

Abstract
Encoding new information in the brain requires changes in synaptic strength. Neuromodulatory transmitters can facilitate synaptic plasticity by modifying the actions and expression of specific signaling cascades, transmitter receptors and their associated signaling complexes, genes, and effector proteins. One critical neuromodulator in the mammalian brain is norepinephrine (NE), which regulates multiple brain functions such as attention, perception, arousal, sleep, learning, and memory. The mammalian hippocampus receives noradrenergic innervation and hippocampal neurons express β-adrenergic receptors, which are known to play important roles in gating the induction of long-lasting forms of synaptic potentiation. These forms of long-term potentiation (LTP) are believed to importantly contribute to long-term storage of spatial and contextual memories in the brain. In this review, we highlight the contributions of noradrenergic signaling in general and β-adrenergic receptors in particular, toward modulating hippocampal LTP. We focus on the roles of NE and β-adrenergic receptors in altering the efficacies of specific signaling molecules such as NMDA and AMPA receptors, protein phosphatases, and translation initiation factors. Also, the roles of β-adrenergic receptors in regulating synaptic “tagging” and “capture” of LTP within synaptic networks of the hippocampus are reviewed. Understanding the molecular and cellular bases of noradrenergic signaling will enrich our grasp of how the brain makes new, enduring memories, and may shed light on credible strategies for improving mental health through treatment of specific disorders linked to perturbed memory processing and dysfunctional noradrenergic synaptic transmission.
Synaptic plasticity is a fundamental property of nervous system function. A neuron's ability to alter the strength of its synaptic connections is thought to be the foundation for multiple processes, including learning and memory. Synaptic plasticity is not a single, unitary cellular process, but rather an extremely diverse phenomenon that encompasses many forms and functions. Synapses are dynamic by nature, and only through elucidation of the mechanisms that govern their organization and reorganization can we hope to fully understand how the brain processes and stores information.
Acting as potent regulatory agents, neuromodulatory transmitters can significantly alter the properties of neurons at cellular and network levels. Specifically, the modulatory role of the noradrenergic system has been extensively characterized, in part because of the system's anatomically ubiquitous connections. Noradrenergic fibers originate mainly in the locus coeruleus (LC) and project widely throughout the forebrain, with dense innervation of the hippocampus, amygdala, and thalamus (Sara 2009). These connections, especially within the hippocampus, strongly modulate synaptic strength and neural network physiology, which lead to significant alterations in learning, memory, attention, and perception.
Noradrenergic receptor signaling
Norepinephrine (NE), the endogenous ligand for noradrenergic receptors, acts through G-protein-coupled receptors that activate distinct downstream signaling cascades depending on the receptor subtype. These can be broadly classified as α1-, α2-, β1-, and β2-adrenergic receptors (Gelinas and Nguyen 2007). Hippocampal pyramidal cells and dentate gyrus granule cells express all four receptor subtypes (Nicholas et al. 1993; Hillman et al. 2005; Guo and Li 2007). Interneurons also express α1- and β-adrenergic receptors (β-ARs), although there are substantial differences in levels of expression of β1 and β2 receptors in different types of interneurons (Papay et al. 2006; Cox et al. 2008). The α adrenergic receptors are coupled to Gq or Gi type G proteins and appear to have a role in both memory and synaptic plasticity in some brain regions (for reviews, see Gelinas and Nguyen 2007; Tully and Bolshakov 2010). In contrast, β-ARs signal via activation of Gs type G proteins, followed by stimulation of adenylyl cyclase and increased production of intracellular cAMP. Cyclic-AMP activates cAMP-dependent protein kinase (PKA) and, indirectly, extracellular signal-regulated protein kinase (ERK) through Rap1 (a GTPase) and B-Raf (a protein kinase) (Schmitt and Stork 2000). In addition, β-ARs can also activate ERK through a G protein-independent mechanism mediated by β-arrestin, an adaptor protein that associates with ligand bound β-ARs and recruits ERK and its upstream activators to the receptor (Luttrell et al. 1999; Shukla et al. 2011). PKA and ERK both have critical roles in long-term memory formation and long-term synaptic plasticity in numerous species, including mammals, and they are thought to have a central role in the memory enhancing effects of β-AR activation (Barros et al. 1999; Kandel 2001; Nguyen and Woo 2003; Sweatt 2004).
β-ARs account for the memory-promoting effects of NE
Heightened states of emotional arousal and attention have a powerful effect on memory formation (McGaugh 2013). This is related, in part, to activation of the brain's noradrenergic system, thereby promoting plasticity in brain regions that mediate enduring behavioral adaptations (Harley 1991; Bouret and Sara 2005). Indeed, acute inhibition of β-ARs with the specific antagonist propranolol significantly impairs the ability of human subjects to remember emotionally charged stories, while having no effect on neutral ones (Cahill et al. 1994; Chamberlain and Robbins 2013). As the amygdala and hippocampus are activated during the acquisition and retrieval of emotional memories, respectively, and as both structures contain β-adrenergic receptors, it is not surprising that propranolol blocked the increased amygdalar and hippocampal activity following the encoding and recall, respectively, of emotional stimuli (Strange and Dolan 2004). Thus, β-ARs are critical for neural processing of emotionally charged memories.
Elucidating the detailed mechanisms by which NE promotes memory processing requires the use of animal models which allow for the direct manipulation of the noradrenergic system and the LC. The LC can enhance processing of polymodal sensory input (Bouret and Sara 2005), primarily through its facilitation of synaptic plasticity. The hippocampus is a major processor of polymodal inputs and is densely innervated by noradrenergic fibers from the LC. Behavioral studies indicate that LC stimulation or intrahippocampal NE injection promotes retrieval of spatial and associative memories, and that this requires activation of β-ARs (Devauges and Sara 1991; Przybyslawski et al. 1999; Sara et al. 1999). Conversely, propranolol inhibits spatial memory (Ji et al. 2003b) and long-term, but not short-term, contextual fear memory (Ji et al. 2003a) when infused into the hippocampus, further implicating β-ARs in hippocampal memory consolidation (but see Murchison et al. 2004, Debiec and Ledoux 2004; Qi et al. 2008 for evidence that propranolol does not alter performance in contextual fear memory). Furthermore, Izquierdo et al. (1998) found that direct NE injection into rodent CA1 selectively enhanced long-term memory of the inhibitory avoidance task. Interestingly, knockout mice lacking NE (dopamine β-hydroxylase null mutants) display impaired contextual fear memory; however, the memory can be rescued by restoration of NE just prior to memory testing (Murchison et al. 2004). This rescue required activation of hippocampal β-ARs, but not α-ARs (Murchison et al. 2004). Hippocampal infusion of isoproterenol (ISO), a β-AR receptor agonist, also rescued memory in NE-knockout mice (Murchison et al. 2004). Thus, β-AR signaling is critical for contextual memory retrieval in NE-knockout mice.
