

Abstract
Obstructive sleep apnea (OSA) is a major source of cardiovascular morbidity and mortality, and represents an increasing burden on health care resources. Understanding underlying pathogenic mechanisms of OSA will ultimately allow for the development of rational therapeutic strategies. In this article, we review current concepts about the pathogenesis of OSA. Specifically, we consider the evidence that the upper airway plays a primary role in OSA pathogenesis and provide a framework for modelling its biomechanical properties and propensity to collapse during sleep. Anatomical and neuromuscular factors that modulate upper airway obstruction are also discussed. Finally, we consider models of periodic breathing, and elaborate generalizable mechanisms by which upper airway obstruction destabilizes respiratory patterns during sleep. In our model, upper airway obstruction triggers a mismatch between ventilatory supply and demand. In this model, trade-offs between maintaining sleep stability or ventilation can account for a full range of OSA disease severity and expression. Recurrent arousals and transient increases in airway patency may restore ventilation between periods of sleep, while alterations in neuromuscular and arousal responses to upper airway obstruction may improve sleep stability at still suboptimal levels of ventilation.
Keywords: Obstructive sleep apnea (OSA), upper airway, inspiratory flow limitation, Starling resistor
Introduction
Obstructive sleep apnea (OSA) is a highly prevalent disease, characterized by upper airway collapse during sleep resulting in recurring arousals and desaturations. Estimates of disease prevalence range between 3% and 10% of the population (1,2). Prevalence has risen with escalating rates of obesity, a major risk factor for OSA (2,3). Significant clinical consequences of the disorder cover a wide spectrum and include daytime hypersomnolence, neurocognitive dysfunction, cardiovascular disease, metabolic dysfunction, respiratory failure, and cor pulmonale (4-16). As a result, OSA represents an increasing burden on health care resources. Understanding underlying pathogenic mechanisms of OSA will ultimately allow for the development of rational therapeutic strategies in an era of personalized medicine.
In this article, we will review current concepts about the pathogenesis of OSA. Specifically, we will consider factors that initiate upper airway obstruction during sleep and examine responses to airway obstruction over the course of the ensuing event. In considering factors responsible for the initiation of an obstructive apnea, we will model the alterations in pharyngeal biomechanics that to lead airway obstruction during sleep. When the pharynx collapses, airflow obstruction elicits neuromuscular responses that can mitigate the obstruction and restore airway patency and ventilation. If these neuromuscular mechanisms are inadequate, additional factors contribute to the development of recurrent periods of airway obstruction and arousals from sleep. In this context, models that predict the likelihood of developing recurrent sleep disordered breathing episodes will be considered. In modelling recurrent sleep disordered breathing episodes, we will elaborate generalizable mechanisms by which upper airway obstruction destabilizes respiratory and sleep-wake patterns.
Upper airway biomechanics
OSA is characterized by recurrent periods of upper airway occlusion during sleep (17). In modeling the biomechanics of pharyngeal airflow obstruction, we consider the fact that the upper airway collapses dynamically during sleep and reopens during wakefulness. Investigators have previously modelled dynamic alterations in patency as a function of transmural pressure across collapsible segments in biologic conduits in the cardiovascular, gastrointestinal, and genitourinary systems (18-24) (Figure 1). In the case of the upper airway, the collapsible segment is bordered by two rigid segments upstream (nasal passages) and downstream (trachea) (Figure 1A) (26). The segments upstream and downstream to the collapsible site have fixed diameters and resistances, RUS and RDS, respectively, and the pressures upstream and downstream are PUS and PDS, respectively.
Figure 1.
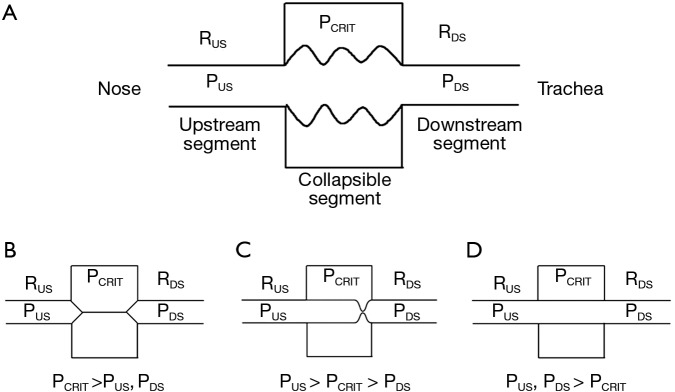
Starling resistor model of the upper airway: (A) A collapsible segment is bordered by rigid upstream (nasal passages) and downstream (trachea) segments. These rigid segments are characterized by intraluminal pressures (PUS and PDS in the upstream and downstream segments, respectively) and resistance to airflow (RUS and RDS, respectively); (B) when PUS and PDS are less than PCRIT, the airway is closed; (C) when PUS is greater than PCRIT, but PDS falls below PCRIT, the airway becomes flow limited on inspiration and can rapidly cycle between an open and closed state; (D) when both PUS and PDS remain above PCRIT, the airway is patent. Adapted from Gleadhill et al. 1991 (25).
Several important features of this model, known as the Starling resistor, are worth emphasizing. When PUS and PDS are less than the critical pressure surrounding the collapsible segment (PCRIT), the transmural pressure is negative, the airway closes and airflow ceases (Figure 1B). Flow can be re-established by raising PUS above PCRIT. If both PUS and PDS are greater than PCRIT, however, transmural pressure remains positive (Figure 1D). Under these conditions, flow through the upper airway is proportional to the pressure gradient across the entire airway, and can be described by the voltage-current relationship in Ohm’s law:
| [1] |
In contrast, when PUS is greater than PCRIT and PDS is less than PCRIT, however, the airway enters a flow-limited condition (Figure 1C). To illustrate the mechanism by which inspiratory flow limitation arises, consider the effects of lowering PDS progressively during inspiration. At the start of inspiration, flow commences upon lowering PDS. As inspiration progresses and PDS falls below PCRIT, the airway begins to collapse. If the upper airway were to occlude, flow would cease transiently. As the upper airway occludes, the pressure immediately upstream of the occlusion would equilibrate with PUS and rise above PCRIT. This increase in pressure would inevitably lead to reopening of the airway. As the airway cycles rapidly between an open and closed state, the pressure at the collapsible segment remains nearly constant at PCRIT. Because pressure in the collapsible segment is constant, airflow also remains constant. Under these circumstances, flow becomes independent of PDS and plateaus at a maximal level (VImax) as PCRIT replaces PDS and becomes the effective back-pressure to inspiratory flow. The level of VImax is therefore governed by the pressure gradient between PUS and PCRIT and the resistance across the upstream segment, according to the following equation:
| [2] |
In this model, decreasing PDS does not cause the upper airway to occlude and cannot account for the development of obstructive apneas during sleep.
Now consider the effects of altering PUS on inspiratory flow in Figure 2. The effects of increasing levels of CPAP on upper airway pressure-flow dynamics is represented. In this figure, CPAP is applied at pressures of 4, 6, 9 and 12 cmH2O. At a low nasal pressure (4 cmH2O), both PUS and PDS are less than PCRIT, the airway occludes and no flow occurs. At an intermediate pressure of 6 cmH2O, PUS exceeds PCRIT and flow is re-established. Nevertheless, PDS still falls below PCRIT over the course of inspiration, resulting in a plateau of mid-inspiratory airflow (flow-limitation) at VImax (see arrows). With further increases in nasal pressure from 6 to 9 cmH2O, inspiratory flow plateaus at a proportionately higher level, as described by Eq. [2], but flow still remains limited. Finally, at nasal pressure of 12 cmH2O, both PUS and PDS have increased sufficiently such that both pressures remain above PCRIT throughout inspiration and flow limitation is abolished. Conversely, decreases in PUS elicit flow limitation and progressive decreases in VImax until flow ceases (the upper airway occludes) when PUS falls below PCRIT. A simple pressure-flow plot (Figure 3) describes this linear relationship between VImax and PUS (Eq. [2]). This relationship can be used to define PCRIT at the zero flow intercept and RUS as the reciprocal of the slope of this line (27,28).
Figure 2.
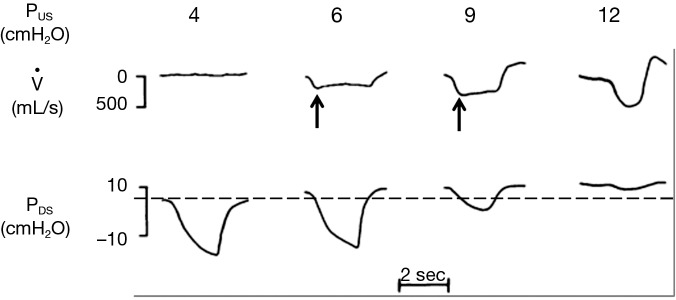
The effects of varying the upstream nasal pressure on inspiratory flow. At PUS of 4 cmH2O, PUS is less than the critical closing pressure, PCRIT (dashed line). As a result, the upper airway is closed, and flow cannot occur. At PUS of 6 and 9 cmH2O, PUS is greater than PCRIT, and flow is established. As inspiration progresses, PDS falls below PCRIT, which causes flow to plateau at maximal inspiratory flow (VImax, arrows), denoting the onset of flow limitation in mid-inspiration. At PUS of 12 cmH2O, both PUS and PDS remain above PCRIT throughout inspiration, and flow limitation is abolished. Adapted from Schwartz et al. 1998 (27).
