

Abstract
Polymorphism and morphology can represent key factors tremendously limiting the bioavailability of active pharmaceutical ingredients (API), in particular, due to solubility issues. Within this work, the generation of a yet unknown surface-induced polymorph (SIP) of the model drug, 5,5-diphenylimidazolidin-2,4-dion (phenytoin), is demonstrated in thin films through altering the crystallization kinetics and the solvent type. Atomic force microscopy points toward the presence of large single-crystalline domains of the SIP, which is in contrast to samples comprising solely the bulk phase, where extended dendritic phenytoin networks are observed. Grazing incidence X-ray diffraction reveals unit cell dimensions of the SIP significantly different from those of the known bulk crystal structure of phenytoin. Moreover, the aqueous dissolution performance of the new polymorph is benchmarked against a pure bulk phase reference sample. Our results demonstrate that the SIP exhibits markedly advantageous drug release performance in terms of dissolution time. These findings suggest that thin-film growth of pharmaceutical systems in general should be explored, where poor aqueous dissolution represents a key limiting factor in pharmaceutical applications, and illustrate the experimental pathway for determining the physical properties of a pharmaceutically relevant SIP.
Short abstract
Crystallization of phenytoin in the proximity of a silica surface allows a new polymorphic form to be induced: a so-called surface-induced phase is observed. AFM and X-ray diffraction reveal differences from the commonly observed bulk phase. The enhanced dissolution rate of samples containing this surface-induced phase demonstrates a new approach that promises access to larger therapeutic variability.
Introduction
Numerous approaches exist for the development of new drug formulations aiming for advanced dissolution properties, including modern techniques like the preparation of water-soluble inclusion complexes,1 self-(micro)emulsifying drug delivery systems,2,3 solid dispersions4,5 and solutions,4 nanosuspensions,6−8 and nano extrusion. However, these approaches frequently turn out to be highly complex and hard to control on a large scale. On the contrary, there exist well-established methods for dissolution profile variation, like polymorph control9 and morphology alteration.10 In the former, the internal structure, i.e., the (crystalline) assembly, of the molecules is changed and, thus, physicochemical properties can be significantly altered. In the latter, the morphology is changed, aiming for the formation of smaller grains, which can, in general, tremendously reduce the dissolution time scale by increasing the surface area.11
In recent years, extensive theoretical work was directed toward identifying and predicting new polymorphic forms of organic molecules, in particular, for compounds employed in pharmaceutical applications.12 In general, defined changes in the experimental protocol, like the choice of solvent type, the concentration of the solution, the temperature, and pressure, can promote switching between polymorphs as different local energy minima become accessible.13 The vast majority of such experiments are performed in bulk solutions. However, the application of solid surfaces as a nucleation or crystallization mediator is a highly promising emerging alternative to assist the formulation of new drugs.14 The presence of a substrate upon nucleation generally results in an entropy reduction of the system, therewith facilitating initial nucleation and subsequent bulk crystal growth. Furthermore, evidence exists that surfaces are able to stabilize metastable phases.15 Even more importantly, organic molecules nucleating in the proximity of a surface may assemble differently to any bulk phase, hence forming so-called surface-induced polymorphs (SIP), also denoted surface-mediated polymorphs, which are not obtained otherwise.16,17
In the field of organic electronics, growth in various SIPs is frequently observed and can alter the electronic properties of the functional film, making polymorphism a decisive factor for the charge transport in a device.17 Several experimental pathways have been reported through which such polymorphs are experimentally achievable.18 The most prominent example is likely the physical vapor deposition of the prototypical organic semiconductor, pentacene, onto silicon dioxide (SiO2) surfaces.17,19 Extensive growth studies have been performed showing that pentacene molecules at the very interface pack in a so-called thin-film phase.20 Macroscopic free-standing single crystals of this very phase are not accessible, as surface interactions are required for its existence; thus, this polymorph is surface-induced. Note that, however, upon subsequent layer growth an increasing and independently growing portion of the bulk phase is observed and nucleation in the SIP is limited to less than 100 nm from the substrate surface.21 A more recent example showing a SIP is that of the soluble organic semiconductor, dihexyl-terthiophene,18 where vacuum deposition and solution processing both revealed SIP formation, thus showing that SIP growth is, indeed, induced by the presence of the substrate and can be independent from the preparation technique. Furthermore, it was demonstrated in this study on dihexyl-terthiophene through well-defined drop casting and spin coating series that the formation of the SIP is kinetically driven rather than a thermodynamic equilibrium state, i.e., the time frame at which crystallization takes place has a decisive impact on the formation of a SIP.