β-adrenergic receptors facilitate long-lasting hippocampal synaptic plasticity
Norepinephrine acting through β-ARs not only enhances memory formation but also has powerful effects on the induction of LTP at excitatory synapses throughout the hippocampus (O'Dell et al. 2010). The effects of noradrenergic receptor activation on LTP induction have been extensively studied at Schaeffer collateral fiber synapses onto pyramidal cells in the hippocampal CA1 region. At these synapses, repeated trains of high-frequency stimulation (HFS; usually 3–4 trains of 100 Hz stimulation) induce a translation- and transcription-dependent potentiation of synaptic strength that can last for 3 or more hours (Stanton and Sarvey 1984; Frey et al. 1988; Nguyen et al. 1994; Kelleher et al. 2004; Sutton and Schuman 2006; Abraham and Williams 2008). This translation/transcription-dependent form of LTP, often called L-LTP, is correlated with the formation of hippocampus-dependent, enduring memories (Abel et al. 1997; Bourtchouladze et al. 1998; Schafe et al. 1999; Scharf et al. 2002; Costa-Mattioli et al. 2007, 2009). Less robust HFS protocols, such as a single train of 100 Hz stimulation, typically induce a shorter-lasting potentiation of synaptic transmission (lasting ∼1–2 h) that does not require translation or transcription. Although noradrenergic receptor activation is not required for the induction of LTP by strong stimulation protocols (Swanson-Park et al. 1999; Murchison et al. 2004), activation of β-ARs potently facilitates the induction of LTP by weaker patterns of synaptic stimulation. For example, β-AR activation enables the induction of protein synthesis-dependent L-LTP by a single 100-Hz train of HFS (Gelinas and Nguyen 2005; Gelinas et al. 2007; Ma et al. 2011). Moreover, θ-pulse stimulation (TPS; 150–900 stimulation pulses delivered at 5–10 Hz), a pattern of synaptic stimulation that alone has little or no lasting effect on synaptic strength, can induce LTP if delivered in the presence of β-AR agonists (Thomas et al. 1996; Katsuki et al. 1997; Moody et al. 1998; Winder et al. 1999; Brown et al. 2000; Gelinas and Nguyen 2005; Havekes et al. 2012; Qian et al. 2012). The induction of LTP by spike-timing-dependent plasticity protocols, where excitatory postsynaptic potentials elicited by presynaptic fiber stimulation are paired with postsynaptic action potentials evoked by current injection through an intracellular recording electrode, is also enhanced by β-AR activation (Lin et al. 2003; Seol et al. 2007; Makino et al. 2011).
The overall picture that emerges from studies of β-AR modulation of LTP in the hippocampus is that β-AR lowers the threshold for LTP induction, enabling the induction of LTP by weak patterns of synaptic activity that alone have no lasting effect on synaptic transmission, and facilitating the induction of L-LTP by patterns of synaptic stimulation that induce shorter lasting, protein synthesis-independent LTP. The relative contribution of β1 versus β2-ARs in the enhancement of LTP induction by NE and ISO is unclear, however an early study found that the induction of LTP by TPS in the presence of ISO was impaired in β1-AR but not β2-AR null mutant mice (Winder et al. 1999), indicating a key role for β1-ARs. In contrast, a subsequent study found that the enhancement of TPS-induced LTP by ISO is impaired in both β1-AR and β2-AR knockout mice (Qian et al. 2012). Moreover, the induction of LTP by TPS in the presence of ISO in hippocampal slices from wild-type mice was blocked by the selective β2-AR antagonist ICI-118551 while the β1-AR selective antagonist CGP-20712 had no effect (Qian et al. 2012). The different outcomes reported by Winder et al. (1999) and Qian et al. (2012) may, in part, be due to strain differences in the mice used for these experiments: the knockouts of Winder et al. (1999) were primarily on an FVL strain background, whereas those used by Qian et al. (2012) were backcrossed to C57BL/6. Moreover, Qian et al. (2012) found that paired-pulse facilitation at Schaffer collateral fiber synapses in the hippocampal CA1 region is reduced in both β1-AR and β2-AR knockout mice, indicating alterations in presynaptic function. In addition, levels of the postsynaptic adaptor protein Shank are increased in β1-AR mutants while levels of NMDA receptor GluN2B subunits are increased in β2-AR mutant mice (Qian et al. 2012). Thus, compensatory and other changes in gene expression associated with null mutations in genes encoding β1 and β2-ARs complicate the interpretation of the plasticity phenotypes in these mutants. Yet another possible contributor to the differential outcomes is quality control of receptor antagonist production by suppliers (Qian et al. 2012). In any event, these studies underscore the sensitivity of noradrenergic receptor studies to genetic and empirical factors that deserve closer scrutiny.
Molecular mechanisms underlying the enhancement of LTP by β-AR activation
At the molecular level, β-AR activation has the ability to regulate the signaling events underlying activity-dependent forms of synaptic plasticity in two distinct ways. As discussed in more detail below, this includes direct effects mediated by phosphorylation of ligand- and voltage-gated ion channels involved in the induction and expression of LTP, as well as regulating proteins responsible for controlling the synthesis of proteins needed for long-term maintenance of LTP. Importantly, one of these direct downstream targets of β-AR signaling is a protein known as inhibitor-1 (Fig. 1; Brown et al. 2000). When phosphorylated by PKA, inhibitor-1 binds to, and suppresses, protein phosphatase 1, which in turn facilitates activation of protein kinases, such as CaMKII, required for induction of LTP (Blitzer et al. 1998). Down-regulation of protein phosphatase signaling thus provides a second mode of modulation whereby β-AR activation can act in a more global, albeit indirect, way to enhance LTP induction by facilitating protein phosphorylation mediated by multiple serine/threonine protein kinases. Consistent with this notion, bath application of membrane permeable inhibitors of protein phosphatase 1 or injection of phosphorylated inhibitor-1 into CA1 pyramidal cells mimics the ability of β-AR activation to enable the induction of LTP by TPS (Thomas et al. 1996; Brown et al. 2000).
Figure 1.
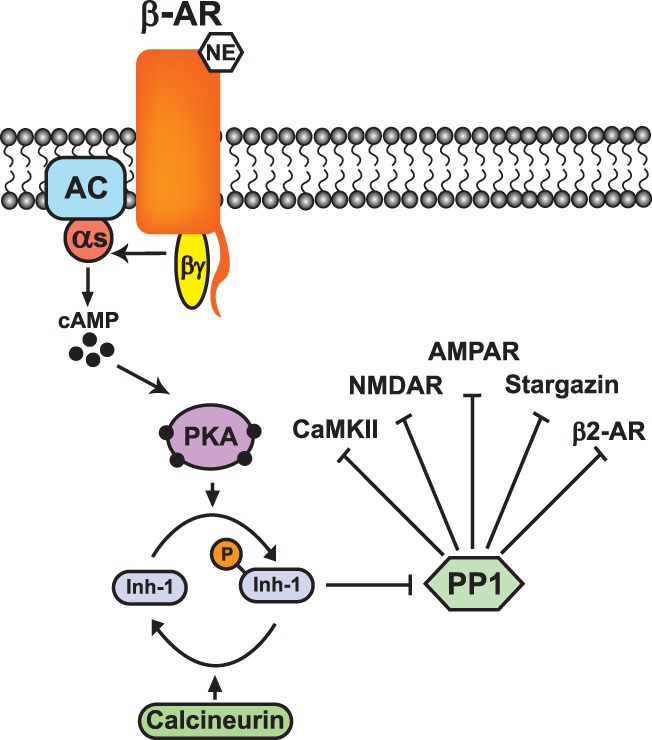
Down-regulation of protein phosphatase 1 following β-AR activation. Protein phosphatase 1 dephosphorylates a number of synaptic proteins with important roles in LTP induction including CaMKII, NMDARs, and stargazin as well as β2-ARs. The activity of protein phosphatase 1 (PP1) at synapses is regulated by inhibitor-1 (Inh-1), a regulatory protein that is phosphorylated by PKA and dephosphorylated by protein phosphatase 2B (calcineurin). Inhibitor-1 phosphorylation by PKA following β-AR activation leads to an inhibition of protein phosphatase 1 thereby facilitating protein phosphorylation needed for LTP induction.
