

Genetic analysis among patients with dilated cardiomyopathy (DCM) is becoming an important part of clinical assessment, as it is in hypertrophic cardiomyopathy (HCM). The genetics of DCM is complex and therefore next-generation sequencing strategies are essential when providing genetic diagnostics. To achieve maximum yield, the diagnostic approach should include comprehensive clinical phenotyping combined with high-quality, high-coverage deep sequencing of DCM-associated genes and clinical variant classification as a basis for defining true yield in genetic testing. Our study has combined a novel sequencing strategy and clinical interpretation to analyse the yield and genotype–phenotype correlations among well-phenotyped Finnish DCM patients.
Keywords: Dilated cardiomyopathy, Genetics, Diagnosis
Abstract
Aims
Despite our increased understanding of the genetic basis of dilated cardiomyopathy (DCM), the clinical utility and yield of clinically meaningful findings of comprehensive next-generation sequencing (NGS)-based genetic diagnostics in DCM has been poorly described. We utilized a high-quality oligonucleotide-selective sequencing (OS-Seq)-based targeted sequencing panel to investigate the genetic landscape of DCM in Finnish population and to evaluate the utility of OS-Seq technology as a novel comprehensive diagnostic tool.
Methods and results
Using OS-Seq, we targeted and sequenced the coding regions and splice junctions of 101 genes associated with cardiomyopathies in 145 unrelated Finnish patients with DCM. We developed effective bioinformatic variant filtering strategy and implemented strict variant classification scheme to reveal diagnostic yield and genotype–phenotype correlations. Implemented OS-Seq technology provided high coverage of the target region (median coverage 410× and 99.42% of the nucleotides were sequenced at least 15× read depth). Diagnostic yield was 35.2% (familial 47.6% and sporadic 25.6%, P = 0.004) when both pathogenic and likely pathogenic variants are considered as disease causing. Of these, 20 (53%) were titin (TTN) truncations (non-sense and frameshift) affecting all TTN transcripts. TTN truncations accounted for 20.6% and 14.6% of the familial and sporadic DCM cases, respectively.
Conclusion
Panel-based, high-quality NGS enables high diagnostic yield especially in the familial form of DCM, and bioinformatic variant filtering is a reliable step in the process of interpretation of genomic data in a clinical setting.
Translational perspective.
Genetic analysis among patients with dilated cardiomyopathy (DCM) is becoming an important part of clinical assessment, as it is in hypertrophic cardiomyopathy (HCM). The genetics of DCM is complex and therefore next-generation sequencing strategies are essential when providing genetic diagnostics. To achieve maximum yield, the diagnostic approach should include comprehensive clinical phenotyping combined with high-quality, high-coverage deep sequencing of DCM-associated genes and clinical variant classification as a basis for defining true yield in genetic testing. Our study has combined a novel sequencing strategy and clinical interpretation to analyse the yield and genotype–phenotype correlations among well-phenotyped Finnish DCM patients.
Introduction
According to the position statement of the European Society of Cardiology (ESC), DCM is defined as left ventricular dilatation and dysfunction in the absence of abnormal loading conditions or coronary artery disease sufficient to cause global systolic impairment. Furthermore, DCM can be divided into familial and non-familial forms.1 Prevalence of DCM is at least 1:2500 and up to 30–50% of DCM cases are familial.2 Dilated cardiomyopathy is the leading cause for heart transplantation and a relatively common cause for heart failure and sudden cardiac death.3,4
In 2012, Herman and colleagues5 published a study showing truncating titin (TTN) mutations in 25% of familial and 18% of sporadic DCM patients. This index report has triggered more thorough and ambitious DCM research by next-generation sequencing (NGS) approaches that enable analysis at a scale once considered impossible. By now DCM-associated mutations have been found in >50 genes.6 Most identified mutations, often unique to families, are inherited in an autosomal dominant fashion, although some can be autosomal recessive, X-linked, or mitochondrial. These genes encode components of the sarcomere, desmosome, cytoskeleton, nuclear lamina, mitochondria, and calcium-handling proteins.7,8 However, many of the genes associated with cardiomyopathies are not specific to a particular type of cardiomyopathy, and it has been suggested that 9.0–12.6% of the patients carry at least two potentially disease-causing mutations.9,10 Penetrance of cardiomyopathies is clearly age and gene dependent but so far poorly understood.5,11 Though whole exome- and genome-level sequencing are required for DCM gene discovery, targeted high-quality NGS strategies are evolving as the primary diagnostic tool in clinical setting.
Fast accumulation of knowledge on cardiomyopathy genetics has started to reshape clinical practices as identification of causative mutation enables effective differential diagnostics and facilitates screening of the family members, but may also help in driving treatment decisions. Evolvement of open-access large reference databases such as the ExAC (http://exac.broadinstitute.org) together with diagnostic and research laboratories sharing variant information with classification into public databases such as ClinVar is already improving the interpretation and utility of genetic data. Due to this development, genetic diagnostics in cardiomyopathies is recommended in international guidelines.12,13 Moreover, genetic screening has shown to be cost-effective in hypertrophic cardiomyopathy,14 but the evaluation of the clinical utility and cost-effectiveness of genetic screening in DCM is still on the way.
Our goal was to investigate the genetic landscape of DCM in Finnish population and to evaluate associations between genotype and phenotype. Furthermore, our goal was to evaluate the utility of OS-Seq technology as a novel comprehensive diagnostic tool.
