being off treatment without any new treatment (Fig. 1C). One patient from the 5 mg/kg/dose group came off treatment due to an initial disease progression, but a delayed response was observed with his PSA decline attaining 50% at 7 months without any new treatment.
Long-term follow-up
This was a Phase Ib study and survival analysis was not a planned protocol endpoint. Nevertheless, since immunotherapies are now known to induce improvements in overall survival even in the absence of objective responses (9,15), survival analysis was carried out post-hoc. The median OS for all patients (n=42) is 23.6 months (95% confidence interval (16) = {16.2, 39.3}) (Fig. 1D).
Analysis on the censor date showed that two of the 42 patients were still living and 40 patients have died. Four of the 5 patients who demonstrated clinical responses have OS greater than the median for the group (OS range: 25 to 100 months). For all patients who demonstrated clinical responses described above as defined by objective tumor response and/or PSA decline of ≥ 50% from baseline, OS ranged from 14 months to 100 months. For the remainder of patients from the same dose cohorts (3 mg/kg/dose – 10 mg/kg/dose) who did not demonstrate clinical responses, OS ranged from 2 months to 86 months. Long OS was observed in patients without clinical responses.
Distribution of patient baseline characteristics with overall survival
Since ipilimumab is FDA approved for treatment of unresectable or metastatic melanoma at the 3 mg/kg/dose and clinical responses were not observed at less than 3 mg/kg/dose in this trial, we chose to evaluate further patients that were treated with at least 3 mg/kg/dose of ipilimumab. Patient characteristics and clinical responses for individual patients treated with ≥ 3 mg/kg/dose and sargramostim at 250 μg/m2/dose are presented in Supplementary Table S3. These patients were divided into two groups using median survival of 23.6 months as the cutoff. Baseline characteristics of patients with long overall survival (LTS, OS range: 25.4 months – 99.7 months) (n = 11) were compared with that of patients with short overall survival (STS, OS range: 1.9 months – 22.4 months) (n = 12) (Supplementary Fig. 1). Patients’ age, baseline PSA levels, LDH levels, or months on study did not correlate with OS (p-values = 0.193, 0.311, 0.277, and 0.100 respectively). The number of patients with ECOG status of 0 or 1, Gleason scores grouped as 3 to 6, or 7 to 9, prior radical prostatectomy, and prior radiation, were not significantly different between the two groups (p-values = 1.00, 0.90, 1.00, and 0.67, respectively). The number of patients with clinical responses as described above and the number of patients who went on to subsequent therapies also did not correlate with OS (p-values = 0.16 and 0.38, respectively).
Treatment increased the levels of Treg cells, CD4 Teff cells and PD-1+ CD4 Teff and PD-1+ CD8 T cells
Where possible, analyses for treatment-induced changes in levels of immune subsets for patients treated with ≥ 3 mg/kg/dose and sargramostim at 250 μg/m2/dose were performed. Distribution and levels of immune subsets from week 4 (cycle 1) or week 8 (cycle 2) of treatment were compared to those of pre-treatment levels at week 0.
The absolute lymphocyte counts were significantly higher compared to those of pre-treatment levels after cycle 1 but not after cycle 2 of treatment (p-values = 0.002 and 0.119, respectively) (Fig. 2A).
Figure 2.

Levels of circulating lymphocytes, CD4 Teff cells and CD8 T cells with treatment. A, Time course of absolute lymphocyte counts for assessed patients at week 0 (pre-treatment), week 4 (cycle 1), and week 8 (cycle 2) of treatment. Connected dots show time course of the same patient. B, Box plots of absolute lymphocyte counts for long-term (L) and short-term (S) survivors at each time point. C and G, Time course of percentages of CD4 Teff cells and CD8 T cells in total lymphocytes, respectively. Connected dots show time course of the same patient. E and I, Time course of absolute counts of CD4 Teff cells and CD8 T cells, respectively. D and H, Box plots of percentage of CD4 Teff cells and CD8 T cells in total lymphocytes, respectively, for long-term (L) and short-term (S) survivors at each time point. F and J, Box plots of absolute counts of CD4 Teff cells and CD8 T cells, respectively, for long-term (L) and short-term (S) survivors at each time point. Whiskers show minimum and maximum levels. * p-value < 0.05, *** p-value < 0.001.