Figure 3.
Relationship between maximal inspiratory flow (VImax ± standard deviation) and upstream nasal pressure (PUS) during NREM sleep. Inspiratory flow limitation in a non-apneic normal individual was induced by sequentially lowering PUS. Under these conditions, VImax is linearly dependent on PUS. The reciprocal of the line slope represents the upstream resistance (RUS). The critical pressure (PCRIT) is the zero-flow intercept, −21 cmH2O (indicated by arrow), indicating total upper airway occlusion. Adapted from Schwartz et al. 1988 (27).
It is important to note that inspiratory airflow limitation exerts two distinct loads on the respiratory system. First, airway resistance increases markedly in the flow limited compared to the non-flow limited state. During non-flow limited breathing (in the absence of upper airway collapse), the combined resistances of the upstream and downstream segments (Eq. [1]) is approximately 1 to 2 cmH2O/L/s, which accounts for approximately half of the total resistance of the respiratory system during tidal breathing. In contrast, resistance of the upstream segment alone (Eq. [2]) during periods of inspiratory airflow limitation increases into the range of 20 to 40 cmH2O/L/s. Second, additional load is imposed on the respiratory system during periods of flow limitation by virtue of the fact that patients continue to exert ever-increasing effort without increasing inspiratory airflow. In essence, a large portion of the pressure generated by the respiratory pump muscles is wasted by dynamic collapse of the upper airway without augmenting ventilation. Thus, increases in airway resistance and dynamic collapse of the upper airway augment work of breathing during periods of inspiratory airflow limitation and/or complete upper airway obstruction.
The role of upper airway obstruction in OSA pathogenesis
Current evidence suggests that disturbances in PCRIT play a primary role in OSA pathogenesis. The role of pharyngeal obstruction in OSA pathogenesis can be considered in light of Koch’s postulates (29), which establish criteria for demonstrating a causal relationship between pathogenic factors that promote upper airway obstruction and the overt polysomnographic manifestation of the disease. These principles require first and foremost that pathogenic factors causing upper airway collapse are associated with OSA. Investigators have examined the association between pharyngeal collapsibility and the clinical manifestations of OSA in several observational studies (25,28,30-35). Elevations in PCRIT have been demonstrated in OSA patients compared to age, sex and body mass index (BMI) matched controls under general anesthesia and neuromuscular blockade (35) as well as during sleep (25).
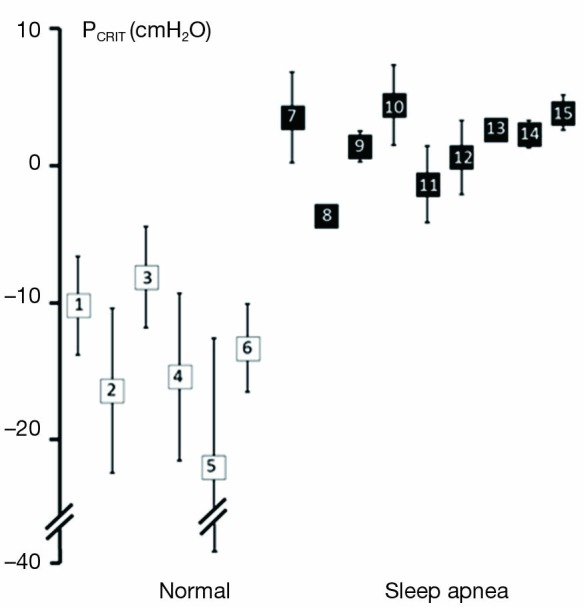
Further strength for the association between upper airway obstruction and OSA pathogenesis is evidenced by studies demonstrating that pharyngeal collapsibility (PCRIT) is both a sensitive (a large proportion of persons with OSA have collapsible upper airways) and specific (a large proportion of normal persons do not have collapsible airways) finding in OSA. High levels of sensitivity and specificity of PCRIT can be inferred from numerous studies that have demonstrated quantitative differences in PCRIT between health and disease (27,28,30-34,36-41). In the aggregate, these studies demonstrated that nearly all persons with OSA have a PCRIT greater than −5 cmH2O, indicating that upper airway collapsibility is a sensitive marker for OSA (Figure 4). In contrast, elevations in PCRIT were nearly absent in normal controls, suggesting that increased pharyngeal collapsibility is also highly specific to OSA.
Figure 4.
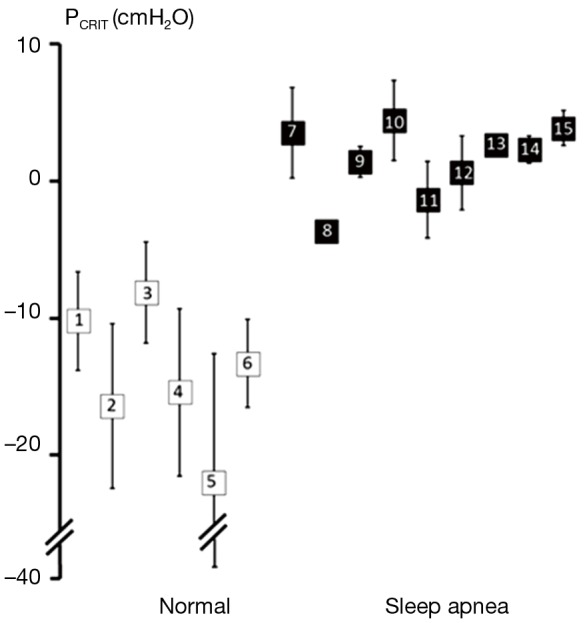
Summary of studies examining upper airway collapsibility (critical closing pressure, PCRIT) in normal individuals and obstructive sleep apnea (OSA) patients. Mean PCRIT (± standard deviation) in non-apneic controls (open squares) and apneic subjects (closed squares) are plotted. PCRIT is elevated in sleep apnea patients compared to normal controls. Furthermore, these studies demonstrate minimal overlap of PCRIT between disease and health, suggesting that an elevated PCRIT is both sensitive and specific for OSA. Source studies, as indicated by enclosed numbers, are: 1. Meurice et al., 1996 (37); 2. Meurice et al., 1996 (36); 3. Issa & Sullivan, 1984 (30); 4. Gold et al., 2002 (33); 5. Philip-Joet et al., 1996 (38); 6. Schwartz et al., 1988 (27); 7. Smith et al., 1988 (28); 8. Ng et al., 2003 (39); 9. Kirkness et al., 2003 (34); 10. Farre et al., 2003 (40); 11. Series et al., 1996 (42); 12. Gold et al., 2002 (33); 13. Sforza et al., 2000 (32); 14. Sforza et al., 1999 (31); 15. Issa & Sullivan, 1984 (30). Adapted from Pack, 2009 (41).
Additional evidence for the primacy of upper airway collapse in OSA pathogenesis is provided by studies demonstrating a dose-response relationship between pharyngeal collapsibility and severity of OSA (Figure 5). As PCRIT rises progressively, increases in severity of upper airway obstruction during sleep have also been observed clinically. Modest elevations in PCRIT have been associated with snoring, whereas moderate elevations in PCRIT to levels between −5 and 0 cmH2O have been associated with sleep disordered breathing characterized primarily by obstructive hypopneas. With further increases in PCRIT (PCRIT becomes positive), periodic obstructive apneas are observed during sleep. Quantitative differences in PCRIT have therefore been associated with graded changes in the severity of airway obstruction during sleep.
Figure 5.
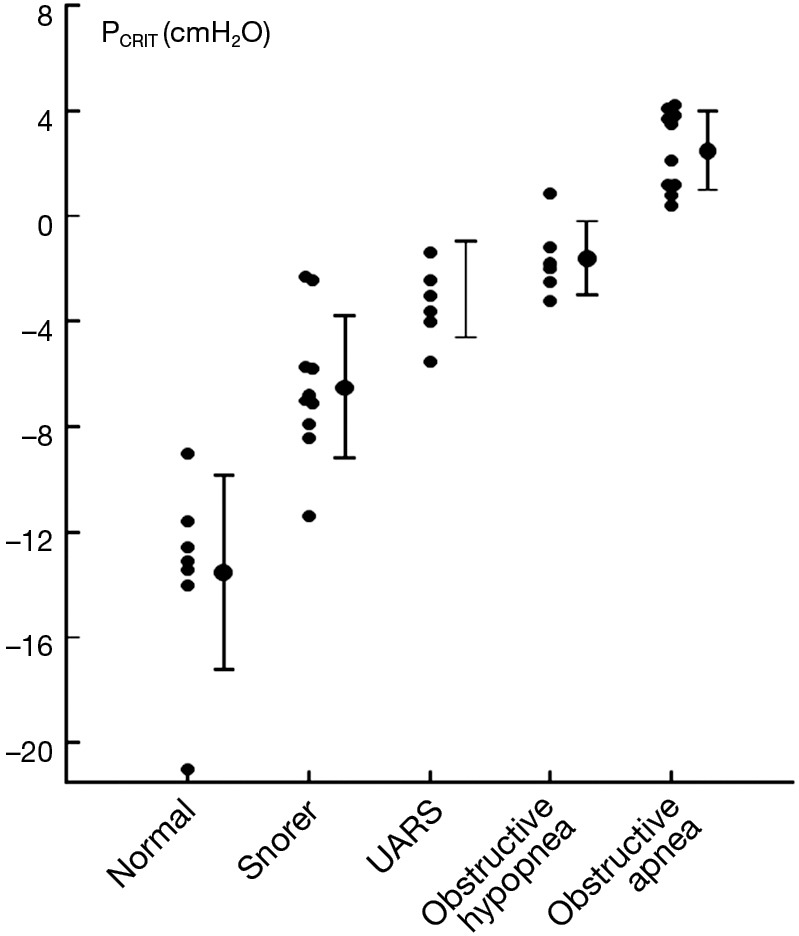
Relationship between PCRIT and clinical disorders characterized by upper airway obstruction during sleep. As PCRIT increases, the severity of upper airway obstruction also increases progressively, suggesting a dose-response relationship between upper airway collapsibility and worsening obstruction in asymptomatic snorers and patients with upper airway resistance syndrome (UARS), obstructive hypopneas, and obstructive apneas. Adapted from Schwartz et al. 1988 and Gleadhill et al. 1991 (25,27).