Methods
Subjects and clinical evaluation
The Finn-DCM study cohort comprised 145 unrelated DCM patients (63 familial and 82 sporadic) of Finnish origin. The patients were recruited between 1999 and 2013 from Helsinki University Hospital, the only tertiary centre in the country to perform cardiac transplantations. All patients fulfilled the following diagnostic criteria: left ventricular (LV) end-diastolic diameter (LVEDD) >27 mm/m2 [modified from original criteria of >117% of the predicted value corrected for age and body surface area (BSA)] and LV systolic dysfunction (LVEF < 45%) in the absence of abnormal loading conditions such as hypertensive heart disease, primary valve disease, or significant coronary artery disease.15 Definition of familial cardiomyopathy was family history of hypertrophic cardiomyopathy (HCM), DCM, or arrhythmogenic right ventricular cardiomyopathy (ARVC), or two or more patients with atrial fibrillation before age 40 or rhythm/conduction disturbances requiring pacemakers in the family. Detailed clinical information was obtained at multiple time points for each subject including family history, anthropometrics, age at presentation, age at death due to cardiomyopathy, symptoms, blood pressure, electrocardiograms, echocardiograms, pacemaker implantation, transplantation, and resuscitation. Written informed consent was obtained from all patients, and the institutional ethical committee of the University of Helsinki approved the study (Dnro 307/13/03/01/11).
ExAC reference population
Exome Aggregation Consortium (http://exac.broadinstitute.org) is an international collaborative effort to aggregate and harmonize exome-sequencing data from a variety of large-scale sequencing projects. It consists of >60 000 individuals including Finnish SISu (Sequencing Initiative Suomi Project) Control Cohorts. To filter common polymorphisms in the Finnish population, identified candidate variants were queried for overlap against the available variants reported in the ExAC and SISu project databases.
Oligonucleotide-selective targeted sequencing of 101 cardiomyopathy-related genes
Genomic DNA was extracted from blood samples and sequenced using OS-Seq. One hundred and one-gene OS-Seq-based targeted sequencing panel with automated target DNA capture and sequencing was applied in the MiSeq sequencer.16 The gene panel (Pan Cardiomyopathy Panel, Blueprint Genetics v.1.0) was designed to screen 2515 consensus coding sequence (CCDS) regions and splice junctions of 51 known genes and 50 candidate genes associated with DCM and implicated in other inherited cardiomyopathies based on Online Mendelian Inheritance in Man database annotations and literature.17,18 Analytic validation of the Pan Cardiomyopathy Panel has been submitted elsewhere. Shortly, the panel had sensitivity of 99.0%, specificity of 100.0%, and positive predictive value of 99.0% to detect single-nucleotide polymorphisms (SNPs) and sensitivity of 100% for indels ranged 1–19 bp.
Bioinformatic data analyses and variant filtering
Raw paired-end sequence reads quality trimming and adapter sequence removal were carried out using Trimmomatic in paired-end mode.19 High-quality reads were mapped to human genome reference sequence (hg19) using Burrows–Wheeler Algorithm (BWA).20 After excluding PCR duplicates, we applied GATK (V.3.1–1)21 base quality score recalibration, indel realignment, and performed SNP and insertion-deletion (INDEL) discovery and genotyping across all samples simultaneously using standard hard filtering parameters (Phred-scaled genotype quality score threshold of 30) according to GATK Best Practices recommendations.22 Identified variants were annotated using Ensembl's Variant Effects Predictor (VEP) tool,23 and the pathogenicity of missense variants was predicted in silico using two ensemble scores from dbNSFP (RadialSVM and LR) based on 10 component scores (SIFT, PolyPhen-2 HDIV, PolyPhen-2 HVAR, GERP++, MutationTaster, Mutation Assessor, FATHMM, LRT, SiPhy, PhyloP) and the maximum frequency observed in the 1000 genomes populations.24 Variant filtering was done using an in-house variant filtering and prioritization strategy (Figure 1). We adopted Blueprint Genetics' variant classification scheme (see Supplementary material online, Table S1), and a team of clinicians and geneticists evaluated and classified all rare variants as ‘pathogenic’, ‘likely pathogenic’, ‘variant of unknown significance’, ‘likely benign’, or ‘benign’. For a detailed description of the data analysis and variant filtering strategy, refer to the Supplementary material online.
Figure 1.

Bioinformatics variants filtering and characterization strategy. RadialSVM prediction, support vector machine (SVM)-based ensemble prediction; LR, logistic regression-based ensemble prediction.
Statistical analyses
The Shapiro–Wilk test was applied to determine whether data are normally distributed. Normally distributed, continuous variables were expressed as mean ± SD and non-parametric as median, and lower and upper quartiles. All categorical variables were depicted using relative frequency distributions. Characteristics of different groups such as patients with or without disease-causing mutation were compared using the χ2 test for categorical variables if appropriate; otherwise, Fisher's exact test was used. Independent samples t-tests combined with Levene's tests were used for comparison between the groups for all continuous variables, and Mann–Whitney U tests were used for non-parametric variables. Differences were considered significant if the two-sided P-value was <0.05. All statistical analyses were done using R statistical software.
Results
Study population
The Finn-DCM cohort consisted of 145 unrelated DCM patients. The mean age at diagnosis was 44.3 ± 12.2 years in familial DCM [n = 63, 44 (69.8%) men] and 46.8 ± 11.7 years in sporadic disease [n = 82, 63 (77.0%) men]. The mean follow-up time was 6.2 years. In 83.9% cases of the familial and 28.9% cases of sporadic DCM at least one family member was studied by echocardiography. Endomyocardial biopsy or pathology report from the explanted heart was available from 50.3% of the patients. Angiography was performed on 78.6% (n = 114) of the patients, and 29.7% (n = 43) have undergone heart transplantation. Details on study population are presented in Table 1.
Table 1.