β-adrenergic receptor modulation of NMDA receptor signaling
In many regions of the brain, including the hippocampus, activation of PKA or β-ARs enhances N-methyl-D-aspartate receptor (NMDAR)-mediated responses (Cerne et al. 1993; Huang et al. 1993; Raman et al. 1996; Blank et al. 1997; Westphal et al. 1999; Bird et al. 2005). Given the central role of NMDAR activation in the induction of LTP, this suggests that modulation of NMDAR function has a pivotal role in the ability of β-AR activation to enhance LTP induction. NMDARs are assembled from four subunits: two obligatory GluN1 subunits and two additional, GluN2A, 2B, 2C, or 2D subunits (Traynelis et al. 2010). In the hippocampus and other forebrain regions, essentially all NMDARs are heteromeric dimers or trimers containing GluN1 subunits and either GluN2A and/or GluN2B subunits (Monyer et al. 1994; Gray et al. 2011). Importantly, all three of these NMDAR subunits are phosphorylated by PKA at sites that regulate NMDAR ion channel function (Fig. 2; Leonard and Hell 1997; Tingley et al. 1997; Murphy et al. 2014). For example, PKA phosphorylation of two sites in the cytoplasmic C-terminal tail of GluN2A subunits (serine 900 and 929) and at serine 1166 in the C-terminus of GluN2B subunits increases channel open probability (Krupp et al. 2002; Maki et al. 2013; Aman et al. 2014). In addition to effects on channel gating, PKA activation also enhances the Ca2+ permeability of NMDAR ion channels (Skeberdis et al. 2006), an effect mediated by phosphorylation of GluN1 subunits at serine 897 and GluN2B subunits at serine 1166 (Aman et al. 2014; Murphy et al. 2014).
Figure 2.
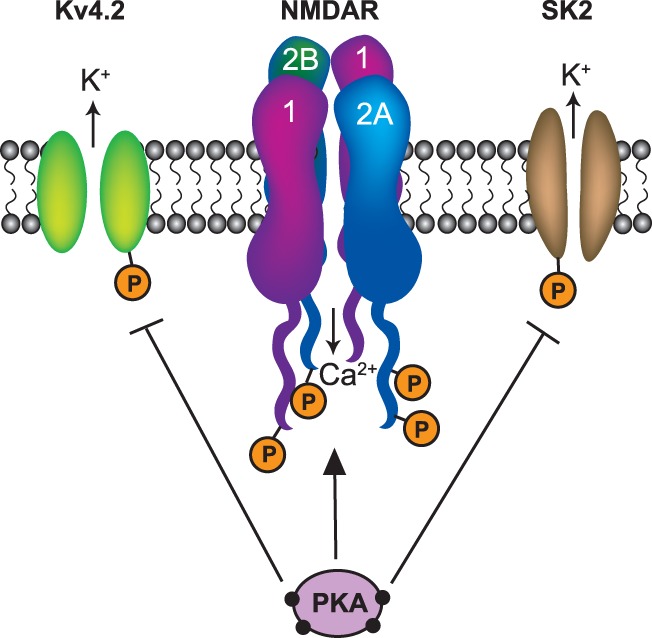
Modulation of NMDAR activity mediated by PKA activation downstream from β-AR activation. PKA activation enhances NMDAR activity by phosphorylating sites in the cytoplasmic C-terminal tails of NMDAR GluN1, GluN2A, and GluN2B subunits. In addition, PKA phosphorylates and inhibits Kv4.2 and SK2 type potassium channels present in dendritic spines, thereby enhancing NMDAR activation by facilitating spine depolarization at active synapses.
Although PKA activation enhances multiple aspects of NMDAR function, it is unclear to what degree direct modulation of NMDARs contributes to the enhancement of LTP by β-AR activation. The induction of LTP by TPS in the presence of ISO is not disrupted in mice expressing truncated GluN2A subunits that lack the entire cytoplasmic C-terminus (Moody et al. 2011). Thus, facilitation of LTP induction by β-AR activation does not require phosphorylation of GluN2A at serine 900 and/or serine 929, suggesting that PKA phosphorylation sites in GluN1 or GluN2B might have a more important role. Consistent with this, β-AR activation increases GluN1 phosphorylation at serine 897 (Murphy et al. 2014) and LTP in the hippocampal CA1 region is strongly reduced in “knock-in” mutant mice expressing GluN1 subunits where serine 897 is replaced with a nonphosphorylatable alanine (Li et al. 2009). However, these mutants also have deficits in basal NMDAR and AMPA receptor-mediated synaptic transmission and the ability of β-AR activation to facilitate LTP induction has not yet been tested. The potential contribution of GluN2B phosphorylation at Ser1166 in the modulation of LTP by β-ARs has also not yet been directly tested. However, β-AR activation induces a robust increase in GluN2B phosphorylation and enhances NMDAR-mediated Ca2+ transients in dendritic spines of hippocampal neurons (Murphy et al. 2014). Thus, PKA-mediated phosphorylation of NMDAR GluN2B subunits may have an especially important role in the modulation of LTP induction by β-ARs.
NMDAR activation is not only dependent on glutamate binding but also requires postsynaptic membrane depolarization to relieve the voltage-dependent Mg2+ ion block of the NMDAR ion channel (Mayer et al. 1984; Nowak et al. 1984). Importantly, β-AR activation has long been known to enhance the excitability of hippocampal CA1 pyramidal cells (for example, see Madison and Nicoll 1982, 1986; Haas and Konnerth 1983; Dunwiddie et al. 1992). Thus, in addition to direct effects on NMDAR ion channel gating and permeability mediated by PKA, β-AR receptor activation could also facilitate NMDAR activation by enhancing membrane depolarization at active synapses (Fig. 2). For example, Kv4.2 subunit-containing A-type potassium channels and small conductance, Ca2+-activated SK2 channels are present in postsynaptic dendritic spines and their activation strongly limits spine depolarization induced by excitatory postsynaptic potentials (Ngo-Anh et al. 2005; Bloodgood and Sabatini 2007; Kim et al. 2007; Kim and Hoffman 2008; Lin et al. 2008; Wang et al. 2014). In addition, the back-propagation of somatically generated action potentials into pyramidal cell dendrites is another potentially important source of postsynaptic depolarization needed for NMDAR activation and LTP induction (Watanabe et al. 2002) and dendritically localized Kv4.2/A-type potassium channels can strongly attenuate action potential back-propagation (Hoffman et al. 1997; Chen et al. 2006). β-AR activation inhibits both SK2 and Kv4.2 potassium channels through effects mediated by PKA and ERK (Hoffman and Johnston 1999; Yuan et al. 2002; Faber et al. 2008), thus providing a mechanism whereby β-AR activation can boost spine depolarization at active synapses and facilitate LTP induction by enhancing NMDAR activation.
β-Adrenergic receptor modulation of AMPA receptors in LTP
AMPA receptors (AMPARs) are generated by the combination of four subunit proteins known as GluA1, GluA2, GluA3, and GluA4 (Traynelis et al. 2010). In the hippocampus, the vast majority (80% or more) of synaptic and extrasynaptic AMPARs are heteromeric receptors containing GluA1 and GluA2 subunits with GluA2/GluA3 subunit-containing receptors accounting for essentially all of the remaining receptors (Wenthold et al. 1996; Lu et al. 2009). Unlike NMDARs, which contain multiple PKA phosphorylation sites, the only site phosphorylated by PKA in GluA1, GluA2, and GluA3 subunits is found at serine 845 in the cytoplasmic C-terminal domain of GluA1 (Traynelis et al. 2010; Lu and Roche 2012). Phosphorylation of this single site, however, regulates multiple aspects of AMPAR function including channel gating (Roche et al. 1996; Banke et al. 2000; Jenkins et al. 2014) and receptor trafficking (Ehlers 2000; Man et al. 2007). Phosphorylation of GluA1 at serine 845 also triggers the insertion of AMPARs into extrasynaptic sites where the receptors appear to be primed for synaptic insertion during LTP induction (Esteban et al. 2003; Oh et al. 2006). Trafficking of AMPARs from this extrasynaptic pool into synapses appears to have a crucial role in LTP (Granger et al. 2013), suggesting that β-AR activation may enhance LTP, at least in part, by increasing the pool of membrane AMPA receptors that are available for synaptic insertion following LTP induction.