We have shown previously that ipilimumab and sargramostim expanded the levels of circulating Treg cells (CD4+CD3+FoxP3+CD127−CD25+) (17). This was also observed for patients who were treated with ≥ 3 mg/kg/dose of ipilimumab and 250 μg/m2/dose of sargramostim after cycle 1 and after cycle 2 of treatment (Fig. 3A, B, C).
Figure 3.

Levels of circulating Treg cells with treatment. A, Gating strategy for Treg cells, CD4 Teff cells and CD8 T cells. B and C, Time course of percentage of Treg cells of total lymphocytes and absolute counts of Treg cells, respectively, for accessed patients at week 0 (pre-treatment), week 4 (cycle 1) and week 8 (cycle 2). Connected dots show time course of the same patient. D and E, Box plots of percentage of Treg cells of total lymphocytes and absolute counts of Treg cells, respectively, for long-term (L) and short-term (S) survivors at each time point. Whiskers show minimum and maximum levels. * p-value < 0.05, ** p-value < 0.01, *** p-valye < 0.001.
The percentages of total lymphocytes and absolute counts of CD4 Teff cells (CD4+CD3+FoxP3−) were significantly higher after one cycle of treatment (Fig. 2C and E). However, for CD8 T cells, only the absolute counts and not the percentage of total lymphocytes were significantly higher after one cycle of treatment (Fig. 2G and I). This difference could be due to the higher levels of absolute lymphocyte counts after one cycle of treatment as described above.
The percentages of CD4 Teff cells that express surface PD-1 were significantly higher after cycle 1 (p-value = 0.0001) and continued to be significantly higher after cycle 2 compared to pre-treatment levels (p-value = 0.0002) (Fig. 4A, B, D). The absolute counts of PD-1+ CD4 Teff cells were also significantly higher after both cycles (p-values = 0.0001 and 0.0002, respectively). The percentages of CD8 T cells that express PD-1 were also significantly higher from pre-treatment levels after cycle 1 (p-value = 0.004) and after cycle 2 of treatment (p-value = 0.005) (Fig. 4A, C, E). The absolute counts of PD-1+ CD8 T cells were also significantly higher after both cycles (p-values = 0.005 and 0.022, respectively).
Figure 4.

PD-1 expression of CD4 Teff cells and CD8 T cells. A, Flow cytometry was used to assess PD-1 expression by CD4 Teff cells and CD8 T cells. Percentage of PD-1+ cells in antibody-stained sample was gated based on isotype-matched controls. Shaded histograms denote isotype controls; open histograms denote stained samples. B and C, Time course of percentages of CD4 Teff and CD8 T cells that express PD-1 respectively. Connected dots show time course of the same patient. D and E, Time course of absolute counts of CD4 Teff and CD8 T cells that express PD-1 respectively. F and G, Box plots of percentage of CD4 Teff and CD8 T cells that express PD-1, respectively, for long-term (L) and short-term (S) survivors at each time point. H and I, Box plots of absolute counts of CD4 Teff and CD8 T cells that express PD-1, respectively, for long-term (L) and short-term (S) survivors at each time point. Whiskers show minimum and maximum levels. * p-value < 0.05, ** p-value < 0.01, *** p-value < 0.001.
Lower levels of pre-existing PD-1+ CD4 Teff cells correlated with longer survival
Next, we investigated in an exploratory manner if immune subsets were related to survival duration with available samples. The levels of immune subsets of patients treated with ≥ 3 mg/kg/dose and sargramostim at 250 μg/m2/dose with long overall survival (LTS, OS range: 25.4 months – 99.7 months) were compared with those of patients with short overall survival (STS, OS range: 1.9 months – 22.4 months).
Distribution of the absolute lymphocyte counts did not differ between LTS and STS at pre-treatment (p-value = 0.201), after cycle 1 (p-value = 0.670), and after cycle 2 of treatment (p-value = 0.779) (Fig. 2B).
Distribution of the percentages of total lymphocytes and the absolute counts of Treg cells did not differ between LTS and STS at pre-treatment (p-values = 0.113 and 0.504, respectively), after cycle 1 (p-values = 0.980 and 0.348, respectively), and after cycle 2 of treatment (p-values = 0.387 and 0.752, respectively) (Fig. 3D, E) (Table 2 and Supplementary Table S4).
Table 2.