Distribution of pathogenic and likely pathogenic variants among tested genes in patients with familial or sporadic dilated cardiomyopathy in the Finnish dilated cardiomyopathy study
| Patients | All (N = 145) | Familial (N = 63) | Sporadic (N = 82) |
|---|---|---|---|
| Males | 107 (73.8%) | 44 (69.8%) | 63 (76.8%) |
| Females | 38 (26.2%) | 19 (30.2%) | 19 (23.2%) |
| Diagnostic yield | |||
| Mutation positive, n (%) | 51 (35.2) | 30 (47.6) | 21 (25.6) |
| Mutation negative, n (%) | 94 (64.8) | 33 (52.4) | 61 (74.4) |
| Causative gene in Finnish dilated cardiomyopathy cohort, N (%) | |||
| Sarcomere genes | |||
| Titin (TTN) | 25 (17.2%) | 13 (20.6%) | 12 (14.6%) |
| β-myosin heavy chain 7 (MYH7) | 1 (0.7%) | 1 (1.6%) | — |
| Troponin T type 2 (TNNT2) | 1 (0.7%) | 1 (1.6%) | — |
| Nuclear lamina | |||
| Lamin A/C (LMNA) | 12 (8.3%) | 9 (14.3%) | 3 (3.7%) |
| Z-disc/related, desmosomal, Ca2+ handling | |||
| Desmoplakin (DSP) | 8 (5.5%) | 4 (6.3%) | 4 (4.9%) |
| RNA-binding motif protein 20 (RBM20) | 2 (1.4%) | 1 (1.6%) | 1 (1.2%) |
| Dystrophin (DMD) | 1 (0.7%) | — | 1 (1.2%) |
| Titin-cap; telethonin (TCAP) | 1 (0.7%) | 1 (1.6%) | — |
| Total | 51 (35.2%) | 30 (47.6%) | 21 (25.6%) |
Sequencing quality
Using genomic DNA isolated from 145 subjects with DCM, we captured and performed oligonucleotide-selective sequencing of 667 kb of genomic region covering the CCDS coding regions and splice junctions of 101 genes. Sequencing depth was evaluated in each target base to demonstrate the efficiency of sequencing. Results from the sequencing coverage analyses are shown as percentage of targeted bases with >1×, >5×, >10×, >15×, >25×, >50×, >100×, and until >500× depth in 145 subjects in the Supplementary material online, Figure S1. The median read depth in the target region across the samples was 410×, and 99.42% of the nucleotides had over 15× read depth. Individual gene-specific sequencing coverage plot is shown in the Supplementary material online, Figure S2.
Variant filtering to identify clinically significant variants
Genotyping with GATK Unified Genotyper multi-sample mode generated a total of 1513 variants present in 145 subjects (234 variants/patient). Excluding synonymous single-nucleotide substitutions, we found 658 exonic and splice-site variants called with phred-scaled quality of ≥400. After excluding common polymorphisms based on variants reported in 1000 Genomes and ESP databases, we obtained 167 rare exonic (131 missense, 13 non-sense, 17 frameshift-indel, and 6 in-frame indel) and 2 splice-site variants as candidates for further analysis (see Supplementary material online, Table S2). In total, 119 patients (82.1%) carried at least one rare variant in the target genes. Sixty-eight patients (46.9%) carried multiple rare variants (see Supplementary material online, Tables S2 and S4). In silico analysis predicted 32.8% (43/131) of the rare missense variants as deleterious. Of the rare variants, 79.2% (134/169) were private. In total, 38 variants were classified as pathogenic or likely pathogenic (see Supplementary material online, Table S3).
Diagnostic yield
Our diagnostic yield in whole Finn-DCM cohort was 35.2% when both pathogenic and likely pathogenic are considered as disease-causing variants. Diagnostic yield was significantly higher in familial vs. sporadic cases (47.6 vs. 25.6%, P = 0.004, Table 1). Based on our classification scheme, 8.3% of the rare variants were pathogenic, 26.2% likely pathogenic, 27.6% variant of unknown significance, 20.0% likely benign, and 17.9% benign. Disease-causing genes included TTN (17.2%); lamin A/C (LMNA) (8.3%); desmoplakin (DSP) (5.5%); RNA-binding motif protein 20 (RBM20) (1.4%); myosin, heavy chain 7 (MYH7), troponin T type 2 (TNNT2), dystrophin (DMD), and titin-cap (TCAP) (0.7%). Distribution of the disease-causing mutations within these genes in Finn-DCM cohort is presented in Table 1.
In total, 64 unique rare TTN variants were found in 48% (71/145) of Finn-DCM cohort. Of the rare TTN variants, 70% (38/64) were missense variants with 11 variants being novel based on ExAC database. As TTN missense variants were considered variants of unknown significance, we focused on truncating mutations. Twenty-one truncating TTN variants were predicted to cause loss of function in 25 patients (17.2%): 11 non-sense variants in 16 patients; 9 frameshift variants in 10 patients; and 1 consensus splice-site variant in one patient (see Supplementary material online, Table S3). Of note, one additional truncating TTN variant (p.Glu4647Lysfs*54) was graded as variant of unknown significance (VUS), because it is quite common and affects only ENST00000360870 (TTN-010) transcript encoding a 5604 amino acid protein. Notably, 67% (14/21) of the truncating TTN mutations were private and not reported in the ExAC database. Truncating TTN mutations in the Finn-DCM cohort were not randomly distributed along the gene; instead, the majority 62% (13/21) of the mutations were located in the large A band region of titin that associates with the thick filament (Figure 2). In addition, 95% (20/21) of the truncating TTN mutations affected all transcripts of TTN gene. Family segregation analysis was performed in five families with truncating TTN mutations, and it revealed positive segregation with the disease in all cases (Figure 3). Disease penetrance increased from 53.8% at age 50 to 84.6% at age 60 and 100.0% at age 70.
Figure 2.
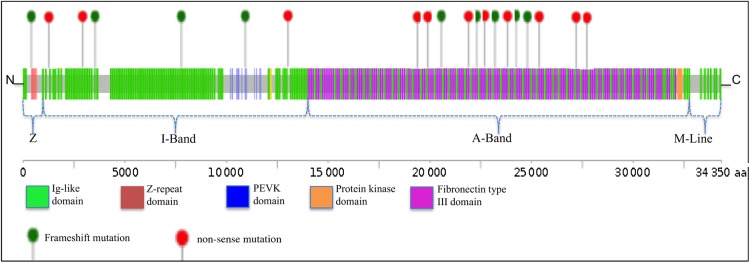
Spatial distribution of titin frameshift and non-sense mutations in Finnish dilated cardiomyopathy cohort. Titin is linearly depicted with its 152 Immunoglobulin-like domains in green and 132 fibronectin type III domains in purple. TTN mutations are shown as lollipops. Depicted are non-sense (red) and frameshift (green) mutations in subjects with dilated cardiomyopathy. Variants are shown relative to the titin Uniprot Sequence identifier Q8WZ42. The dashed lines below the protein schematic indicate the location of variants within the sarcomere.