Consistent with the notion that AMPARs are an important downstream target of β-AR signaling at synapses, GluA1 subunits are readily phosphorylated at serine 845 following β-AR activation (Vanhoose and Winder 2003; Vanhoose et al. 2006; Tenorio et al. 2010; Moody et al. 2011; Gray et al. 2014). Indeed, through protein interactions mediated by the scaffolding protein PSD-95 and transmembrane AMPAR regulatory proteins (TARPs), GluA1 subunit-containing AMPARs form a complex with β2-adrenergic receptors (Fig. 3; Joiner et al. 2010). This complex also contains PKA and adenylyl cyclase (Joiner et al. 2010), which are recruited into the complex by A kinase anchoring proteins (AKAPs), a family of adaptor proteins that can interact with PSD-95 (Colledge et al. 2000). Thus, β-adrenergic receptors, AMPARs, PKA, and adenylyl cyclase assemble into a large, multicomponent signaling complex that can enable rapid and localized AMPAR phosphorylation following β-AR activation. In cultured hippocampal neurons, peptide inhibitors that disrupt the association of either β2-ARs or AMPARs with PSD-95 block the increase in GluA1 serine 845 phosphorylation induced by β-AR activation (Joiner et al. 2010). Moreover, GluA1 serine 845 phosphorylation induced by β-AR activation in hippocampal neurons is disrupted in mice with a null mutation in the AKAP5 gene, which encodes AKAP79/150 (Zhang et al. 2013). The induction of LTP by TPS in the presence of ISO is also abolished in AKAP5 mutants (Zhang et al. 2013) and in hippocampal slices from wild-type mice that are exposed to membrane-permeant peptide inhibitors that disrupt PKA binding to AKAPs (Havekes et al. 2012). Thus, β-AR signaling complexes are crucial for both AMPAR phosphorylation and facilitation of LTP induction. The induction of LTP by TPS in the presence of ISO is also disrupted in AKAP12 mutant mice (Havekes et al. 2012). Basal levels of GluA1 serine 845 phosphorylation appear to be normal in these mutants and the deficits in β-AR enhancement of LTP may stem from disruption of ERK signaling downstream of β-AR activation (Havekes et al. 2012). Together, these findings indicate that targeting of PKA and other signaling molecules to β-AR signaling complexes is critical for the enhancement of LTP by β-AR agonists, perhaps by providing a mechanism that ensures rapid and efficient phosphorylation of AMPAR GluA1 subunits and other signaling molecules following β-AR activation.
Figure 3.
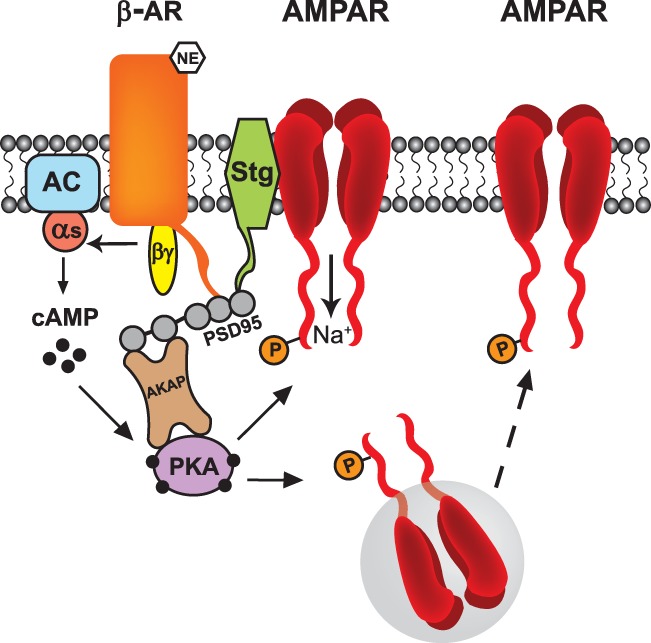
β-AR signaling complexes and regulation of AMPARs. The scaffolding protein PSD-95 interacts with the AMPAR-associated protein stargazin (stg), β-ARs, and AKAPs to form a signaling complex. PKA is recruited into the complex via interactions with AKAPs, thus enabling rapid and efficient phosphorylation of AMPARs following β-AR activation. Phosphorylation of AMPARs by PKA enhances ion channel open probability and facilitates the trafficking of AMPAR to the plasma membrane.
Perhaps the strongest evidence implicating AMPAR modulation in the mechanisms underlying β-AR enhancement of LTP comes from experiments using genetically modified mice with knock-in mutations that disrupt specific GluA1 phosphorylation sites. For example, the induction of LTP in the hippocampal CA1 region by TPS in the presence of NE is abolished in mutant mice expressing GluA1 subunits where serine 845 and serine 831 (a site phosphorylated by protein kinase C and CaMKII) have been replaced with nonphosphorylatable alanines (Hu et al. 2007). Strikingly, the ability of systemic epinephrine injections to enhance contextual fear learning is also disrupted in these mutants, suggesting that AMPAR phosphorylation has a key role in the enhancement of memory formation by emotional arousal as well as in β-AR modulation of LTP (Hu et al. 2007). Other experiments have found that the β-AR facilitation of LTP induction at excitatory synapses onto pyramidal cells in visual cortex (Seol et al. 2007) and hippocampal CA1 (Qian et al. 2012) is also abolished in mice expressing GluA1 subunits with a single amino acid substitution that specifically blocks serine 845 phosphorylation. This indicates that serine 845 is required for β-adrenergic modulation of LTP induction. Finally, LTP induction is enhanced and the effects of β-AR activation on LTP are occluded in knock-in mutants where serine 845 (and 831) is replaced with aspartate to mimic phosphorylation (Makino et al. 2011).
Given the broad impact that β-AR activation has on multiple components of the molecular mechanisms underlying LTP, the fact that disruption of a single phosphorylation site in one protein can completely disrupt β-AR enhancement of LTP induction seems surprising. One possibility is that modulation of AMPAR function represents a key point of convergence for all of these signaling pathways. In other words, β-AR activation-induced increases in neuronal excitability and NMDAR activation, as well as modulation of protein kinase and protein phosphatase signaling, all ultimately lead to an enhancement of LTP induction by regulating AMPAR phosphorylation. This AMPAR-centric view has recently been challenged, however, by results indicating that levels of AMPAR GluA1 subunit phosphorylation at serine 845 in the hippocampus are extremely low (Hosokawa et al. 2015). In these experiments, quantitative biochemical approaches were used to measure the stoichiometry of AMPAR subunit phosphorylation at different sites. Remarkably, this analysis revealed that in the hippocampus <0.02% of GluA1 is basally phosphorylated at serine 845. Assuming that synapses contain ∼25 GluA1/GluA2 hetero-tetrameric receptors (Nusser et al. 1998; Sheng and Hoogenraad 2007), and thus 50 GluA1 subunits, the low levels of GluA1 phosphorylation at serine 845 found by Hosokawa et al. (2015) indicate that, on average, an AMPAR phosphorylated at S845 is found in only ∼1 out of every 100 synapses. Although β-AR activation can induce a two- to fourfold increase in GluA1 serine 845 phosphorylation (for examples, see Vanhoose and Winder 2003; Vanhoose et al. 2006; Tenorio et al. 2010; Moody et al. 2011; Gray et al. 2014), such low basal levels of phosphorylation suggest that even following β-AR activation only 0.04%–0.08% of all GluA1 subunits will be phosphorylated at serine 845. Importantly, although the vanishingly low levels of basal GluA1 phosphorylation have raised doubts about the role of AMPAR phosphorylation in synaptic plasticity, an earlier study using different approaches found that basal levels of GluA1 serine 845 phosphorylation are much higher (15% or more) (Oh et al. 2006). Clearly, more work is needed to reconcile the disparate findings coming from physiological and biochemical studies of the role of AMPAR phosphorylation in mediating the modulation of LTP induction by β-ARs.
