

Abstract
Granulomas, organized aggregates of immune cells, are a defining feature of tuberculosis (TB). Granuloma formation is implicated in the pathogenesis of a variety of inflammatory disorders. However, the tuberculous granuloma has been assigned the role of a host protective structure which “walls-off” mycobacteria. Work conducted over the past decade has provided a more nuanced view of its role in pathogenesis. On the one hand, pathogenic mycobacteria accelerate and exploit granuloma formation for their expansion and dissemination by manipulating host immune responses to turn leukocyte recruitment and cell death pathways in their favor. On the other hand, granuloma macrophages can preserve granuloma integrity by exerting a microbicidal immune response, thus preventing an even more rampant expansion of infection in the extracellular milieu. Even this host-beneficial immune response required to maintain the bacteria intracellular must be tempered, as an overly vigorous immune response can also cause granuloma breakdown, thereby directly supporting bacterial growth extracellularly. This review will discuss how mycobacteria manipulate inflammatory responses to drive granuloma formation and will consider the roles of the granuloma in pathogenesis and protective immunity, drawing from clinical studies of TB in humans and from animal models—rodents, zebrafish, and nonhuman primates. A deeper understanding of TB pathogenesis and immunity in the granuloma could suggest therapeutic approaches to abrogate the host-detrimental aspects of granuloma formation to convert it into the host-beneficial structure that it has been thought to be for nearly a century.
Tuberculosis-causing bacteria have evolved a variety of virulence mechanisms to evade and co-opt immune responses in the host. Most of these processes occur in the context of the granuloma—an organized aggregate of immune cells.
Pathogenic mycobacteria are exquisitely adapted to surviving in their hosts, and they have evolved a variety of virulence mechanisms to evade and co-opt immune responses, which allow them to infect the host, replicate within it, and transmit to new hosts. A major hurdle that mycobacteria must face is that for much of their life within the host, they reside within macrophages and dendritic cells (DCs), phagocytes that play pivotal roles in immunity to tuberculosis (TB) (Philips and Ernst 2012). For instance, to fulfill its life cycle, Mycobacterium tuberculosis, which causes human TB, must gain access to growth-permissive phagocytes, avoid microbicidal phagocytes, dampen productive innate and adaptive immune responses, and finally, exit the intracellular environment so as to gain access to the lung airspace where it can become aerosolized and infect new hosts. How do mycobacteria achieve these remarkable feats? Most of these processes do not occur in isolated macrophages, but rather in the context of the granuloma, a cellular aggregate primarily consisting of several types of infected and uninfected phagocytes (macrophages, monocytes, DCs, and neutrophils) and T lymphocytes (Ramakrishnan 2012). A better understanding of TB pathogenesis then requires tackling its epicenter, the granuloma. This review will provide an overview of our current understanding of the granuloma, its formation, maturation, maintenance, and role in disease outcome, highlighting recent findings and new concepts emerging from studies in animal models (Table 1) and clinical studies of human TB.
Table 1.
Animal models of tuberculous granuloma formation
| Advantages | Disadvantages | Reference(s) | |
|---|---|---|---|
| Animal models of M. tuberculosis Rabbit | Forms lesions similar to humans, including necrotic mature granulomas | Few immunological reagents Few genetic tools |
Flynn 2006; Helke et al. 2006 |
| Guinea pig | Forms nonnecrotic mature granulomas | Few immunological reagents Few genetic tools Rarely forms necrotic granulomas |
Flynn 2006; Helke et al. 2006 |
| Mouse | Many immunological reagents Numerous genetic tools Well-characterized immune cell subsets and immunodominant M. tuberculosis epitopes Isogenic strains enable cell transfer studies Amenable to intravital microscopy |
Most commonly used mouse strains form loose, nonnecrotic granulomas Persistent infection differs from human disease |
Flynn 2006; Helke et al. 2006; Egen et al. 2008; Ernst 2012 |
| Nonhuman primate (cynomolgus and rhesus macaques) | Similarity to humans Heterogeneous disease outcomes from active to latent disease Heterogeneous lesions, including necrotic mature granulomas Amenable to noninvasive imaging techniques |
Few genetic tools Expensive |
Flynn 2006; Helke et al. 2006; Lin et al. 2014 |
| Animal models of M. marinum | |||
| Zebrafish | Natural host of M. marinum Forms lesions similar to humans, including necrotic mature granulomas Genetically tractable Larvae are transparent, and thus well-suited for intravital microscopy Large clutch sizes enable high-throughput studies (e.g., genetic and pharmacological screens) |
No lungs Poorly defined immune cell subsets Few immunological reagents |
Swaim et al. 2006; Takaki et al. 2012; Ramakrishnan 2014 |
| Leopard frog | Natural host of M. marinum Asymptomatic infection resembles latency in humans Forms nonnecrotic mature granulomas |
Poorly defined immune cell subsets Few immunological reagents Few genetic tools |
Ramakrishnan et al. 1997 |
LEADING UP TO THE GRANULOMA
Poor Microbicidal Macrophages Transport M. tuberculosis to Granuloma Initiation Sites in Deeper Tissues
Human epidemiological studies and animal studies have suggested that TB begins with the inhalation of as few as one to three M. tuberculosis bacilli and their deposition in the lower respiratory tract, where alveolar macrophages are thought to engulf the bacteria and transport them from the airspace across the lung epithelium into the parenchyma (Fig. 1) (Wells et al. 1948; Bates et al. 1965; Dannenberg 1993; Wolf et al. 2007). Experiments in zebrafish larvae modeling these early interactions show that macrophages recruited to the site of infection engulf the mycobacteria and transport them into deeper tissues (Clay et al. 2007). The observation that macrophage depletion prevents tissue dissemination underscores the role of macrophages in transporting the bacteria from their site of pathogen entry into distal tissues, a step that is required for development and transmission of TB.
Figure 1.
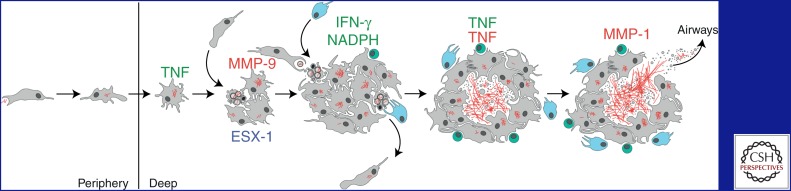
Overview of tuberculosis infection development. Host factors that are protective (green) or detrimental (red) are listed above the drawings. Note that the tumor necrosis factor (TNF) can have both beneficial and pathogenic roles. Macrophages (gray) engulf extracellular M. tuberculosis bacilli (red) in the alveolar space and transport them into deeper sites in the lung. Mycobacteria replicate within minimally microbicidal macrophages that antagonize bacterial growth via TNF-dependent mechanisms. Mycobacteria induce infected macrophage apoptosis and expression of host MMP9 in an ESX-1-dependent manner. Newly recruited macrophages engulf infected cell debris, contributing to granuloma expansion. Some of these newly infected macrophages can exit the primary granuloma and establish secondary granulomas in distal tissues. Neutrophils (light blue) can also scavenge dying infected cells and kill bacteria through an NADPH-dependent mechanism. M. tuberculosis-specific T cells (green) arrive at the granuloma and produce interferon-γ (IFN-γ) to enhance the microbicidal activity of macrophages. TNF excess and strong T-cell immunity can lead to macrophage necrosis and release of mycobacteria into the extracellular space, where they can grow relatively unchecked. Subsequent induction of MMP1 causes granuloma cavitation and release of mycobacteria into the airways.
Our laboratory recently identified a mechanism in mice and zebrafish by which M. tuberculosis and the closely related pathogen Mycobacterium marinum evade immune recognition via the Toll-like receptor (TLR)-MyD88 pathway to recruit and infect permissive macrophages that transport them across the epithelium (Cambier et al. 2014). These mycobacteria use the cell-wall-associated lipid phthiocerol dimycoceroserate (PDIM) to mask underlying PAMPs (pathogen-associated molecular patterns), thereby avoiding the recruitment of microbicidal macrophages through TLR-MyD88-dependent pathways (Cambier et al. 2014). The surface-associated phenolic glycolipid (PGL), a molecule that is structurally related to PDIMs (Onwueme et al. 2005), induces expression of the macrophage-monocyte chemokine CCL2 to recruit and infect mycobacterium-permissive CCR2-expressing macrophages. These newly infected macrophages then migrate into deeper tissues where they can incite granuloma formation.
Mycobacteria Induce Granuloma Formation in the Context of Innate Immunity
Real-time visualization of the events following M. marinum infection of zebrafish larvae shows that within 2–3 d of the infected macrophage reaching deeper tissues, granulomas can form around it, through a second wave of macrophage migration (Davis et al. 2002). These structures are not simple macrophage aggregates, but bona fide granulomas as evidenced by two features—first, the participating macrophages undergo the cardinal “epithelioid” transformation described for mature tuberculous granulomas in humans and mammalian animal models (Spector 1969; Adams 1976; Williams and Williams 1983; Bouley et al. 2001; Ramakrishnan 2012), and second, these structures induce the expression of mycobacterial granuloma-activated genes that are expressed in the mature granulomas of adult frogs but not in axenic cultures or in cultured macrophages (Ramakrishnan et al. 2000; Davis et al. 2002). Indeed, within the same larvae, these genes are not activated when the bacteria infect individual macrophages, but only when they participate in granuloma formation (Davis et al. 2002).
The finding that the tuberculous granuloma is an innate immune-cell-derived structure was made possible mainly by the unprecedented temporal and spatial resolution afforded by real-time studies in the transparent zebrafish larva. It was the beginning of the new understanding that the granuloma, at least in its incipient stages, is a structure built by mycobacteria for their expansion and dissemination, thus challenging the long-held view that granulomas are strictly host-protective structures.