In pharmaceutical science, however, scarce data exist on the formation of SIPs. Although there are numerous excellent solution-based growth studies14,22 showing, indeed, that growth alteration occurs in the proximity of a surface, typically, only large crystals of already known polymorphs were hitherto observed. This is, however, likely brought about the fact that most of these studies lack the experimental tools necessary to unambiguously identify the nature of a SIP, if present. First of all, homogeneous flat samples are required on which atomic force microscopy (AFM) measurements can be performed to identify morphology alteration. Such samples then further allow for specular X-ray diffraction in order to identify the crystallographic netplanes parallel to the substrate’s surface.23 Frequently, such experiments reveal a single peak series (diffraction from one set of netplanes (hkl) and its higher order reflections), which is indicative of a unique crystalline orientation (texture) in the film. To gain knowledge regarding netplanes that are strongly tilted or even perpendicular with respect to the surface, grazing incidence X-ray diffraction (GIXD) is the method of choice, which is applicable even on layers as thin as a single monolayer.24,25 This technique allowed, for instance, three groups16,17,20 to solve independently the crystal structure of the pentacene thin-film phase,26 almost at the same time.
In the present work, we demonstrate the applicability of these experimental tools on a model system relevant in the field of pharmaceutical science. As model drug, 5,5-diphenyl-2,4-imidazolidinedione (phenytoin) is used. Phenytoin is applicable in various fields, as it has anticonvulsive, antiepileptic, and antiarrhythmic effects in the human organism and is typically applied via solid oral dosage forms, like capsules, or in parenteral formulations. For the long-term treatment of epilepsy, topical routes (e.g., transdermal patches) are also possible. Besides this, phenytoin has been recently investigated extensively, as it shows very promising film-forming properties.11,27−29 In this study, phenytoin is solution-processed onto silicon dioxide surfaces, and the films are investigated by combining AFM with specular X-ray diffraction and GIXD. By altering the preparation parameters, we induce growth in a yet unknown SIP and compare this system to samples comprising the bulk phase of phenytoin. Finally, marked differences in the dissolution profiles of SIP and bulk phase samples are identified and discussed in detail.
Materials and Methods
5,5-Diphenylimidazolidin-2,4-dion (or phenytoin) was purchased from Sigma-Aldrich (Germany) and used without any modification/purification. Spectral grade tetrahydrofuran (THF) (Fluka, Germany) and ethanol (EtOH) (Aldrich, Germany) were used as solvents. Solutions of 1 mg/mL were prepared, treated in an ultrasonic bath, and stirred until use.
Conventional glass slides (Roth, Germany) cut into 1.25 × 1.25 cm2 pieces served as substrates. The surfaces were cleaned in acetone followed by immersion into a 0.1 mol NaOH solution for 30 min, rinsed with Milli-Q water, and finally dried under a nitrogen stream prior to use. Thin films of phenytoin were prepared via drop casting. A 35 μL drop of the solution was placed on the substrate, where special care was taken for an exact leveling of the surface to guarantee a homogeneous drying process. Furthermore, a cover was placed over the sample in order to reduce the solvent evaporation rate, resulting in a drying time of about 30 min for the EtOH samples and 10 min for THF samples. While sample preparation from EtOH was performed under ambient conditions at 22 °C, the THF solution was dropped onto glass slides within an oven held at 35 °C, as this turned out to be necessary for obtaining the SIP.
Crystallographic investigations were performed via X-ray diffraction. Specular X-ray diffraction scans were taken with an Empyrean reflectometer (Panalytical, Netherlands). The device was equipped with a copper sealed tube, a Goebel mirror, primary and secondary slit systems, and a 3D-PIXcel detector. Angular scans are represented in reciprocal space as I(qz),30 where qz is the out-of-plane component of the scattering vector. In situ temperature-dependent specular X-ray diffraction scans were done using the DHS900 (Anton Paar GmbH, Austria) under an ambient atmosphere. Grazing incidence X-ray diffraction (GIXD) experiments were performed at the ID10 beamline at the ESRF (Grenoble, France) using a wavelength of 0.56 Å and a Pilatus 300 K detector; three individual images were recorded and added at slightly shifted detector positions to account for blind detector areas. The detector images were transferred into reciprocal space using the X-ray tool-library xrayutilities;31 for data evaluation and figure generation, the software PyGID was used.30
The optical morphological investigations of the sample were performed using an Olympus BX51 optical microscope. A FlexAFM atomic force microscope with an Easyscan 2 controller (all from Nanosurf, Switzerland) in noncontact mode was used to investigate the morphology on smaller length scales. For AFM measurements, TAP190 cantilevers (Budgetsensors, Bulgaria) were used. The measurements were corrected for typical artifacts like line displacement or plane inclination and are illustrated using the software package Gwyddion.32
Time-dependent drug release from the surface was investigated via a nonstandard dissolution experiment. Standard dissolution tests are typically performed following the United States Pharmacopeia (USP), which involves the usage of a proper USP apparatus, which consists of standardized vessels and controlled flow control.33 However, these setups typically require large amounts of liquid media for the performance of controlled dissolution experiments. Because the amount of phenytoin deposited onto a single glass slide was small (35 μg), a homemade setup was applied, as this allowed much lower quantities of the dissolution media to be used. This test consisted of conventional sealed glass containers fixed on a horizontal shaker (IKA yellow line RS 10 control, IKA Werke GmbH & Co. KG, Germany, 130 rpm at 25 °C). As dissolution media, 10 mL of Milli-Q water was used. Note that this amount of water is required to guarantee sink conditions, meaning that the concentration gradient in the vessel is of minor importance for the experiment. Samples of 1 mL of dissolution media were used and placed back into the dissolution vessel after the quantification procedure using standard quartz cuvettes (Hellma, Müllheim, Germany). An absorption wavelength of 210 nm was set at the nanophotometer (Implen, Munich, Germany). Values given in this article are mean values of three samples; error bars are given as standard deviation but are, for most of the data points, smaller than the data marker.