The role of ERK signaling in β-adrenergic receptor modulation of LTP
In addition to the canonical cAMP/PKA pathway, β-AR activation is likely to engage other downstream signaling pathways that contribute to the potent modulatory effects of these receptors on LTP induction. For example, β-AR activation triggers activation of the ERK family of MAP kinases in hippocampal neurons (Roberson et al. 1999; Winder et al. 1999) and pharmacological inhibitors of MEK, the upstream protein kinase responsible for phosphorylating and activating ERK, block the induction of LTP by TPS delivered in the presence of ISO (Winder et al. 1999; Giovannini et al. 2001). Importantly, the ability of TPS protocols to induce LTP is highly dependent on complex spike-like postsynaptic action potential bursting (Thomas et al. 1998). Thus, the ERK-mediated down-regulation of dendritic potassium channels and enhancement of action potential back-propagation in dendrites induced by β-AR activation (Yuan et al. 2002) is likely to have a key role in the enhancement of TPS-induced LTP by β-ARs. Moreover, as discussed below, ERK-mediated changes in protein synthesis are also likely to have a significant role in the β-AR modulation of LTP. A number of important questions remain, however, regarding the role of ERK signaling in mediating the modulatory effects of β-AR activation on LTP induction. For example, although ERK can facilitate cAMP/PKA signaling by phosphorylating and inhibiting some forms of type-4 phosphodiesterases (Hoffmann et al. 1999; Song et al. 2013), the potential contribution of this mechanism to β-AR modulation of LTP has not been tested. In addition, although ERK activation by β-arrestin-dependent β-AR signaling appears to contribute to behavioral learning (Liu et al. 2015), the role of this G-protein-independent form of β-AR signaling in modulating LTP has not yet been investigated.
β-adrenergic receptor modulation of protein synthesis in LTP
Although β-AR activation activates signaling pathways leading to phosphorylation of synaptic proteins known to have key roles in the induction and early phases of LTP maintenance, activation of these receptors also enhances the induction of long-lasting, protein synthesis-dependent LTP by both modest high-frequency synaptic stimulation and TPS (Gelinas and Nguyen 2005; Gelinas et al. 2007; Ma et al. 2011). This indicates that regulation of protein synthesis also contributes to the enhancement of LTP by β-AR activation.
One important process where various signals and transmitters can act to regulate protein synthesis is at the level of translation initiation, a rate-limiting step in the translation of most species of mRNA. Here, different protein kinases act to phosphorylate eukaryotic initiation factors (eIFs) involved in the assembly of translation initiation complexes that promote mRNA binding to ribosomal proteins (Costa-Mattioli et al. 2009). For example, two key protein kinases, ERK and mammalian target of rapamycin (mTOR), participate in the formation of the eukaryotic initiation factor 4F (eIF4F) complex (Kelleher et al. 2004; Klann et al. 2004; Gelinas et al. 2007; Tsokas et al. 2007). The eIF4F initiation complex is assembled from the initiation factors eIF4A, 4E, and 4G (Fig. 4). In the basal state, formation of this complex is restrained by the binding of eIF4E to the inhibitory protein 4E-binding protein (4E-BP) (Banko et al. 2005). Phosphorylation of 4E-BP by mTOR triggers the release of eIF4E which is then able to associate with eIF4G and form the eIF4F initiation complex (Fig. 4). In addition, ERK can phosphorylate and activate the protein kinase Mnk1 (MAPK signal-integrating kinase-1), which in turn phosphorylates 4E to further enhance translation. Importantly, several of these steps are known to be involved in activity-dependent changes in protein synthesis involved in L-LTP (for reviews, see Kelleher et al. 2004; Klann et al. 2004). For example, blocking 4E binding to 4G disrupts consolidation of associative fear memory (Hoeffer et al. 2011) and impairs translation-dependent LTP (Hoeffer et al. 2013). These same sites of regulation also appear to have a crucial role in the enhancement of L-LTP induction by β-AR activation (Fig. 4). The induction of L-LTP by weaker, more modest patterns of HFS delivered in the presence of ISO is blocked by ERK and mTOR inhibitors and is associated with increased phosphorylation of the Mnk1 substrate, eIF4E, and the mTOR substrate, p70-S6 kinase (Gelinas et al. 2007; Ma et al. 2011). Moreover, β-AR activation also enables activity-dependent increases in 4E-BP phosphorylation, as well as increases in eIF4F complex formation (Gelinas et al. 2007). Interestingly, ISO induces a robust increase in dendritic levels of phosphorylated 4E-BP in hippocampal pyramidal cells (Gelinas et al. 2007), suggesting that β-AR regulates activity-dependent changes in local dendritic protein synthesis.
Figure 4.
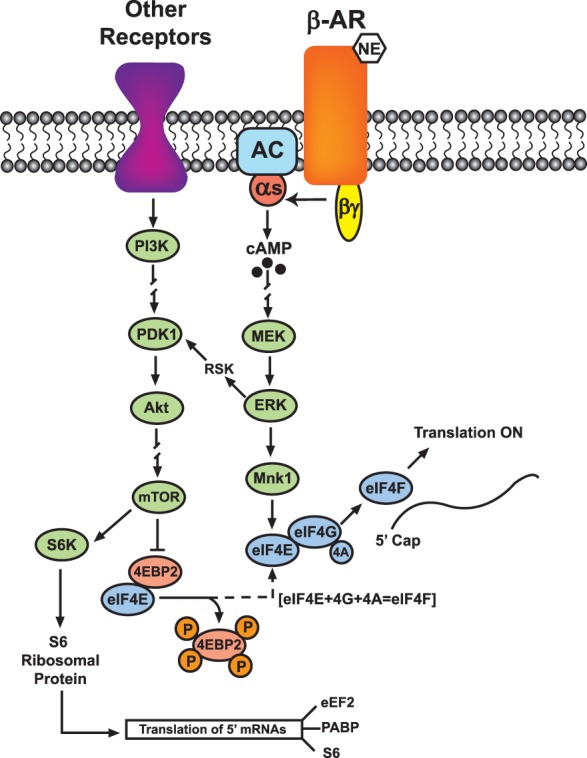
Translational control by ERK and mTOR. Activation of β-ARs during synaptic stimulation promotes translation initiation through ERK and mTOR pathways. mTOR phosphorylates and inhibits 4E-BP2 (4E-binding protein 2), releasing eukaryotic initiation factor 4E (eIF4E) from repression by 4E-BP2. eIF4E assists translation initiation by binding to eIF4G to form the initiation complex eIF4F. mTOR also activates S6 kinase (S6K), which phosphorylates ribosomal protein S6 to increase synthesis of translation regulatory proteins such as eukaryotic elongation factor 2 (eEF2), poly(A) binding protein (PABP), and S6 itself. ERK may cross-talk with the mTOR pathway via ribosomal S6 kinase (RSK), phosphoinositide-dependent kinase-1 (PDK1), and protein kinase-B (Akt). Diagram is simplified.