Studies inducing experimental upper airway collapse during sleep also implicate pharyngeal obstruction in OSA pathogenesis. Indeed, manipulating nasal pressure recapitulates the entire OSA disease spectrum. With the application of subatmospheric nasal pressure during sleep, stable flow limited breathing and snoring were observed in healthy test subjects (27). Further reductions in nasal pressure resulted in recurrent obstructive hypopneas and apneas, which occurred at a rate of ~20-40 episodes per hour and were associated with oxyhemoglobin desaturations and arousals. Continuous application of subatmospheric nasal pressure during sleep also caused alterations in sleep architecture, with increases in stage 1 and stage 2 sleep, and decreases in stage 3/4 and REM sleep compared to baseline (43). Moreover, when study participants were subjected to two consecutive nights of experimentally induced OSA, multiple daytime sleep latency times fell markedly, indicating that excessive daytime somnolence had developed. Thus, experimental evidence suggests that airway collapse alone is sufficient to cause OSA.
Conversely, OSA can be treated with interventions designed to restore upper airway patency, further fulfilling Koch’s postulate that upper airway collapse is necessary for disease pathogenesis. In fact, treatments that decrease PCRIT (e.g., weight loss or uvulopalatopharyngoplasty) lead to improvements in OSA and to resolution of disease when PCRIT falls below −5 cmH2O (27,44). At this level of PCRIT, a transmural pressure of 5 cmH2O provides adequate airflow to stabilize breathing patterns during sleep. Similarly, a positive transmural pressure can be induced by increasing PUS, leading to resolution of upper airway obstruction. With application of progressively increasing nasal pressure during CPAP titration, upper airway obstruction and recurrent obstructive apneas and hyponeas are reversed. As nasal pressure increases, episodic obstructive apneas give way to hypopneas when PUS rises above PCRIT. Further increases in nasal pressure stabilize respiratory patterns, leading to regular snoring and ultimately to the resolution of flow-limitation altogether as CPAP pressures rise to therapeutic levels.
Finally, the relationship between upper airway collapsibility and OSA is biologically plausible, as exemplified by several approaches to modelling upper airway obstruction. First, airway obstruction could be due to increasing airway resistance that is produced by narrowing of an otherwise rigid structure. In this model, no matter how narrow the tube, flow still remains dependent on downstream pressure. Although flow is reduced when resistance is high, this model cannot account for the development of inspiratory flow limitation (or snoring) in which airflow becomes independent of downstream pressure. Second, investigators have postulated that airflow obstruction could be due to increased pharyngeal compliance. Under these circumstances, reductions in downstream pressure during inspiration can produce increases in linear airflow velocity along streamlines, which further decrease intraluminal pressure (Bernoulli’s principle). As intraluminal pressure falls, the airway collapses at its most compliant region [generally located in the velopharynx (35)], forming a choke point with a discontinuity in pressure between the upstream and downstream segments. Elevations in pharyngeal compliance, therefore, can account for the development of inspiratory flow limitation. As the airway wall becomes infinitely compliant, however, its collapsibility is determined by the tissue pressure surrounding the airway rather than the compliance of the airway wall itself. Under these circumstances, the surrounding critical pressure determines airway patency and the severity of airflow obstruction (the Starling resistor, see Figure 1). In fact, empiric evidence suggests that the pharynx is a highly compliant structure that approximates the behavior of a Starling resistor, in which flow limitation develops because surrounding tissue pressures produce a constant back pressure to airflow.
Determinants of upper airway collapsibility
Elevations in PCRIT can be attributed to passive structural defects in the upper airway and disturbances in neuromuscular control (45,46). Utilizing specialized physiologic techniques, investigators have separated structural from neuromuscular components by measuring PCRIT during sleep under conditions of reduced (passive) and elevated (active) neuromuscular activity, respectively (35,47,48). They demonstrated that airway collapsibility was elevated in OSA patients under passive conditions, suggesting underlying anatomic defects in OSA patients compared to age, sex and BMI matched normal controls (see Figure 6A). These OSA patients also exhibited blunted active responses to airway obstruction compared to controls, indicating concomitant deficits in pharyngeal neuromuscular control (48). Disturbances in neuromuscular control remained even in those OSA patients whose structural loads were comparable to those of normal controls (see Figure 6B). These findings suggest that elevations in PCRIT in OSA patients are due to defects in both upper airway structural and neuromuscular control (Figure 7), and those disturbances in both play a pivotal role in OSA pathogenesis. In fact, OSA can only develop when neuromuscular responses do not adequately mitigate the obstruction caused by excess pharyngeal mechanical loads.
Figure 6.
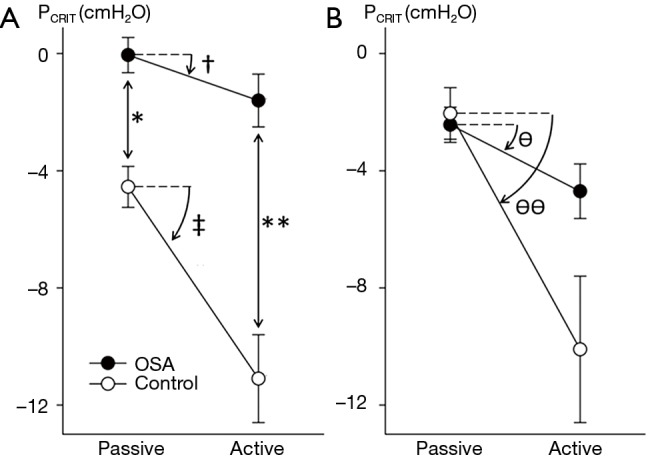
Structural and neuromechanical defects in obstructive sleep apnea (OSA) patients: upper airway critical pressure (PCRIT ± SD) measurements in patients with obstructive sleep apnea (closed circles) and age-, sex-, and body mass index-matched normal controls (open circles) under passive and active conditions. (A) Sleep apnea patients compared to controls demonstrated elevations in both passive PCRIT (*) and active PCRIT (**) compared to controls. Furthermore, under active conditions, OSA patients produced lesser reductions in PCRIT compared to normal controls (see † vs. ‡); (B) among subjects who were matched by structural loads (passive PCRIT), sleep apnea patients lowered the active PCRIT less than normal asymptomatic controls [ΔPCRIT 2.2 cmH2O ±2.2 (ϴ) vs. 8.0±7.4 (ϴϴ), respectively]. Adapted from Patil et al. 2007 (48).
Figure 7.
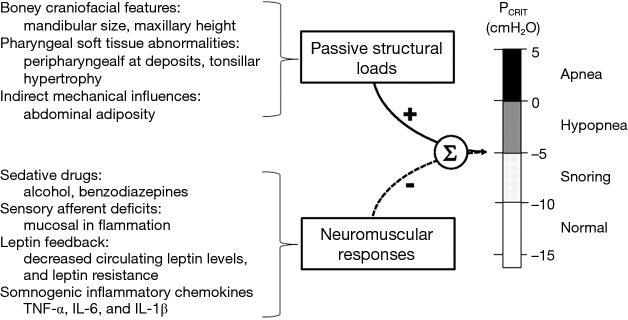
Upper airway collapsibility is the result of the sum of passive structural loads, which increase PCRIT, and neuromuscular responses, which decrease PCRIT and stabilize an inherently collapsible upper airway. Increased airway collapsibility therefore requires defects in both upper airway structure, and neuromuscular responses. Alterations in boney craniofacial features (e.g., retrognathia, decreased maxillary height) and soft tissues (e.g., peripharyngeal fat deposits and tonsillar hypertrophy) may account for structural loads. Defects in neuromuscular responses may arise from sedative drugs, sensory afferent deficits, alterations in leptin levels and/or leptin responses, or somnogenic inflammatory chemokines that may be related to obesity. Adapted from Patil et al. 2007 (48).