Results and Discussion
As is typical in organic thin-film preparation, the resulting morphologies are significantly altered by the choice of solvent and/or the specific processing conditions. For phenytoin thin films, a variety of morphologies can be obtained, as recently shown.11,25 In these previous studies, EtOH and acetone were used for film preparation, which, however, left the microstructure of the different morphologies, that is, the crystal polymorph, unchanged.11,27 An example of such a morphology obtained from EtOH solution is illustrated in Figure 1a. This AFM height micrograph shows the presence of extended dendrites of several hundred micrometers in length on the glass substrate (Figure 1a), with adjacent branches inclined by about 45° with respect to each other. Furthermore, the dendrites are strongly extended along their obvious growth direction but are thin in their cross-section; a breadth of ca. 1 μm is observed for most branches over the entire sample.
Figure 1.

Atomic force microscopy height images of a phenytoin sample prepared from EtOH solution (a) or from THF solution (b). A reflection mode optical microscopy image taken under crossed polarizers of the THF sample is shown in (c). The dotted lines indicate the grain boundaries of the surface-induced phase, and arrows indicate the position of trenches in between two common areas.
In contrast, homogeneous films also can be obtained by using tetrahydrofuran (THF) as solvent (Figure 1b), but they have a strongly altered morphology (compare Figure 1 panel a to panels b and c). In the AFM image (Figure 1b), the structures appear to be of dendritic type; however, the width of the branches is now strongly increased as compared to that of the previous example: a typical width of 15 μm is observed here for individual branches (Figure 1b,c). In one growth direction, a lot of vacancies between adjacent dendrites are observed, whereas in the other direction (90° rotated), the film is nearly fully covered. As all morphologies result from a common center and are mirror symmetric, this suggests that these structures consist of single domains. Interestingly, the structures on a larger scale appear to be squeezed hexagons, as indicated by dotted areas in Figure 1c. At the outer borders of these hexagons, less material is forming a trench (indicated by arrows). Next to this material-depleted area, the growth of phenytoin continues, which seems to follow the order within these clearly present hexagons. The color of this very adjacent area and the morphology seem to be very similar, which typically means that both sides of the trench have a similar extension from the surface (thickness). However, the outer edge of these structures has a more arbitrary shape, i.e., it is not hexagonal. Adjacent domains seem to interrupt the crystal formation and thus the hexagon shape on a larger scale.
To gain further insight into crystallographic differences between samples prepared from EtOH and THF, X-ray diffraction experiments were performed. From previous reports, it is already known that the preparation of phenytoin films from EtOH solutions results in phenytoin growing its known bulk structure11,27 with lattice constants of a = 0.62 nm, b = 1.36 nm, and c = 1.55 nm.34 The specular X-ray diffraction spectra of such a sample is shown in Figure 2 with peaks located at qz = 8.07, 16.14, and 24.21 nm–1. A comparison with calculated peak positions of the bulk polymorph shows that these values agree well with the 002 Bragg peak and its higher order (004 and 006) reflections. Notably, compared to an ideal (simulated) powder pattern, various peaks are missing (cf. Figure 2). By nature, a specular scan allows only reflections arising from netplanes that are parallel to the substrate surface to be assessed. Peaks missing in specular X-ray diffraction data thus point toward a preferential orientation of the crystallites; e.g., on isotropic substrates, an orientational (fiber) texture23 occurs, as is found here for our EtOH sample with a common contact plane (020) to the substrate surface.
Figure 2.

Specular X-ray diffraction scans of phenytoin samples prepared from THF (bottom) and EtOH (center) solutions compared to a calculated spectra of an ideal powder of the bulk polymorph (top).
Samples prepared via drop casting from a THF solution at a slightly elevated temperature of 35 °C reveal a distinct behavior in the specular X-ray diffraction measurements (Figure 2). While some small peaks still indicate the presence of (some) bulk phase crystallites, significantly stronger peaks occur at qz = 8.97, 17.94, and 26.91 nm–1. The positions of these new peaks are different from those found for the EtOH sample, which might suggest a different contact plane (texture). However, the peak positions are not explainable from any other theoretical position of the bulk phase on the basis of the calculated powder pattern. Thus, this sample must contain a different phenytoin polymorph (or pseudopolymorph, see below), i.e., a surface-induced phase (SIP). As its peak intensities are much larger compared to the bulk phase reflections in the sample, the ratio of this new phase is likely much higher in the thin film. Furthermore, the absence of peaks other than higher order reflections indicates, in analogy to the case with EtOH, evidence for the presence of defined texture also for the SIP.