INSIDE THE GRANULOMA
Mycobacteria Use the Granuloma as a Vehicle for Expansion through Intercellular Spread Enhanced by the RD1/ESX-1 Virulence Locus
If mycobacterial interactions with innate immunity induce granulomas, is there a specific mycobacterial determinant involved? The answer to this came from a serendipitous observation on the pattern of infection with an ESX-1-deficient M. marinum strain (Volkman et al. 2004) and was recently confirmed in vitro in a human lung tissue model of M. tuberculosis granuloma formation (Parasa et al. 2014). ESX-1 is a type VII secretion system that is expressed by pathogenic mycobacteria, including M. tuberculosis, Mycobacterium bovis, and M. marinum, and is strongly implicated in M. tuberculosis virulence (Simeone et al. 2009). In fact, a molecular explanation for the reduced virulence of the Bacillus Calmette–Guérin (BCG) live attenuated vaccine strain of M. bovis came from genomic and genetic studies that linked BCG’s attenuation to the loss of the region of difference 1 (RD1) locus that contains this secretion system (Behr et al. 1999; Pym et al. 2002; Hsu et al. 2003; Guinn et al. 2004). After ascertaining that the M. marinum ΔRD1 mutant was attenuated in adult leopard frogs and zebrafish, which are two of its natural hosts, an exploratory study comparing wild-type and ΔRD1 M. marinum infection in zebrafish larvae was conducted to try to identify the cellular basis of ΔRD1’s attenuation in vivo (Volkman et al. 2004; Swaim et al. 2006). If granulomas were strictly host-protective, one would expect attenuated mycobacteria to show accelerated or at least unimpaired granuloma formation. Instead, ΔRD1 M. marinum infection resulted in more delayed and smaller granuloma formation than wild-type mycobacteria (Volkman et al. 2004). Importantly, the defective granuloma formation was in the face of grossly normal growth of the ΔRD1 M. marinum strain in the individual macrophages; despite becoming heavily infected with time, they simply did not aggregate (Volkman et al. 2004). The normal intracellular growth of ΔRD1 M. marinum was in contrast to a different M. marinum mutant in another secreted virulence protein, Erp, famous for being the first mycobacterial virulence determinant to be identified by molecular genetics (Berthet et al. 1998). Δerp M. marinum displayed an intracellular growth defect in individual zebrafish larval macrophages, thus was compromised at an earlier step than ΔRD1 (Fig. 1) (Cosma et al. 2006). Despite having no intracellular growth defect, ΔRD1 was as attenuated as Δerp for overall growth in the animals.
The finding that ΔRD1 infection had a primary defect at the step of granuloma formation and resulted in lower bacterial numbers suggested that the granuloma expands rather than restricts bacterial numbers (Volkman et al. 2004). Macrophage aggregation might impact bacterial numbers by facilitating the spread of bacteria to uninfected macrophages that are recruited to the aggregates. If so, then aggregate formation in wild-type infection should correlate with a dramatic increase in the number of infected macrophages and bacterial burdens, a hypothesis made eminently testable by the feasibility of direct and serial observation of granuloma formation combined with infected macrophage enumeration in the larvae. Indeed, the number of infected macrophages did not change significantly until aggregates formed and only on aggregation did the number of infected macrophages increase dramatically. Similarly, the number of viable bacteria also did not increase until after aggregation occurred 3–5 d postinfection. Collectively, these data suggested that granuloma formation driven by the ESX-1 system promotes intercellular bacterial spread, thereby providing the bacteria with a means to expand the infection niche.
In fact, the idea that the granuloma might benefit mycobacteria first came from earlier studies in chronically infected adult frogs and zebrafish with mature granulomas (Cosma et al. 2004). When these animals were superinfected with an isogenic virulent strain that was differentially marked, the new strain was found to preferentially migrate rapidly within host macrophages into the preexisting granulomas, including their necrotic centers. This trafficking phenomenon was pathogen-specific, as superinfecting Salmonella was largely excluded from granulomas. The superinfecting strain grew robustly within the preformed granulomas, suggesting that the granuloma is a microenvironment of impaired immunity that supports mycobacterial growth. A follow-up study in mice showed that superinfecting M. tuberculosis also migrates to mature lung granulomas (Cosma et al. 2008). These experiments also indicate that even mature granulomas are open structures that allow cellular influx, challenging the notion that they serve as physical barriers which “wall-off” mycobacteria.
Cellular Basis of the Granuloma’s Role in Bacterial Expansion
The granuloma drives bacterial expansion by coordinately inducing infected macrophage apoptosis and uninfected macrophage recruitment to nascent granulomas (Fig. 1) (Davis and Ramakrishnan 2009). Time-lapse microscopy showed that newly recruited macrophages migrate within the growing granuloma and engulf the apoptotic debris of infected macrophages together with their bacterial contents. Quantitative analyses of infected cell death and phagocytosis by new recruits revealed that during wild-type M. marinum infection, ∼80% of the infected granuloma macrophages die within 24 h and that on average, two or three new recruits become infected for every single dying macrophage. In contrast, ΔRD1 M. marinum induces the death of ∼50% fewer infected macrophages as wild-type mycobacteria and recruits ∼25% the number of uninfected macrophages, and only ∼50% of these new recruits become infected. The rate of bacterial growth estimated by measuring fluorescence area was proportional to the increase in number of infected macrophages, suggesting that the process of infected cell death and rephagocytosis could explain most, if not all, of granuloma expansion. Because the kinetics of cell death and recruitment are slower during ΔRD1 M. marinum infection, these observations might also largely explain the cellular basis of attenuation of ΔRD1 mycobacteria in vivo. Rather than inhibiting macrophage responses that are generally protective against nonpathogenic mycobacteria (exemplified by case of the attenuated BCG/RD1 mutant) (Adams 1976; Dannenberg 2003), virulent mycobacteria have evolved mechanisms (i.e., their ESX-1 secretion system) for accelerating these responses, ultimately molding them into tools for infection development. Studies in adult mice and zebrafish suggest that the ESX-1 locus continues to play a role in mature granuloma formation/maintenance in the context of adaptive immunity, as the attenuated infection with RD1-mutant bacteria is associated with loose, poorly developed granulomas (Sherman et al. 2004; Volkman et al. 2004; Swaim et al. 2006).
Mechanistic Basis of Bacterial ESX-1 Induced Granuloma Formation: Co-Option of Host MMP9
An initial clue to the mechanism of RD1-induced granuloma formation came from the finding that the RD1 mutant’s aggregation defect could be rescued by superinfecting the animals with wild-type bacteria marked with a different fluorescent protein; the superinfecting strain infected individual macrophages that were then able to cause the aggregation of the ΔRD1-infected macrophages that had remained isolated before (Volkman et al. 2004). Moreover, at least one wild-type-infected macrophage was required to serve as a nidus for each aggregate, suggesting that the presence of a macrophage infected with RD1-competent bacteria creates a chemotactic gradient that recruits macrophages (Volkman et al. 2004).
A better understanding of this chemotactic pathway came from a differential screen for host genes that are induced during wild-type but not ΔRD1 M. marinum infection in zebrafish larvae (Volkman et al. 2010). Transcriptional induction of matrix metalloproteinase-9 (MMP9) was greater with wild-type than the ΔRD1 bacteria, and this difference was further amplified when MMP9-dependent gelatinase activity was assayed in vivo; wild-type infection induced robust MMP9-dependent gelatinase activity whereas ΔRD1 infection had barely any activity. Genetic knockdown of mmp9 in wild-type M. marinum-infected fish phenocopied the granuloma defect observed during ΔRD1 M. marinum infection and concomitantly reduced bacterial burdens and improved host survival (Volkman et al. 2010). The cellular pattern of ESX-1-dependent MMP9 induction was intriguing: It was more prominent in the epithelial cells surrounding the granuloma than in the granuloma macrophages themselves. Because ESX-1-dependent MMP9 induction occurred even in macrophage-depleted larvae, it appeared to be a consequence of the direct interaction between the bacterial secretion system and the epithelial cells rather than via signals from the infected macrophages. Furthermore, the ESX-1-dependent MMP9 induction could be attributed to ESAT-6, a small secreted protein substrate of the ESX-1 secretion system that has long been identified as one of the earliest proteins to be recognized by the T-cell response in humans (giving it its name early secretory antigenic target of T cells of 6 kDa size that abbreviates to ESAT-6) (Andersen et al. 1995; Ravn et al. 1999).
Compositely, the zebrafish findings suggested a model for granuloma formation in which ESAT-6 induces MMP9 in epithelial cells neighboring the nascent granuloma. The details of this mechanism and the relative importance of macrophage versus epithelial cell MMP9 in pathogenesis are yet to be worked out. ESX-1-dependent induction of MMP9 in the epithelium might benefit mycobacteria by providing a means of signal amplification. For example, a single infected macrophage might be sufficient to induce MMP9 in multiple epithelial cells and thus jumpstart the recruitment of uninfected macrophages. The mechanisms by which ESX-1 induces MMP9 and how epithelium-derived MMP9 promotes macrophage recruitment to nascent granulomas remain unknown and are under active study. As to how MMP9 induces macrophage chemotaxis, a potential mechanism of action might involve enhancing the activity of CCL7 or other chemokines via proteolysis of the amino terminus (Parks et al. 2004). CCL7 is an appealing candidate because it is a substrate of MMP9 in vivo (Greenlee et al. 2006), is produced by macrophages in response to mycobacterial infection (Scott and Flynn 2002), and can recruit monocytes from the bloodstream (Soehnlein and Lindbom 2010). Other nonmutually exclusive mechanisms could involve remodeling of the extracellular matrix at the site of infection through the release of a sequestered chemoattractant (e.g., glycan-bound chemokines) (Sarris et al. 2012) or degradation of a physical barrier (e.g., α-dystroglycan proteolysis along basement membranes) (Parks et al. 2004; Agrawal et al. 2006).
Again, the zebrafish findings all have parallels in humans. MMP9 levels are elevated in human TB (Price et al. 2003; Sheen et al. 2009), and in lung TB, it is expressed prominently in the epithelium surrounding the granulomas in addition to the macrophages themselves (Elkington et al. 2007). Among pleural TB patients, pleural fluid MMP9 concentrations are higher in those patients with granulomatous pleural disease than in those with nongranulomatous disease (Sheen et al. 2009). Among tuberculous meningitis patients, increased MMP9 levels in the cerebrospinal fluid are associated with worse outcomes (Price et al. 2003). In a mouse model of M. tuberculosis aerosol infection, MMP9 knockout mice recruited fewer macrophages to the lung and formed smaller granulomas, features that were associated with reduced lung bacterial loads as early as 2 wk postinfection and continuing through the 120-d assay period (Taylor et al. 2006). These data implicate MMP9 and granuloma formation in pathogenesis similar to the zebrafish and human data. It is perplexing then that the authors instead designated MMP9 as a host resistance factor that acts by promoting granuloma formation; this misinterpretation of the data at hand so as to align the conclusions with the protective granuloma model may attest to how entrenched the model is among TB researchers. Given the broad conservation of the MMP9-induced susceptibility, identifying the mycobacterial and host factors involved in macrophage recruitment to nascent granulomas would be a key step in designing targeted therapies to reduce the kinetics of granuloma formation just to the point where it is no longer host-detrimental.
Macrophage Apoptosis in the Granuloma: Protective or Pathogenic?