Anatomic alterations
Investigators have identified a variety of anatomic factors that contribute to increases in airway collapsibility. Various craniofacial features related to either skeletal morphology or pharyngeal soft tissue may predispose to upper airway collapse. Mandibular size, maxillary height, and hyoid position have been associated with risk for OSA (32,35,49-55). Decreased velopharyngeal area, and tonsillar hypertrophy are soft tissue features that have been associated with increased upper airway collapsibility (35,56). In general, these anatomical variants are thought to increase PCRIT by restricting the size of the boney enclosure around the pharynx and/or increasing the amount of soft tissue contained therein (57).
Obesity and especially abdominal adiposity are also important anatomical risk factors for upper airway obstruction during sleep. The upper airways of obese individuals are more susceptible to collapse (44) and PCRIT increases 1.0 and 1.7 cmH2O per 10 kg/m2 BMI increase in women and men, respectively (58). Increased fat deposition around the pharynx and airway narrowing (59-61) may increase the extraluminal tissue pressure and augment upper airway collapsibility (62). In addition, lung volumes are decreased in obese persons, leading to decreased caudal traction on the upper airway and an increased critical closing pressure (63-67). These reductions in caudal traction are most pronounced in patients with abdominal adiposity, which can decrease lung volume nearly to the level of residual volume (68-71). Conversely, improvements in OSA with weight loss are likely due to reductions in surrounding tissue pressure and increases in caudal traction, both of which decrease PCRIT (44).
Disturbances in neuromuscular control
Although structural defects play a clear role in the pathogenesis of OSA, these defects may only account for one-third of the variability in OSA severity (72), leaving neuromuscular responses accounting for much of the balance of OSA variability. OSA patients appear to be especially dependent on neuromuscular activity to maintain airway patency and ventilation during sleep (73). This activity varies markedly with reduction in pharyngeal dilator tone at sleep onset that predispose to airway obstruction (74). Reductions in neuromuscular tone are also suspected to contribute to increased OSA severity during REM compared to NREM sleep in selected patients, and particularly in women and children (75-78). Neuromuscular responses are also influenced by pharmacologic modulators of sleep-wake state (79). Alcohol, sedative medications and hypnotics may decrease active responses to upper airway occlusion, and contribute to upper airway obstruction during sleep. Benzodiazepines have been demonstrated to prolong obstructive apneas and hypopneas (80). The effects of opiate medications on upper airway collapsibility have not been well studied. Nevertheless, blockade of opioid receptors has been demonstrated to decrease PCRIT, which suggests that narcotic medications may increase susceptibility to pharyngeal occlusion (37).
Current evidence also suggests that endogenous neurohumoral agents can modulate upper airway neuromuscular responses. Neurohormonal modulation of pharyngeal neuromuscular activation may in part account for differences in prevalence and severity of OSA between men and women. When matched by BMI, age, and passive mechanical loads, women demonstrated increased neuromuscular compensation and lower disease burden during NREM sleep compared to men (81). Sex differences in neuromuscular control may well be due elevations in circulating leptin levels in women compared to men (82,83). With weight loss, PCRIT falls by 6.2 cmH2O per 10 kg/m2 decrease in BMI in apneic men (44), which is greater than the above noted 1.7 cmH2O increase in passive PCRIT per 10 kg/m2 increase in BMI attributable to weight gain (58). These observations suggest that obesity may increase upper airway collapsibility through alterations in pharyngeal neuromuscular responses in addition to imposing increased anatomical loads. Investigations have demonstrated elevations in circulating levels of somnogenic inflammatory chemokines, specifically TNF-α, TNF-α receptor I, IL-6, and IL-1β in association with obesity, which may account for the decreases in neuromuscular activity in obesity and OSA patients (84-97).
Pharyngeal neuromuscular activity is also controlled by chemical and mechanical reflexes. Hypercapnia is also a potent stimulator of upper airway neuromuscular activity, which decreases PCRIT (98-100). Hypocapnia, on the other hand, produces a relatively passive state, and is associated with elevations in PCRIT. The upper airway demonstrates decreased collapsibility during expiration compared to inspiration due to phasic activation of pharyngeal muscles (101). Phasic volume feedback, which is mediated by pulmonary stretch receptors, can also inhibit upper airway neuromuscular activity and increase PCRIT (102,103). Pharyngeal sensory afferents can detect intraluminal negative pressure swings during airflow obstruction, and recruit neuromuscular activity (104). Pharyngeal sensory inhibition with topical analgesics has been demonstrated to decrease these neuromuscular responses to upper airway obstruction (105,106). Similarly, mucosal inflammation may blunt local afferents and neuromuscular responses to upper airway obstruction, leading to worsening upper airway obstruction during sleep (107).
Responses in respiratory timing can further augment reflex responses to upper airway obstruction and stabilize ventilation during sleep. The inspiratory duty cycle (IDC), which is the ratio of inspiratory time to total respiratory cycle time, is the most significant determinant of ventilation during periods of inspiratory flow limitation (81,108,109) (see Figure 8). In response to pharyngeal obstruction during sleep, IDC increases nearly immediately with resultant increases in ventilation (108,110,111). In contrast, respiratory rate does not significantly alter minute ventilation in the flow-limited state. Thus, increases in pharyngeal neuromuscular activation and IDC can help maintain ventilation during periods of inspiratory flow limitation and will stabilize respiratory patterns accordingly (see below).
Figure 8.
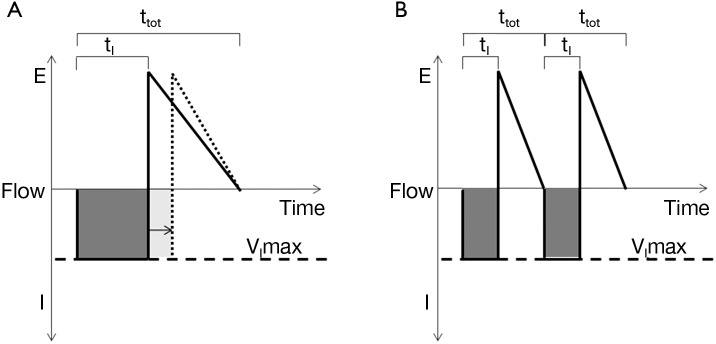
Flow profiles during inspiratory flow limitation. Positive flow represents expiration [E], whereas negative flow represents inspiration [I]. (A) During inspiratory flow limitation, minute ventilation (dark shaded area) can be approximated by the product of maximal inspiratory flow (VImax, dashed line) and inspiratory time (tI). Minute ventilation can be increased by increasing inspiratory duty cycle (IDC) (tI/ttot), as demonstrated by the arrow and dotted line; (B) in contrast, increasing the respiratory rate does not increase minute ventilation (the sum of dark shaded areas in B is equal to A) when tI/ttot remains constant. Adapted from Schneider et al. 2009 (108).
Modeling the oscillatory patterns in OSA
Thus far, we have examined factors that promote airway collapse at the onset of obstructive sleep disordered breathing events. To explain the repetitive nature of OSA events, investigators have cast the respiratory system as a closed-loop control system and have elucidated fundamental determinants of these oscillatory patterns. Crowell and colleagues first described the influence of circulatory delay on the periodic breathing. By inducing feedback delay between the central circulation and chemoreceptors in the dog, they reproduced the periodic respiratory pattern of Cheyne-Stokes respirations (112). Cherniack and colleagues highlighted the effects of hypocapnia which lead to apnea and subsequent periodic breathing patterns (113). They further expanded on our understanding of ventilatory control and periodic breathing with the development mathematical models that featured chemosensitivity as a predictor of unstable respiratory patterns (114,115). Khoo and colleagues extended this methodology by incorporating transitions in sleep-wake state and arousal phenomena into mathematical models of periodic breathing (116). In these models, chemosensitivity and ventilatory efficiency were summarized by a singular term, loop gain, to describe the propensity towards oscillations in respiratory patterns. Younes and colleagues manipulated loop gain experimentally with proportional assist ventilators, and demonstrated that elevations in loop gain can cause periodic breathing (117). In cross-sectional studies by Wellman and colleagues, loop gain was identified as one of several possible determinants of sleep apnea pathogenesis (118,119). Studies by these investigators have also suggested that pharmacological manipulation of loop gain and arousal threshold can ameliorate OSA (120-122).
Two key assumptions underlie traditional approaches to mathematical models of periodic breathing patterns. The first is that ventilatory responses change in proportion to alterations in ventilatory demand. For instance, hypoventilation, which results in elevations in CO2, would lead to proportionate increases in ventilatory drive. The second is that the mechanical components of the respiratory system and the control of ventilation remain relatively constant across cycles of periodic breathing. These two principles—linearity and time-invariance (123)—define linear control systems, which can be described by mathematical models that accurately predict smooth sinusoidal oscillations as those observed in Cheyne-Stokes respirations.