Comparison of T-cell subsets between LTS and STS
| T-cell subsetsa | LTSb Medianc (Ranged) |
STSb Medianc (Ranged) |
p-valuee |
|---|---|---|---|
|
| |||
| Week 0 (pre-treatment) | |||
| Total CD4 T cells (CD4+CD3+) | 41.0 (32.8 – 59.6) | 48.3 (33.6 – 71.8) | 0.203 |
| CD4 Teff cells (CD4+CD3+FoxP3−) | 38.9 (30.1 – 56.2) | 45.2 (30.4 – 66.8) | 0.263 |
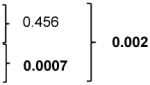
| PD-1+ (PD-1+CD4+CD3+FoxP3−) | 10.1 (5.4 – 14.5) | 22.0 (12.8 – 42.3) | 0.0007 |
| Treg (CD4+CD3+FoxP3+CD127−CD25+) | 1.6 (0.7 – 2.8) | 2.0 (1.23 – 3.9) | 0.113 |
| Total CD8 T cells (CD4−CD3+) | 24.5 (5.03 – 42.2) | 18.3 (6.71 – 50.7) | 0.461 |
| PD-1+ (PD-1+CD4−CD3+) | 15.0 (5.31 – 28.0) | 22.2 (8.4 – 33.0) | 0.246 |
| Week 4 (cycle 1) | |||
| Total CD4 T cells (CD4+CD3+) | 58.1 (43.3 – 62.3) | 58.3 (45.0 – 62.0) | 0.942 |
| CD4 Teff cells (CD4+CD3+FoxP3−) | 53.3 (41.1 – 54.7) | 51.9 (32.1 – 59.0) | 0.805 |
| PD-1+ (PD-1+CD4+CD3+FoxP3−) | 18.8 (7.12 – 35.4) | 28.1 (13.6 – 49.5) | 0.055 |
| Treg (CD4+CD3+FoxP3+CD127−CD25+) | 3.0 (1.9 – 4.2) | 2.7 (1.7 – 6.0) | 0.980 |
| Total CD8 T cells (CD4−CD3+) | 20.6 (5.1 – 32.2) | 16.1 (8.6 – 22.0) | 0.555 |
| PD-1+ (PD-1+CD4−CD3+) | 16.5 (6.49 – 33.0) | 26.7 (14.1 – 38.7) | 0.246 |
| Week 8 (cycle 2) | |||
| Total CD4 T cells (CD4+CD3+) | 54.5 (38.5 – 65.6) | 55.4 (29.8 – 70.1) | 0.931 |
| CD4 Teff cells (CD4+CD3+FoxP3−) | 50.8 (36.1 – 61.1) | 50.4 (26.8 – 63.1) | 0.920 |
| PD-1+ (PD-1+CD4+CD3+FoxP3−) | 18.3 (8.91 – 34.5) | 31.1 (17.2 – 51.2) | 0.054 |
| Treg (CD4+CD3+FoxP3+CD127−CD25+) | 2.5 (1.4 – 3.6) | 3.1 (1.5 – 4.1) | 0.387 |
| Total CD8 T cells (CD4−CD3+) | 15.4 (5.8 – 31.7) | 18.6 (13.2 – 52.6) | 0.671 |
| PD-1+ (PD-1+CD4−CD3+) | 21.2 (5.93 – 30.3) | 26.5 (14.7 – 40.4) | 0.228 |
T-cell subsets are defined by immune markers as indicated in the table;
Not all 23 patients have PBMCs available at all time points. Pre-treatment, n = 8 for LTS and n = 12 for STS; cycle 1, n = 7 for LTS and n = 9 for STS; cycle 2, n = 7 for LTS and n = 8 for STS;
Median values of total CD4 T cells, total CD8 T cells, CD4 Teff cells and Treg cells are % of total lymphocytes. Median values of PD-1 positive cells are % of the respective parent gate;
Values in brackets () are range of each data set;
Mann-Whitney test;
Bold-faced characters highlight p-value ≤ 0.05; LTS, long-term survivors; STS, short-term survivors
Distribution of the percentages of total lymphocytes and absolute counts of CD4 Teff cells also did not differ between LTS and STS at pre-treatment (p-values = 0.263 and 0.841, respectively), after cycle 1 (p-values = 0.805 and 0.745, respectively), and after cycle 2 of treatment (p-values = 0.920 and 0.845, respectively) (Fig. 2D, F) (Table 2 and Supplementary Table S4). The percentages of total lymphocytes and absolute counts of CD8 T cells also did not correlate with survival at pre-treatment (p-values = 0.461 and 0.304, respectively), after cycle 1 (p-values = 0.555 and 0.670, respectively), and after cycle 2 of treatment (p-values = 0.671 and 0.835, respectively) (Fig. 2H, J) (Table 2 and Supplementary Table S4).