An intriguing feature of β-AR modulation of LTP is that the LTP-enhancing effects of β-AR activation persist for long periods of time after transient β-AR activation (Hu et al. 2007; Seol et al. 2007; Tenorio et al. 2010). This suggests that β-AR activation induces a form of metaplasticity (see Abraham and Bear 1996; Abraham 2008) that converts synapses into a long-lasting, highly plastic state. Transient β-AR activation increases neuronal excitability and elevates AMPAR phosphorylation for an hour or more (Dunwiddie et al. 1992; Vanhoose and Winder 2003; Tenorio et al. 2010), suggesting that persistent down-regulation of potassium channels and/or changes in AMPAR trafficking might be involved. However, β-AR modulation of protein synthesis is likely to have a central role in this form of metaplasticity. For example, weak high-frequency stimulation induces L-LTP even when delivered 1 h after transient activation of β-ARs (Tenorio et al. 2010). Strikingly, the metaplastic enhancement of LTP was blocked when protein synthesis inhibitors were applied during β-AR activation but not when protein synthesis inhibitors were applied during HFS delivered 1 h after β-AR activation. These findings are consistent with the induction of a “silent” (i.e., ISO application does not have any lasting effect on baseline synaptic transmission) up-regulation of translation triggered by β-ARs that primes synapses for future plasticity. It remains to be determined which proteins are up-regulated during β-AR-mediated metaplasticity. Given that the endogenous β-AR ligand, NE, is secreted during novelty, arousal, and spatial exploration (Sara 2009), the enduring reduction in the threshold for L-LTP induction triggered by β-AR activation may create an ideal synaptic state for facilitating memory formation. NE may both enhance immediate encoding of ongoing experience as well as assist with incorporating temporally disparate aspects of that experience into coherent memories, processes that are prime targets for metaplastic mechanisms.
β-adrenergic receptor activation and synaptic tagging
A neuron in the CNS typically receives thousands of synaptic contacts, and yet, changes in synaptic strength can be input-specific and are spatially restricted. L-LTP requires protein synthesis, the products of which may be transported in a cell-wide manner. In order to preserve the input specificity of L-LTP, a mechanism is needed to mark, or tag, active synapses. These tagged synapses can then capture, and utilize, newly synthesized proteins (Frey and Morris 1997, 1998). Transient potentiation resulting from weaker synaptic activation could be prolonged to resemble L-LTP if paired with established L-LTP induced at separate inputs (Frey and Morris 1997). Thus, heterosynaptic transfer of L-LTP (i.e., expression of L-LTP at synapses experiencing weak activation that normally leads only to transient potentiation) can occur through tagging and capture of plasticity-related proteins (PRPs) (for review, see Redondo and Morris 2011). In principle, neuromodulatory receptors may enhance memory processing across spatially distinct synaptic networks by expanding the temporal window for synaptic tagging and capture.
The mechanism responsible for how synapses are selected for use in memory formation is a key question in the field. Initial data attempting to address this question revealed that synaptic tagging and capture (STC) of PRPs generated by strong HFS protocols (usually consisting of multiple trains of 100 Hz stimulation) might enable neural networks to enhance memory formation over time while maintaining the synaptic specificity that is presumed necessary for retaining mnemonic fidelity. Importantly, the incorporation of synapses into a memory through STC is subject to the degradation rate of both PRPs and synaptic tags (Frey and Morris 1998; Young and Nguyen 2005). Additionally, neuromodulators can affect the production of PRPs as well as the setting of synaptic tags at activated synapses (Moncada and Viola 2007; Navakkode et al. 2010; Wang et al. 2010). For example, β-AR activation coincident with HFS initiates the synthesis of PRPs which facilitates heterosynaptic transfer of L-LTP (Connor et al. 2011a,b). Pairing HFS with ISO facilitated the induction of L-LTP that could be captured by an independent set of synapses activated with weaker TPS delivered 30 min prior to or after HFS in the presence of ISO. Heterosynaptic capture was prevented by blocking β-ARs or by genetic suppression of PKA activation (Young et al. 2006), suggesting that PKA plays a role in capturing PRPs at recently stimulated synapses (Young et al. 2006; see also Connor et al. 2011b). Preventing GluA2 endocytosis by applying a peptide inhibitor of endocytosis during TPS at a “weak” pathway extended the time window for heterosynaptic transfer of L-LTP, suggesting that processes governing AMPAR endocytosis may be rate-limiting for capture of L-LTP (Connor et al. 2011b). Inhibiting β-ARs with propranolol, or blocking translation during HFS, also prevented heterosynaptic transfer of L-LTP, likely by preventing PRP synthesis (Connor et al. 2011b). However, shifting application of emetine (a translation inhibitor) to overlap with TPS did not block capture of L-LTP (Connor et al. 2011b), indicating that the critical period of PRP synthesis necessary for enabling subsequent capture of L-LTP occurred during ISO plus HFS treatment. These results are consistent with β-AR activation as helping to trigger PRP synthesis that might bolster synapse-based association of events over time.
Understanding how β-ARs contribute to synaptic tagging and metaplasticity may have clinical relevance in light of data demonstrating altered β-AR-dependent heterosynaptic LTP in a Fragile-X Syndrome (FXS) mouse model (Connor et al. 2011a). Fmr1 knockout mice lack fragile-X mental retardation protein (FMRP), which acts as a repressor of translation at synapses (Laggerbauer et al. 2001). Inducing heterosynaptic LTP in Fmr1 KO mice yielded LTP that was sensitive to inhibition of ERK, but not to inhibition of mTOR (Connor et al. 2011a). The mTOR inhibitor rapamycin failed to block the induction of homosynaptic β-AR-dependent LTP in Fmr1 KO mouse hippocampus, consistent with an overproduction of PRPs in the absence of FMRP. Taken together, these data suggest that targeting β-ARs or their downstream effectors may provide novel therapeutic avenues for treating specific disorders that may be causally linked to altered synaptic plasticity.
β-adrenergic receptors are a potential therapeutic target for treating memory disorders
Alterations in noradrenergic neurotransmission may contribute to a number of disorders including Alzheimer's disease, post-traumatic stress disorder (PTSD), attention deficit disorder, and depression (Berridge and Waterhouse 2003). For example, degeneration of noradrenergic neurons in the locus coeruleus occurs early in Alzheimer's disease (AD) (Szot et al. 2006; Grudzien et al. 2007; Takahashi et al. 2015) and soluble oligomers of amyloid beta (Aβ) proteins can induce a rapid down-regulation and internalization of β2-ARs (Wang et al. 2011; Li et al. 2013). In addition, activation of β2-ARs has been shown to prevent the inhibition of LTP by soluble Aβ at excitatory synapses in both the dentate gyrus (Wang et al. 2009) and the hippocampal CA1 region (Li et al. 2013). Activation of β2-ARs may also have a key role in the ability of environmental enrichment to reduce the effects of Aβ peptides on synaptic plasticity (Li et al. 2013). Thus, enhancing noradrenergic receptor signaling may represent a useful approach for treatment of early cognitive impairment in AD (Chalermpalanupap et al. 2013).