Rather than smooth oscillations in airflow, OSA appears to be characterized by abrupt transitions in upper airway patency and ventilation, which suggest a striking departure from the principles of linearity and time-invariance (see Figure 9). Remmers et al. demonstrated that obstructive apneas are characterized by the development of dynamic pharyngeal obstruction during sleep with prompt re-opening upon arousal, suggesting marked state-dependence in pharyngeal neuromuscular activity and upper airway collapsibility (17). In fact, upper airway patency is suddenly restored at apnea termination as it shifts from a flow-limited state to a non-flow-limited state. The abrupt termination of these sleep disordered breathing events can be modelled as a switch in an electrical analog of sleep disordered breathing (see Figure 10). When this shift occurs, ventilation can once again track ventilatory drive. Ventilatory drive, in turn, responds to changes in mechanical and chemical afferent inputs, which detect differences between ventilatory supply and demand (left and right sides of Figure 10). At sleep onset, however, the upper airway transitions to a flow limited state. Under these circumstances, ventilation is determined by the degree of upper airway patency rather than ventilatory drive. If ventilatory supply no longer matches demand, ventilatory drive progressively increases (see marker ① in Figure 11). These increases can be associated with alterations in upper airway patency and ventilatory timing that help mitigate the obstruction and/or restore ventilation (marker ② in Figure 11). If neuromuscular and ventilatory timing mechanisms do not adequately restore ventilation, ventilatory supply-demand mismatch continues, leading to progressive increases in ventilatory drive. Once drive exceeds a threshold, arousal is triggered (marker ③ in Figure 11), relieving pent-up ventilatory demand as the airway transitions to a non-flow-limited state (maker ④ in Figure 11). In fact, repetitive transitions have been demonstrated when ventilation decreases by more than 12-20% of baseline or VImax falls below 250 mL/s (81,124). The aforementioned model suggests that state-dependent alterations in upper airway patency and resulting mismatch between ventilatory supply and demand play pivotal roles in OSA pathogenesis.
Figure 9.
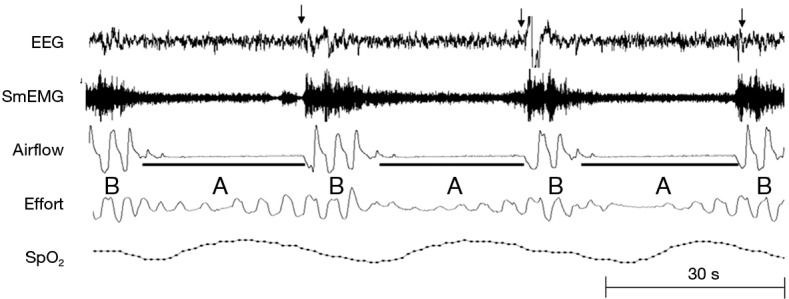
A nocturnal polysomnogram of a patient with sleep apnea demonstrates recurrent apneas (black bars) and arousals (arrows). Rather than smooth oscillations in respiratory patterns, airflow is characterized by abrupt transitions between periods of (A) upper airway obstruction (apneas) during sleep and (B) upper airway patency during arousals.
Figure 10.
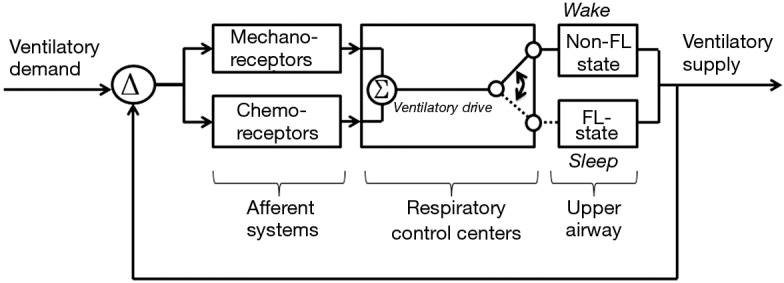
Upper airway transition model for obstructive sleep apnea (OSA). In OSA, the upper airway alternates between states of flow limitation during sleep, and non-flow-limited breathing during wakefulness. During sleep, a flow limited upper airway caps ventilatory supply, which dissociates from ventilatory drive (see dashed line). When ventilatory supply does not meet demand, differences (Δ) are detected by mechanical and chemical receptors and respiratory control centers, leading to increases in ventilatory drive. Increases in ventilatory drive during periods of flow limitation trigger arousal from sleep with restoration of upper airway patency.
Figure 11.
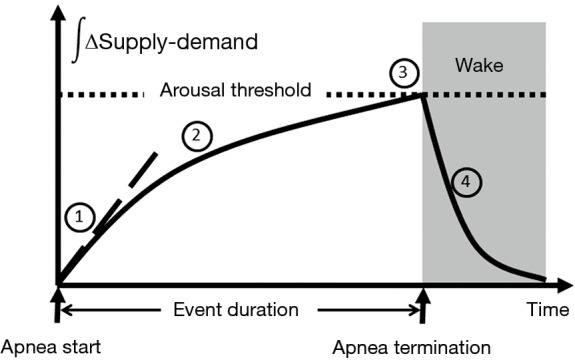
Ventilatory supply-demand dynamics. 1. At the start of an obstructive event, cumulative supply-demand difference (see Δ in Figure 10) is initially determined by the degree of upper airway obstruction (supply) and CO2 production (demand); 2. rate of accumulation may be decreased by neuromuscular responses and increase in ventilatory efficiency as CO2 rises; 3. when supply-demand difference meets a threshold, arousal occurs and terminates the apnea; 4. pent-up ventilatory demand is relieved due to improved upper airway patency during the arousal.
In this model, several factors can influence the overall evolution and severity of obstructive breathing episodes. In Figure 11, we plot the cumulative difference between ventilatory supply and demand over time during an obstructive sleep disordered breathing event. In general, the duration of sleep disordered breathing events is governed by the time required for cumulative supply-demand mismatch to reach the arousal threshold. At the onset of sleep disordered breathing episodes, the initial shortfall in ventilation is determined by the severity of upper airway obstruction. As ventilation falls, CO2 rises in proportion to the decrease in alveolar ventilation and the metabolic rate (CO2 production). Metabolic rate, in turn, is determined by body mass and composition, food sources (e.g., respiratory quotient), basal metabolic rate, sex and work of breathing at rest. Inspiratory flow limitation can further increase the work of breathing and thereby widen ventilatory supply-demand mismatch. Under these circumstances, minute ventilation must increase well above that in the non-flow limit state to satisfy the additional ventilatory demand and stabilize breathing patterns. Supply-demand dynamics can also be affected by ventilatory efficiency. Decreases in ventilatory efficiency due to increased dead space and cardiopulmonary disease will accelerate the development of supply-demand differences. Along with the severity of upper airway obstruction, excess metabolic demand and underlying cardiopulmonary disease can increase the overall severity of sleep disordered breathing. In contrast, sleep disordered breathing events may be prolonged by factors that slow the development of supply-demand differences, including compensatory neuromuscular mechanisms that mitigate upper airway obstruction and augment ventilation. Sleep disordered breathing events can also be prolonged by raising the arousal threshold with sleep deprivation (125) or hypnotic agents (121,122). Because upper airway neuromuscular control is highly dependent upon sleep wake state, increasing arousal threshold will tend to increase the degree of hypoventilation and oxyhemoglobin desaturation during sleep, as well (79). On the other hand, lowering the arousal threshold may shorten events and increase sleep disruption while minimizing alterations in gas exchange. Thus, the arousal threshold and ventilatory supply-demand dynamics can interact to modulate the overall polysomnographic expression of OSA.
Conclusions
Upper airway obstruction is essential in the pathogenesis of OSA. OSA is largely absent in those individuals without an inherently collapsible upper airway on a structural basis. When PCRIT exceeds −5 cmH2O, the risk for OSA markedly increases. The appearance of OSA features parallels the rise in PCRIT, increasing from simple snoring, to cyclic hypopneas, and then to fully occlusive apneas. These features are recapitulated in normal persons when upper airway obstruction is induced, and are abolished in OSA patients when airway patency is restored. Therefore, upper airway obstruction alone constitutes both a necessary and sufficient condition for the development of OSA.
Once the airway has collapsed, several factors modify the response to airway obstruction, and affect the ultimate expression of sleep disordered breathing. Neuromuscular responses preserve ventilation and protect against the development of OSA. When neuromuscular compensatory mechanisms are insufficient for a given structural load, ventilatory demand and ventilation dissociate and repeated sleep disordered breathing events ensue. Trade-offs between sleep stability and ventilation can result in a full range of OSA severity and expression. Recurrent arousals and transient increases in airway patency may restore ventilation between periods of sleep, while alterations in neuromuscular responses to upper airway obstruction may improve sleep stability at still suboptimal levels of ventilation.
Acknowledgements
None.