Studies of granuloma development in the zebrafish suggest that infected macrophage apoptosis plays a role in expanding infection (Davis and Ramakrishnan 2009). Detailed observations of the growing granuloma show that the uninfected macrophages recruited through RD1-MMP9 signaling remain motile within the granuloma as if moving in the context of a gradientless signal. A second signal emanating from dead or dying macrophages appears to draw these macrophages to them, and their encased bacteria are then phagocytosed to create new infected cells. Indeed, RD1-competent mycobacteria also induce more apoptotic death of infected macrophages in the granuloma (Volkman et al. 2004; Davis and Ramakrishnan 2009), consistent with studies showing that the RD1-encoded virulence determinant ESAT-6 can induce multiple programs associated with cell death in vitro (Guinn et al. 2004; Derrick and Morris 2007; van der Wel et al. 2007; Choi et al. 2010; Mishra et al. 2010; Aguilo et al. 2013). It would be predicted that the coordinated acceleration of new macrophage recruitment and infected macrophage apoptosis would be required to maximize bacterial expansion in the granuloma. However, because both processes are dependent on the same locus, RD1, the specific contribution of bacterially induced apoptosis in expanding infection has been difficult to tease out. Engineering host MMP9 deficiency dissociates the two processes; it reduces macrophage recruitment to the granuloma and virulence without affecting apoptosis of infected macrophages (Volkman et al. 2010). This finding suggests that RD1-mediated macrophage apoptosis is not sufficient for macrophage recruitment but does not tell us if its acceleration further expands infection.
Further confounding the role of apoptosis, pathogenic mycobacteria express virulence genes that inhibit apoptosis of cultured macrophages—nuoG, secA2, and pknE. nuoG encodes a subunit of the type I NADH dehydrogenase complex, secA2 encodes a mycobacterial secretion system, and pknE encodes a serine/threonine kinase (Braunstein et al. 2003; Hinchey et al. 2007; Velmurugan et al. 2007; Briken and Miller 2008; Jayakumar et al. 2008). Pathogenic mycobacteria deficient in any of these genes show growth defects in cultured macrophages and in mice or zebrafish (Braunstein et al. 2003; Hinchey et al. 2007; Velmurugan et al. 2007; Jayakumar et al. 2008; Sullivan et al. 2012; Watkins et al. 2012; van der Woude et al. 2014). However, a direct link between their attenuation and impaired inhibition of macrophage apoptosis has not been made. It is not known if these bacterial mutants induce more apoptosis in vivo, much less if inhibition of apoptosis by targeting the corresponding host pathways restores their virulence. These experiments would be particularly important because NuoG, SecA2, and PknE participate broadly in cellular processes whose disruption might be responsible for their attenuation.
Based on in vitro studies, it has been proposed that apoptosis is a host strategy that is detrimental to the infecting mycobacteria, in contrast to necrosis, which favors bacterial growth (Fratazzi et al. 1997; Oddo et al. 1998; Keane et al. 2002; Gan et al. 2008; Behar et al. 2011). Some of these studies have caveats: the use of genetically undefined mutant strains and/or additional agents to induce specific apoptotic death pathways that might override mycobacterially induced pathways (Molloy et al. 1994; Fratazzi et al. 1997; Oddo et al. 1998; Keane et al. 2000; Gan et al. 2008). In terms of apoptotic pathways, apoptosis induced by Fas ligand or ATP have opposite effects on intracellular mycobacterial viability (Lammas et al. 1997). ESAT-6-induced apoptotic cell death per se may use bacterium-sparing pathways. Even if apoptosis is detrimental to mycobacteria, the rapid rephagocytosis of the dead macrophages in the context of the granuloma may nullify any bactericidal effects of apoptosis. In summary, it is possible that mycobacteria inhibit or induce macrophage apoptosis depending on the stage of infection. Mycobacteria might benefit from inhibiting macrophage apoptosis during the earliest stages of infection, when they rely on these cells to access deeper tissues, and then induce macrophage apoptosis to expand within the granuloma.
Neutrophils in the Granuloma: Protective or Pathogenic?
In early zebrafish granulomas, in contrast to arriving macrophages, neutrophils arriving at the granuloma may help control infection (Fig. 1) (Yang et al. 2012). Their recruitment to the granulomas is via signals from the dying macrophages, which they engulf to become infected just as the macrophages do. A subset of neutrophils kills the ingested mycobacteria in an NADPH oxidase-dependent manner. Experimentally preventing neutrophil entry into granulomas by sequestering neutrophils in hematopoietic tissues enhanced mycobacterial expansion and granuloma growth. In the context of the granuloma, neutrophil clearance of infected dying macrophages appears to be protective via two mechanisms: (1) directly lowering mycobacterial loads, and (2) simultaneously reducing the kinetics of intercellular spread into uninfected macrophages and their subsequent death. Work in other animal models and in humans has also identified a protective role of neutrophils in early TB. Neutrophil depletion in mice increased bacterial burdens. This effect could be mitigated by neutralization of interleukin-10 (IL-10) or administration of IL-12, interventions that likely enhanced the microbicidal activity of macrophages (Pedrosa et al. 2000). In rats, neutrophil depletion exacerbated TB infection, whereas transient neutrophilia enhanced control (Sugawara et al. 2004). Humans with chronic granulomatous disease (CGD), a genetic disorder caused by NADPH deficiency and characterized by severely impaired neutrophil oxidative burst (Amulic et al. 2012), are susceptible to mycobacterial infections, including TB (Lee et al. 2008; Dogru et al. 2010). Finally, work in mice suggests an indirect role for how neutrophils might be host-protective. DCs can engulf the apoptotic remnants of infected neutrophils and migrate to the draining lymph node, where they support M. tuberculosis–specific T-cell priming (Blomgran et al. 2012). However, the relative importance of this mode of antigen transfer on T-cell priming is unclear given that migratory DCs can also transfer unprocessed, soluble M. tuberculosis antigens to lymph node–resident DCs through a mechanism not involving apoptosis (Srivastava and Ernst 2014).
As with macrophages, the role of neutrophils in TB protection is confounded by studies showing they are pathological. In mouse TB infection, aggressive neutrophil responses to M. tuberculosis have been documented in genetically susceptible mice (Eruslanov et al. 2005); in cases of suboptimal macrophage immune responses, such as during CARD9 (Dorhoi et al. 2010) or miR-223 deficiency (Dorhoi et al. 2013); in the context of impaired IFN-γ-mediated immunoregulation (Desvignes and Ernst 2009; Nandi and Behar 2011), and as a consequence of pathogenic Th17 responses (Cruz et al. 2006, 2010). Together, these studies assign a pathological role to neutrophils.
So it is likely that neutrophils, similar to macrophages, play a nuanced role in TB pathogenesis; they may participate in protective immunity during early infection but may cause immunopathology in advanced stages of the disease. Both roles are potentially relevant in human TB where neutrophils are readily detected in the blood of patients with active TB by a signature IFN gene expression profile (Berry et al. 2010), in the cerebrospinal fluid of tuberculous meningitis (Thwaites et al. 2002; Lowe et al. 2012) an a in pulmonary TB, where they can be abundant in both early granulomas and late cavitary granulomas (Canetti 1955; Eum et al. 2010).
EXITING THE GRANULOMA
Egress of Infected Macrophages from the Primary Granuloma
At least in the innate stages of granuloma development, even as uninfected macrophages are being recruited to feed the granuloma, some newly infected macrophages exit the nascent granuloma and establish secondary granulomas, thus disseminating the infection within the host (Fig. 1) (Davis and Ramakrishnan 2009). Indeed, close temporal monitoring of the spread of infection from a primary granuloma focus tightly linked departing macrophages to the spread of infection. A similar phenomenon was recently documented in a transplantation model of TB in mice, where infected DCs were observed to exit the primary granuloma and spread the infection into distal tissues (Schreiber et al. 2011). Although the mechanism of infected macrophage egress remains unclear, it does appear to be the main mode of establishing multiple infection foci early in infection. It is conceivably a mycobacterial bet-hedging strategy because different granulomas within the same individual can expand or contract from the earliest stages of infection as shown by close monitoring of guinea pigs, zebrafish, and monkeys and surmised from detailed histopathological studies of the lungs of infected humans in the prechemotherapy era (Balasubramanian et al. 1994; Davis and Ramakrishnan 2009; Adams et al. 2011; Lin et al. 2014).
Exiting the Intracellular Environment through Macrophage Necrosis
Although mycobacteria exploit macrophages to infect and spread within a host, they enhance their transmission to a new host by becoming extracellular (Fig. 1). Mycobacteria are notoriously adapted to intracellular growth, so the vast majority of mycobacterial pathogenesis research has focused on mechanisms of intracellular survival. However, it has long been known that in humans, necrotic granulomas, called caseating granulomas for their cheesy appearance on macroscopic examination, thought to result from the necrotic breakdown of infected macrophages, are a major contributor to morbidity and transmission (Rich 1946; Canetti 1955; Grosset 2003; Ramakrishnan 2012). Detailed histological studies of tuberculous organs obtained from autopsies in the prechemotherapy era revealed that areas of caseation in early granulomas were associated with more numerous bacteria than noncaseating lesions (Canetti 1955). Corroborating these old human studies, work in M. marinum-infected adult zebrafish (Swaim et al. 2006), as well in M. tuberculosis-infected macaques (Lin et al. 2014), similarly find that caseation is associated from early on with high bacterial burdens. Virulent M. tuberculosis grown in culture acquires a corded appearance characterized by the formation of long, intertwined serpentine structures (Koch 1882). This in vitro phenotype correlates well with virulence (Middlebrook et al. 1947) and has been described to occur in vivo under conditions in which mycobacteria grow extracellularly (Clay et al. 2007, 2008). A recent study of Mycobacterium abscessus infection in zebrafish suggests that the cording morphology helps mycobacteria avoid macrophage-mediated immunity; uninfected macrophages that were recruited to sites of bacterial cording failed to rephagocytose the bacterial cords, perhaps because they were too large to be engulfed (Bernut et al. 2014). Finally, the bacterial RD1 virulence locus promotes necrosis of mature granulomas, either by postapoptotic necrosis or through yet-undefined independent mechanisms (Volkman et al. 2004; Swaim et al. 2006; Ramakrishnan 2012). These observations suggest that extracellular survival may be an important part of mycobacterium’s ability to grow to high numbers in the compartment most conducive for transmission, a critical survival trait for a pathogen.
Previous studies could not discern whether high bacterial burdens promote necrosis, and the numerous bacteria released can simply survive in the necrotic debris, or whether the necrotic debris and/or extracellular milieu actually promote bacterial growth. Work in the zebrafish larvae has shed light on this issue and has provided evidence that although the early granuloma promotes growth, mycobacterial growth is further enhanced in the extracellular milieu when bacteria are released after macrophage necrosis, or if macrophages are absent altogether (Clay et al. 2007, 2008; Tobin et al. 2010, 2012; Ramakrishnan 2012; Roca and Ramakrishnan 2013). Several immune mechanisms have been implicated in necrosis.