However, distribution of the percentages of surface PD-1+ CD4 Teff cells and absolute counts of PD-1+ CD4 Teff at pre-treatment was significantly lower in LTS compared to that in STS (p-values = 0.0007 and 0.003, respectively) (Fig. 4F, H) (Table 2 and Supplementary Table S4).
After treatment, the distribution of the percentages of surface PD-1+ CD4 Teff cells and absolute counts of PD-1+ CD4 Teff were not significantly different between STS or LTS after cycle 1 (p-values = 0.055 and 0.090, respectively) and after cycle 2 (p-values = 0.054 and 0.150, respectively) (Fig. 4F, H).
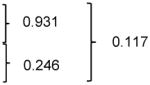
Distributions of the percentages and absolute counts of surface PD-1+ CD8 T cells between STS and LTS did not differ at pre-treatment (p-value = 0.246 and > 0.999, respectively) or at any time point after treatment (Fig. 4A, G, I) (Table 2 and Supplementary Table S4).
Comparison of the distribution of percentages of surface PD-1+ CD4 T cells in mCRPC patients with cancer-free male controls revealed that STS have significantly higher levels of PD-1+ CD4 T cells compared to those in cancer-free male controls (p-value = 0.002). There was no significant difference in the distribution of the percentages of PD-1+ CD8 T-cell levels between cancer-free male controls and patients with STS or LTS (Table 3).
Table 3.
Comparison of pre-treatment T-cell subsets between cancer-free male controls and LTS or STS
| T-cell subsetsa | Groupsb | Medianc (Ranged) | p-valuee | |||
|---|---|---|---|---|---|---|
|
| ||||||
| PD-1+ CD4 T eff cells | Cancer-free controls | 11.7 (6.67 – 14.30) |
|
|||
| LTS week 0 | 10.1 (5.4 – 14.5) | |||||
| STS week 0 | 22.2 (8.4 – 33.0) | |||||
|
| ||||||
| PD-1+ CD8 T cells | Cancer-free controls | 13.7 (8.7 – 27.5) |
|
|||
| LTS week 0 | 15.0 (5.31 – 28.0) | |||||
| STS week 0 | 22.2 (8.4 – 33.0) | |||||
T-cell subsets are defined by immune markers as indicated in the table;
cancer-free male controls, n = 7; LTS, n = 8; STS, n = 12;
Median values of PD-1 positive cells are % of the respective parent gate;
Values in brackets () are range of each data set;
Mann-Whitney test;
Bold-faced characters highlight p-value ≤ 0.017 (Bonferroni correction); LTS, long-term survivors; STS, short-term survivors
Discussion
Treatment of mCRPC patients with ipilimumab and sargramostim in this study revealed several findings. First, delayed response by PSA decline can be observed. Second, a small number of patients continued to experience durable responses off treatment without additional treatment. Third, a subset of patients had longer OS than expected for this disease with or without a clinical response to study treatment. Durable benefit and the potential for long-term survival were similarly observed for treatment of melanoma with ipilimumab and treatment of prostate cancer with sipuleucel-T and PROSTVAC-VF (9,15). Treatment kinetics of ipilimumab differ from those observed with radiation and chemotherapy and could be explained by the mechanism of action of immunotherapy (18). Immunotherapy targets the immune system, which subsequently targets the tumor. Presumably, the immune system is capable of generating long-lived memory cells to sustain clinical response beyond the duration of treatment. The immune system may also slow tumor growth without reducing tumor size, resulting in longer OS without accompanying clinical responses.
Identifying patients most likely to benefit before start of treatment is a significant unmet clinical need. We hypothesized that expression of immune checkpoint markers in T cells could differentiate patients with long and short overall survival. In cancer patients, higher levels of immune checkpoint markers in T cells could be indicative of endogenous T-cell activation and perhaps tumor-induced immune suppression. We found that study treatment-induced increased levels of Treg cells, CD4 Teff cells, CD8 T cells, and surface PD-1 expression on CD4 Teff cells and CD8 T cells, which are consistent with activating T cells in vivo. These observations may also explain the increased effectiveness of combined CTLA-4 and PD-1 blockade therapies (19), as CTLA-4 blockade would increase the levels of PD-1 on CD4 and CD8 Teff cells during treatment. However, the increased levels of these cells examined after one and two cycles of ipilimumab and sargramostim were not associated with clinical outcome. While the absolute lymphocyte counts of melanoma patients were reported to correlate with OS after two ipilimumab doses (20), this relationship was not observed with the subset of prostate cancer patients that were analyzed.