Footnotes
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- 1.Punjabi NM. The epidemiology of adult obstructive sleep apnea. Proc Am Thorac Soc 2008;5:136-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Peppard PE, Young T, Barnet JH, et al. Increased prevalence of sleep-disordered breathing in adults. Am J Epidemiol 2013;177:1006-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Young T, Peppard PE, Gottlieb DJ. Epidemiology of obstructive sleep apnea: a population health perspective. Am J Respir Crit Care Med 2002;165:1217-39. [DOI] [PubMed] [Google Scholar]
- 4.Yaggi HK, Concato J, Kernan WN, et al. Obstructive sleep apnea as a risk factor for stroke and death. N Engl J Med 2005;353:2034-41. [DOI] [PubMed] [Google Scholar]
- 5.Punjabi NM, Sorkin JD, Katzel LI, et al. Sleep-disordered breathing and insulin resistance in middle-aged and overweight men. Am J Respir Crit Care Med 2002;165:677-82. [DOI] [PubMed] [Google Scholar]
- 6.Marin JM, Carrizo SJ, Kogan I. Obstructive sleep apnea and acute myocardial infarction: clinical implications of the association. Sleep 1998;21:809-15. [PubMed] [Google Scholar]
- 7.Marin JM, Carrizo SJ, Vicente E, et al. Long-term cardiovascular outcomes in men with obstructive sleep apnoea-hypopnoea with or without treatment with continuous positive airway pressure: an observational study. Lancet 2005;365:1046-53. [DOI] [PubMed] [Google Scholar]
- 8.Nieto FJ, Young TB, Lind BK, et al. Association of sleep-disordered breathing, sleep apnea, and hypertension in a large community-based study. Sleep Heart Health Study. JAMA 2000;283:1829-36. [DOI] [PubMed] [Google Scholar]
- 9.Peppard PE, Young T, Palta M, et al. Prospective study of the association between sleep-disordered breathing and hypertension. N Engl J Med 2000;342:1378-84. [DOI] [PubMed] [Google Scholar]
- 10.Punjabi NM, Ahmed MM, Polotsky VY, et al. Sleep-disordered breathing, glucose intolerance, and insulin resistance. Respir Physiol Neurobiol 2003;136:167-78. [DOI] [PubMed] [Google Scholar]
- 11.Punjabi NM, Shahar E, Redline S, et al. Sleep-disordered breathing, glucose intolerance, and insulin resistance: the Sleep Heart Health Study. Am J Epidemiol 2004;160:521-30. [DOI] [PubMed] [Google Scholar]
- 12.Drager LF, Bortolotto LA, Lorenzi MC, et al. Early signs of atherosclerosis in obstructive sleep apnea. Am J Respir Crit Care Med 2005;172:613-8. [DOI] [PubMed] [Google Scholar]
- 13.Yaffe K, Laffan AM, Harrison SL, et al. Sleep-disordered breathing, hypoxia, and risk of mild cognitive impairment and dementia in older women. JAMA 2011;306:613-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nieto FJ, Peppard PE, Young T, et al. Sleep-disordered breathing and cancer mortality: results from the Wisconsin Sleep Cohort Study. Am J Respir Crit Care Med 2012;186:190-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Drager LF, Jun JC, Polotsky VY. Metabolic consequences of intermittent hypoxia: relevance to obstructive sleep apnea. Best Pract Res Clin Endocrinol Metab 2010;24:843-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Drager LF, Lopes HF, Maki-Nunes C, et al. The impact of obstructive sleep apnea on metabolic and inflammatory markers in consecutive patients with metabolic syndrome. PLoS One 2010;5:e12065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Remmers JE, deGroot WJ, Sauerland EK, et al. Pathogenesis of upper airway occlusion during sleep. J Appl Physiol Respir Environ Exerc Physiol 1978;44:931-8. [DOI] [PubMed] [Google Scholar]
- 18.Permutt S, Riley RL. Hemodynamics of collapsible vessels with tone: the vascular waterfall. J Appl Physiol 1963;18:924-32. [DOI] [PubMed] [Google Scholar]
- 19.Permutt S, Howell JB, Proctor DF, et al. Effect of lung inflation on static pressure-volume characteristics of pulmonary vessels. J Appl Physiol 1961;16:64-70. [DOI] [PubMed] [Google Scholar]
- 20.Pride NB, Permutt S, Riley RL, et al. Determinants of maximal expiratory flow from the lungs. J Appl Physiol 1967;23:646-62. [DOI] [PubMed] [Google Scholar]
- 21.Guyton AC, Granger HJ, Coleman TG. Autoregulation of the total systemic circulation and its relation to control of cardiac output and arterial pressure. Circ Res 1971;28:Suppl 1:93-7. [PubMed] [Google Scholar]
- 22.Shi G, Ergun GA, Manka M, et al. Lower esophageal sphincter relaxation characteristics using a sleeve sensor in clinical manometry. Am J Gastroenterol 1998;93:2373-9. [DOI] [PubMed] [Google Scholar]
- 23.Claridge M, Shuttleworth KE. The dynamics of obstructed micturition. Invest Urol 1964;2:188-99. [PubMed] [Google Scholar]
- 24.West JB, Dollery CT, Naimark A. Distribution of blood flow in isolated lung; relation to vascular and alveolar pressures. J Appl Physiol 1964;19:713-24. [DOI] [PubMed] [Google Scholar]
- 25.Gleadhill IC, Schwartz AR, Schubert N, et al. Upper airway collapsibility in snorers and in patients with obstructive hypopnea and apnea. Am Rev Respir Dis 1991;143:1300-3. [DOI] [PubMed] [Google Scholar]
- 26.Gold AR, Schwartz AR. The pharyngeal critical pressure. The whys and hows of using nasal continuous positive airway pressure diagnostically. Chest 1996;110:1077-88. [DOI] [PubMed] [Google Scholar]
- 27.Schwartz AR, Smith PL, Wise RA, et al. Induction of upper airway occlusion in sleeping individuals with subatmospheric nasal pressure. J Appl Physiol (1985) 1988;64:535-42. [DOI] [PubMed] [Google Scholar]
- 28.Smith PL, Wise RA, Gold AR, et al. Upper airway pressure-flow relationships in obstructive sleep apnea. J Appl Physiol (1985) 1988;64:789-95. [DOI] [PubMed] [Google Scholar]
- 29.Evans AS. Causation and disease: the Henle-Koch postulates revisited. Yale J Biol Med 1976;49:175-95. [PMC free article] [PubMed] [Google Scholar]
- 30.Issa FG, Sullivan CE. Upper airway closing pressures in obstructive sleep apnea. J Appl Physiol Respir Environ Exerc Physiol 1984;57:520-7. [DOI] [PubMed] [Google Scholar]
- 31.Sforza E, Petiau C, Weiss T, et al. Pharyngeal critical pressure in patients with obstructive sleep apnea syndrome. Clinical implications. Am J Respir Crit Care Med 1999;159:149-57. [DOI] [PubMed] [Google Scholar]
- 32.Sforza E, Bacon W, Weiss T, et al. Upper airway collapsibility and cephalometric variables in patients with obstructive sleep apnea. Am J Respir Crit Care Med 2000;161:347-52. [DOI] [PubMed] [Google Scholar]
- 33.Gold AR, Marcus CL, Dipalo F, et al. Upper airway collapsibility during sleep in upper airway resistance syndrome. Chest 2002;121:1531-40. [DOI] [PubMed] [Google Scholar]
- 34.Kirkness JP, Eastwood PR, Szollosi I, et al. Effect of surface tension of mucosal lining liquid on upper airway mechanics in anesthetized humans. J Appl Physiol (1985) 2003;95:357-63. [DOI] [PubMed] [Google Scholar]
- 35.Isono S, Remmers JE, Tanaka A, et al. Anatomy of pharynx in patients with obstructive sleep apnea and in normal subjects. J Appl Physiol (1985) 1977;82:1319-26. [DOI] [PubMed] [Google Scholar]
- 36.Meurice JC, Marc I, Carrier G, et al. Effects of mouth opening on upper airway collapsibility in normal sleeping subjects. Am J Respir Crit Care Med 1996;153:255-9. [DOI] [PubMed] [Google Scholar]
- 37.Meurice JC, Marc I, Sériès F. Effects of naloxone on upper airway collapsibility in normal sleeping subjects. Thorax 1996;51:851-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Philip-Joët F, Marc I, Sériès F. Effects of genioglossal response to negative airway pressure on upper airway collapsibility during sleep. J Appl Physiol (1985) 1996;80:1466-74. [DOI] [PubMed] [Google Scholar]
- 39.Ng AT, Gotsopoulos H, Qian J, et al. Effect of oral appliance therapy on upper airway collapsibility in obstructive sleep apnea. Am J Respir Crit Care Med 2003;168:238-41. [DOI] [PubMed] [Google Scholar]
- 40.Farré R, Rigau J, Montserrat JM, et al. Static and dynamic upper airway obstruction in sleep apnea: role of the breathing gas properties. Am J Respir Crit Care Med 2003;168:659-63. [DOI] [PubMed] [Google Scholar]
- 41.Pack AI. Sleep apnea: Pathogenesis, diagnosis and treatment. London: Informa Healthcare, 2009. [Google Scholar]
- 42.Sériès F, Côté C, Simoneau JA, et al. Upper airway collapsibility, and contractile and metabolic characteristics of musculus uvulae. FASEB J 1996;10:897-904. [DOI] [PubMed] [Google Scholar]