Foamy Macrophages
Lipid-laden foamy macrophages are thought to contribute to caseum formation. In vitro granuloma models suggest that foamy macrophages form through a host transcription-dependent process involving peroxisome proliferator γ (PPARγ) and testicular receptor 4 (TR4) and induced by mycobacterial oxygenated mycolic acids, which leads to the accumulation of low-density lipoprotein-derived lipids (Peyron et al. 2008; Mahajan et al. 2012). ESX-1-competent mycobacteria induce the foamy phenotype by diverting glycolitic metabolism toward ketone body synthesis and inducing the expression of the antilipolytic G-protein-coupled receptor GPR109A (Singh et al. 2012). These macrophages are common in caseating granulomas, typically around the edges of the necrotic core, but rarely present in nonnecrotic lesions (Caceres et al. 2009). Foamy macrophages are poorly bactericidal compared with their nonfoamy counterparts, attributed in part to increased M2-type macrophage activation and IL-10 production (Mahajan et al. 2012) and impaired autophagy and lysosomal acidification (Singh et al. 2012). In fact, recent work indicates that M1-type macrophage polarization inhibits foamy macrophage development. Activation of the vitamin D pathway, which is induced on IFN-γ-mediated stimulation (Fabri et al. 2011), prevents lipid accumulation by reducing PPARγ expression (Salamon et al. 2014).
Bacteria can be found inside lipid bodies and contain lipid inclusions, suggesting that they can use host lipids as nutrient sources (Peyron et al. 2008; Russell et al. 2009). The lipid composition of the caseum reflects that of foamy macrophages, suggesting a link between foamy macrophages and caseum formation. It has been proposed that foamy macrophages contribute to caseum formation by undergoing necrosis, thereby spilling their intracellular contents (Russell et al. 2009). The lipids released into the extracellular space might serve as a nutrient source for enhanced mycobacterial growth in an environment of impaired cell-mediated immunity.
Tumor Necrosis Factor Deficiency as a Cause of Granuloma Macrophage Necrosis
In general, the failure of the infected macrophage to control bacterial replication caused by tumor necrosis factor (TNF) deficiency results in their accelerated necrotic death and release of bacteria (Fig. 1) (Clay et al. 2008; Tobin et al. 2010). Numerous lines of evidence have established the importance of the proinflammatory cytokine TNF in immunity to M. tuberculosis. Early studies in mice showed that TNF deficiency enhances mycobacterial growth and disease progression (Flynn et al. 1995), and they have been corroborated by studies in humans showing that therapeutic neutralization of TNF for the treatment of autoimmune diseases such as rheumatoid arthritis and noninfectious inflammatory disorders such as Crohn’s disease increases TB susceptibility (Keane 2005). TNF signaling deficiency in mice produces disorganized tuberculous lesions teeming with extracellular mycobacteria (Flynn et al. 1995). The stark morphological differences between the organized granulomas of TNF-sufficient mice and the disorganized, necrotic structures of TNF-deficient mice helped establish the idea that TNF participates in granuloma formation and supported the notion that granulomas are host-protective structures formed by the immune system (Ramakrishnan 2012). However, the sequential monitoring of granuloma development that is possible in zebrafish larvae provides a more nuanced view of TNF involvement in immunity to TB and granuloma formation (Clay et al. 2008). Larvae rendered deficient in TNF-signaling via knockdown of TNF Receptor 1 showed increased intramacrophage mycobacterial loads and, unexpectedly, accelerated granuloma formation. The accelerated kinetics of granuloma formation are likely caused by an increased abundance of ESX-1 substrates in the extracellular space, which would induce MMP9 expression on the epithelial cells surrounding the infected macrophages. These heavily infected granuloma macrophages eventually became necrotic, releasing mycobacteria into the extracellular milieu. These observations suggest that TNF is not required for granuloma formation per se, but rather preserves granuloma integrity by inhibiting mycobacterial growth and preventing macrophage death. A live imaging study in mice showed that TNF blockade in established granulomas led to their disintegration (Egen et al. 2008), adding credence to the idea that TNF is critical for granuloma maintenance. Consistent with the zebrafish work, a study in cynomolgus macaques showed that TNF inhibition increased mycobacterial burdens and extrapulmonary dissemination of the infection without impeding granuloma formation (Lin et al. 2010). During active disease, granulomas in the treated group became more invasive, extending into adjacent vessels and airways. Strikingly, TNF inhibition did not appear to disrupt the architecture of established granulomas, but secondary granulomas in adjacent tissue and in distal organs nonetheless formed (Lin et al. 2010). These findings are in agreement with the observations in humans undergoing anti-TNF therapies who can develop organized caseating granulomas (Garcia Vidal et al. 2005; Iliopoulos et al. 2006). Because only some of the macrophages in established granulomas are infected, TNF deficiency would only cause the necrosis of these, thus forming a necrotic core within a cellular granuloma that would facilitate an exit strategy for the bacteria.
TNF Excess as a Cause of Granuloma Necrosis
Unexpectedly, excessive TNF in the granuloma is also host detrimental by inducing macrophage necrosis, but through a different mechanism from TNF deficiency (Fig. 1). Excess TNF caused excessive inflammation leading to Receptor-interacting serine/threonine kinase (RIPK) 1- and RIPK3-dependent necrosis of granuloma macrophages via production of mitochondrial reactive oxygen species (ROS) (Tobin et al. 2012; Roca and Ramakrishnan 2013). Accordingly, and in stark contrast to TNF-deficiency, macrophages experiencing an excess of TNF signaling initially have enhanced control of intracellular mycobacterial growth because of the ROS. But this ROS then causes cell necrosis through multiple mechanisms rendering the bacteria extracellular.
T Cells and Granuloma Necrosis
Like phagocytes, CD4+ T cells can also play a dual role in TB protection and pathology, and have also been implicated in granuloma necrosis (Fig. 1). HIV-positive individuals with low CD4+ T-cell counts in the blood are less likely to have cavitary TB lesions than individuals with normal CD4+ T-cell frequencies and are less likely to transmit disease (Kwan and Ernst 2011). A recent study documented that human T-cell epitopes in the M. tuberculosis complex are highly conserved, suggesting that pathogenic mycobacteria benefit from T-cell recognition (Comas et al. 2010; Ernst 2012). The mechanism by which CD4+ T cells contribute to necrosis is unclear, but it might involve an overly exuberant mycobacteria-specific response that induces the necrotic death of infected cells. It is plausible that the specific effector is TNF, and this might explain the widespread incidence of necrotic granulomas among patients with advanced TB. The effects of excessive TNF production might be present, albeit at a smaller scale, in the normal spectrum of TB lesions in nongenetically susceptible individuals and might contribute to caseum formation in established granulomas.
Cytotoxic T lymphocyte antigen-4 (CTLA-4) and program death-1 (PD-1), two members of the CD28 family of receptors (Greenwald et al. 2005), have been implicated in determining whether a T-cell response in TB is host-protective or host-detrimental. These receptors are induced during T-cell activation and recognize ligands decorating the surface of antigen-presenting cells (APCs). Engagement of these receptors dampens stimulatory signals transduced through the T-cell antigen receptor (TCR) signaling complex on TCR binding to cognate peptide:major histocompatibility complex (p:MHC) molecules displayed by the APCs (Greenwald et al. 2005). Although these receptors exert critical roles in preventing autoimmunity by restraining autoreactive T-cell responses, their engagement in the context of several infections and cancers is host-detrimental because it inhibits the development of robust pathogen- or tumor-specific T-cell immunity (Peggs et al. 2009; Fife and Pauken 2011; Odorizzi and Wherry 2012; Kamphorst and Ahmed 2013). A study of pulmonary TB in a West African cohort identified a single nucleotide polymorphism (SNP) in the 3′-untranslated region of CTLA4 that correlated with increased disease severity (Thye et al. 2009). Patients bearing the +6230G allele had larger and more numerous lesions than those with an A/A genotype at the same position, and the +6230G individuals appeared to have more cavitary granulomas. Interestingly, this same SNP has been associated with several autoimmune diseases (Ueda et al. 2003), suggesting that the susceptibility in TB is due to a dysfunctional T-cell response. The effects of CTLA-4 inhibition on mouse models of TB remain largely unexplored. A study of BCG infection found that CTLA-4 blockade accelerated the kinetics of T-cell activation and enhanced IFN-γ production but did not alter bacterial burdens or tissue pathology (Kirman et al. 1999). It remains to be determined if CTLA-4 blockade during M. tuberculosis infection elicits a similar response. At the very least, this study shows that CTLA-4 exerts an inhibitory function during a mycobacterial infection. Nonetheless, studies of PD-1 in a mouse model of TB make a compelling case for the involvement of inhibitory receptors in preventing T-cell immunopathology in TB (Lazar-Molnar et al. 2010; Barber et al. 2011). PD-1 deficiency enhanced M. tuberculosis-specific CD4+ T-cell expansion, increased the levels of proinflammatory cytokines in the lungs, including TNF, caused extensive tissue necrosis, and precipitated death (Lazar-Molnar et al. 2010; Barber et al. 2011). Again, TNF is a plausible culprit for widespread necrosis observed in the context of PD-1 deficiency. It will be interesting to determine if genetic polymorphisms in the TNF pathway, PD-1, and other inhibitory coreceptors or their ligands modulate susceptibility to TB in humans and, specifically, development of cavitary granulomas.
Cavitary Granulomas as Vehicles for TB Transmission
The formation of cavities that harbor very large numbers of bacteria greatly facilitates aerosol transmission in the advanced stages of human lung TB (Fig. 1). Patients harboring radiologically apparent cavitary lesions are more likely to have burdens of M. tuberculosis in their expectorated sputum and to transmit TB than those that do not have as advanced lesions (Palaci et al. 2007). Individual cavitary lesions may contain 107–109 bacilli (Canetti 1965). The high level of bacterial replication observed in cavitary granulomas may also contribute to the selection of genetic resistance to antitubercular drugs (Howard et al. 1949).
Recent work argues that the pathogenesis of cavitation is distinct from necrosis and that although necrosis is a prerequisite for cavitation, it is not sufficient. The collagenase matrix metalloproteinase-1 (MMP1) has been implicated in the final steps of granuloma cavitation through the degradation of the fibrous extracellular matrix surrounding the granuloma (Fig. 1) (Elkington et al. 2011c). SNPs in MMP1 that increase protein abundance or enzymatic activity have been associated with TB hypersusceptibility (Ganachari et al. 2010; Wang et al. 2010). M. tuberculosis infection in humans induces MMP1 expression in epithelial cells surrounding the granuloma and in leukocytes (Elkington et al. 2005). Overexpression of human MMP1 in mice infected with M. tuberculosis induced granuloma breakdown (Elkington et al. 2011a). Although the specific mycobacterial signals that induce MMP1 (Ganachari et al. 2010) have not been identified, it has been noted that M. bovis BCG is a poor inducer compared with virulent M. tuberculosis (Elkington et al. 2005), suggesting that MMP1 induction might share similarities with that of MMP9. The factors that induce cavitation could be therapeutically intercepted to reduce tissue destruction and minimize transmission (Elkington et al. 2011b).
