

Abstract
Uncontrollable stress has been recognized to influence the hippocampus at various levels of analysis. Behaviorally, human and animal studies have found that stress generally impairs various hippocampal-dependent memory tasks. Neurally, animal studies have revealed that stress alters ensuing synaptic plasticity and firing properties of hippocampal neurons. Structurally, human and animal studies have shown that stress changes neuronal morphology, suppresses neuronal proliferation, and reduces hippocampal volume. Since the inception of stress research nearly 80 years ago, much focus has been on the varying levels of hypothalamic-pituitary-adrenal (HPA) axis neuroendocrine hormones, namely glucocorticoids, as mediators of the myriad stress effects on the hippocampus and as contributing factors to stress-associated psychopathologies such as post-traumatic stress disorder (PTSD). However, reports of glucocorticoid-produced alterations in hippocampal functioning vary widely across studies. This review provides a brief history of stress research, examines how the glucocorticoid hypothesis emerged and guides contemporary stress research, and considers alternative approaches to understanding the mechanisms underlying stress effects on hippocampal functioning.
Stress is a ubiquitous life experience that influences daily behaviors and well-being of the organisms, and in extreme circumstances, contributes to numerous psychopathologies in humans, including anxiety, PTSD, depression, schizophrenia, and drug use relapse (Widiger and Clark 2000). A common thread binding these disorders appears to be stress-associated alterations in cognitive processes, such as learning and memory (McEwen and Sapolsky 1995). In recent decades, human and animal research has shown that the hippocampus, a medial temporal lobe structure implicated in the formation of stable declarative (or explicit) memories, is highly susceptible to stress (Fig. 1; Kim and Diamond 2002). One prevailing view that has steered both basic and clinical stress research is that corticosteroids—synthesized and secreted by the hypothalamic-pituitary-adrenal (HPA) axis in response to stress (Fig. 2)—are mediators of stress effects on the hippocampus (for other neurochemical hypotheses of stress, see Maier 1984). For example, some human studies have sought to relate the severity of PTSD to cortisol levels (e.g., Yehuda et al. 1990). Here, we critically review the evidence that supports and conflicts with the glucocorticoid hypothesis of stress and consider the alternative hypotheses that have been proposed. From here on, the term stress will be used to signify uncontrollable (unpredictable and inescapable) stress (Kim and Diamond 2002), and stress effects will be focused mainly on the hippocampal physiology and mnemonic functions.
Figure 1.
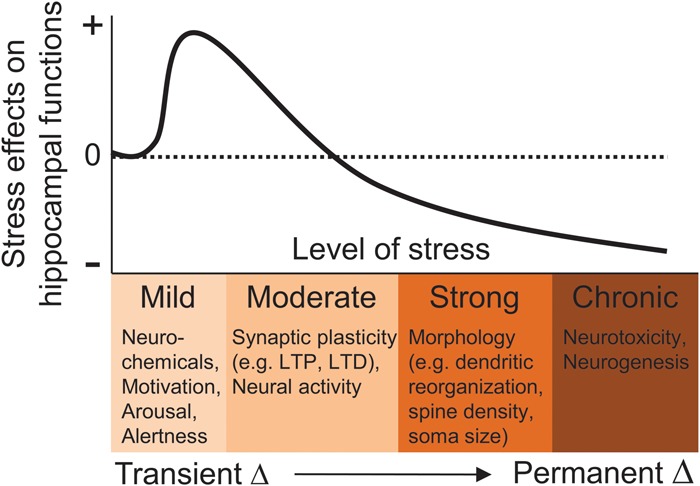
Biological effects of stress on the hippocampus. As the severity (intensity, duration) of stress increases, alterations in neurochemicals, synaptic plasticity, neural activity, cytoarchitecture, and neurogenesis occur in the hippocampus that can influence subsequent cognitive functions, such as learning and memory, and contribute to psychopathologies. + and − represent an increase and decrease in hippocampal functioning, respectively. Adapted from Kim and Yoon (1998).
Figure 2.
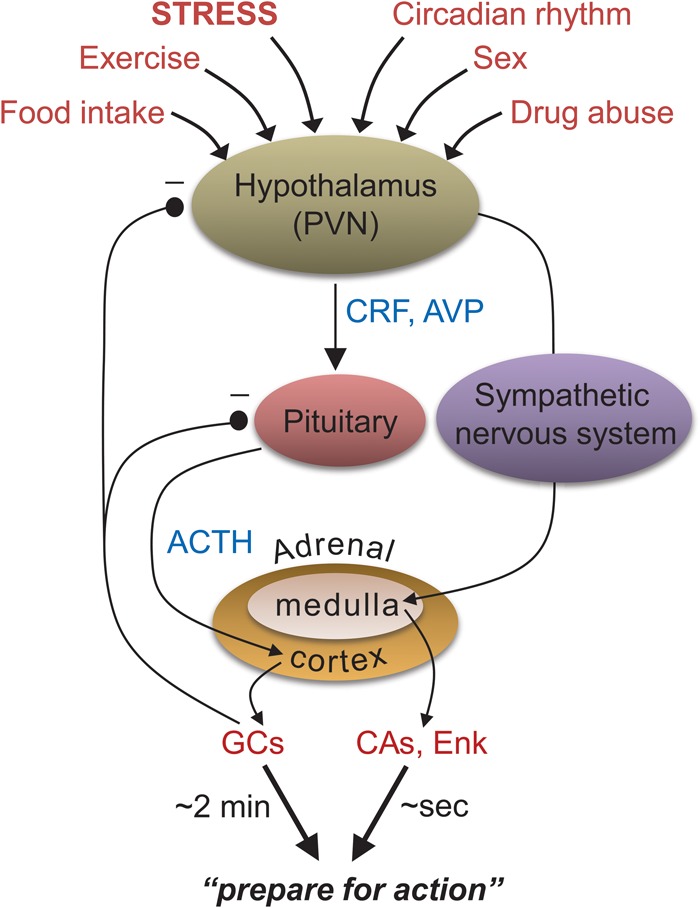
The hypothalamic-pituitary-adrenal (HPA) axis. A major neuroendocrine system associated with various bodily and behavioral activities. None of these compounds or structures responds uniquely to stress. (PVN) paraventricular nucleus of hypothalamus, (CRF) corticotropin-releasing factor, (AVP) arginine-vasopressin, (ACTH) adrenocorticotropic hormone, (GCs) glucocorticoids, (CAs) catecholamines, (Enk) enkephalins.