Additional information on the crystallographic structure of the SIP is obtained via GIXD, as this technique allows information to be assessed on netplanes that are normal (or nearly normal) to the substrate surface. The top of Figure 3 shows the GIXD pattern of the THF sample, revealing two different crystallographic features. First, ring-like structures over the entire space map are observed. A comparison with the known bulk phase reveals that these rings are a result of randomly oriented phenytoin crystallites grown in the bulk polymorph. The second, clearly dominating set of features is a number of high-intensity Bragg spots along qp = 10.3, 15.4, and 20.6 nm–1. Importantly, the separation of these spots along qz is 4.5 nm–1, which is exactly half of the value of the peak observed at 9 nm–1 in the specular X-ray diffraction experiment (Figure 2). This gives full confidence that the Bragg spots in the GIXD pattern observed here indeed correspond to the SIP.
Figure 3.
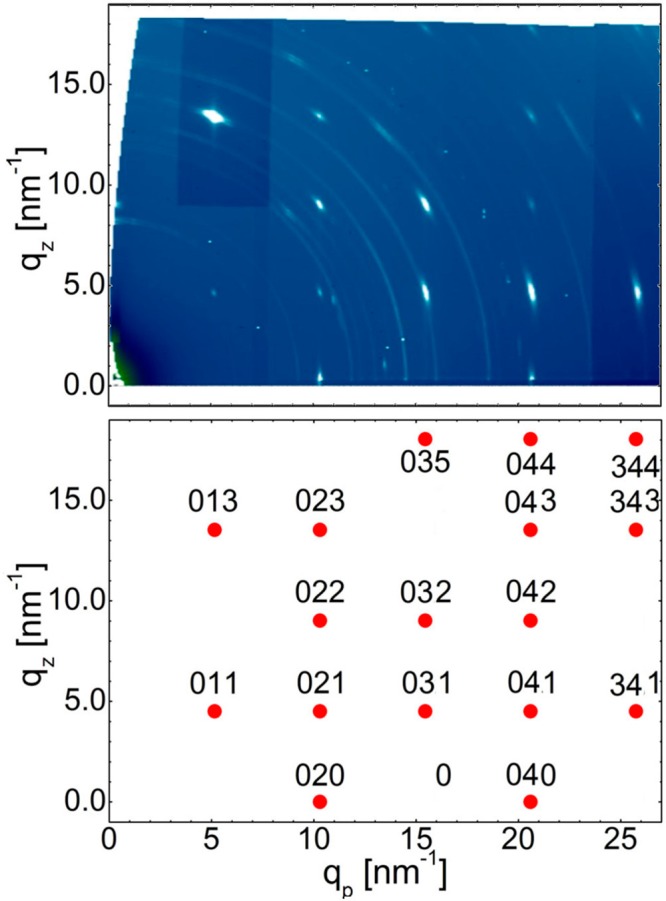
Top: Experimental grazing incidence X-ray diffraction data of the THF sample containing randomly distributed bulk-phase crystallites (rings) and sharp spots originating from the SIP. Bottom: Corresponding indexation of the spots on the basis of the SIP unit cell parameters derived within this work (see text).
For unit cell determination, each Bragg spot needs to be described by a certain set of Miller indices (hkl), which is achieved by a proper choice of the unit cell parameters. As the spots show clear equidistant separation in both the qz and qp directions, all unit cell angles are most likely 90°. An orthorhombic unit cell of a = 0.61 nm, b = 1.22 nm, and c = 1.395 nm can well-describe the observed diffraction pattern. In this indexation, the specular diffraction peaks correspond to the 002 and 004 Bragg peaks. This means that the SIP film is grown in a (001) fiber texture, i.e., this very netplane is the contact plane to the substrate.
From the different diffraction characteristics of the two polymorphs forming in the THF sample, the following conclusions can now be drawn. For the bulk phase, the diffraction pattern yields intensity distributed on rings, which indicates randomly oriented crystallites more or less unaffected by the presence of the substrate surface. This indicates that crystallites adopting the bulk polymorph structure most likely already form in the THF bulk solution and remain on the substrate after solvent removal. This, then, results in a random orientation of these crystallites. Although both phases nucleate from the same parent solution, the observed defined spots of the SIP in the GIXD pattern show that the degree of order in this phase is high; most of these crystallites must be well-aligned with respect to the surface forming a fiber texture. Therefore, it is obvious that the surface has a decisive impact on the crystal alignment in this case and, likewise, on the choice of polymorph, which finally justifies our labeling of the new phase as a SIP of phenytoin. This perception is further supported by the fact that, at this stage, there is also no evidence that the SIP can be formed in any bulk experiment despite significant experimental efforts on this question: variations of the supersaturating degree, the solvent evaporation rate, the temperature, or the solvent type did not allow the SIP to be formed in the bulk. A consideration of the differences of both situations in terms of the Hansen-solubility parameter35,36 shows that phenytoin dissolved much better in THF compared to that in EtOH.28 At the interface, the relative affinity of EtOH to interact with the silica surface is higher compared to that of THF. So, from this follows that a high solubility with a surface affinity poor solvent might be responsible for the occurrence of this surface-induced phase.