Figure 3.

Pedigrees of the patients with truncating TTN mutations. All family members with available DNA samples were included. Index patients are marked with an arrow. All family members with dilated cardiomyopathy carried truncating TTN mutation. Disease penetrance increased from 53.8% at age 50 to 84.6% at age 60 and 100.0% at age 70.
Recurrent variants in multiple dilated cardiomyopathy patients
To further elucidate the role of the disease-causing variants identified in more than one Finn-DCM family, the allele frequencies were compared with the largest available genetically matched control population (Finnish controls in the ExAC study) using Fishers' exact test. All of the identified four variants were significantly enriched in the Finn-DCM cohort (Table 3).
Table 3.
Pathogenic and likely pathogenic variants present in multiple DCM families
| Gene | Transcript | Genomic location | Nucleotide change | Amino acid change | Calls (N) | ExAC (N) | P-value |
|---|---|---|---|---|---|---|---|
| LMNA | ENST00000368300 | 1:156100478 | c.427T>C | p.Ser143Pro | 3 | 0/3372 | 7.3E−05 |
| DSP | ENST00000379802 | 6:7583804 | c.6310delA | p. Thr2104Glnfs*12 | 6 | 2/3372 | 1.5E−07 |
| TTN | ENST00000589042 | 2:179453719 | c.62733G>A | p.Trp20911* | 3 | 0/3372 | 7.3E−05 |
| TTN | ENST00000589042 | 2:179419765 | c.88421G>A | p.Trp29474* | 3 | 0/3372 | 7.3E−05 |
DCM, dilated cardiomyopathy.
Clinical characteristics and genotype–phenotype correlations
The clinical characteristics of the patients with vs. without a mutation were not statistically different (Table 1). Male patients had larger LVEDD (72 ± 10 vs. 64 ± 8 mm, P < 0.001) and lower EF (23 ± 8 vs. 28 ± 9%, P = 0.003) than females and a trend of more frequent cardiac transplantation (33 vs. 16%, P = 0.10). Otherwise, gender had no significant effect on age at onset, arrhythmias, resuscitation, and use of pacemakers.
We compared clinical characteristics between different mutation groups: TTN, LMNA, DSP, and others. Other mutation group included two cases with RBM20 mutations, one mutation in each of MYH7, TNNT2, TCAP, and DMD. Left ventricular end-diastolic diameter was larger in patients with TTN vs. LMNA mutations (70 ± 8 vs. 60 ± 10, P = 0.03) (Table 1). In the LMNA group, pacemaker implantation, history of resuscitation (resuscitation or appropriate ICD shock), and atrial fibrillation were more frequent than in some of the other groups (Table 2). Age of disease onset (fulfilling DCM diagnostic criteria) seemed lower in the other mutation group compared with the LMNA (P = 0.04), DSP (P = 0.03), and TTN groups (P = 0.10) (Table 2), but it should be noted that especially in LMNA cardiomyopathy disease manifestation such as conduction abnormalities and arrhythmias exist commonly before LV dilatation and impaired systolic function. There was no significant difference in reaching the composite end point (cardiac transplantation and death from cardiac causes) among mutation positive vs. negative patients (P = 0.34, Figure 4A) or patients with disease-causing mutations in TTN vs. LMNA genes (P = 0.58, Figure 4B).
Table 2.
Clinical presentation of dilated cardiomyopathy in relation to familial vs. sporadic presentation, mutation status, and causative gene in the Finnish dilated cardiomyopathy cohort (n = 145)
| Familial (n = 63) | Sporadic (n = 82) | P | Mutation-positive (n = 50) | No mutation (n = 95) | P | TTN (n = 25) | LMNA (n = 12) | DSP (n = 7) | Other (n = 6) | |
|---|---|---|---|---|---|---|---|---|---|---|
| Age at diagnosis (years) | 44.3 ± 12.2 | 46.8 ± 11.7 | 0.21 | 43.7 ± 11.8 | 46.7 ± 11.9 | 0.12 | 43.4 ± 11.4 | 46.7 ± 13.3*d | 49.0 ± 4.9*d | 31.7 ± 9.0*b,c |
| BMI (kg/m2) | 26.0 ± 4.3 | 25.6 ± 3.9 | 0.51 | 24.9 ± 3.9 | 26.2 ± 4.2 | 0.04 | 25.3 ± 3.8 | 23.1 ± 1.5 | 25.6 ± 6.1 | 25.6 ± 4.2 |
| LVEF (%) | 25.2 ± 10.2 | 23.0 ± 7.5 | 0.14 | 24.9 ± 10.4 | 23.5 ± 7.9 | 0.31 | 23.1 ± 9.3 | 27.3 ± 10.1 | 29.7 ± 10.6 | 23.3 ± 14.6 |
| LVEDD index (mm/m2) | 35.1 ± 5.2 | 36.9 ± 5.1 | 0.05 | 36.2 ± 3.7 | 35.3 ± 5.7 | 0.48 | 36.1 ± 3.7*b | 33.4 ± 5.5*a | 37.3 ± 7.4 | 36.0 ± 5.2 |
| Alive (%) | 79.4% | 85.4% | 0.23 | 79.6% | 84.4% | 0.52 | 88.0%*b | 58.3%*a | 85.7% | 83.3% |
| None | 50.0% | 56.1% | 0.38 | 57.1% | 52.1% | 0.79 | 68.0%*b | 8.3%*a,b,c | 85.7%*b | 66.7%*b |
| Pacemaker | 11.1% | 3.7% | 10.2% | 5.2% | 0.0%*b | 41.7%*a,c | 0.0%*b | 0.0%*b | ||
| ICD | 19.0% | 17.1% | 16.3% | 18.8% | 20.0% | 8.3% | 14.3% | 16.7% | ||
| CRT | 7.9% | 6.1% | 6.1% | 7.3% | 4.0% | 8.3% | 0.0% | 16.7% | ||
| CRT-D | 11.1% | 17.1% | 10.2% | 16.7% | 8.0% | 33.3% | 0.0% | 0.0% | ||
| Any ICD (ICD + CRT-D) | 30.1% | 34.2% | 26.5% | 35.5% | 28.0% | 41.6% | 14.3% | 16.7% | ||
| Angiography | 76.2% | 80.5% | 0.55 | 75.5% | 80.2% | 0.67 | 84.0% | 75.0% | 57.1% | 66.7% |
| Resuscitated (%) | 22.2% | 17.1% | 0.54 | 20.4% | 18.8% | 0.44 | 12.0%*b | 41.7%*a | 28.6% | 16.7% |
| Transplantation (%) | 27.0% | 31.7% | 0.59 | 32.7% | 28.1% | 0.70 | 28.0% | 41.7% | 28.6% | 33.3% |
| Age at transplantation | 49.7 ± 10.3 | 46.9 ± 10.4 | 0.39 | 45.9 ± 8.8 | 49.2 ± 11.1 | 0.32 | 46.7 ± 6.8 | 44.8 ± 11.2 | 55.0 ± 5.7 | 37.0 ± 4.2 |
| Atrial fibrillation (%) | 47.6% | 37.8% | 0.24 | 51.0% | 37.5% | 0.16 | 40.0%*b | 83.3%*a,c | 28.6%*b | 50.0% |