The scientific study of stress is widely associated with Hans Selye's (1936) report that dissimilar acute nocuous agents (stressors) produce stereotyped physiological effects (i.e., enlargement of the adrenal gland, shrinkage of the thymus, spleen lymph nodes, and gastric ulceration), which he termed “general adaptation syndrome” consisting of an alarm reaction stage, adaptation stage, and exhaustion stage (Fig. 3). In brief, this hypothesis posits that stress responses serve adaptive functions during the alarm and adaptation stages, but once the chemical substrates that normally fend off external insults are depleted (in severe and/or prolonged stress conditions), bodily functions become susceptible to diseases. However, the search for HPA axis-associated “stress compounds” that exhaust with unmitigated stress ultimately turned out to be unsubstantiated (e.g., Kosten et al. 1984).
Figure 3.
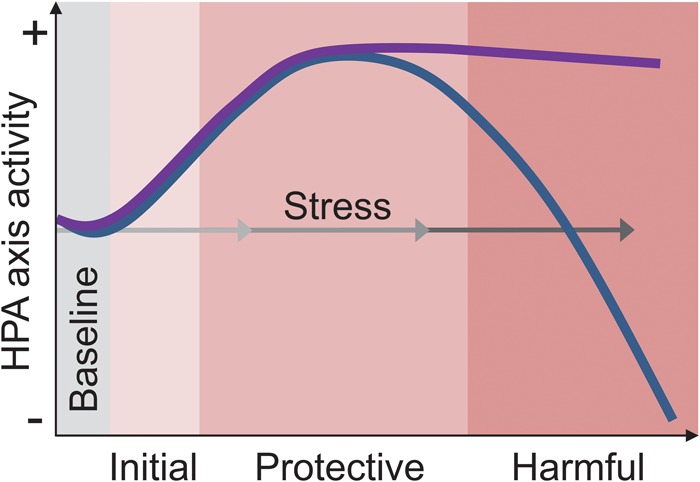
Two HPA axis-based models of stress. Selye's (1936) general adaptation syndrome proposed that the depletion of neuroendocrine factors with persistent stress makes the body susceptible to diseases (blue line). In contrast, current views hypothesize that abnormally elevated and/or long-lasting neuroendocrine activity causes harm to the body (purple line). The three gray-gradient arrows represent mild, moderate, and severe stress.
Succeeding Selye's notion of the general adaptation syndrome are the current glucocorticoid-based hypotheses (see McEwen and Sapolsky 1995). Here, the focus still remains on the HPA axis but instead, the glucocorticoid hypothesis proposes that it is the sustained elevation of the adrenal hormones, and not depletion as Selye originally proposed, that causes the detrimental effects of stress, somewhat analogous to autoimmune disease, where an overactive immune system damages the body. The glucocorticoid hypothesis of stress is particularly attractive with respect to hippocampal functioning because the hippocampus is densely concentrated with receptors for corticosteroids (cortisol in human, corticosterone in rodent; CORT), participates in glucocorticoid-mediated negative feedback of the HPA axis (McEwen and Sapolsky 1995), and is consequently susceptible to heightened CORT action. Hence, corticoids are often considered prima facie causes of stress effects on the hippocampus (Fig. 3).
Stress and memory
One of the earliest scientific accounts of the relationship between stress, learning, and psychopathology comes from Pavlov (1927), who reported that the use of difficult discriminating conditioned stimuli (e.g., circle versus ellipse) or intense/unpredictable stimuli deteriorated conditioned (learned) responses and caused behavioral disturbances in animals, which he described as “experimental neurosis.” Similarly, Yerkes and Dodson (1908) described an inverted-U function of decreased learning in difficult tasks as a function of shock intensity (see Diamond et al. 2007). In the late 1960s, Seligman, Maier, and Overmaier reported that dogs (and later cats, rats, fish, and humans) exposed to inescapable, but not escapable, shocks were impaired in subsequent escape-avoidance response learning, a phenomenon they termed learned helplessness (for review, see Maier and Seligman 1976). They proposed that when an organism internalizes that its action has no bearing on the aversive outcome (stressor), this learning produces changes in the cognitive, emotional, and motivational systems that impede subsequent learning. The learned helplessness phenomenon, where stress-associated learning negatively influences subsequent learning, in essence, is the negative counterpart to the concept of meta-learning in humans, which Maudsley (1979) described as “the process by which learners become aware of and increasingly in control of habits of perception, inquiry, learning, and growth that they have internalized.” In recent decades, much research has shown that stressful experiences can alter hippocampal mnemonic functioning in animals and humans.