In order to investigate the (meta)stability of the SIP, temperature-dependent in situ specular X-ray diffraction scans were performed. For this experiment, a sample was chosen for which the SIP and bulk phase are simultaneously well-observed by their respective Bragg reflections (002SIP and 020bulk); the results are shown in Figure 4. For the experiment at room temperature, the peak positions are identical with those of the previous sample (cf. Figure 2). As the temperature increases, the peaks position start to shift toward smaller qz values (Figure 4a), which, in turn, means that an expansion of the unit cell’s c-axis takes place. Interestingly, besides the shift in peak position, an increase in intensity is also observed (Figure 4b). Both effects continue until a temperature of 118 °C is reached. There, the peak position is at its minimum value of 8.95 nm–1, which translates into a total expansion of the (002) netplanes by 0.004 nm in real space and that of the c-axis by 0.008 nm (1% larger than at room temperature). Comparing these results to the concomitant expansion of the bulk phase in this temperature range shows a very similar behavior (0.6% expansion); the difference is well within the error margin of the experiment. Comparing the intensity development of the two phases shows that the SIP peak intensity increases by a factor of 2.4 while the bulk-peak intensity remains essentially constant (Figure 4b). From this follows that during this moderate heating more diffraction intensity of the SIP into the 002SIP develops, which might be either a rearrangement of crystallites, crystal growth along the surface normal, or defect healing, all of which might account for such a change in intensity.
Figure 4.
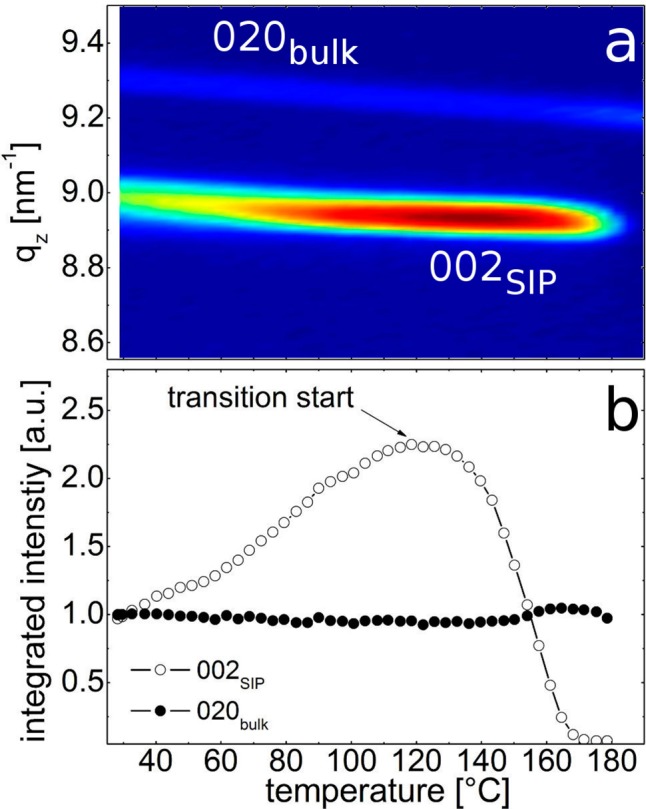
In situ temperature-dependent specular X-ray diffraction scans of a phenytoin sample established from THF solution (a) and the extracted peak intensities of the two different phases normalized to the initial value (b). A common abscissa is used for comparability.
Finally, at 118 °C the intensity of the SIP starts to decrease rapidly, with its Bragg peak positions now remaining unaffected by further increases of the temperature. This typically means that a solid–solid phase transition has occurred (note that the melting point of phenytoin is 300 °C). As the intensity of the bulk peak slightly increases at this stage (black curve in Figure 4b), it is likely that a transition into the bulk phase now takes place. However, as the intensity increase is low, we assume that this transition occurs either into randomly ordered bulk phase or into another texture, which is not accessible by specular X-ray diffraction. As we were not able to regenerate the SIP on cooling, it is assumed that the SIP is of monotropic nature, i.e., it cannot be reversibly transited back and forth.