- 43.King ED, O'Donnell CP, Smith PL, et al. A model of obstructive sleep apnea in normal humans. Role of the upper airway. Am J Respir Crit Care Med 2000;161:1979-84. [DOI] [PubMed] [Google Scholar]
- 44.Schwartz AR, Gold AR, Schubert N, et al. Effect of weight loss on upper airway collapsibility in obstructive sleep apnea. Am Rev Respir Dis 1991;144:494-8. [DOI] [PubMed] [Google Scholar]
- 45.Brouillette RT, Thach BT. A neuromuscular mechanism maintaining extrathoracic airway patency. J Appl Physiol Respir Environ Exerc Physiol 1979;46:772-9. [DOI] [PubMed] [Google Scholar]
- 46.Brouillette RT, Thach BT. Control of genioglossus muscle inspiratory activity. J Appl Physiol Respir Environ Exerc Physiol 1980;49:801-8. [DOI] [PubMed] [Google Scholar]
- 47.Isono S, Tanaka A, Sho Y, et al. Advancement of the mandible improves velopharyngeal airway patency. J Appl Physiol (1985) 1995;79:2132-8. [DOI] [PubMed] [Google Scholar]
- 48.Patil SP, Schneider H, Marx JJ, et al. Neuromechanical control of upper airway patency during sleep. J Appl Physiol (1985) 2007;102:547-56. [DOI] [PubMed] [Google Scholar]
- 49.Clark RW, Schmidt HS, Schuller DE. Sleep-induced ventilatory dysfunction in Down's syndrome. Arch Intern Med 1980;140:45-50. [PubMed] [Google Scholar]
- 50.Strome M. Obstructive sleep apnea in Down syndrome children: a surgical approach. Laryngoscope 1986;96:1340-2. [DOI] [PubMed] [Google Scholar]
- 51.Southall DP, Stebbens VA, Mirza R, et al. Upper airway obstruction with hypoxaemia and sleep disruption in Down syndrome. Dev Med Child Neurol 1987;29:734-42. [DOI] [PubMed] [Google Scholar]
- 52.Stebbens VA, Dennis J, Samuels MP, et al. Sleep related upper airway obstruction in a cohort with Down's syndrome. Arch Dis Child 1991;66:1333-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cakirer B, Hans MG, Graham G, et al. The relationship between craniofacial morphology and obstructive sleep apnea in whites and in African-Americans. Am J Respir Crit Care Med 2001;163:947-50. [DOI] [PubMed] [Google Scholar]
- 54.Chi L, Comyn FL, Keenan BT, et al. Heritability of craniofacial structures in normal subjects and patients with sleep apnea. Sleep 2014;37:1689-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Trois MS, Capone GT, Lutz JA, et al. Obstructive sleep apnea in adults with Down syndrome. J Clin Sleep Med 2009;5:317-23. [PMC free article] [PubMed] [Google Scholar]
- 56.Marcus CL, McColley SA, Carroll JL, et al. Upper airway collapsibility in children with obstructive sleep apnea syndrome. J Appl Physiol (1985) 1994;77:918-24. [DOI] [PubMed] [Google Scholar]
- 57.Watanabe T, Isono S, Tanaka A, et al. Contribution of body habitus and craniofacial characteristics to segmental closing pressures of the passive pharynx in patients with sleep-disordered breathing. Am J Respir Crit Care Med 2002;165:260-5. [DOI] [PubMed] [Google Scholar]
- 58.Kirkness JP, Schwartz AR, Schneider H, et al. Contribution of male sex, age, and obesity to mechanical instability of the upper airway during sleep. J Appl Physiol (1985) 2008;104:1618-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schwab RJ, Gupta KB, Gefter WB, et al. Upper airway and soft tissue anatomy in normal subjects and patients with sleep-disordered breathing. Significance of the lateral pharyngeal walls. Am J Respir Crit Care Med 1995;152:1673-89. [DOI] [PubMed] [Google Scholar]
- 60.Schwab RJ, Gefter WB, Hoffman EA, et al. Dynamic upper airway imaging during awake respiration in normal subjects and patients with sleep disordered breathing. Am Rev Respir Dis 1993;148:1385-400. [DOI] [PubMed] [Google Scholar]
- 61.Davies RJ, Stradling JR. The relationship between neck circumference, radiographic pharyngeal anatomy, and the obstructive sleep apnoea syndrome. Eur Respir J 1990;3:509-14. [PubMed] [Google Scholar]
- 62.Kairaitis K, Howitt L, Wheatley JR, et al. Mass loading of the upper airway extraluminal tissue space in rabbits: effects on tissue pressure and pharyngeal airway lumen geometry. J Appl Physiol (1985) 2009;106;106:887-92. [DOI] [PubMed] [Google Scholar]
- 63.Squier SB, Patil SP, Schneider H, et al. Effect of end-expiratory lung volume on upper airway collapsibility in sleeping men and women. J Appl Physiol (1985) 2010;109:977-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rowley JA, Permutt S, Willey S, et al. Effect of tracheal and tongue displacement on upper airway airflow dynamics. J Appl Physiol (1985) 1996;80:2171-8. [DOI] [PubMed] [Google Scholar]
- 65.Thut DC, Schwartz AR, Roach D, et al. Tracheal and neck position influence upper airway airflow dynamics by altering airway length. J Appl Physiol (1985) 1993;75:2084-90. [DOI] [PubMed] [Google Scholar]
- 66.Sériès F, Marc I. Influence of lung volume dependence of upper airway resistance during continuous negative airway pressure. J Appl Physiol (1985) 1994;77:840-4. [DOI] [PubMed] [Google Scholar]
- 67.Sériès F, Cormier Y, Couture J, et al. Changes in upper airway resistance with lung inflation and positive airway pressure. J Appl Physiol (1985) 1990;68:1075-9. [DOI] [PubMed] [Google Scholar]
- 68.Sharp JT, Henry JP, Sweany SK, et al. Effects of mass loading the respiratory system in man. J Appl Physiol 1964;19:959-66. [DOI] [PubMed] [Google Scholar]
- 69.Hoffstein V, Mateika S. Differences in abdominal and neck circumferences in patients with and without obstructive sleep apnoea. Eur Respir J 1992;5:377-81. [PubMed] [Google Scholar]
- 70.Shinohara E, Kihara S, Yamashita S, et al. Visceral fat accumulation as an important risk factor for obstructive sleep apnoea syndrome in obese subjects. J Intern Med 1997;241:11-8. [DOI] [PubMed] [Google Scholar]
- 71.Isono S. Obesity and obstructive sleep apnoea: mechanisms for increased collapsibility of the passive pharyngeal airway. Respirology 2012;17:32-42. [DOI] [PubMed] [Google Scholar]
- 72.Younes M. Contributions of upper airway mechanics and control mechanisms to severity of obstructive apnea. Am J Respir Crit Care Med 2003;168:645-58. [DOI] [PubMed] [Google Scholar]
- 73.Mezzanotte WS, Tangel DJ, White DP. Influence of sleep onset on upper-airway muscle activity in apnea patients versus normal controls. Am J Respir Crit Care Med 1996;153:1880-7. [DOI] [PubMed] [Google Scholar]
- 74.Fogel RB, White DP, Pierce RJ, et al. Control of upper airway muscle activity in younger versus older men during sleep onset. J Physiol 2003;553:533-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Marcus CL, Moreira GA, Bamford O, et al. Response to inspiratory resistive loading during sleep in normal children and children with obstructive apnea. J Appl Physiol (1985) 1999;87:1448-54. [DOI] [PubMed] [Google Scholar]
- 76.Resta O, Carpanano GE, Lacedonia D, et al. Gender difference in sleep profile of severely obese patients with obstructive sleep apnea (OSA). Respir Med 2005;99:91-6. [DOI] [PubMed] [Google Scholar]
- 77.Goh DY, Galster P, Marcus CL. Sleep architecture and respiratory disturbances in children with obstructive sleep apnea. Am J Respir Crit Care Med 2000;162:682-6. [DOI] [PubMed] [Google Scholar]
- 78.O'Connor C, Thornley KS, Hanly PJ. Gender differences in the polysomnographic features of obstructive sleep apnea. Am J Respir Crit Care Med 2000;161:1465-72. [DOI] [PubMed] [Google Scholar]
- 79.Horner RL, Hughes SW, Malhotra A. State-dependent and reflex drives to the upper airway: basic physiology with clinical implications. J Appl Physiol (1985) 2014;116:325-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Berry RB, Kouchi K, Bower J, et al. Triazolam in patients with obstructive sleep apnea. Am J Respir Crit Care Med 1995;151:450-4. [DOI] [PubMed] [Google Scholar]
- 81.Chin CH, Kirkness JP, Patil SP, et al. Compensatory responses to upper airway obstruction in obese apneic men and women. J Appl Physiol (1985) 2012;112:403-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Polotsky M, Elsayed-Ahmed AS, Pichard L, et al. Effects of leptin and obesity on the upper airway function. J Appl Physiol (1985) 2012;112:1637-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Shapiro SD, Chin CH, Kirkness JP, et al. Leptin and the control of pharyngeal patency during sleep in severe obesity. J Appl Physiol (1985) 2014;116:1334-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Fang J, Wang Y, Krueger JM. Effects of interleukin-1 beta on sleep are mediated by the type I receptor. Am J Physiol 1998;274:R655-60. [DOI] [PubMed] [Google Scholar]