The vast majority of studies have reported that exposures to stress or elevated levels of CORT impair performance on memory tasks dependent on the hippocampus (McEwen and Sapolsky 1995; Kim and Diamond 2002). In human studies, individuals diagnosed with PTSD and depression are impaired in various verbal recall tests (Bremner et al. 2000). The evidence for the direct role of CORT is based on findings of memory deficits in patients with Cushing's syndrome (with chronic hypercortisolemia; Starkman et al. 1992), and in healthy subjects administered with CORT (Newcomer et al. 1994). Similarly, rodent studies have shown that exposures to stress and injections of high doses of CORT produce deficits in spatial memory tasks that involve the hippocampus (de Quervain et al. 1998). Memory impairments have also been reported in transgenic mice with elevated CORT due to central overexpression of corticotropin-releasing factor (CRF; Heinrichs et al. 1996). Recent findings of stress altering the firing properties of place cells in the hippocampus (e.g., Kim et al. 2007), which are thought to support spatial navigation and memory (O'Keefe and Dostrovsky 1971), are consistent with the stress effects on spatial memory tasks.
However, stress does not produce global memory deficits as it has been shown to enhance cerebellum-dependent eyeblink conditioning (Beylin and Shors 2003) and amygdala- and hippocampus-dependent contextual fear conditioning, which is thought to be due to prior stress augmenting glucocorticoid release during training (Cordero et al. 2003). Interestingly, the same stress that impairs hippocampal memory tasks seems to enhance the relative use of competing nonhippocampal (e.g., the caudate-dependent stimulus-response) memory tasks in rats and humans (Kim et al. 2001; Schwabe and Wolf 2012). At present, it is unknown whether these differing effects on memory are indirect, i.e., the result of stress decreasing the hippocampus’ ability to interact (e.g., compete) with other brain–memory systems, or direct facilitatory effects on nonhippocampal memory systems.
Stress and hippocampal plasticity
Long-term potentiation (LTP), resulting from a brief high-frequency stimulation of afferent fibers initially demonstrated in the hippocampus, has characteristics desirable of a synaptic model of memory, such as rapid induction, longevity, stimulation threshold requirement (cooperativity), strengthening by repetition, input specificity, and associativity (see Bliss and Collingridge 1993). The first evidence linking LTP and stress was reported by Thompson, Levine and colleagues who found impairments in the Schaffer collateral/commissural-CA1 LTP in hippocampal slices prepared from rats that experienced 30 intermittent tailshocks during 30 min of restraint (Foy et al. 1987). Importantly, CA1 LTP was relatively normal in hippocampal slices from rats that received the same amount of tailshocks but were able to terminate them voluntarily (Shors et al. 1989), indicating that, similar to learned helplessness, the LTP impairment is largely due to the psychological, rather than physical, qualities of stress.
Stress-associated LTP impairments have also been demonstrated in the dentate gyrus (e.g., Shors and Dryver 1994) and CA3 regions of the hippocampus (e.g., Pavlides et al. 2002). Other studies have found time-dependent, biphasic effects of stress on LTP—an initial enhancing effect followed by a longer-lasting, suppressing effect on LTP (Akirav and Richter-Levin 1999). Stress has also been reported to enhance LTP induced by θ-burst stimulation but impair LTP induced by high-frequency stimulation. These findings indicate that differences in stress paradigms, in vitro versus in vivo recordings, and stimulation patterns produce a more complex picture of stress effects on LTP.
The discovery of a relationship between stress and hippocampal LTP is significant because it offers both a testable synaptic mechanism that may explain stress effects on memory and a “neurophysiological marker” to compare behavioral results from studies that use different stress paradigms. For example, not all putative stress paradigms would lead to LTP impairments in the hippocampus, and this would provide insights into the types of memory that may be influenced by different stress procedures. In theory, LTP alone is insufficient to provide an effective synaptic model of memory as decreases in synaptic strength are needed to normalize synaptic transmission and prevent LTP-inducing memory experiences from saturating synapses in the hippocampus. Long-term depression (LTD), characterized by a decrease in synaptic strength following different (i.e., low frequency) stimulation patterns of afferent fibers (Dudek and Bear 1992), is believed to work in conjunction with LTP to process and store information. The dynamic relationship between LTP and LTD with respect to stress is supported by findings that the same stress that impaired LTP enhanced LTD in the CA1 hippocampus, and that these effects are mediated via the activation of the N-methyl-d-aspartate (NMDA) receptors (Kim et al. 1996).
The opposing effects of stress on synaptic plasticity may be due to the possibility that during the stress experience, as the subject develops learned helplessness, LTP or LTP-like changes take place and saturate hippocampal synapses such that the hippocampus becomes unable to support further LTP, while the range for LTD has increased (Kim et al. 1996). An alternative possibility is that stress produces a “metaplastic” effect, such that the threshold for synaptic plasticity is biased toward LTD over LTP induction (Fig. 4; see Abraham and Tate 1997; Kim and Yoon 1998). At present, there is no definitive evidence supporting either hypothesis.