In the following, the relevance of this new polymorph of phenytoin on future pharmaceutical applications is demonstrated by its superior dissolution behavior. Drug dissolution testing, in general, provides information on in vitro drug release, which is crucial for drug development. Such data are relatable to in vivo pharmacodynamics data in order to develop an appropriate drug formulation design.33 For the sake of comparability, identical amounts of phenytoin from both EtOH and THF solutions were deposited on equally sized substrates; after deposition, each substrate surface contained 35 μg of phenytoin that was dissolved in the same amount of dissolution media (10 mL of Milli-Q water). Note that the usage of water rather than any buffered salt solution minimizes solution/surface interactions; for the latter, it can be expected that ions strongly enhance dissolution due to their surface activity. Furthermore, it should be noted that results within this study are fully comparable, but, due to the nonstandard methods, caution in comparing these data with literature data needs to be advised. Nevertheless, the resulting release of phenytoin over time for both samples is depicted in Figure 5 on linear (a) and on double logarithmic (b) scales. The EtOH sample reveals a strong increase in concentration with time; zero is set at the first measurement point, 1 min after the start of the experiment. A mass increase (slope) of 1.1 μg/min is observed up to 9 min. After ca. 10 min, the dissolution rate decreases, and a second, roughly linear regime is visible, with the slope being significantly smaller there. The double logarithmic representation reveals that, after about 30 min, the amount released per time interval decreases further, until reaching a maximum of 35 μg after 420 min. This last value is the maximum amount of the sample released.
Figure 5.
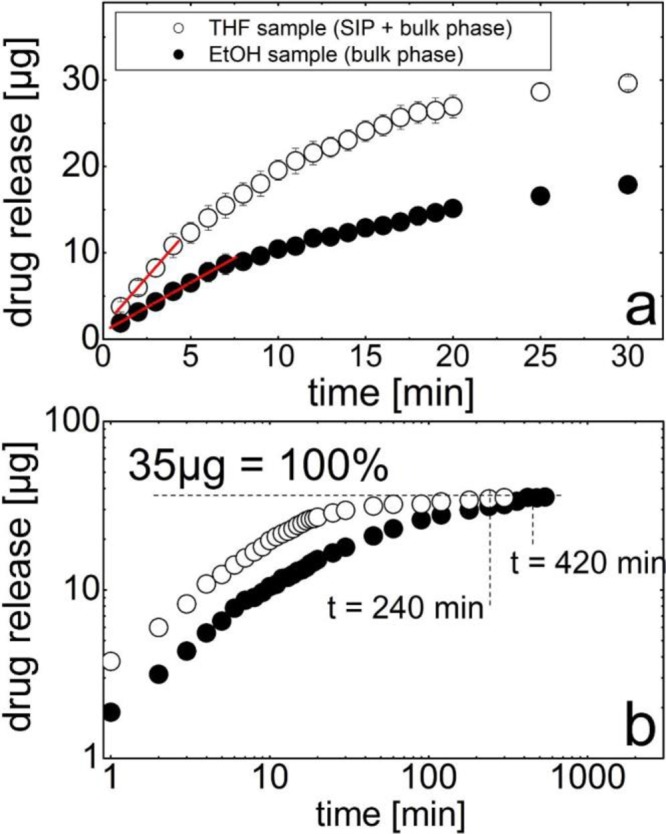
Phenytoin release as a function of time for two samples represented on a linear scale for a short time period (a) and the entire experimental data on a double logarithmic scale (b).
Inspecting the dissolution behavior of the THF sample comprising dominantly the SIP (see above) shows distinct behavior as compared to that of the EtOH sample. Initially, fast release is apparent, with the slope now being steeper at 2.3 μg/min. This represents a more than 2 times faster release after initial contact with the dissolution media compared to that of the EtOH sample. At around 5 min, the rate at which phenytoin is released into the media reduces, and the dissolution rate decreases continuously until all of the phenytoin is released. This is different from EtOH, which shows linear regimes instead, indicating constant dissolution rates. The maximum amount of 35 μg is finally reached after 240 min (Figure 5b), which is drastically shorter. These results demonstrate that the new polymorph is significantly more effective in being released from the sample surface.
Differences in dissolution profiles typically result from various parameters. While diffusion coefficients and concentration gradients might be almost identical for both samples, a larger surface area means, according to the Whitney–Noyes equation,33 a higher dissolution rate. The morphologies present in Figure 1 show that more and smaller crystallites are present in the EtOH sample. This means that the surface area is larger compared to that of the THF sample, where larger crystallites are present. However, interestingly, the dissolution experiments show even faster dissolution for the THF sample, suggesting that the surface area difference is not dominant here. Another parameter that affects dissolution behavior is the solvent boundary layer, i.e., the capability of the solvent to assemble in the proximity of a crystal, which thus can affect solvent–crystal interactions. A higher dissolution rate might suggest that the SIP reduces the thickness of this boundary layer, providing the ability to transition faster from the solid into the dissolved state.