SUMMARY: A NEW ROLE FOR THE GRANULOMA FROM A HISTORICAL PERSPECTIVE
Work over the last decade has revised the role of the granuloma from host-protective structure to one that substantially benefits mycobacterial expansion. The protective granuloma model was based on several lines of circumstantial evidence from traditional animal models in which only single endpoint analyses of tissue histology and bacterial burdens were possible (Ramakrishnan 2012). In particular, the surmise that granuloma formation was dependent on adaptive immunity, the development of which is accompanied by a plateau in bacterial load contributed to this thinking (Andersen 1997; Saunders and Cooper 2000; North and Jung 2004; Urdahl et al. 2011). The idea has been that adaptive immunity is required for granuloma formation, which in turn restricts bacterial growth. In this model, mycobacterium-specific T cells migrate to the site of infection and interact with infected macrophages through the secretion of cytokines such as IFN-γ and TNF that induce granuloma formation while enhancing macrophage microbicidal functions. Whereas IFN-γ and TNF can be produced by innate immune cells, T cells are potent producers of these cytokines and have been shown to be critical for controlling mycobacterial infection in animal models and humans (Szabo et al. 2003; Al-Muhsen and Casanova 2008; Kwan and Ernst 2011; Philips and Ernst 2012; Tubo and Jenkins 2014). Mice deficient in CD4+ T cells, IFN-γ signaling, or TNF signaling show disorganized macrophage aggregates that become necrotic (Kindler et al. 1989; Flynn et al. 1993, 1995; Bean et al. 1999; Caruso et al. 1999; Scanga et al. 2000; Flynn and Chan 2001; Roach et al. 2002; Algood et al. 2005; Stenger 2005; Chakravarty et al. 2008; Wallis and Schluger 2010). These observations associated granuloma formation with a protective immune response to TB.
The finding that granuloma formation can occur in the sole context of innate immunity was instrumental in starting to appreciate the granuloma’s role in TB pathogenesis. Indeed, granuloma formation is accelerated by the very mycobacteria that it was said to “wall-off,” turning a fundamentally host-beneficial process into a bacterium-beneficial one. Therefore, slowing the kinetics of granuloma formation may in fact benefit the host by allowing processes such as macrophage apoptosis to play out in the host’s favor. Furthermore, maintaining macrophage cellularity by preventing their necrosis is also favorable to the host by keeping mycobacteria from the extracellular environment, which is even more growth-permissive than the intracellular milieu.
In summary then, the granuloma, as constructed and deconstructed by the pathogenic mycobacteria, is a pathological entity from its inception when it promotes intracellular bacterial spread to its advanced stages when it promotes extracellular bacterial growth and transmission. An understanding of which processes are pathogenic, and to what extent, may allow their modulation so as to render the tuberculous granuloma into a structure that promotes immunity without immunopathology.
ACKNOWLEDGMENTS
We thank C.J. Cambier for critical review. This work was funded by grants from the National Institutes of Health (NIH) (T32-AI055396 to A.J.P; A154503 and A136396 to L.R). L.R. is a recipient of the NIH Director’s Pioneer Award.
Footnotes
Editors: Stefan H.E. Kaufmann, Eric J. Rubin, and Alimuddin Zumla
Additional Perspectives on Tuberculosis available at www.perspectivesinmedicine.org
REFERENCES
- Adams DO. 1976. The granulomatous inflammatory response. A review. Am J Pathol 84: 164–192. [PMC free article] [PubMed] [Google Scholar]
- Adams KN, Takaki K, Connolly LE, Wiedenhoft H, Winglee K, Humbert O, Edelstein PH, Cosma CL, Ramakrishnan L. 2011. Drug tolerance in replicating mycobacteria mediated by a macrophage-induced efflux mechanism. Cell 145: 39–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agrawal S, Anderson P, Durbeej M, van Rooijen N, Ivars F, Opdenakker G, Sorokin LM. 2006. Dystroglycan is selectively cleaved at the parenchymal basement membrane at sites of leukocyte extravasation in experimental autoimmune encephalomyelitis. J Exp Med 203: 1007–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilo JI, Alonso H, Uranga S, Marinova D, Arbues A, de Martino A, Anel A, Monzon M, Badiola J, Pardo J, et al. 2013. ESX-1-induced apoptosis is involved in cell-to-cell spread of Mycobacterium tuberculosis. Cell Microbiol 15: 1994–2005. [DOI] [PubMed] [Google Scholar]
- Al-Muhsen S, Casanova JL. 2008. The genetic heterogeneity of mendelian susceptibility to mycobacterial diseases. J Allergy Clin Immunol 122: 1043–1051; quiz 1052–1043. [DOI] [PubMed] [Google Scholar]
- Algood HM, Lin PL, Flynn JL. 2005. Tumor necrosis factor and chemokine interactions in the formation and maintenance of granulomas in tuberculosis. Clin Infect Dis 41: S189–S193. [DOI] [PubMed] [Google Scholar]
- Amulic B, Cazalet C, Hayes GL, Metzler KD, Zychlinsky A. 2012. Neutrophil function: From mechanisms to disease. Annu Rev Immunol 30: 459–489. [DOI] [PubMed] [Google Scholar]
- Andersen P. 1997. Host responses and antigens involved in protective immunity to Mycobacterium tuberculosis. Scand J Immunol 45: 115–131. [DOI] [PubMed] [Google Scholar]
- Andersen P, Andersen AB, Sorensen AL, Nagai S. 1995. Recall of long-lived immunity to Mycobacterium tuberculosis infection in mice. J Immunol 154: 3359–3372. [PubMed] [Google Scholar]
- Balasubramanian V, Wiegeshaus EH, Taylor BT, Smith DW. 1994. Pathogenesis of tuberculosis: Pathway to apical localization. Tuber Lung Dis 75: 168–178. [DOI] [PubMed] [Google Scholar]
- Barber DL, Mayer-Barber KD, Feng CG, Sharpe AH, Sher A. 2011. CD4 T cells promote rather than control tuberculosis in the absence of PD-1-mediated inhibition. J Immunol 186: 1598–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates JH, Potts WE, Lewis M. 1965. Epidemiology of primary tuberculosis in an industrial school. N Engl J Med 272: 714–717. [DOI] [PubMed] [Google Scholar]
- Bean AG, Roach DR, Briscoe H, France MP, Korner H, Sedgwick JD, Britton WJ. 1999. Structural deficiencies in granuloma formation in TNF gene-targeted mice underlie the heightened susceptibility to aerosol Mycobacterium tuberculosis infection, which is not compensated for by lymphotoxin. J Immunol 162: 3504–3511. [PubMed] [Google Scholar]
- Behar SM, Martin CJ, Booty MG, Nishimura T, Zhao X, Gan HX, Divangahi M, Remold HG. 2011. Apoptosis is an innate defense function of macrophages against Mycobacterium tuberculosis. Mucosal Immunol 4: 279–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behr MA, Wilson MA, Gill WP, Salamon H, Schoolnik GK, Rane S, Smal PM. 1999. Comparative genomics of BCG vaccines by whole-genome DNA microarray. Science 284: 1520–1523. [DOI] [PubMed] [Google Scholar]
- Bernut A, Herrmann JL, Kissa K, Dubremetz JF, Gaillard JL, Lutfalla G, Kremer L. 2014. Mycobacterium abscessus cording prevents phagocytosis and promotes abscess formation. Proc Natl Acad Sci 111: E943–E952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry MP, Graham CM, McNab FW, Xu Z, Bloch SA, Oni T, Wilkinson KA, Banchereau R, Skinner J, Wilkinson RJ, et al. 2010. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature 466: 973–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthet FX, Lagranderie M, Gounon P, Laurent-Winter C, Ensergueix D, Chavarot P, Thouron F, Maranghi E, Pelicic V, Portnoi D, et al. 1998. Attenuation of virulence by disruption of the Mycobacterium tuberculosis erp gene. Science 282: 759–762. [DOI] [PubMed] [Google Scholar]
- Blomgran R, Desvignes L, Briken V, Ernst JD. 2012. Mycobacterium tuberculosis inhibits neutrophil apoptosis, leading to delayed activation of naive CD4 T cells. Cell Host Microbe 11: 81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouley DM, Ghori N, Mercer KL, Falkow S, Ramakrishnan L. 2001. Dynamic nature of host-pathogen interactions in Mycobacterium marinum granulomas. Infect Immun 69: 7820–7831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braunstein M, Espinosa BJ, Chan J, Belisle JT, Jacobs WR Jr. 2003. SecA2 functions in the secretion of superoxide dismutase A and in the virulence of Mycobacterium tuberculosis. Mol Microbiol 48: 453–464. [DOI] [PubMed] [Google Scholar]
- Briken V, Miller JL. 2008. Living on the edge: Inhibition of host cell apoptosis by Mycobacterium tuberculosis. Future Microbiol 3: 415–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caceres N, Tapia G, Ojanguren I, Altare F, Gil O, Pinto S, Vilaplana C, Cardona PJ. 2009. Evolution of foamy macrophages in the pulmonary granulomas of experimental tuberculosis models. Tuberculosis (Edinb) 89: 175–182. [DOI] [PubMed] [Google Scholar]
- Cambier CJ, Takaki KK, Larson RP, Hernandez RE, Tobin DM, Urdahl KB, Cosma CL, Ramakrishnan L. 2014. Mycobacteria manipulate macrophage recruitment through coordinated use of membrane lipids. Nature 505: 218–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canetti G. 1955. The tubercle bacillus in the pulmonary lesion of man; histobacteriology and its bearing on the therapy of pulmonary tuberculosis. Springer, New York. [Google Scholar]
- Canetti G. 1965. Present aspects of bacterial resistance in tuberculosis. Am Rev Respir Dis 92: 687–703. [DOI] [PubMed] [Google Scholar]
- Caruso AM, Serbina N, Klein E, Triebold K, Bloom BR, Flynn JL. 1999. Mice deficient in CD4 T cells have only transiently diminished levels of IFN-γ, yet succumb to tuberculosis. J Immunol 162: 5407–5416. [PubMed] [Google Scholar]