We found that lower pre-treatment levels of surface PD-1 on CD4 Teff cells (PD-1+CD4+CD3+FoxP3−) were correlated positively with longer survival in this study. Consistent with our data, lower baseline PD-1+Tim-3− CD4 memory T cells (CD45RA−CD62L−CCR7−) was reported to correlate significantly with longer survival in a Phase I trial combining ipilimumab and PROSTVAC vaccine (21,22). We also observed that levels of PD-1+ CD4 Teff cells in patients with short OS were higher than those in male controls who did not have a diagnosis of prostate cancer. Therefore, higher baseline levels of immune checkpoint molecules in circulating lymphocytes from cancer patients may reflect the presence of tumor-reactive T cells and that also serve to maintain the lymphocytes in a tolerant state (23). Notably, the ipilimumab plus PROSTVAC vaccine study and our study both revealed associations of inhibitory immune markers with survival on CD4 rather than CD8 T cells. This suggests that there may be differential effects of PD-1 in CD4 and CD8 T cells, and further studies of PD-1+ CD4 T cells in cancer patients are required.
Since all patients received ipililmumab and sargramostim treatment, it is unclear at present whether these findings are specific to this combined regimen and/or to prostate cancer. It is also unclear whether expression levels of PD-1 on CD4 Teff cells are prognostic or predictive biomarkers. It will be interesting to see if the same or different observations would be found in clinical trials with ipilimumab with or without sargramostim. The data reported in this study are intended to be hypothesis generating. Prospective clinical trials will be needed to examine formally whether low PD-1 levels in circulating CD4 Teff cells could be potential biomarkers for CTLA-4 blockade and GM-CSF combination immunotherapy.
Supplementary Material
Acknowledgments
Grant Support: Peter Michael Foundation (SK), National Cancer Institute R01-CA136753 (LF), UCSF Prostate SPORE (EJS, and LF), and Prostate Cancer Foundation (EJS, AH). Equipment support was provided by the UCSF Clinical and Translational Science Institute NIH NCRR UL1 RR024131.
Ipilimumab was provided by CTEP. Sargramostim was provided by Sanofi.
Footnotes
Disclosure of Potential Conflicts of Interest: LF has received research support from Bristol-Myers Squibb.
Authors’ Contributions:
Conception and design: S.S. Kwek, L. Fong
Development of methodology: S.S. Kwek, L. Fong
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): S.S. Kwek, J. Lewis, A. Harzstark, S, Greaney, A. Lin, C. Ryan, E.J. Small, L. Fong
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): S.S. Kwek, J. Lewis, L. Zhang, V. Weinberg
Writing, review, and/or revision of the manuscript: S.S. Kwek, J. Lewis, L. Zhang, V. Weinberg, L. Fong
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): S.S. Kwek, J. Lewis, A. Harzstark, A. Lin
Study supervision: L. Fong
References
- 1.Walunas TL, Lenschow DJ, Bakker CY, Linsley PS, Freeman GJ, Green JM, et al. CTLA-4 can function as a negative regulator of T cell activation. Immunity. 1994;1:405–13. doi: 10.1016/1074-7613(94)90071-x. [DOI] [PubMed] [Google Scholar]
- 2.Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med. 1995;182:459–65. doi: 10.1084/jem.182.2.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271:1734–6. doi: 10.1126/science.271.5256.1734. [DOI] [PubMed] [Google Scholar]
- 4.Kwon ED, Hurwitz AA, Foster BA, Madias C, Feldhaus AL, Greenberg NM, et al. Manipulation of T cell costimulatory and inhibitory signals for immunotherapy of prostate cancer. Proc Natl Acad Sci U S A. 1997;94:8099–103. doi: 10.1073/pnas.94.15.8099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.http://www.accessdata.fda.gov/drugsatfda_docs/label/2011/125377s0000lbl.pdf.