Besides an explanation based on the Whitney–Noyes equation, changes in the polymorphic form can also account for dissolution differences. The exact assembly of the phenytoin molecules in the SIP unit cell is, however, not accessible from the present data in terms of a full structure solution. In any case, a slightly different molecular arrangement can tremendously change the relative importance of H-bonding and van der Waals interaction within organic solids; thus, the different crystal structures of the two polymorphs might readily account for the alteration in dissolution found here.
Overall, our present set of experiments clearly evidences the different dissolution behavior of two types of samples. While some explanations for this behavior can be suggested, it is likely that a combination of various aspects is responsible for the different behavior, requiring additional work to resolve this issues in more detail.
Conclusions
Drop casting phenytoin on solid surfaces from a THF solution instead of a EtOH solution induces growth in a new polymorph (SIP) with significantly altered crystallographic and morphological properties, which translates into changes that are beneficial for dissolution behavior. As this specific polymorph was hitherto impossible to achieve via different experimental approaches in bulk solutions, we regard this form as being mediated by the presence of the substrate, whereby the solvent has, in addition, a decisive impact on its formation. Overall, the significant increase in dissolution rate and, therefore, improved drug release from the surface open up a new and promising pathway for the development of novel drug formulations. While the preparation of thin films on model substrates allows for the fundamental identification and characterization of such a new polymorph, future work will now be directed toward generating this structure also on nanoparticles and materials suitable for patches, aiming for its use in pharmaceutical applications.
Acknowledgments
The work was funded by the Austrian Science Fund (FWF) [P25541-N19]. The authors thank NAWI-Graz for support. The GIXD experiments were performed at beamline ID10 at the European Synchrotron Radiation Facility (ESRF), Grenoble, France. We acknowledge the ESRF for the provision of synchrotron radiation facilities, and we thank Oleg Konovalov and Giovanni Li Destri for assistance with using beamline ID10.
The authors declare no competing financial interest.
References
- Sansone F.; Barboso S.; Casnati A.; Sciotto D.; Ungaro R. Tetrahedron Lett. 1999, 40, 4741. 10.1016/S0040-4039(99)00838-2. [DOI] [Google Scholar]
- Neslihan Gursoy R.; Benita S. Biomed. Pharmacother. 2004, 58, 173. 10.1016/j.biopha.2004.02.001. [DOI] [PubMed] [Google Scholar]
- Pouton C. W. Eur. J. Pharm. Sci. 2000, 11, S93. 10.1016/S0928-0987(00)00167-6. [DOI] [PubMed] [Google Scholar]
- Leuner C.; Dressman J. Eur. J. Pharm. Biopharm. 2000, 50, 47. 10.1016/S0939-6411(00)00076-X. [DOI] [PubMed] [Google Scholar]
- Schrank S.; Kann B.; Saurugger E.; Ehmann H.; Werzer O.; Windbergs M.; Glasser B. J.; Zimmer A.; Khinast J.; Roblegg E. Mol. Pharmaceutics 2014, 11, 599. 10.1021/mp4005782. [DOI] [PubMed] [Google Scholar]
- Pouton C. W. Eur. J. Pharm. Sci. 2006, 29, 278. 10.1016/j.ejps.2006.04.016. [DOI] [PubMed] [Google Scholar]
- Rabinow B. E. Nat. Rev. Drug Discovery 2004, 3, 785. 10.1038/nrd1494. [DOI] [PubMed] [Google Scholar]
- Khinast J.; Baumgartner R.; Roblegg E. AAPS PharmSciTech 2013, 14, 601. 10.1208/s12249-013-9946-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pudipeddi M.; Serajuddin A. T. J. Pharm. Sci. 2005, 94, 929. 10.1002/jps.20302. [DOI] [PubMed] [Google Scholar]
- Nokhodchi A.; Bolourtchian N.; Dinarvand R. Int. J. Pharm. 2003, 250, 85. 10.1016/S0378-5173(02)00488-X. [DOI] [PubMed] [Google Scholar]
- Werzer O.; Baumgartner R.; Zawodzki M.; Roblegg E. Mol. Pharmaceutics 2014, 11, 610. 10.1021/mp4006479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price S. L. Adv. Drug Delivery Rev. 2004, 56, 301. 10.1016/j.addr.2003.10.006. [DOI] [PubMed] [Google Scholar]