- Chakravarty SD, Zhu G, Tsai MC, Mohan VP, Marino S, Kirschner DE, Huang L, Flynn J, Chan J. 2008. Tumor necrosis factor blockade in chronic murine tuberculosis enhances granulomatous inflammation and disorganizes granulomas in the lungs. Infect Immun 76: 916–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi HH, Shin DM, Kang G, Kim KH, Park JB, Hur GM, Lee HM, Lim YJ, Park JK, Jo EK, et al. 2010. Endoplasmic reticulum stress response is involved in Mycobacterium tuberculosis protein ESAT-6-mediated apoptosis. FEBS Lett 584: 2445–2454. [DOI] [PubMed] [Google Scholar]
- Clay H, Davis JM, Beery D, Huttenlocher A, Lyons SE, Ramakrishnan L. 2007. Dichotomous role of the macrophage in early Mycobacterium marinum infection of the zebrafish. Cell Host Microbe 2: 29–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clay H, Volkman HE, Ramakrishnan L. 2008. Tumor necrosis factor signaling mediates resistance to mycobacteria by inhibiting bacterial growth and macrophage death. Immunity 29: 283–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comas I, Chakravartti J, Small PM, Galagan J, Niemann S, Kremer K, Ernst JD, Gagneux S. 2010. Human T cell epitopes of Mycobacterium tuberculosis are evolutionarily hyperconserved. Nat Genet 42: 498–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosma CL, Humbert O, Ramakrishnan L. 2004. Superinfecting mycobacteria home to established tuberculous granulomas. Nat Immunol 5: 828–835. [DOI] [PubMed] [Google Scholar]
- Cosma CL, Klein K, Kim R, Beery D, Ramakrishnan L. 2006. Mycobacterium marinum Erp is a virulence determinant required for cell wall integrity and intracellular survival. Infect Immun 74: 3125–3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosma CL, Humbert O, Sherman DR, Ramakrishnan L. 2008. Trafficking of superinfecting mycobacterium organisms into established granulomas occurs in mammals and is independent of the Erp and ESX-1 mycobacterial virulence loci. J Infect Dis 198: 1851–1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz A, Khader SA, Torrado E, Fraga A, Pearl JE, Pedrosa J, Cooper AM, Castro AG. 2006. Cutting edge: IFN-γ regulates the induction and expansion of IL-17-producing CD4 T cells during mycobacterial infection. J Immunol 177: 1416–1420. [DOI] [PubMed] [Google Scholar]
- Cruz A, Fraga Ag, Fountain JJ, Rangel-Moreno J, Torrado E, Saraiva M, Pereira DR, Randall TD, Pedrosa J, Cooper AM, et al. 2010. Pathological role of interleukin 17 in mice subjected to repeated BCG vaccination after infection with Mycobacterium tuberculosis. J Exp Med 207: 1609–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dannenberg AM Jr. 1993. Immunopathogenesis of pulmonary tuberculosis. Hosp Pract (Off Ed) 28: 51–58. [DOI] [PubMed] [Google Scholar]
- Dannenberg AM Jr. 2003. Macrophage turnover, division and activation within developing, peak and “healed” tuberculous lesions produced in rabbits by BCG. Tuberculosis (Edinb) 83: 251–260. [DOI] [PubMed] [Google Scholar]
- Davis JM, Ramakrishnan L. 2009. The role of the granuloma in expansion and dissemination of early tuberculous infection. Cell 136: 37–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis JM, Clay H, Lewis JL, Ghori N, Herbomel P, Ramakrishnan L. 2002. Real-time visualization of mycobacterium-macrophage interactions leading to initiation of granuloma formation in zebrafish embryos. Immunity 17: 693–702. [DOI] [PubMed] [Google Scholar]
- Derrick SC, Morris SL. 2007. The ESAT6 protein of Mycobacterium tuberculosis induces apoptosis of macrophages by activating caspase expression. Cell Microbiol 9: 1547–1555. [DOI] [PubMed] [Google Scholar]
- Desvignes L, Ernst JD. 2009. Interferon-γ-responsive nonhematopoietic cells regulate the immune response to Mycobacterium tuberculosis. Immunity 31: 974–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dogru D, Kiper N, Ozcelik U, Yalcin E, Tezcan I. 2010. Tuberculosis in children with congenital immunodeficiency syndromes. Tuberk Toraks 58: 59–63. [PubMed] [Google Scholar]
- Dorhoi A, Desel C, Yeremeev V, Pradl L, Brinkmann V, Mollenkopf HJ, Hanke K, Gross O, Ruland J, Kaufmann SH. 2010. The adaptor molecule CARD9 is essential for tuberculosis control. J Exp Med 207: 777–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorhoi A, Iannaccone M, Farinacci M, Fae KC, Schreiber J, Moura-Alves P, Nouailles G, Mollenkopf HJ, Oberbeck-Muller D, Jorg S, et al. 2013. MicroRNA-223 controls susceptibility to tuberculosis by regulating lung neutrophil recruitment. J Clin Invest 123: 4836–4848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egen JG, Rothfuchs AG, Feng CG, Winter N, Sher A, Germain RN. 2008. Macrophage and T cell dynamics during the development and disintegration of mycobacterial granulomas. Immunity 28: 271–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkington PT, O’Kane CM, Friedland JS. 2005. The paradox of matrix metalloproteinases in infectious disease. Clin Exp Immunol 142: 12–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkington PT, Green JA, Emerson JE, Lopez-Pascual LD, Boyle JJ, O’Kane CM, Friedland JS. 2007. Synergistic up-regulation of epithelial cell matrix metalloproteinase-9 secretion in tuberculosis. Am J Respir Cell Mol Biol 37: 431–437. [DOI] [PubMed] [Google Scholar]
- Elkington P, Shiomi T, Breen R, Nuttall RK, Ugarte-Gil CA, Walker NF, Saraiva L, Pedersen B, Mauri F, Lipman M, et al. 2011a. MMP-1 drives immunopathology in human tuberculosis and transgenic mice. J Clin Invest 121: 1827–1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkington PT, D’Armiento JM, Friedland JS. 2011b. Tuberculosis immunopathology: The neglected role of extracellular matrix destruction. Sci Transl Med 3: 71–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkington PT, Ugarte-Gil CA, Friedland JS. 2011c. Matrix metalloproteinases in tuberculosis. Eur Respir J 38: 456–464. [DOI] [PubMed] [Google Scholar]
- Ernst JD. 2012. The immunological life cycle of tuberculosis. Nat Rev Immunol 12: 581–591. [DOI] [PubMed] [Google Scholar]
- Eruslanov EB, Lyadova IV, Kondratieva TK, Majorov KB, Scheglov IV, Orlova MO, Apt AS. 2005. Neutrophil responses to Mycobacterium tuberculosis infection in genetically susceptible and resistant mice. Infect Immun 73: 1744–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eum SY, Kong JH, Hong MS, Lee YJ, Kim JH, Hwang SH, Cho SN, Via LE, Barry CE III. 2010. Neutrophils are the predominant infected phagocytic cells in the airways of patients with active pulmonary TB. Chest 137: 122–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabri M, Stenger S, Shin DM, Yuk JM, Liu PT, Realegeno S, Lee HM, Krutzik SR, Schenk M, Sieling PA, et al. 2011. Vitamin D is required for IFN-γ-mediated antimicrobial activity of human macrophages. Sci Transl Med 3: p104ra102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fife BT, Pauken KE. 2011. The role of the PD-1 pathway in autoimmunity and peripheral tolerance. Ann N Y Acad Sci 1217: 45–59. [DOI] [PubMed] [Google Scholar]
- Flynn JL. 2006. Lessons from experimental Mycobacterium tuberculosis infections. Microbes Infect 8: 1179–1188. [DOI] [PubMed] [Google Scholar]
- Flynn JL, Chan J. 2001. Immunology of tuberculosis. Annu Rev Immunol 19: 93–129. [DOI] [PubMed] [Google Scholar]
- Flynn JL, Chan J, Triebold KJ, Dalton DK, Stewart TA, Bloom BR. 1993. An essential role for interferon γ in resistance to Mycobacterium tuberculosis infection. J Exp Med 178: 2249–2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn JL, Goldstein MM, Chan J, Triebold KJ, Pfeffer K, Lowenstein CJ, Schreiber R, Mak TW, Bloom BR. 1995. Tumor necrosis factor-α is required in the protective immune response against Mycobacterium tuberculosis in mice. Immunity 2: 561–572. [DOI] [PubMed] [Google Scholar]
- Fratazzi C, Arbeit RD, Carini C, Remold HG. 1997. Programmed cell death of Mycobacterium avium serovar 4-infected human macrophages prevents the mycobacteria from spreading and induces mycobacterial growth inhibition by freshly added, uninfected macrophages. J Immunol 158: 4320–4327. [PubMed] [Google Scholar]
- Gan H, Lee J, Ren F, Chen M, Kornfeld H, Remold HG. 2008. Mycobacterium tuberculosis blocks crosslinking of annexin-1 and apoptotic envelope formation on infected macrophages to maintain virulence. Nat Immunol 9: 1189–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganachari M, Ruiz-Morales JA, Gomez de la Torre Pretell JC, Dinh J, Granados J, Flores-Villanueva PO. 2010. Joint effect of MCP-1 genotype GG and MMP-1 genotype 2G/2G increases the likelihood of developing pulmonary tuberculosis in BCG-vaccinated individuals. PLoS ONE 5: e8881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia Vidal C, Rodriguez Fernandez S, Martinez Lacasa J, Salavert M, Vidal R, Rodriguez Carballeira M, Garau J. 2005. Paradoxical response to antituberculous therapy in infliximab-treated patients with disseminated tuberculosis. Clin Infect Dis 40: 756–759. [DOI] [PubMed] [Google Scholar]
- Greenlee KJ, Corry DB, Engler DA, Matsunami RK, Tessier P, Cook RG, Werb Z, Kheradmand F. 2006. Proteomic identification of in vivo substrates for matrix metalloproteinases 2 and 9 reveals a mechanism for resolution of inflammation. J Immunol 177: 7312–7321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwald RJ, Freeman GJ, Sharpe AH. 2005. The B7 family revisited. Annu Rev Immunol 23: 515–548. [DOI] [PubMed] [Google Scholar]
- Grosset J. 2003. Mycobacterium tuberculosis in the extracellular compartment: An underestimated adversary. Antimicrob Agents Chemother 47: 833–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guinn KM, Hickey MJ, Mathur SK, Zakel KL, Grotzke JE, Lewinsohn DM, Smith S, Sherman DR. 2004. Individual RD1-region genes are required for export of ESAT-6/CFP-10 and for virulence of Mycobacterium tuberculosis. Mol Microbiol 51: 359–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helke KL, Mankowski JL, Manabe YC. 2006. Animal models of cavitation in pulmonary tuberculosis. Tuberculosis (Edinb) 86: 337–348. [DOI] [PubMed] [Google Scholar]