Figure 4.
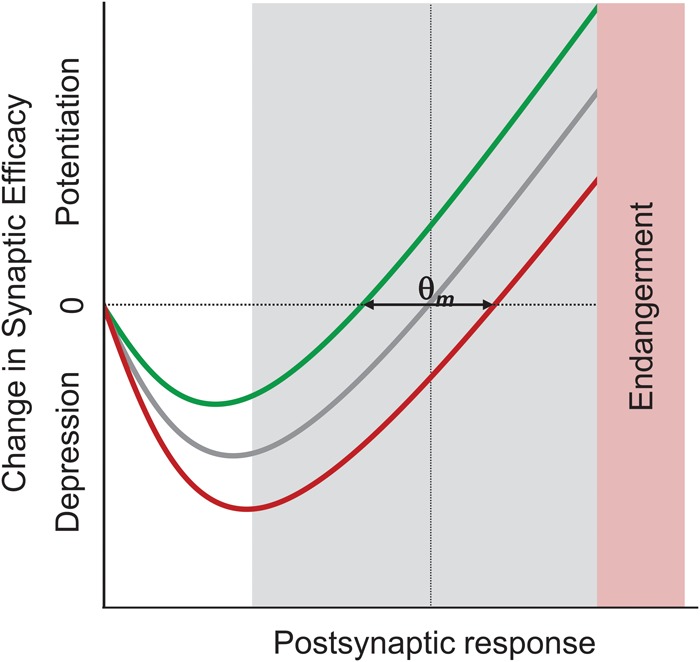
A metaplasticity model of stress effects on hippocampal synaptic plasticity. Based on the metaplasticity hypothesis, stress and glucocorticoids have been proposed to shift the dynamic range of depression and potentiation (represented by the modification threshold, θm) to the right such that subsequent synaptic plasticity favors LTD over LTP. The shaded area represents the physiological range of θm associated with the varying endogenous state (e.g., circadian rhythms, age, sex) of the animal. From Kim and Yoon (1998).
Stress effects on hippocampal morphology
Prolonged and/or traumatic stressors have also been shown to cause morphological changes in the hippocampus. In particular, and with significant clinical implications, human brain imaging studies have reported that PTSD patients had smaller hippocampal volume, which correlated with deficits in verbal memory (e.g., Bremner et al. 1995). Although a subsequent monozygotic twin study has suggested that reduced hippocampal volume is a predisposing factor for the development of PTSD following trauma (Gilbertson et al. 2002), a recent longitudinal MRI study in rats demonstrated that chronic restraint stress does cause a reduction in hippocampal volume from the prestress size (Lee et al. 2009). These effects are likely mediated by stress effects on neuronal morphology and/or neurogenesis in the hippocampus. Chronic restraint stress transiently reduces the number of dendritic spines and branches of pyramidal neurons in the CA3 (Conrad et al. 1999) and suppresses the production of new granule neurons in the dentate gyrus (Schoenfeld and Gould 2012) regions of the hippocampus. These structural changes to the hippocampus following stress appear to correspond to the deficits in spatial navigation and episodic memory produced by stress (Kim and Diamond 2002; Vyas et al. 2002).
Glucocorticoid hypothesis of stress: compatible and incompatible evidence
As mentioned in the Introduction, much basic and clinical stress research has focused on the varying levels of HPA axis neuroendocrine hormones, namely glucocorticoids, as causal factors of stress effects on the hippocampus (McEwen and Sapolsky 1995). Both the high-affinity Type-I mineralocorticoid receptors (MR) and the lower-affinity Type-II glucocorticoid receptors (GR) are abundant in the hippocampus, and there appears to be a dual relationship between the level of CORT and LTP, where both low (via adrenalectomy) and high (via acute administration) levels of CORT are associated with impaired LTP (Diamond et al. 1992). Subsequent studies have reported that selective activation of MRs increased LTP while further activation of GRs decreased LTP and enhanced LTD (e.g., Pavlides et al. 1995; see Vaher et al. 1994 for parallel effects on behaviors). These findings suggest that low levels of CORT enhance LTP through preferential stimulation of the high-affinity MRs, but during stress, CORT becomes high enough to saturate the low-affinity receptors, leading to the impairment of LTP (McEwen and Sapolsky 1995). In support, bath application of CORT has been reported to reduce CA1 LTP in hippocampal slices (e.g., Alfarez et al. 2002).
Prolonged exposures to CORT via injection, implantable pellets, or drinking water have also been shown to cause morphological and molecular changes, reduce neurogenesis, and impair synaptic plasticity in the hippocampus, physiological outcomes which are thought to precipitate hippocampus-dependent memory impairments and anxiety- and depression-like behaviors (Sterner and Kalynchuk 2010). These long-term effects of chronic CORT elevation on the hippocampus have been hypothesized to occur via epigenetic mechanisms (i.e., DNA methylation and histone modification) in the HPA axis (e.g., McEwen et al. 2012).
Recently, intra-hippocampal infusion of CORT has been shown to induce PTSD-like memory impairment; i.e., low levels of freezing to a shock-predicting context, but high levels of freezing to a nonshock-predicting discrete cue (Kaouane et al. 2012), implicating glucocorticoids in mediating detrimental effects of stress. CORT has also been shown to interact with catecholamines in the basolateral amygdala to intensify long-term memory consolidation of emotional events (Schwabe et al. 2012). However, CRF1 receptor-deficient mice (deficient in CORT elevation) showed normal fear learning (Tovote et al. 2005) and displayed decreased remote, but not recent, contextual fear memory (Thoeringer et al. 2012).