BMI, body mass index; LVEF, left ventricular ejection fraction; LVEDD index, left ventricular end-diastolic diameter indexed to body surface area; ICD, implantable cardioverter defibrillator; CRT, cardiac resynchronization therapy; CRT-D, pacemaker with CRT + ICD. All data are presented percentages or mean ± SD. Resuscitated includes both resuscitated and those with appropriate shocks by ICD. Other mutation group includes two RBM20 mutations, one in each MYH7, TNNT2, TCAP, and DMD.
*Mean significant difference compared with other mutation group (aTTN, bLMNA, cDSP, and dother).
Figure 4.

Kaplan–Meier curve demonstrating freedom from composite endpoint (cardiac transplantation and death from cardiac causes). Panel A shows Kaplan–Meier curve for patients with and without detected disease-causing mutation and Panel B shows comparison between patients with TTN vs. LMNA mutations. Importantly, many patients are 40–60 years old at a moment and have not reached any endpoint. Hatch marks indicate patient age at last follow-up date in Finnish dilated cardiomyopathy study.
Discussion
Multiple genes can cause inherited DCM. Due to incomplete knowledge of the genes involved in DCM coupled with variation in population structure and genetics, genetic characterization of DCM has been a challenging task. This becomes more challenging in a bottlenecked population like the Finnish population, as such populations have a smaller spectrum of rare variation. To investigate the genetic landscape of DCM in the Finnish population, to evaluate associations between genotype and phenotype, and to evaluate the utility of OS-Seq technology as a novel comprehensive diagnostic tool, we developed and utilized a high-quality 101-gene OS-Seq-based targeted resequencing panel. In this study, we show excellent sequencing performance and clinical utility of a large cardiomyopathy targeted sequencing panel in genetic diagnosis of patients with DCM. Using rigorous clinically oriented variant classification and interpretation strategy, we obtained an unmatched diagnostic yield of 35.2% (51/145) in the Finn-DCM cohort. Diagnostic yield was significantly higher in familial 47.6% (30/63) vs. sporadic 25.6% (21/82) DCM. We also demonstrate the capability of bioinformatic filtering using appropriate reference population coupled with in silico prediction in improving variant interpretation when utilizing large NGS panels in diagnostics. Finally our study confirmed prominent role of TTN in DCM and genotype–phenotype association showed that LMNA cardiomyopathy is clinically distinct from DCM with other genetic origin.
In clinical diagnostic setting, sensitivity of the NGS panel is an important parameter affecting diagnostic yield. Haas et al. published recently an NGS cardiomyopathy panel with a median coverage of 2526×, which is higher than 410× in our study.10 Both panels have sensitivity of 99.0% in HapMap (NA12878) validation, but our accuracy was significantly better (100.00 vs. 91.3%). Most probably this reflects more uniform sequencing coverage in our study. Uniform sequencing depth is also an important element for affordable genetics diagnostics. Moreover, Haas et al. did not provide data of indel validation, which is critical as NGS strategies can have significant differences in specificity and sensitivity for indels. Indel detection efficiency is critical when aiming for high diagnostic yield in DCM as over 1000 cardiomyopathy-associated indel variants are listed in public and commercial databases such as ClinVar and Human Gene Mutation Database (HGMD).
In addition to sequencing quality, another equally important factor determining diagnostic hit rate is variant classification and interpretation. In many studies, relatively loose frequency criteria have been utilized to claim disease-associating variants.5,10 In Finn-DCM study, 89% (34/38) of the variants classified as pathogenic or likely pathogenic were not present in large control population (ExAC) and all except two patients carried only one known disease-causing variant, which is in-line with autosomal dominant inheritance pattern of familial DCM but opposite to that reported by Haas et al. In their DCM cohort, Haas et al. reported that 73.2% of the patients had ‘known’, ‘likely’, or ‘potential’ disease-causing mutation, and 46% of the patients were found to have ‘known’ disease-causing mutation in cardiomyopathy or chanellopathy genes, but only 16% of the patients had ‘known’ DCM mutation. Moreover, variant interpretation in the study by Haas et al. was based on variants reported in HMGD, which is known to contain variants classified as pathogenic, although some of them are increasingly being identified as benign polymorphisms with the advent of NGS. We randomly selected 10 variants from Haas et al. study classified as potential disease causing (Classes 1a-III) variants and queried their frequency from the ExAC database. Interestingly, 80% of the variants were present in over 9 (allele frequency >0.015%) subjects in the ExAC database with an average of 263 carriers (allele frequency >0.44%) in the database. This prevalence in the population makes the variants highly unlikely to be disease causing.