- Moulton B.; Zaworotko M. J. Chem. Rev. (Washington, DC, U. S.) 2001, 101, 1629. 10.1021/cr9900432. [DOI] [PubMed] [Google Scholar]
- Diao Y.; Myerson A. S.; Hatton T. A.; Trout B. L. Langmuir 2011, 27, 5324. 10.1021/la104351k. [DOI] [PubMed] [Google Scholar]
- Ehmann H. M.; Werzer O. Cryst. Growth Des. 2014, 14, 3680. 10.1021/cg500573e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H.; Inaba K.; Sato N. Appl. Phys. Lett. 2007, 90, 181930. 10.1063/1.2736193. [DOI] [Google Scholar]
- Nabok D.; Puschnig P.; Ambrosch-Draxl C.; Werzer O.; Resel R.; Smilgies D. M. Phys. Rev. B: Condens. Matter Mater. Phys. 2007, 76, 235322. 10.1103/PhysRevB.76.235322. [DOI] [Google Scholar]
- Wedl B.; Resel R.; Leising G.; Kunert B.; Salzmann I.; Oehzelt M.; Koch N.; Vollmer A.; Duhm S.; Werzer O.; Gbabode G.; Sferrazza M.; Geerts Y. RSC Adv. 2012, 2, 4404. 10.1039/c2ra20272g. [DOI] [Google Scholar]
- Salzmann I.; Moser A.; Oehzelt M.; Breuer T.; Feng X.; Juang Z.-Y.; Nabok D.; Della Valle R. G.; Duhm S.; Heimel G.; Brillante A.; Venuti E.; Bilotti I.; Christodoulou C.; Frisch J.; Puschnig P.; Draxl C.; Witte G.; Müllen K.; Koch N. ACS Nano 2012, 6, 10874. 10.1021/nn3042607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiefer S.; Huth M.; Dobrinevski A.; Nickel B. J. Am. Chem. Soc. 2007, 129, 10316. 10.1021/ja0730516. [DOI] [PubMed] [Google Scholar]
- Mayer A. C.; Kazimirov A.; Malliaras G. G. Phys. Rev. Lett. 2006, 97, 105503. 10.1103/PhysRevLett.97.105503. [DOI] [PubMed] [Google Scholar]
- Aizenberg J.; Black A. J.; Whitesides G. M. Nature 1999, 398, 495–498 10.1038/19047. [DOI] [Google Scholar]
- Birkholz M.Thin Film Analysis by X-ray Scattering; Wiley-VCH: Weinheim, 2006. [Google Scholar]
- Smits E. C. P.; Mathijssen S. G. J.; van Hal P. A.; Setayesh S.; Geuns T. C. T.; Mutsaers K. A. H. A.; Cantatore E.; Wondergem H. J.; Werzer O.; Resel R.; Kemerink M.; Kirchmeyer S.; Muzafarov A. M.; Ponomarenko S. A.; de Boer B.; Blom P. W. M.; de Leeuw D. M. Nature 2008, 455, 956. 10.1038/nature07320. [DOI] [Google Scholar]
- Musumeci C.; Salzmann I.; Bonacchi S.; Röthel C.; Duhm S.; Koch N.; Samorì P. Adv. Funct. Mater. 2015, 25, 2501. 10.1002/adfm.201403773. [DOI] [Google Scholar]
- Fritz S. E.; Martin S. M.; Frisbie C. D.; Ward M. D.; Toney M. F. J. Am. Chem. Soc. 2004, 126, 4084. 10.1021/ja049726b. [DOI] [PubMed] [Google Scholar]
- Ehmann H. M.; Baumgartner R.; Reischl D.; Roblegg E.; Zimmer A.; Resel R.; Werzer O. Cryst. Growth Des. 2015, 15, 326. 10.1021/cg501391j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehmann H. M. A.; Baumgartner R.; Kunert B.; Zimmer A.; Roblegg E.; Werzer O. J. Phys. Chem. C 2014, 118, 12855. 10.1021/jp502330e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehmann H. M. A.; Kellner T.; Werzer O. CrystEngComm 2014, 16, 4950. 10.1039/c4ce00424h. [DOI] [Google Scholar]
- Moser A.; Werzer O.; Flesch H. G.; Koini M.; Smilgies D. M.; Nabok D.; Puschnig P.; Ambrosch-Draxl C.; Schiek M.; Rubahn H. G.; Resel R. Eur. Phys. J.: Spec. Top. 2009, 167, 59. 10.1140/epjst/e2009-00937-4. [DOI] [Google Scholar]
- Kriegner D.; Wintersberger E.; Stangl J. J. Appl. Crystallogr. 2013, 46, 1162. 10.1107/S0021889813017214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nečas D.; Klapetek P. Open Phys. 2012, 10, 181–188 10.2478/s11534-011-0096-2. [DOI] [Google Scholar]
- Pharmaceutical Preformulation and Formulation: A Practical Guide from Candidate Drug Selection to Commercial Dosage Form; Gibson M., Ed.; Informa Healthcare: New York, 2009. [Google Scholar]
- Camerman A.; Camerman N. Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem. 1971, 27, 2205. 10.1107/S0567740871005570. [DOI] [Google Scholar]
- Hansen C.Hansen Solubility Parameters: A User’s Handbook, 2nd ed.; CRC Press: Boca Raton, FL, 2007. [Google Scholar]
- Ehmann H. M.; Zimmer A.; Roblegg E.; Werzer O. Cryst. Growth Des. 2014, 14, 1386–1391 10.1021/cg401859p. [DOI] [PMC free article] [PubMed] [Google Scholar]