Once again, we emphasize that high CORT levels do not necessarily indicate high levels of stress (Fig. 2). If stress can indeed be reduced to changes in the level of corticosteroids, it follows that removing CORT elevation during stress and directly raising the CORT level in naïve animals should preclude and simulate stress effects, respectively. However, there are behavioral and neurophysiological studies that do not support this notion of a linear relationship between CORT level and stress magnitude (e.g., Shors et al. 1989; Woodson et al. 2003). Instead, the impairing effects of high levels of CORT on cognitive functions depend on the psychological conditions during which they are generated. For example, recent studies have found that while both stress and environmental enrichment significantly and comparably elevate CORT levels, they yield opposite effects on hippocampal neurogenesis (e.g., Schoenfeld and Gould 2012). Likewise, high CORT levels through aversive (exposure to a cat) but not appetitive (exposure to a female rat) conditions impair memory (Woodson et al. 2003). It is crucial to recognize the fact that CORT and other neurochemicals implicated in stress have multifaceted functions and none are known to respond uniquely to stress (i.e., lacks specificity), and thus no single one is likely to be a sufficient mediator of stress effects (Koolhaas et al. 2011). For example, if both stress and exercise elevate CORT but produce opposite physiological effects, in vitro hippocampal tissues challenged with bath-applied CORT are not representative of the hippocampus in animals challenged with stress. Although HPA axis hormones are highly responsive to stress, they are involved in regulating broad cellular metabolic processes, and thus to refer to them as stress hormones is a misnomer.
Systems-level analysis of stress effects on hippocampus
In recent years, the amygdala and prefrontal cortex (PFC) have been implicated in the development of stress effects on the hippocampus. Both structures have extensive sensory-defense circuit connections required to perform stress operations as they receive sensory inputs from various brain regions (e.g., thalamus and sensory cortex) and project to various structures associated with defensive responses (e.g., the mPFC is interconnected with the amygdala which projects to the bed nucleus of stria terminalis for activating stress hormones, the periaqueductal gray for protective behavior, and the lateral hypothalamus for sympathetic activation; see LeDoux 1996).
Earlier studies have found that amygdalar lesions prevent a range of bodily and behavioral functions impacted by stress (e.g., gastric erosions; Henke 1981). In agreement, amygdalar lesions and inactivation (via the GABAA receptor agonist muscimol) also blocked stress effects on hippocampal LTP and spatial memory in rats (Kim et al. 2001, 2005). Since immediate, post-stress muscimol infusions into the amygdala failed to prevent stress effects on LTP and memory, it appears the critical time window of amygdalar activity is during, and not after, stress (Kim et al. 2005). It should be mentioned that amygdalar lesions/inactivation blocked stress effects on hippocampal LTP and memory despite the increase in CORT levels (see also Shors et al. 1989). Recently, electrical stimulation of the amygdala was found to be sufficient in suppressing CA1 LTP (Vouimba and Richter-Levin 2005) and altering place cell activity (Kim et al. 2012). These findings suggest that the amygdala is a critical component of the central stress mechanism affecting hippocampal functioning.
The amygdala, which enhances glucocorticoid secretion in response to stress (LeDoux 1996) itself undergoes stress-induced changes in LTP and cytoarchitecture. In contrast to the hippocampus, stress enhances LTP and increases dendritic arborizations and spines of neurons in the amygdala, and these changes have been proposed as mechanisms underlying stress-associated anxiety disorders (Vyas et al. 2002). At present, however, it is unknown whether neurophysiological changes in the amygdala are necessary for stress-induced changes in the hippocampus or vice versa.
The PFC is another brain structure susceptible to stress effects (e.g., impairments in LTP, morphological changes in PFC neurons; Maroun and Richter-Levin 2003) and implicated in regulating the stress effects on the hippocampus. In particular, the medial PFC (mPFC) projects to both the amygdala and hippocampus through which the mPFC's “executive” level processing (Maier et al. 2006) can influence the hippocampus during stress. Consistent with this view, mPFC activity closely correlates with inhibition (or extinction) of aversively motivated behavior (see Maren and Quirk 2004). Together, these findings suggest that stress is comprised of multiple components and is unlikely to be represented by simple changes in neurochemical levels, and therefore a systems-level approach is required to understand the detrimental effects of uncontrollable stress on the hippocampus (Fig. 5).
Figure 5.