Clinical exome sequencing strategies can also result in a false-negative test result meaning a known genetic cause was missed in the analysis. This is due to inadequate depth and breadth of sequencing coverage at clinically relevant locations. Recently, Park et al.25 examined 57 exome sequencing data for adequacy in the detection of potentially pathogenic variant locations in the 56 genes described in the ACMG incidental findings recommendation. Despite high mean coverage between 74× and 120×, only 83–87% of the bases were covered at ≥20×. Of note, the clinical exome data had inadequate coverage for >50% of HGMD variant locations in 7/56 ACMG genes with a recommendation to report even as incidental findings.26 This probably explains why targeted NGS panels outperform exome sequencing in clinical diagnostics.
Gudkova et al.27 demonstrated a patient whose cardiomyopathy phenotype changed from HCM to restrictive and finally to DCM. Limited data exist on late-stage HCM developing to so call burnout physiology and resemblance to DCM.28 Furthermore, the incidence of this phenomenon is unknown. Our study did not reveal any typical HCM-associated mutations, suggesting that at least in our DCM cohort the incidence of HCM with burnout physiology is extremely low. In addition to potential phenotypic overlap between HCM and DCM, there is increasing evidence of overlap between ARVC and DCM. Cardiac desmosomal genes such as desmoglein 2 (DSG2), desmocollin-2 (DSC2), junction plakoglobin (JUP), plakophilin-2 (PKP2), and DSP were originally reported in association with ARVC, but segregating mutations in these genes are increasingly found in patients with classical DCM.29,30 In one of these studies, 16% of DCM patients undergoing heart transplantation harboured known desmosomal ARVC mutations, and these patients were clinically and histologically indistinguishable from classical DCM.29 In our study, we identified three likely pathogenic truncating DSP mutations in eight patients. These mutations were absent or extremely rare among controls. The truncating mutation p.Thr2104Glnfs*12 in DSP gene is especially interesting as we identified it in six DCM patients. Of note, three out of six patients harbouring this mutation have familial DCM, but no samples were available for segregation analysis. We have previously identified this mutation in a clinical setting in two other Finnish patients suffering either from DCM or ventricular fibrillation at childhood. In ExAC, the two out of four carriers of this mutation among over 60 000 individuals are Finnish. Originally this mutation has been described in two reports to cause early childhood-onset multiorgan disease with severe DCM combined with skin, hair, and tooth abnormalities.31,32 In both reports, the patients were compound heterozygous for two DSP mutations: both were carriers of the p.Thr2104Glnfs*12 mutation but harboured also another mutation in the DSP gene. Interestingly, in the study by Mahoney et al.32 the patient had a Finnish mother. In the other report, the family came from Swedish west coast, an area with large population of Finnish immigrants in the past. Based on previous reports, our experience from diagnostics and our results in this study, the p.Thr2104Glnfs*12 in DSP could represent the first Finnish DSP founder mutation associated with DCM disease.
In 2012, Herman et al.5 identified TTN truncating mutations in 25% of familial DCM and 18% of sporadic DCM, whereas diagnostic hit rate was a bit lower (19 and 11%, respectively) in recently published European DCM cohort.10 Of note, the European DCM cohort in the study by Haas et al. does not include any Finnish DCM patients. In our Finn-DCM cohort, 17.2% of the patients had truncating TTN mutation. This accounts for 20.6 and 14.6% of the familial and sporadic DCM cases, respectively. This is slightly lower compared with the frequency reported by Herman et al. However, 95% of the truncating TTN mutations classified as disease causing in our study affect all transcripts of TTN, but this was not taken into consideration in the other index studies. Clinical practice has shown that co-segregation of truncating TTN variant is lower if it is present only in short TTN isoform/transcripts. Importantly, we performed family segregation analysis in five families with truncating TTN variants affecting all TTN transcripts (p.Trp29474*, p.Ser25617Cysfs*18, p.Ala486Serfs*26, p.Gln25732*, and p.Ile20447*) and found complete segregation. Disease penetrance increased from 53.8% at age 50 to 84.6% at age 60 and 100.0% at age 70.
Patients with TTN truncating mutations had clinical manifestations, morbidity, and mortality that were indistinct from those with DSP and other gene mutation groups. In contrast, TTN truncating mutation carriers had clinically and statistically larger LVEDD but lower mortality, less pacemakers, resuscitation, and atrial fibrillation compared with LMNA mutation carriers.
Although recent studies have identified same disease-causing variants in multiple DCM families,33,34 majority of the disease-causing variants in our Finn-DCM cohort were unique to a family. We identified four variants present in multiple families (total 15 patients): Ser143Pro in LMNA, Thr2104Glnfs*12 in DSP, Trp20911* and Trp29474* in TTN; and absent or very rare in control populations (Table 3). Of note, 73% (11/15) of the patients with a recurrent variant carried no other disease-causing variant, 20% (3/15) carried a VUS, whereas only one patient had another likely pathogenic variant.
Our study demonstrates that comprehensive targeted sequencing can provide genetic diagnosis for a substantial proportion of DCM patients. The diagnostic yield is in our experience very similar to what is seen in practice with HCM patients. We suggest that the genetic aetiology of DCM patients should be sought for more vigorously. Despite the relatively high diagnostic yield, gene discovery with whole exome or genome is still needed as several families with DCM are still left without a diagnosis, suggesting that new genes are to be found. Several mutation-negative families from our cohort are already enrolled into exome sequencing to reveal novel genetic mechanisms underlying the pathogenesis of DCM.
Limitations
We were unable to perform comprehensive co-segregation analysis in many cases due to sporadic DCM and low number of family member samples in familial cases. We hope that future follow-ups and studies will bring light to this limitation.
Supplementary material
Supplementary material is available at European Heart Journal online.