- Hinchey J, Lee S, Jeon BY, Basaraba RJ, Venkataswamy MM, Chen B, Chan J, Braunstein M, Orme IM, Derrick SC, et al. 2007. Enhanced priming of adaptive immunity by a proapoptotic mutant of Mycobacterium tuberculosis. J Clin Invest 117: 2279–2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard WL, Maresh F, Mueller EE, Yanitelli SA, Woodruff GF. 1949. The role of pulmonary cavitation in the development of bacterial resistance to streptomycin. Am Rev Tuberc 59: 391–401. [DOI] [PubMed] [Google Scholar]
- Hsu T, Hingley-Wilson SM, Chen B, Chen M, Dai AZ, Morin PM, Marks CB, Padiyar J, Goulding C, Gingery M, et al. 2003. The primary mechanism of attenuation of bacillus Calmette-Guerin is a loss of secreted lytic function required for invasion of lung interstitial tissue. Proc Natl Acad Sci 100: 12420–12425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliopoulos A, Psathakis K, Aslanidis S, Skagias L, Sfikakis PP. 2006. Tuberculosis and granuloma formation in patients receiving anti-TNF therapy. Int J Tuberc Lung Dis 10: 588–590. [PubMed] [Google Scholar]
- Jayakumar D, Jacobs WR Jr, Narayanan S. 2008. Protein kinase E of Mycobacterium tuberculosis has a role in the nitric oxide stress response and apoptosis in a human macrophage model of infection. Cell Microbiol 10: 365–374. [DOI] [PubMed] [Google Scholar]
- Kamphorst AO, Ahmed R. 2013. Manipulating the PD-1 pathway to improve immunity. Curr Opin Immunol 25: 381–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keane J. 2005. TNF-blocking agents and tuberculosis: New drugs illuminate an old topic. Rheumatology (Oxford) 44: 714–720. [DOI] [PubMed] [Google Scholar]
- Keane J, Remold HG, Kornfeld H. 2000. Virulent Mycobacterium tuberculosis strains evade apoptosis of infected alveolar macrophages. J Immunol 164: 2016–2020. [DOI] [PubMed] [Google Scholar]
- Keane J, Shurtleff B, Kornfeld H. 2002. TNF-dependent BALB/c murine macrophage apoptosis following Mycobacterium tuberculosis infection inhibits bacillary growth in an IFN-γ independent manner. Tuberculosis (Edinb) 82: 55–61. [DOI] [PubMed] [Google Scholar]
- Kindler V, Sappino AP, Grau GE, Piguet PF, Vassalli P. 1989. The inducing role of tumor necrosis factor in the development of bactericidal granulomas during BCG infection. Cell 56: 731–740. [DOI] [PubMed] [Google Scholar]
- Kirman J, McCoy K, Hook S, Prout M, Delahunt B, Orme I, Frank A, Le Gros G. 1999. CTLA-4 blockade enhances the immune response induced by mycobacterial infection but does not lead to increased protection. Infect Immun 67: 3786–3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch R. 1882. The aetiology of tuberculosis. National Tuberculosis Association, New York. [Google Scholar]
- Kwan CK, Ernst JD. 2011. HIV and tuberculosis: A deadly human syndemic. Clin Microbiol Rev 24: 351–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammas DA, Stober C, Harvey CJ, Kendrick N, Panchalingam S, Kumararatne DS. 1997. ATP-induced killing of mycobacteria by human macrophages is mediated by purinergic P2Z(P2X7) receptors. Immunity 7: 433–444. [DOI] [PubMed] [Google Scholar]
- Lazar-Molnar E, Chen B, Sweeney KA, Wang EJ, Liu W, Lin J, Porcelli SA, Almo SC, Nathenson SG, Jacobs WR Jr. 2010. Programmed death-1 (PD-1)-deficient mice are extraordinarily sensitive to tuberculosis. Proc Natl Acad Sci 107: 13402–13407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee PP, Chan KW, Jiang L, Chen T, Li C, Lee TL, Mak PH, Fok SF, Yang X, Lau YL. 2008. Susceptibility to mycobacterial infections in children with X-linked chronic granulomatous disease: A review of 17 patients living in a region endemic for tuberculosis. Pediatr Infect Dis J 27: 224–230. [DOI] [PubMed] [Google Scholar]
- Lin PL, Myers A, Smith L, Bigbee C, Bigbee M, Fuhrman C, Grieser H, Chiosea I, Voitenek NN, Capuano SV, et al. 2010. Tumor necrosis factor neutralization results in disseminated disease in acute and latent Mycobacterium tuberculosis infection with normal granuloma structure in a cynomolgus macaque model. Arthritis Rheum 62: 340–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin PL, Ford CB, Coleman MT, Myers AJ, Gawande R, Ioerger T, Sacchettini J, Fortune SM, Flynn JL. 2014. Sterilization of granulomas is common in active and latent tuberculosis despite within-host variability in bacterial killing. Nat Med 20: 75–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe DM, Redford PS, Wilkinson RJ, O’Garra A, Martineau AR. 2012. Neutrophils in tuberculosis: Friend or foe? Trends Immunol 33: 14–25. [DOI] [PubMed] [Google Scholar]
- Mahajan S, Dkhar HK, Chandra V, Dave S, Nanduri R, Janmeja AK, Agrewala JN, Gupta P. 2012. Mycobacterium tuberculosis modulates macrophage lipid-sensing nuclear receptors PPARγ and TR4 for survival. J Immunol 188: 5593–5603. [DOI] [PubMed] [Google Scholar]
- Middlebrook G, Dubos RJ, Pierce C. 1947. Virulence and morphological characteristics of mammalian tubercle bacilli. J Exp Med 86: 175–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra BB, Moura-Alves P, Sonawane A, Hacohen N, Griffiths G, Moita LF, Anes E. 2010. Mycobacterium tuberculosis protein ESAT-6 is a potent activator of the NLRP3/ASC inflammasome. Cell Microbiol 12: 1046–1063. [DOI] [PubMed] [Google Scholar]
- Molloy A, Laochumroonvorapong P, Kaplan G. 1994. Apoptosis, but not necrosis, of infected monocytes is coupled with killing of intracellular bacillus Calmette-Guerin. J Exp Med 180: 1499–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nandi B, Behar SM. 2011. Regulation of neutrophils by interferon-γ limits lung inflammation during tuberculosis infection. J Exp Med 208: 2251–2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North RJ, Jung YJ. 2004. Immunity to tuberculosis. Annu Rev Immunol 22: 599–623. [DOI] [PubMed] [Google Scholar]
- Oddo M, Renno T, Attinger A, Bakker T, MacDonald HR, Meylan PR. 1998. Fas ligand-induced apoptosis of infected human macrophages reduces the viability of intracellular Mycobacterium tuberculosis. J Immunol 160: 5448–5454. [PubMed] [Google Scholar]
- Odorizzi PM, Wherry EJ. 2012. Inhibitory receptors on lymphocytes: Insights from infections. J Immunol 188: 2957–2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onwueme KC, Vos CJ, Zurita J, Ferreras JA, Quadri LE. 2005. The dimycocerosate ester polyketide virulence factors of mycobacteria. Prog Lipid Res 44: 259–302. [DOI] [PubMed] [Google Scholar]
- Palaci M, Dietze R, Hadad DJ, Ribeiro FK, Peres RL, Vinhas SA, Maciel EL, do Valle Dettoni V, Horter L, Boom WH, et al. 2007. Cavitary disease and quantitative sputum bacillary load in cases of pulmonary tuberculosis. J Clin Microbiol 45: 4064–4066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parasa VR, Rahman MJ, Ngyuen Hoang AT, Svensson M, Brighenti S, Lerm M. 2014. Modeling Mycobacterium tuberculosis early granuloma formation in experimental human lung tissue. Dis Model Mech 7: 281–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks WC, Wilson CL, Lopez-Boado YS. 2004. Matrix metalloproteinases as modulators of inflammation and innate immunity. Nat Rev Immunol 4: 617–629. [DOI] [PubMed] [Google Scholar]
- Pedrosa J, Saunders BM, Appelberg R, Orme IM, Silva MT, Cooper AM. 2000. Neutrophils play a protective nonphagocytic role in systemic Mycobacterium tuberculosis infection of mice. Infect Immun 68: 577–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peggs KS, Quezada SA, Allison JP. 2009. Cancer immunotherapy: Co-stimulatory agonists and co-inhibitory antagonists. Clin Exp Immunol 157: 9–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyron P, Vaubourgeix J, Poquet Y, Levillain F, Botanch C, Bardou F, Daffe M, Emile JF, Marchou B, Cardona PJ, et al. 2008. Foamy macrophages from tuberculous patients’ granulomas constitute a nutrient-rich reservoir for M. tuberculosis persistence. PLoS Pathog 4: e1000204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philips JA, Ernst JD. 2012. Tuberculosis pathogenesis and immunity. Annu Rev Pathol 7: 353–384. [DOI] [PubMed] [Google Scholar]
- Price NM, Gilman RH, Uddin J, Recavarren S, Friedland JS. 2003. Unopposed matrix metalloproteinase-9 expression in human tuberculous granuloma and the role of TNF-α-dependent monocyte networks. J Immunol 171: 5579–5586. [DOI] [PubMed] [Google Scholar]
- Pym AS, Brodin P, Brosch R, Huerre M, Cole ST. 2002. Loss of RD1 contributed to the attenuation of the live tuberculosis vaccines Mycobacterium bovis BCG and Mycobacterium microti. Mol Microbiol 46: 709–717. [DOI] [PubMed] [Google Scholar]
- Ramakrishnan L. 2012. Revisiting the role of the granuloma in tuberculosis. Nat Rev Immunol 12: 352–366. [DOI] [PubMed] [Google Scholar]
- Ramakrishnan L. 2013. The zebrafish guide to tuberculosis immunity and treatment. Cold Spring Harb Symp Quant Biol 78: 179–192. [DOI] [PubMed] [Google Scholar]
- Ramakrishnan L, Valdivia RH, McKerrow JH, Falkow S. 1997. Mycobacterium marinum causes both long-term subclinical infection and acute disease in the leopard frog (Rana pipiens). Infect Immun 65: 767–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramakrishnan L, Federspiel NA, Falkow S. 2000. Granuloma-specific expression of mycobacterium virulence proteins from the glycine-rich PE-PGRS family. Science 288: 1436–1439. [DOI] [PubMed] [Google Scholar]
- Ravn P, Demissie A, Eguale T, Wondwosson H, Lein D, Amoudy HA, Mustafa AS, Jensen AK, Holm A, Rosenkrands I, et al. 1999. Human T cell responses to the ESAT-6 antigen from Mycobacterium tuberculosis. J Infect Dis 179: 637–645. [DOI] [PubMed] [Google Scholar]