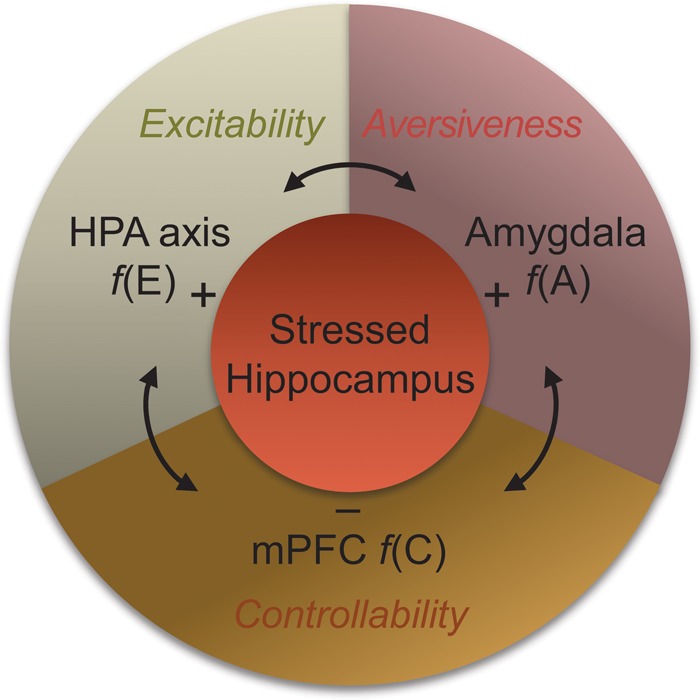
A systems-level model of stress. The complexity of stress is operationally defined to comprise heightened arousal or excitability [f(E)], perceived aversiveness [f(A)], and decreased controllability [f(C)], with the HPA axis, the amygdala, and the mPFC hypothesized as biological counterparts, respectively (Kim and Diamond 2002). The interaction between these components is thought to mediate stress effects on the hippocampus (and other brain structures).
Summary
Toward the twilight of his illustrious scientific career, Selye (1973) stated “stress is not that which causes a secretion by the adrenal cortex of its hormones (the corticoids). ACTH, the adrenal-stimulating pituitary hormone, can discharge these hormones without producing any evidence of stress.” However, contemporary stress research continues to be largely driven by the view that aberrant and/or prolonged levels of HPA axis hormones (e.g., glucocorticoids) are responsible for producing various neurobiological changes in the hippocampus that can potentially influence mnemonic functioning (e.g., McEwen and Sapolsky 1995) and underlie psychopathologies, such as PTSD (e.g., Yehuda et al. 1990). Although this approach has generated a wealth of information on hippocampal intracellular signaling cascades, synaptic plasticity, structural changes, cell death, and neurogenesis, it illuminates neither the psychological nor the biological complexity of stress. Furthermore, it is important to recognize that some aspects of stress effects on the hippocampus (and stress disorders in humans) may not show structural changes in neurons or alterations in particular chemical levels, but rather that stress may subtly alter neural computation of the brain, which can only be discovered through in vivo recording studies in behaving animals. Hence, it may be a time for stress research to shift its focus from the usual neurochemical emphasis to systems-level and neural computation approaches to capture the multifaceted nature of stress.
Acknowledgments
This work was supported by NIH grants MH64457 and MH099073 (J.J.K.). The authors declare no competing financial interests.
Footnotes
Article is online at http://www.learnmem.org/cgi/doi/10.1101/lm.037291.114.
References
- Abraham WC, Tate WP. 1997. Metaplasticity: a new vista across the field of synaptic plasticity. Prog Neurobiol 52: 303–323. [DOI] [PubMed] [Google Scholar]
- Akirav I, Richter-Levin G. 1999. Biphasic modulation of hippocampal plasticity by behavioral stress and basolateral amygdala stimulation in the rat. J Neurosci 19: 10530–10535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfarez DN, Wiegert O, Joëls M, Krugers HJ. 2002. Corticosterone and stress reduce synaptic potentiation in mouse hippocampal slices with mild stimulation. Neuroscience 115: 1119–1126. [DOI] [PubMed] [Google Scholar]
- Beylin AV, Shors TJ. 2003. Glucocorticoids are necessary for enhancing the acquisition of associative memories after acute stressful experience. Horm Behav 43: 124–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL. 1993. A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361: 31–39. [DOI] [PubMed] [Google Scholar]
- Bremner JD, Randall P, Scott TM, Bronen RA, Seibyl JP, Southwick SM, Delaney RC, McCarthy G, Charney DS, Innis RB. 1995. MRI-based measurement of hippocampal volume in patients with combat-related posttraumatic stress disorder. Am J Psychiatry 152: 973–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bremner JD, Narayan M, Anderson ER, Staib LH, Miller HL, Charney DS. 2000. Hippocampal volume reduction in major depression. Am J Psychiatry 157: 115–118. [DOI] [PubMed] [Google Scholar]
- Conrad CD, LeDoux JE, Magarinos AM, McEwen BS. 1999. Repeated restraint stress facilitates fear conditioning independently of causing hippocampal CA3 dendritic atrophy. Behav Neurosci 113: 902–913. [DOI] [PubMed] [Google Scholar]
- Cordero MI, Venero C, Kruyt ND, Sandi C. 2003. Prior exposure to a single stress session facilitates subsequent contextual fear conditioning in rats. Evidence for a role of corticosterone. Horm Behav 44: 338–345. [DOI] [PubMed] [Google Scholar]
- de Quervain DJ, Roozendaal B, McGaugh JL. 1998. Stress and glucocorticoids impair retrieval of long-term spatial memory. Nature 394: 787–790. [DOI] [PubMed] [Google Scholar]
- Diamond DM, Bennett MC, Fleshner M, Rose GM. 1992. Inverted-U relationship between the level of peripheral corticosterone and the magnitude of hippocampal primed burst potentiation. Hippocampus 2: 421–430. [DOI] [PubMed] [Google Scholar]