Funding
This work was supported by grants from the Sigrid Jusélius Foundation; Finnish Foundation for Pediatric Research; Finnish Foundation for Cardiovascular Research; Finnish Cultural Foundation; Aarne Koskelo Foundation; Ida Montin Foundation; Clinical Research funding of Turku University Hospital (E.V.O.); Helsinki University Hospital [TYH2014208, TYH2012120, TYH7106, TYH200921, TYH4241, TYH2205, and TYH0231]; and Blueprint Genetics. Funding to pay the Open Access publication charges for this article was provided by Blueprint Genetics.
Conflict of interest: Minor: J.T., P.S., M.G., J.W.K.*, T.P.A.*, and S.M.*. *Co-founders of Blueprint Genetics, which offers gene diagnostics for cardiomyopathies.
Acknowledgements
We thank Sini Weckström, RN, for her invaluable contribution to this work and all the patients and their family members for their participation in this study.
References
- 1.Elliott P, Andersson B, Arbustini E, Bilinska Z, Cecchi F, Charron P, Dubourg O, Kuhl U, Maisch B, McKenna WJ, Monserrat L, Pankuweit S, Rapezzi C, Seferovic P, Tavazzi L, Keren A. Classification of the cardiomyopathies: a position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J 2008;29:270–276. [DOI] [PubMed] [Google Scholar]
- 2.Towbin JA, Bowles NE. The failing heart. Nature 2002;415:227–233. [DOI] [PubMed] [Google Scholar]
- 3.Codd MB, Sugrue DD, Gersh BJ, Melton LJ., III Epidemiology of idiopathic dilated and hypertrophic cardiomyopathy. A population-based study in Olmsted County, Minnesota, 1975–1984. Circulation 1989;80:564–572. [DOI] [PubMed] [Google Scholar]
- 4.John R, Rajasinghe HA, Chen JM, Weinberg AD, Sinha P, Mancini DM, Naka Y, Oz MC, Smith CR, Rose EA, Edwards NM. Long-term outcomes after cardiac transplantation: an experience based on different eras of immunosuppressive therapy. Ann Thorac Surg 2001;72:440–449. [DOI] [PubMed] [Google Scholar]
- 5.Herman DS, Lam L, Taylor MR, Wang L, Teekakirikul P, Christodoulou D, Conner L, DePalma SR, McDonough B, Sparks E, Teodorescu DL, Cirino AL, Banner NR, Pennell DJ, Graw S, Merlo M, Di Lenarda A, Sinagra G, Bos JM, Ackerman MJ, Mitchell RN, Murry CE, Lakdawala NK, Ho CY, Barton PJ, Cook SA, Mestroni L, Seidman JG, Seidman CE. Truncations of titin causing dilated cardiomyopathy. N Engl J Med 2012;366:619–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hershberger RE, Hedges DJ, Morales A. Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat Rev Cardiol 2013;10:531–547. [DOI] [PubMed] [Google Scholar]
- 7.Ahmad F, Seidman JG, Seidman CE. The genetic basis for cardiac remodeling. Ann Rev Genomics Hum Genet 2005;6:185–216. [DOI] [PubMed] [Google Scholar]
- 8.Dellefave L, McNally EM. The genetics of dilated cardiomyopathy. Curr Opin Cardiol 2010;25:198–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kelly M, Semsarian C. Multiple mutations in genetic cardiovascular disease: a marker of disease severity? Circ Cardiovasc Genet 2009;2:182–190. [DOI] [PubMed] [Google Scholar]
- 10.Haas J, Frese KS, Peil B, Kloos W, Keller A, Nietsch R, Feng Z, Muller S, Kayvanpour E, Vogel B, Sedaghat-Hamedani F, Lim WK, Zhao X, Fradkin D, Kohler D, Fischer S, Franke J, Marquart S, Barb I, Li DT, Amr A, Ehlermann P, Mereles D, Weis T, Hassel S, Kremer A, King V, Wirsz E, Isnard R, Komajda M, Serio A, Grasso M, Syrris P, Wicks E, Plagnol V, Lopes L, Gadgaard T, Eiskjaer H, Jorgensen M, Garcia-Giustiniani D, Ortiz-Genga M, Crespo-Leiro MG, Deprez RH, Christiaans I, van Rijsingen IA, Wilde AA, Waldenstrom A, Bolognesi M, Bellazzi R, Morner S, Bermejo JL, Monserrat L, Villard E, Mogensen J, Pinto YM, Charron P, Elliott P, Arbustini E, Katus HA, Meder B. Atlas of the clinical genetics of human dilated cardiomyopathy. Eur Heart J 2014;10:1123–1135. [DOI] [PubMed] [Google Scholar]
- 11.van Rijsingen IA, Nannenberg EA, Arbustini E, Elliott PM, Mogensen J, Hermans-van Ast JF, van der Kooi AJ, van Tintelen JP, van den Berg MP, Grasso M, Serio A, Jenkins S, Rowland C, Richard P, Wilde AA, Perrot A, Pankuweit S, Zwinderman AH, Charron P, Christiaans I, Pinto YM. Gender-specific differences in major cardiac events and mortality in lamin A/C mutation carriers. Eur J Heart Fail 2013;15:376–384. [DOI] [PubMed] [Google Scholar]
- 12.Ashley EA, Hershberger RE, Caleshu C, Ellinor PT, Garcia JG, Herrington DM, Ho CY, Johnson JA, Kittner SJ, Macrae CA, Mudd-Martin G, Rader DJ, Roden DM, Scholes D, Sellke FW, Towbin JA, Van Eyk J, Worrall BB, American Heart Association Advocacy Coordinating Committee. Genetics and cardiovascular disease: a policy statement from the American Heart Association. Circulation 2012;126:142–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gollob MH, Blier L, Brugada R, Champagne J, Chauhan V, Connors S, Gardner M, Green MS, Gow R, Hamilton R, Harris L, Healey JS, Hodgkinson K, Honeywell C, Kantoch M, Kirsh J, Krahn A, Mullen M, Parkash R, Redfearn D, Rutberg J, Sanatani S, Woo A. Recommendations for the use of genetic testing in the clinical evaluation of inherited cardiac arrhythmias associated with sudden cardiac death: Canadian Cardiovascular Society/Canadian Heart Rhythm Society joint position paper. Can J Cardiol 2011;27:232–245. [DOI] [PubMed] [Google Scholar]