Alterations in noradrenergic transmission have also been implicated in the underlying pathology of post-traumatic stress disorder (PTSD), with chronic PTSD patients exhibiting elevated levels of urinary and plasma norepinephrine levels and exacerbated noradrenergic responses to psychological stressors (McFall et al. 1990; Yehuda et al. 1992; Strawn and Geracioti 2008). PTSD is characterized by hyper-arousal, avoidance behavior, and recurrence of intrusive memories (Steckler and Risbrough 2012), though precisely how norepinephrine contributes to symptomology in PTSD remains unclear. Blocking β-ARs with propranolol inhibits the enhancement of memory formation by emotionally charged stimuli in healthy human subjects (Cahill et al. 1994; Lonergan et al. 2013), suggesting that excessive noradrenergic receptor activation during traumatic experiences might contribute to the development of PTSD. Moreover, memories are actively updated during recollection and may recapitulate the cellular and molecular processes of de novo memory formation before being stored again, in a process known as “reconsolidation” (Nader et al. 2000). β-AR activation can facilitate reconsolidation of fear memories, suggesting that heightened levels of noradrenergic receptor activation could also lead to facilitated reconsolidation of traumatic memories (Debiec et al. 2011). Importantly, during reconsolidation, memories become labile and can be disrupted by a number of different pharmacological manipulations (Tronson and Taylor 2007), including inhibition of β-ARs (Przybyslawski et al. 1999; Debiec and Ledoux 2004; Debiec et al. 2011). This suggests that blocking β-ARs during memory reconsolidation might offer a potentially beneficial treatment for PTSD. Indeed, propranolol inhibits reconsolidation in healthy human subjects (Kindt et al. 2009; Schwabe et al. 2012) and post-trauma treatments with propranolol can significantly reduce symptoms in patients suffering from PTSD (Vaiva et al. 2003; Brunet et al. 2011). Other studies, however, found little, if any, therapeutic benefit of propranolol treatment in trauma patients (Stein et al. 2007; McGhee et al. 2009; Hoge et al. 2012). There are numerous differences between these studies that could account for these conflicting results including dose, time to first treatment, duration of treatment, method of recall inducement, and the level of trauma experienced by patients. Moreover, work in animal models suggests that the ability of propranolol to inhibit memory reconsolidation may be limited to specific types of memory (e.g., Muravieva and Alberini 2010). Thus, more research is required to validate the broad applicability of β-AR blockers as a treatment for PTSD.
Concluding remarks: a buffet of possibilities
We have given a selective overview of the cellular and molecular mechanisms by which the noradrenergic neuromodulatory system regulates synaptic plasticity in the hippocampus. Clearly, multiple targets for NE's actions exist. These include cell surface NMDARs and AMPARs, protein kinases and phosphatases, and translation initiation factors. Continued integration of biochemical, cellular electrophysiological, and genetic approaches will be needed to address key questions that can further define, at the synaptic level, the roles of β-ARs and NE in sculpting complex brain functions such as learning and memory. For example, does β-AR trafficking occur during learning? If so, does such trafficking collaborate or synergize with the well-documented dynamics of AMPARs and NMDARs? How much do β-ARs contribute toward basal versus HFS-evoked levels of AMPAR phosphorylation? Do these levels reflect a dynamic interplay between protein kinases and phosphatases anchored at AMPARs? Which particular mRNAs show increased translation rates following activation of β-ARs during L-LTP? This is an important question, as it ultimately will address the issue of which proteins are synthesized de novo and subsequently incorporated into existing networks of effector proteins. Can β-ARs in hippocampal neurons signal to the nucleus to trigger transcriptional changes and/or epigenetic modifications that contribute to L-LTP?
It will also be important to use computational, systems, and behavioral approaches, to address the noradrenergic system's roles in gating behavioral and neuronal network adaptations. A prime directive of the noradrenergic system is to reorganize neural network activity in a way that enables rapid behavioral adaptation in the face of environmental challenges. Although our review has not covered systems neurophysiology and behavioral neuroscience in depth, it is clear that the endpoint of all of the molecular and cellular machineries discussed here is the generation of productive behavioral actions. A major challenge for researchers is to elucidate how β-ARs cooperate with other neuromodulatory receptors to fine-tune goal-directed behaviors in an intact organism. Research on synaptic tagging and capture of plasticity-related proteins at the cellular level has advanced to the stage where we can now seek out exact behavioral “equivalents,” and consequences, of tagging and capture. Genetic block or enhancement of expression of specific “tags” (e.g., β-ARs, AKAPs, CaMKII, or AMPAR subunits such as GluA1), or block of capture of proteins, in definable neural networks can be used to gauge the behavioral consequences of such manipulations. Also, the translational potential (“bench to bedside”) of β-AR blockers, functional modifiers of AKAPs, or AMPAR agonists, as treatments for PTSD and dementia-associated memory loss, needs to be explored at the interface between systems neurophysiology and behavioral neuroscience. It may turn out that multiple strategies, enveloping pharmacological, and behavioral therapies, will be optimal for treating these very complex disorders. Given the ubiquitous influences of the noradrenergic system on a broad range of brain functions such as arousal, perception, attention, learning, and memory, we have no doubts about the challenges, but also the important opportunities, that lie ahead of us.
Acknowledgments
Research funding was received from National Institute of Mental Health (T.J.O.) and the Canadian Institutes of Health Research (P.V.N.). S.A.C. was supported by a Queen Elizabeth II scholarship from the University of Alberta, and a Graduate Scholarship from the Natural Sciences and Engineering Research Council of Canada. R.G. was supported by an ARCS Foundation Scholar award. P.V.N. is a Scientist of the Alberta Heritage Foundation.
Footnotes
Article is online at http://www.learnmem.org/cgi/doi/10.1101/lm.031088.113.