- Diamond DM, Campbell AM, Park CR, Halonen J, Zoladz PR. 2007. The temporal dynamics model of emotional memory processing: a synthesis on the neurobiological basis of stress-induced amnesia, flashbulb and traumatic memories, and the Yerkes-Dodson law. Neural Plast 2007: 60803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek SM, Bear MF. 1992. Homosynaptic long-term depression in area CA1 of hippocampus and effects of N-methyl-D-aspartate receptor blockade. Proc Natl Acad Sci 89: 4363–4367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foy MR, Stanton ME, Levine S, Thompson RF. 1987. Behavioral stress impairs long-term potentiation in rodent hippocampus. Behav Neural Biol 48: 138–149. [DOI] [PubMed] [Google Scholar]
- Gilbertson MW, Shenton ME, Ciszewski A, Kasai K, Lasko NB, Orr SP, Pitman RK. 2002. Smaller hippocampal volume predicts pathologic vulnerability to psychological trauma. Nat Neurosci 5: 1242–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinrichs SC, Stenzel-Poore MP, Gold LH, Battenberg E, Bloom FE, Koob GF, Vale WW, Pich EM. 1996. Learning impairment in transgenic mice with central overexpression of corticotropin-releasing factor. Neuroscience 74: 303–311. [DOI] [PubMed] [Google Scholar]
- Henke PG. 1981. Attenuation of shock-induced ulcers after lesions in the medial amygdala. Physiol Behav 27: 143–146. [DOI] [PubMed] [Google Scholar]
- Kaouane N, Porte Y, Vallée M, Brayda-Bruno L, Mons N, Calandreau L, Marighetto A, Piazza PV, Desmedt A. 2012. Glucocorticoids can induce PTSD-like memory impairments in mice. Science 335: 1510–1513. [DOI] [PubMed] [Google Scholar]
- Kim JJ, Diamond DM. 2002. The stressed hippocampus, synaptic plasticity and lost memories. Nat Rev Neurosci 3: 453–462. [DOI] [PubMed] [Google Scholar]
- Kim JJ, Yoon KS. 1998. Stress: metaplastic effects in the hippocampus. Trends Neurosci 21: 505–509. [DOI] [PubMed] [Google Scholar]
- Kim JJ, Foy MR, Thompson RF. 1996. Behavioral stress modifies hippocampal plasticity through N-methyl-D-aspartate receptor activation. Proc Natl Acad Sci 93: 4750–4753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JJ, Lee HJ, Han JS, Packard MG. 2001. Amygdala is critical for stress-induced modulation of hippocampal long-term potentiation and learning. J Neurosci 21: 5222–5228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JJ, Koo JW, Lee HJ, Han JS. 2005. Amygdalar inactivation blocks stress-induced impairments in hippocampal long-term potentiation and spatial memory. J Neurosci 25: 1532–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JJ, Lee HJ, Welday AC, Song E, Cho J, Sharp PE, Jung MW, Blair HT. 2007. Stress-induced alterations in hippocampal plasticity, place cells, and spatial memory. Proc Natl Acad Sci 104: 18297–18302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EJ, Kim ES, Park M, Cho J, Kim JJ. 2012. Amygdalar stimulation produces alterations on firing properties of hippocampal place cells. J Neurosci 32: 11424–11434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koolhaas JM, Bartolomucci A, Buwalda B, de Boer SF, Flügge G, Korte SM, Meerlo P, Murison R, Olivier B, Palanza P, et al. 2011. Stress revisited: a critical evaluation of the stress concept. Neurosci Biobehav Rev 35: 1291–1301. [DOI] [PubMed] [Google Scholar]
- Kosten TR, Jacobs S, Mason JW. 1984. The dexamethasone suppression test during bereavement. J Nerv Ment Dis 172: 359–360. [DOI] [PubMed] [Google Scholar]
- LeDoux J. 1996. Emotional networks and motor control: a fearful view. Prog Brain Res 107: 437–446. [DOI] [PubMed] [Google Scholar]
- Lee T, Jarome T, Li SJ, Kim JJ, Helmstetter FJ. 2009. Chronic stress selectively reduces hippocampal volume in rats: a longitudinal magnetic resonance imaging study. Neuroreport 20: 1554–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier SF. 1984. Learned helplessness and animal models of depression. Prog Neuropsychopharmacol Biol Psychiatry 8: 435–446. [PubMed] [Google Scholar]
- Maier SF, Seligman ME. 1976. Learned helplessness: theory and evidence. J Exp Psychol 105: 3–46. [Google Scholar]
- Maier SF, Amat J, Baratta MV, Paul E, Watkins LR. 2006. Behavioral control, the medial prefrontal cortex, and resilience. Dialogues Clin Neurosci 8: 397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maren S, Quirk GJ. 2004. Neuronal signalling of fear memory. Nat Rev Neurosci 5: 844–852. [DOI] [PubMed] [Google Scholar]
- Maroun M, Richter-Levin G. 2003. Exposure to acute stress blocks the induction of long-term potentiation of the amygdala-prefrontal cortex pathway in vivo. J Neurosci 23: 4406–4409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maudsley DB. 1979. A theory of meta-learning and principles of facilitation: an organismic perspective. University of Toronto, Toronto. [Google Scholar]
- McEwen BS, Sapolsky RM. 1995. Stress and cognitive function. Curr Opin Neurobiol 5: 205–216. [DOI] [PubMed] [Google Scholar]