- 14.Wordsworth S, Leal J, Blair E, Legood R, Thomson K, Seller A, Taylor J, Watkins H. DNA testing for hypertrophic cardiomyopathy: a cost-effectiveness model. Eur Heart J 2010;31:926–935. [DOI] [PubMed] [Google Scholar]
- 15.Henry WL, Gardin JM, Ware JH. Echocardiographic measurements in normal subjects from infancy to old age. Circulation 1980;62:1054–1061. [DOI] [PubMed] [Google Scholar]
- 16.Myllykangas S, Buenrostro JD, Natsoulis G, Bell JM, Ji HP. Efficient targeted resequencing of human germline and cancer genomes by oligonucleotide-selective sequencing. Nat Biotechnol 2011;29:1024–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.OMIM. Online Mendelian Inheritance in Man, OMIM®. Baltimore, MD: McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University; http://omim.org/ . [Google Scholar]
- 18.Pruitt KD, Harrow J, Harte RA, Wallin C, Diekhans M, Maglott DR, Searle S, Farrell CM, Loveland JE, Ruef BJ, Hart E, Suner MM, Landrum MJ, Aken B, Ayling S, Baertsch R, Fernandez-Banet J, Cherry JL, Curwen V, Dicuccio M, Kellis M, Lee J, Lin MF, Schuster M, Shkeda A, Amid C, Brown G, Dukhanina O, Frankish A, Hart J, Maidak BL, Mudge J, Murphy MR, Murphy T, Rajan J, Rajput B, Riddick LD, Snow C, Steward C, Webb D, Weber JA, Wilming L, Wu W, Birney E, Haussler D, Hubbard T, Ostell J, Durbin R, Lipman D. The consensus coding sequence (CCDS) project: Identifying a common protein-coding gene set for the human and mouse genomes. Genome Res 2009;19:1316–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 2014;30:2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010;26:589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010;20:1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 2011;43:491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McLaren W, Pritchard B, Rios D, Chen Y, Flicek P, Cunningham F. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics 2010;26:2069–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu X, Jian X, Boerwinkle E. dbNSFP v2.0: a Database of Human Non-synonymous SNVs and Their Functional Predictions and Annotations. Hum Mutat 2013;34:E2393–E2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Park JY, Clark P, Londin E, Sponziello M, Kricka LJ, Fortina P. Clinical exome performance for reporting secondary genetic findings. Clin Chem 2015;61:213–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Green RC, Berg JS, Grody WW, Kalia SS, Korf BR, Martin CL, McGuire AL, Nussbaum RL, O'Daniel JM, Ormond KE, Rehm HL, Watson MS, Williams MS, Biesecker LG, American College of Medical G, Genomics. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med 2013;15:565–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gudkova A, Kostareva A, Sjoberg G, Smolina N, Turalchuk M, Kuznetsova I, Rybakova M, Edstrom L, Shlyakhto E, Sejersen T. Diagnostic challenge in desmin cardiomyopathy with transformation of clinical phenotypes. Pediatr Cardiol 2013;34:467–470. [DOI] [PubMed] [Google Scholar]
- 28.Karkkainen S, Helio T, Jaaskelainen P, Miettinen R, Tuomainen P, Ylitalo K, Kaartinen M, Reissell E, Toivonen L, Nieminen MS, Kuusisto J, Laakso M, Peuhkurinen K. Two novel mutations in the beta-myosin heavy chain gene associated with dilated cardiomyopathy. Eur J Heart Fail 2004;6:861–868. [DOI] [PubMed] [Google Scholar]
- 29.Garcia-Pavia P, Syrris P, Salas C, Evans A, Mirelis JG, Cobo-Marcos M, Vilches C, Bornstein B, Segovia J, Alonso-Pulpon L, Elliott PM. Desmosomal protein gene mutations in patients with idiopathic dilated cardiomyopathy undergoing cardiac transplantation: a clinicopathological study. Heart 2011;97:1744–1752. [DOI] [PubMed] [Google Scholar]
- 30.Elliott P, O'Mahony C, Syrris P, Evans A, Rivera Sorensen C, Sheppard MN, Carr-White G, Pantazis A, McKenna WJ. Prevalence of desmosomal protein gene mutations in patients with dilated cardiomyopathy. Circu Cardiovasc Genet 2010;3:314–322. [DOI] [PubMed] [Google Scholar]
- 31.Vahlquist A, Virtanen M, Hellstrom-Pigg M, Dragomir A, Ryberg K, Wilson NJ, Ostman--Smith I, Lu L, McGrath JA, Smith FJ. A Scandinavian case of skin fragility, alopecia and cardiomyopathy caused by DSP mutations. Clin Exp Dermatol 2014;39:30–34. [DOI] [PubMed] [Google Scholar]
- 32.Mahoney MG, Sadowski S, Brennan D, Pikander P, Saukko P, Wahl J, Aho H, Heikinheimo K, Bruckner-Tuderman L, Fertala A, Peltonen J, Uitto J, Peltonen S. Compound heterozygous desmoplakin mutations result in a phenotype with a combination of myocardial, skin, hair, and enamel abnormalities. J Invest Dermatol 2010;130:968–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kamisago M, Sharma SD, DePalma SR, Solomon S, Sharma P, McDonough B, Smoot L, Mullen MP, Woolf PK, Wigle ED, Seidman JG, Seidman CE. Mutations in sarcomere protein genes as a cause of dilated cardiomyopathy. N Engl J Med 2000;343:1688–1696. [DOI] [PubMed] [Google Scholar]
- 34.Li D, Morales A, Gonzalez-Quintana J, Norton N, Siegfried JD, Hofmeyer M, Hershberger RE. Identification of novel mutations in RBM20 in patients with dilated cardiomyopathy. Clin Transl Sci 2010;3:90–97. [DOI] [PMC free article] [PubMed] [Google Scholar]