- Rich AR. 1946. Pathogenesis of tuberculosis. Charles C. Thomas, Springfield, IL. [Google Scholar]
- Roach DR, Bean AG, Demangel C, France MP, Briscoe H, Britton WJ. 2002. TNF regulates chemokine induction essential for cell recruitment, granuloma formation, and clearance of mycobacterial infection. J Immunol 168: 4620–4627. [DOI] [PubMed] [Google Scholar]
- Roca FJ, Ramakrishnan L. 2013. TNF dually mediates resistance and susceptibility to mycobacteria via mitochondrial reactive oxygen species. Cell 153: 521–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell DG, Cardona PJ, Kim MJ, Allain S, Altare F. 2009. Foamy macrophages and the progression of the human tuberculosis granuloma. Nat Immunol 10: 943–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salamon H, Bruiners N, Lakehal K, Shi L, Ravi J, Yamaguchi KD, Pine R, Gennaro ML. 2014. Cutting edge: Vitamin D regulates lipid metabolism in Mycobacterium tuberculosis infection. J Immunol 193: 30–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarris M, Masson JB, Maurin D, Van der Aa LM, Boudinot P, Lortat-Jacob H, Herbomel P. 2012. Inflammatory chemokines direct and restrict leukocyte migration within live tissues as glycan-bound gradients. Curr Biol 22: 2375–2382. [DOI] [PubMed] [Google Scholar]
- Saunders BM, Cooper AM. 2000. Restraining mycobacteria: Role of granulomas in mycobacterial infections. Immunol Cell Biol 78: 334–341. [DOI] [PubMed] [Google Scholar]
- Scanga CA, Mohan VP, Yu K, Joseph H, Tanaka K, Chan J, Flynn JL. 2000. Depletion of CD4(+) T cells causes reactivation of murine persistent tuberculosis despite continued expression of interferon γ and nitric oxide synthase 2. J Exp Med 192: 347–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber HA, Harding JS, Hunt O, Altamirano CJ, Hulseberg PD, Stewart D, Fabry Z, Sandor M. 2011. Inflammatory dendritic cells migrate in and out of transplanted chronic mycobacterial granulomas in mice. J Clin Invest 121: 3902–3913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott HM, Flynn JL. 2002. Mycobacterium tuberculosis in chemokine receptor 2-deficient mice: Influence of dose on disease progression. Infect Immun 70: 5946–5954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheen P, O’Kane CM, Chaudhary K, Tovar M, Santillan C, Sosa J, Caviedes L, Gilman RH, Stamp G, Friedland JS. 2009. High MMP-9 activity characterises pleural tuberculosis correlating with granuloma formation. Eur Respir J 33: 134–141. [DOI] [PubMed] [Google Scholar]
- Sherman DR, Guinn KM, Hickey MJ, Mathur SK, Zakel KL, Smith S. 2004. Mycobacterium tuberculosis H37Rv: ΔRD1 is more virulent than M. bovis bacille Calmette-Guerin in long-term murine infection. J Infect Dis 190: 123–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simeone R, Bottai D, Brosch R. 2009. ESX/type VII secretion systems and their role in host-pathogen interaction. Curr Opin Microbiol 12: 4–10. [DOI] [PubMed] [Google Scholar]
- Singh V, Jamwal S, Jain R, Verma P, Gokhale R, Rao KV. 2012. Mycobacterium tuberculosis-driven targeted recalibration of macrophage lipid homeostasis promotes the foamy phenotype. Cell Host Microbe 12: 669–681. [DOI] [PubMed] [Google Scholar]
- Soehnlein O, Lindbom L. 2010. Phagocyte partnership during the onset and resolution of inflammation. Nat Rev Immunol 10: 427–439. [DOI] [PubMed] [Google Scholar]
- Spector WG. 1969. The granulomatous inflammatory exudate. Int Rev Exp Pathol 8: 1–55. [PubMed] [Google Scholar]
- Srivastava S, Ernst JD. 2014. Cell-to-cell transfer of M. tuberculosis antigens optimizes CD4 T cell priming. Cell Host Microbe 15: 741–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenger S. 2005. Immunological control of tuberculosis: Role of tumour necrosis factor and more. Ann Rheum Dis 64: iv24–iv28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugawara I, Udagawa T, Yamada H. 2004. Rat neutrophils prevent the development of tuberculosis. Infect Immun 72: 1804–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan JT, Young EF, McCann JR, Braunstein M. 2012. The Mycobacterium tuberculosis SecA2 system subverts phagosome maturation to promote growth in macrophages. Infect Immun 80: 996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaim LE, Connolly LE, Volkman HE, Humbert O, Born DE, Ramakrishnan L. 2006. Mycobacterium marinum infection of adult zebrafish causes caseating granulomatous tuberculosis and is moderated by adaptive immunity. Infect Immun 74: 6108–6117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo SJ, Sullivan BM, Peng SL, Glimcher LH. 2003. Molecular mechanisms regulating Th1 immune responses. Annu Rev Immunol 21: 713–758. [DOI] [PubMed] [Google Scholar]
- Takaki K, Cosma CL, Troll MA, Ramakrishnan L. 2012. An in vivo platform for rapid high-throughput antitubercular drug discovery. Cell Rep 2: 175–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor JL, Hattle JM, Dreitz SA, Troudt JM, Izzo LS, Basaraba RJ, Orme IM, Matrisian LM, Izzo AA. 2006. Role for matrix metalloproteinase 9 in granuloma formation during pulmonary Mycobacterium tuberculosis infection. Infect Immun 74: 6135–6144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thwaites GE, Chau TT, Stepniewska K, Phu NH, Chuong LV, Sinh DX, White NJ, Parry CM, Farrar JJ. 2002. Diagnosis of adult tuberculous meningitis by use of clinical and laboratory features. Lancet 360: 1287–1292. [DOI] [PubMed] [Google Scholar]
- Thye T, Scarisbrick G, Browne EN, Chinbuah MA, Gyapong J, Osei I, Owusu-Dabo E, Niemann S, Rusch-Gerdes S, Meyer CG, et al. 2009. CTLA4 autoimmunity-associated genotype contributes to severe pulmonary tuberculosis in an African population. PLoS ONE 4: e6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobin DM, Vary JC Jr, Ray JP, Walsh GS, Dunstan SJ, Bang ND, Hagge DA, Khadge S, King MC, Hawn TR, et al. 2010. The lta4 h locus modulates susceptibility to mycobacterial infection in zebrafish and humans. Cell 140: 717–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobin DM, Roca FJ, Oh SF, McFarland R, Vickery TW, Ray JP, Ko DC, Zou Y, Bang ND, Chau TT, et al. 2012. Host genotype-specific therapies can optimize the inflammatory response to mycobacterial infections. Cell 148: 434–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tubo NJ, Jenkins MK. 2014. CD4+ T cells: Guardians of the phagosome. Clin Microbiol Rev 27: 200–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda H, Howson JM, Esposito L, Heward J, Snook H, Chamberlain G, Rainbow DB, Hunter KM, Smith AN, Di Genova G, et al. 2003. Association of the T-cell regulatory gene CTLA4 with susceptibility to autoimmune disease. Nature 423: 506–511. [DOI] [PubMed] [Google Scholar]
- Urdahl KB, Shafiani S, Ernst JD. 2011. Initiation and regulation of T-cell responses in tuberculosis. Mucosal Immunol 4: 288–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Wel N, Hava D, Houben D, Fluitsma D, van Zon M, Pierson J, Brenner M, Peters PJ. 2007. M. tuberculosis and M. leprae translocate from the phagolysosome to the cytosol in myeloid cells. Cell 129: 1287–1298. [DOI] [PubMed] [Google Scholar]
- van der Woude AD, Stoop EJ, Stiess M, Wang S, Ummels R, van Stempvoort G, Piersma SR, Cascioferro A, Jimenez CR, Houben EN, et al. 2014. Analysis of SecA2-dependent substrates in Mycobacterium marinum identifies protein kinase G (PknG) as a virulence effector. Cell Microbiol 16: 280–295. [DOI] [PubMed] [Google Scholar]
- Velmurugan K, Chen B, Miller JL, Azogue S, Gurses S, Hsu T, Glickman M, Jacobs WR Jr, Porcelli SA, Briken V. 2007. Mycobacterium tuberculosis nuoG is a virulence gene that inhibits apoptosis of infected host cells. PLoS Pathog 3: e110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkman HE, Clay H, Beery D, Chang JC, Sherman DR, Ramakrishnan L. 2004. Tuberculous granuloma formation is enhanced by a mycobacterium virulence determinant. PLoS Biol 2: e367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkman HE, Pozos TC, Zheng J, Davis JM, Rawls JF, Ramakrishnan L. 2010. Tuberculous granuloma induction via interaction of a bacterial secreted protein with host epithelium. Science 327: 466–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallis RS, Schluger NW. 2010. Pulmonary infectious complications of tumor necrosis factor blockade. Infect Dis Clin North Am 24: 681–692. [DOI] [PubMed] [Google Scholar]
- Wang CH, Lin HC, Lin SM, Huang CD, Liu CY, Huang KH, Hsieh LL, Chung KF, Kuo HP. 2010. MMP-1(-1607G) polymorphism as a risk factor for fibrosis after pulmonary tuberculosis in Taiwan. Int J Tuberc Lung Dis 14: 627–634. [PubMed] [Google Scholar]
- Watkins BY, Joshi SA, Ball DA, Leggett H, Park S, Kim J, Austin CD, Paler-Martinez A, Xu M, Downing KH, et al. 2012. Mycobacterium marinum SecA2 promotes stable granulomas and induces tumor necrosis factor α in vivo. Infect Immun 80: 3512–3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells WF, Ratcliffe HL, Grumb C. 1948. On the mechanics of droplet nuclei infection; quantitative experimental air-borne tuberculosis in rabbits. Am J Hyg 47: 11–28. [DOI] [PubMed] [Google Scholar]
- Williams GT, Williams WJ. 1983. Granulomatous inflammation—A review. J Clin Pathol 36: 723–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf AJ, Linas B, Trevejo-Nunez GJ, Kincaid E, Tamura T, Takatsu K, Ernst JD. 2007. Mycobacterium tuberculosis infects dendritic cells with high frequency and impairs their function in vivo. J Immunol 179: 2509–2519. [DOI] [PubMed] [Google Scholar]
- Yang CT, Cambier CJ, Davis JM, Hall CJ, Crosier PS, Ramakrishnan L. 2012. Neutrophils exert protection in the early tuberculous granuloma by oxidative killing of mycobacteria phagocytosed from infected macrophages. Cell Host Microbe 12: 301–312. [DOI] [PMC free article] [PubMed] [Google Scholar]