- McEwen BS, Eiland L, Hunter RG, Miller MM. 2012. Stress and anxiety: structural plasticity and epigenetic regulation as a consequence of stress. Neuropharmacology 62: 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newcomer JW, Craft S, Hershey T, Askins K, Bardgett ME. 1994. Glucocorticoid-induced impairment in declarative memory performance in adult humans. J Neurosci 14: 2047–2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Keefe J, Dostrovsky J. 1971. The hippocampus as a spatial map. Preliminary evidence from unit activity in the freely-moving rat. Brain Res 34: 171–175. [DOI] [PubMed] [Google Scholar]
- Pavlides C, Watanabe Y, Magariños AM, McEwen BS. 1995. Opposing roles of type I and type II adrenal steroid receptors in hippocampal long-term potentiation. Neuroscience 68: 387–394. [DOI] [PubMed] [Google Scholar]
- Pavlides C, Nivón LG, McEwen BS. 2002. Effects of chronic stress on hippocampal long-term potentiation. Hippocampus 12: 245–257. [DOI] [PubMed] [Google Scholar]
- Pavlov IP. 1927. Conditioned reflexes: an investigation of the physiological activity of the cerebral cortex. Oxford UP, London. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenfeld TJ, Gould E. 2012. Stress, stress hormones, and adult neurogenesis. Exp Neurol 233: 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwabe L, Wolf OT. 2012. Stress modulates the engagement of multiple memory systems in classification learning. J Neurosci 32: 11042–11049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwabe L, Joëls M, Roozendaal B, Wolf OT, Oitzl MS. 2012. Stress effects on memory: an update and integration. Neurosci Biobehav Rev 36: 1740–1749. [DOI] [PubMed] [Google Scholar]
- Selye H. 1936. A syndrome produced by diverse nocuous agents. Nature 138: 32. [DOI] [PubMed] [Google Scholar]
- Selye H. 1973. The evolution of the stress concept. Am Sci 61: 692–699. [PubMed] [Google Scholar]
- Shors TJ, Dryver E. 1994. Effect of stress and long-term potentiation (LTP) on subsequent LTP and the θ burst response in the dentate gyrus. Brain Res 666: 232–238. [DOI] [PubMed] [Google Scholar]
- Shors TJ, Seib TB, Levine S, Thompson RF. 1989. Inescapable versus escapable shock modulates long-term potentiation in the rat hippocampus. Science 244: 224–226. [DOI] [PubMed] [Google Scholar]
- Starkman MN, Gebarski SS, Berent S, Schteingart DE. 1992. Hippocampal formation volume, memory dysfunction, and cortisol levels in patients with Cushing's syndrome. Biol Psychiatry 32: 756–765. [DOI] [PubMed] [Google Scholar]
- Sterner EY, Kalynchuk LE. 2010. Behavioral and neurobiological consequences of prolonged glucocorticoid exposure in rats: relevance to depression. Prog Neuropsychopharmacol Biol Psychiatry 34: 777–790. [DOI] [PubMed] [Google Scholar]
- Thoeringer CK, Henes K, Eder M, Dahlhoff M, Wurst W, Holsboer F, Deussing JM, Moosmang S, Wotjak CT. 2012. Consolidation of remote fear memories involves corticotropin-releasing hormone (CRH) receptor type 1-mediated enhancement of AMPA receptor GluR1 signaling in the dentate gyrus. Neuropsychopharmacology 37: 787–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovote P, Meyer M, Ronnenberg A, Ogren SO, Spiess J, Stiedl O. 2005. Heart rate dynamics and behavioral responses during acute emotional challenge in corticotropin-releasing factor receptor 1-deficient and corticotropin-releasing factor-overexpressing mice. Neuroscience 134: 1113–1122. [DOI] [PubMed] [Google Scholar]
- Vaher PR, Luine VN, Gould E, McEwen BS. 1994. Effects of adrenalectomy on spatial memory performance and dentate gyrus morphology. Brain Res 656: 71–78. [DOI] [PubMed] [Google Scholar]
- Vouimba RM, Richter-Levin G. 2005. Physiological dissociation in hippocampal subregions in response to amygdala stimulation. Cereb cortex 15: 1815–1821. [DOI] [PubMed] [Google Scholar]
- Vyas A, Mitra R, Shankaranarayana Rao BS, Chattarji S. 2002. Chronic stress induces contrasting patterns of dendritic remodeling in hippocampal and amygdaloid neurons. J Neurosci 22: 6810–6818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widiger TA, Clark LA. 2000. Toward DSM-V and the classification of psychopathology. Psychol Bull 126: 946–963. [DOI] [PubMed] [Google Scholar]
- Woodson JC, Macintosh D, Fleshner M, Diamond DM. 2003. Emotion-induced amnesia in rats: working memory-specific impairment, corticosterone-memory correlation, and fear versus arousal effects on memory. Learn Mem 10: 326–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yehuda R, Southwick SM, Nussbaum G, Wahby V, Giller EL Jr, Mason JW. 1990. Low urinary cortisol excretion in patients with posttraumatic stress disorder. J Nerv Ment Dis 178: 366–369. [DOI] [PubMed] [Google Scholar]
- Yerkes RM, Dodson JD. 1908. The relation of strength of stimulus to rapidity of habit-formation. J Comp Neurol 18: 459–482. [Google Scholar]