- 85.Krueger JM, Fang J, Hansen MK, et al. Humoral Regulation of Sleep. News Physiol Sci 1998;13:189-94. [DOI] [PubMed] [Google Scholar]
- 86.Fang J, Wang Y, Krueger JM. Mice lacking the TNF 55 kDa receptor fail to sleep more after TNFalpha treatment. J Neurosci 1997;17:5949-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Krueger JM, Fang J, Taishi P, et al. Sleep. A physiologic role for IL-1 beta and TNF-alpha. Ann N Y Acad Sci 1998;856:148-59. [DOI] [PubMed] [Google Scholar]
- 88.Takahashi S, Kapás L, Fang J, et al. Somnogenic relationships between tumor necrosis factor and interleukin-1. Am J Physiol 1999;276:R1132-40. [DOI] [PubMed] [Google Scholar]
- 89.Krueger JM, Obál FJ, Fang J, et al. The role of cytokines in physiological sleep regulation. Ann N Y Acad Sci 2001;933:211-21. [DOI] [PubMed] [Google Scholar]
- 90.Punjabi NM, Beamer BA. C-reactive protein is associated with sleep disordered breathing independent of adiposity. Sleep 2007;30:29-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Shearer WT, Reuben JM, Mullington JM, et al. Soluble TNF-alpha receptor 1 and IL-6 plasma levels in humans subjected to the sleep deprivation model of spaceflight. J Allergy Clin Immunol 2001;107:165-70. [DOI] [PubMed] [Google Scholar]
- 92.Churchill L, Rector DM, Yasuda K, et al. Tumor necrosis factor alpha: activity dependent expression and promotion of cortical column sleep in rats. Neuroscience 2008;156:71-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chin K, Shimizu K, Nakamura T, et al. Changes in intra-abdominal visceral fat and serum leptin levels in patients with obstructive sleep apnea syndrome following nasal continuous positive airway pressure therapy. Circulation 1999;100:706-12. [DOI] [PubMed] [Google Scholar]
- 94.Ip MS, Lam KS, Ho C, et al. Serum leptin and vascular risk factors in obstructive sleep apnea. Chest 2000;118:580-6. [DOI] [PubMed] [Google Scholar]
- 95.Ryan S, Taylor CT, McNicholas WT. Predictors of elevated nuclear factor-kappaB-dependent genes in obstructive sleep apnea syndrome. Am J Respir Crit Care Med 2006;174:824-30. [DOI] [PubMed] [Google Scholar]
- 96.Vgontzas AN, Papanicolaou DA, Bixler EO, et al. Sleep apnea and daytime sleepiness and fatigue: relation to visceral obesity, insulin resistance, and hypercytokinemia. J Clin Endocrinol Metab 2000;85:1151-8. [DOI] [PubMed] [Google Scholar]
- 97.Yokoe T, Minoguchi K, Matsuo H, et al. Elevated levels of C-reactive protein and interleukin-6 in patients with obstructive sleep apnea syndrome are decreased by nasal continuous positive airway pressure. Circulation 2003;107:1129-34. [DOI] [PubMed] [Google Scholar]
- 98.Schwartz AR, Thut DC, Brower RG, et al. Modulation of maximal inspiratory airflow by neuromuscular activity: effect of CO2. J Appl Physiol (1985) 1993;74:1597-605. [DOI] [PubMed] [Google Scholar]
- 99.Seelagy MM, Schwartz AR, Russ DB, et al. Reflex modulation of airflow dynamics through the upper airway. J Appl Physiol (1985) 1994;76:2692-700. [DOI] [PubMed] [Google Scholar]
- 100.Rowley JA, Williams BC, Smith PL, et al. Neuromuscular activity and upper airway collapsibility. Mechanisms of action in the decerebrate cat. Am J Respir Crit Care Med 1997;156:515-21. [DOI] [PubMed] [Google Scholar]
- 101.Schneider H, Boudewyns A, Smith PL, et al. Modulation of upper airway collapsibility during sleep: influence of respiratory phase and flow regimen. J Appl Physiol (1985) 2002;93:1365-76. [DOI] [PubMed] [Google Scholar]
- 102.van Lunteren E, Strohl KP, Parker DM, et al. Phasic volume-related feedback on upper airway muscle activity. J Appl Physiol Respir Environ Exerc Physiol 1984;56:730-6. [DOI] [PubMed] [Google Scholar]
- 103.Kuna ST. Inhibition of inspiratory upper airway motoneuron activity by phasic volume feedback. J Appl Physiol (1985) 1986;60:1373-9. [DOI] [PubMed] [Google Scholar]
- 104.Horner RL, Innes JA, Morrell MJ, et al. The effect of sleep on reflex genioglossus muscle activation by stimuli of negative airway pressure in humans. J Physiol 1994;476:141-51. [PMC free article] [PubMed] [Google Scholar]
- 105.Kimoff RJ, Cheong TH, Olha AE, et al. Mechanisms of apnea termination in obstructive sleep apnea. Role of chemoreceptor and mechanoreceptor stimuli. Am J Respir Crit Care Med 1994;149:707-14. [DOI] [PubMed] [Google Scholar]
- 106.Kimoff RJ, Sforza E, Champagne V, et al. Upper airway sensation in snoring and obstructive sleep apnea. Am J Respir Crit Care Med 2001;164:250-5. [DOI] [PubMed] [Google Scholar]
- 107.Kimoff RJ, Hamid Q, Divangahi M, et al. Increased upper airway cytokines and oxidative stress in severe obstructive sleep apnoea. Eur Respir J 2011;38:89-97. [DOI] [PubMed] [Google Scholar]
- 108.Schneider H, Krishnan V, Pichard LE, et al. Inspiratory duty cycle responses to flow limitation predict nocturnal hypoventilation. Eur Respir J 2009;33:1068-76. [DOI] [PubMed] [Google Scholar]
- 109.Tagaito Y, Schneider H, O'Donnell CP, et al. Ventilating with tracheal gas insufflation and periodic tracheal occlusion during sleep and wakefulness. Chest 2002;122:1742-50. [DOI] [PubMed] [Google Scholar]
- 110.Jordan AS, Wellman A, Heinzer RC, et al. Mechanisms used to restore ventilation after partial upper airway collapse during sleep in humans. Thorax 2007;62:861-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hoshino Y, Ayuse T, Kurata S, et al. The compensatory responses to upper airway obstruction in normal subjects under propofol anesthesia. Respir Physiol Neurobiol 2009;166:24-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Crowell JW, Guyton AC, Moore JW. Basic oscillating mechanism of Cheyne-Stokes breathing. Am J Physiol 1956;187:395-8. [DOI] [PubMed] [Google Scholar]
- 113.Cherniack NS, Longobardo GS, Levine OR, et al. Periodic breathing in dogs. J Appl Physiol 1966;21:1847-54. [DOI] [PubMed] [Google Scholar]
- 114.Longobardo GS, Gothe B, Goldman MD, et al. Sleep apnea considered as a control system instability. Respir Physiol 1982;50:311-33. [DOI] [PubMed] [Google Scholar]
- 115.Longobardo GS, Cherniack NS, Damokosh-Giordano A. Possible optimization of respiratory controller sensitivity. Ann Biomed Eng 1980;8:143-58. [DOI] [PubMed] [Google Scholar]
- 116.Khoo MC, Gottschalk A, Pack AI. Sleep-induced periodic breathing and apnea: a theoretical study. J Appl Physiol (1985) 1991;70:2014-24. [DOI] [PubMed] [Google Scholar]
- 117.Younes M, Ostrowski M, Thompson W, et al. Chemical control stability in patients with obstructive sleep apnea. Am J Respir Crit Care Med 2001;163:1181-90. [DOI] [PubMed] [Google Scholar]
- 118.Wellman A, Eckert DJ, Jordan AS, et al. A method for measuring and modeling the physiological traits causing obstructive sleep apnea. J Appl Physiol (1985) 2011; 110:1627-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Eckert DJ, White DP, Jordan AS, et al. Defining phenotypic causes of obstructive sleep apnea. Identification of novel therapeutic targets. Am J Respir Crit Care Med 2013;188:996-1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wellman A, Malhotra A, Jordan AS, et al. Effect of oxygen in obstructive sleep apnea: role of loop gain. Respir Physiol Neurobiol 2008;162:144-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Eckert DJ, Owens RL, Kehlmann GB, et al. Eszopiclone increases the respiratory arousal threshold and lowers the apnoea/hypopnoea index in obstructive sleep apnoea patients with a low arousal threshold. Clin Sci (Lond) 2011;120:505-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Eckert DJ, Malhotra A, Wellman A, et al. Trazodone increases the respiratory arousal threshold in patients with obstructive sleep apnea and a low arousal threshold. Sleep 2014;37:811-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Khoo MCK. Physiological control systems: analysis, simulation, and estimation. New York: IEEE Press, 2000. [Google Scholar]
- 124.McGinley BM, Schwartz AR, Schneider H, et al. Upper airway neuromuscular compensation during sleep is defective in obstructive sleep apnea. J Appl Physiol (1985) 2008;105:197-205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Brooks D, Horner RL, Kimoff RJ, et al. Effect of obstructive sleep apnea versus sleep fragmentation on responses to airway occlusion. Am J Respir Crit Care Med 1997;155:1609-17. [DOI] [PubMed] [Google Scholar]