- 6.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–23. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Robert C, Thomas L, Bondarenko I, O’Day S, Weber J, Garbe C, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364:2517–26. doi: 10.1056/NEJMoa1104621. [DOI] [PubMed] [Google Scholar]
- 8.Prieto PA, Yang JC, Sherry RM, Hughes MS, Kammula US, White DE, et al. CTLA-4 blockade with ipilimumab: long-term follow-up of 177 patients with metastatic melanoma. Clin Cancer Res. 2012;18:2039–47. doi: 10.1158/1078-0432.CCR-11-1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McDermott D, Lebbe C, Hodi FS, Maio M, Weber JS, Wolchok JD, et al. Durable benefit and the potential for long-term survival with immunotherapy in advanced melanoma. Cancer Treat Rev. 2014;40:1056–64. doi: 10.1016/j.ctrv.2014.06.012. [DOI] [PubMed] [Google Scholar]
- 10.Hodi FS, Lee S, McDermott DF, Rao UN, Butterfield LH, Tarhini AA, et al. Ipilimumab plus sargramostim vs ipilimumab alone for treatment of metastatic melanoma: a randomized clinical trial. JAMA. 2014;312:1744–53. doi: 10.1001/jama.2014.13943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kwon ED, Drake CG, Scher HI, Fizazi K, Bossi A, van den Eertwegh AJ, et al. Ipilimumab versus placebo after radiotherapy in patients with metastatic castration-resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184–043): a multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol. 2014;15:700–12. doi: 10.1016/S1470-2045(14)70189-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fong L, Kwek SS, O’Brien S, Kavanagh B, McNeel DG, Weinberg V, et al. Potentiating endogenous antitumor immunity to prostate cancer through combination immunotherapy with CTLA4 blockade and GM-CSF. Cancer Res. 2009;69:609–15. doi: 10.1158/0008-5472.CAN-08-3529. [DOI] [PubMed] [Google Scholar]
- 13.Bubley GJ, Carducci M, Dahut W, Dawson N, Daliani D, Eisenberger M, et al. Eligibility and response guidelines for phase II clinical trials in androgen-independent prostate cancer: recommendations from the Prostate-Specific Antigen Working Group. J Clin Oncol. 1999;17:3461–7. doi: 10.1200/JCO.1999.17.11.3461. [DOI] [PubMed] [Google Scholar]
- 14.Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000;92:205–16. doi: 10.1093/jnci/92.3.205. [DOI] [PubMed] [Google Scholar]
- 15.Cheng ML, Fong L. Beyond sipuleucel-T: immune approaches to treating prostate cancer. Curr Treat Options Oncol. 2014;15:115–26. doi: 10.1007/s11864-013-0267-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aad G, Abbott B, Abdallah J, Abdelalim AA, Abdesselam A, Abdinov O, et al. Search for diphoton events with large missing transverse energy in 7 TeV proton-proton collisions with the ATLAS detector. Physical Rev Letters. 2011;106:121803. doi: 10.1103/PhysRevLett.106.121803. [DOI] [PubMed] [Google Scholar]
- 17.Kavanagh B, O’Brien S, Lee D, Hou Y, Weinberg V, Rini B, et al. CTLA4 blockade expands FoxP3+ regulatory and activated effector CD4+ T cells in a dose-dependent fashion. Blood. 2008;112:1175–83. doi: 10.1182/blood-2007-11-125435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Singh BH, Gulley JL. Immunotherapy and therapeutic vaccines in prostate cancer: an update on current strategies and clinical implications. Asian J Andrology. 2014;16:364–71. doi: 10.4103/1008-682X.122585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369:122–33. doi: 10.1056/NEJMoa1302369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ku GY, Yuan J, Page DB, Schroeder SE, Panageas KS, Carvajal RD, et al. Single-institution experience with ipilimumab in advanced melanoma patients in the compassionate use setting: lymphocyte count after 2 doses correlates with survival. Cancer. 2010;116:1767–75. doi: 10.1002/cncr.24951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jochems C, Tucker JA, Tsang KY, Madan RA, Dahut WL, Liewehr DJ, et al. A combination trial of vaccine plus ipilimumab in metastatic castration-resistant prostate cancer patients: immune correlates. Cancer Immunol Immunother. 2014;63:407–18. doi: 10.1007/s00262-014-1524-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Madan RA, Mohebtash M, Arlen PM, Vergati M, Rauckhorst M, Steinberg SM, et al. Ipilimumab and a poxviral vaccine targeting prostate-specific antigen in metastatic castration-resistant prostate cancer: a phase 1 dose-escalation trial. Lancet Oncol. 2012;13:501–8. doi: 10.1016/S1470-2045(12)70006-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baitsch L, Baumgaertner P, Devevre E, Raghav SK, Legat A, Barba L, et al. Exhaustion of tumor-specific CD8(+) T cells in metastases from melanoma patients. J Clin Invest. 2011;121:2350–60. doi: 10.1172/JCI46102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.