References
- Abel T, Nguyen PV, Barad M, Deuel TA, Kandel ER, Bourtchouladze R. 1997. Genetic demonstration of a role for PKA in the late phase of LTP and in hippocampus-based long-term memory. Cell 88: 615–626. [DOI] [PubMed] [Google Scholar]
- Abraham WC. 2008. Metaplasticity: tuning synapses and networks for plasticity. Nat Rev Neurosci 9: 387. [DOI] [PubMed] [Google Scholar]
- Abraham WC, Bear MF. 1996. Metaplasticity: the plasticity of synaptic plasticity. Trends Neurosci 19: 126–130. [DOI] [PubMed] [Google Scholar]
- Abraham WC, Williams JM. 2008. LTP maintenance and its protein synthesis-dependence. Neurobiol Learn Mem 89: 260–268. [DOI] [PubMed] [Google Scholar]
- Aman TK, Maki BA, Ruffino TJ, Kasperek EM, Popescu GK. 2014. Separate intramolecular targets for protein kinase A control N-methyl-D-aspartate receptor gating and Ca2+ permeability. J Biol Chem 289: 18805–18817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banke TG, Bowie D, Lee H, Huganir RL, Schousboe A, Traynelis SF. 2000. Control of GluR1 AMPA receptor function by cAMP-dependent protein kinase. J Neurosci 20: 89–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banko JL, Poulin F, Hou L, DeMaria CT, Sonenberg N, Klann E. 2005. The translation repressor 4E-BP2 is critical for eIF4F complex formation, synaptic plasticity, and memory in the hippocampus. J Neurosci 25: 9581–9590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barros DM, Izquierdo LA, Sant'Anna MK, Quevedo J, Medina JH, McGaugh JL, Izquierdo I. 1999. Stimulators of the cAMP cascade reverse amnesia induced by intra-amygdala but not intrahippocampal KN-62 administration. Neurobiol Learn Mem 71: 94–103. [DOI] [PubMed] [Google Scholar]
- Berridge CW, Waterhouse BD. 2003. The locus coeruleus-noradrenergic system: modulation of behavioral state and state-dependent cognitive processes. Brain Res Brain Res Rev 42: 33–84. [DOI] [PubMed] [Google Scholar]
- Bird GC, Lash LL, Han JS, Zou X, Willis WD, Neugebauer V. 2005. Protein kinase A-dependent enhanced NMDA receptor function in pain-related synaptic plasticity in rat amygdala neurones. J Physiol 564: 907–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blank T, Nijholt I, Teichert U, Kugler H, Behrsing H, Fienberg A, Greengard P, Spiess J. 1997. The phosphoprotein DARPP-32 mediates cAMP-dependent potentiation of striatal N-methyl-D-aspartate responses. Proc Natl Acad Sci 94: 14859–14864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blitzer RD, Connor JH, Brown GP, Wong T, Shenolikar S, Iyengar R, Landau EM. 1998. Gating of CaMKII by cAMP-regulated protein phosphatase activity during LTP. Science 280: 1940–1942. [DOI] [PubMed] [Google Scholar]
- Bloodgood BL, Sabatini BL. 2007. Nonlinear regulation of unitary synaptic signals by CaV(2.3) voltage-sensitive calcium channels located in dendritic spines. Neuron 53: 249–260. [DOI] [PubMed] [Google Scholar]
- Bouret S, Sara SJ. 2005. Network reset: a simplified overarching theory of locus coeruleus noradrenaline function. Trends Neurosci 28: 574–582. [DOI] [PubMed] [Google Scholar]
- Bourtchouladze R, Abel T, Berman N, Gordon R, Lapidus K, Kandel ER. 1998. Different training procedures recruit either one or two critical periods for contextual memory consolidation, each of which requires protein synthesis and PKA. Learn Mem 5: 365–374. [PMC free article] [PubMed] [Google Scholar]
- Brown GP, Blitzer RD, Connor JH, Wong T, Shenolikar S, Iyengar R, Landau EM. 2000. Long-term potentiation induced by θ frequency stimulation is regulated by a protein phosphatase-1-operated gate. J Neurosci 20: 7880–7887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet A, Poundja J, Tremblay J, Bui E, Thomas E, Orr SP, Azzoug A, Birmes P, Pitman RK. 2011. Trauma reactivation under the influence of propranolol decreases posttraumatic stress symptoms and disorder: 3 open-label trials. J Clin Psychopharmacol 31: 547–550. [DOI] [PubMed] [Google Scholar]
- Cahill L, Prins B, Weber M, McGaugh JL. 1994. β-Adrenergic activation and memory for emotional events. Nature 371: 702–704. [DOI] [PubMed] [Google Scholar]
- Cerne R, Rusin KI, Randic M. 1993. Enhancement of the N-methyl-D-aspartate response in spinal dorsal horn neurons by cAMP-dependent protein kinase. Neurosci Lett 161: 124–128. [DOI] [PubMed] [Google Scholar]
- Chalermpalanupap T, Kinkead B, Hu WT, Kummer MP, Hammerschmidt T, Heneka MT, Weinshenker D, Levey AI. 2013. Targeting norepinephrine in mild cognitive impairment and Alzheimer's disease. Alzheimers Res Ther 5: 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain SR, Robbins TW. 2013. Noradrenergic modulation of cognition: therapeutic implications. J Psychopharmacol 27: 694–718. [DOI] [PubMed] [Google Scholar]
- Chen X, Yuan LL, Zhao C, Birnbaum SG, Frick A, Jung WE, Schwarz TL, Sweatt JD, Johnston D. 2006. Deletion of Kv4.2 gene eliminates dendritic A-type K+ current and enhances induction of long-term potentiation in hippocampal CA1 pyramidal neurons. J Neurosci 26: 12143–12151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colledge M, Dean RA, Scott GK, Langeberg LK, Huganir RL, Scott JD. 2000. Targeting of PKA to glutamate receptors through a MAGUK-AKAP complex. Neuron 27: 107–119. [DOI] [PubMed] [Google Scholar]
- Connor SA, Hoeffer CA, Klann E, Nguyen PV. 2011a. Fragile X mental retardation protein regulates heterosynaptic plasticity in the hippocampus. Learn Mem 18: 207–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor SA, Wang YT, Nguyen PV. 2011b. Activation of β-adrenergic receptors facilitates heterosynaptic translation-dependent long-term potentiation. J Physiol 589: 4321–4340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa-Mattioli M, Gobert D, Stern E, Gamache K, Colina R, Cuello C, Sossin W, Kaufman R, Pelletier J, Rosenblum K, et al. 2007. eIF2α phosphorylation bidirectionally regulates the switch from short- to long-term synaptic plasticity and memory. Cell 129: 195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa-Mattioli M, Sossin WS, Klann E, Sonenberg N. 2009. Translational control of long-lasting synaptic plasticity and memory. Neuron 61: 10–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox DJ, Racca C, LeBeau FE. 2008. β-Adrenergic receptors are differentially expressed in distinct interneuron subtypes in the rat hippocampus. J Comp Neurol 509: 551–565. [DOI] [PubMed] [Google Scholar]
- Debiec J, Ledoux JE. 2004. Disruption of reconsolidation but not consolidation of auditory fear conditioning by noradrenergic blockade in the amygdala. Neuroscience 129: 267–272. [DOI] [PubMed] [Google Scholar]
- Debiec J, Bush DE, LeDoux JE. 2011. Noradrenergic enhancement of reconsolidation in the amygdala impairs extinction of conditioned fear in rats—a possible mechanism for the persistence of traumatic memories in PTSD. Depress Anxiety 28: 186–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devauges V, Sara SJ. 1991. Memory retrieval enhancement by locus coeruleus stimulation: evidence for mediation by β-receptors. Behav Brain Res 43: 93–97. [DOI] [PubMed] [Google Scholar]
- Dunwiddie TV, Taylor M, Heginbotham LR, Proctor WR. 1992. Long-term increases in excitability in the CA1 region of rat hippocampus induced by β-adrenergic stimulation: possible mediation by cAMP. J Neurosci 12: 506–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers MD. 2000. Reinsertion or degradation of AMPA receptors determined by activity-dependent endocytic sorting. Neuron 28: 511–525. [DOI] [PubMed] [Google Scholar]
- Esteban JA, Shi SH, Wilson C, Nuriya M, Huganir RL, Malinow R. 2003. PKA phosphorylation of AMPA receptor subunits controls synaptic trafficking underlying plasticity. Nat Neurosci 6: 136–143. [DOI] [PubMed] [Google Scholar]
- Faber ES, Delaney AJ, Power JM, Sedlak PL, Crane JW, Sah P. 2008. Modulation of SK channel trafficking by β adrenoceptors enhances excitatory synaptic transmission and plasticity in the amygdala. J Neurosci 28: 10803–10813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey U, Morris RG. 1997. Synaptic tagging and long-term potentiation. Nature 385: 533–536. [DOI] [PubMed] [Google Scholar]
- Frey U, Morris RG. 1998. Weak before strong: dissociating synaptic tagging and plasticity-factor accounts of late-LTP. Neuropharmacology 37: 545–552. [DOI] [PubMed] [Google Scholar]
- Frey U, Krug M, Reymann KG, Matthies H. 1988. Anisomycin, an inhibitor of protein synthesis, blocks late phases of LTP phenomena in the hippocampal CA1 region in vitro. Brain Res 452: 57–65. [DOI] [PubMed] [Google Scholar]
- Gelinas JN, Nguyen PV. 2005. β-adrenergic receptor activation facilitates induction of a protein synthesis-dependent late phase of long-term potentiation. J Neurosci 25: 3294–3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelinas JN, Nguyen PV. 2007. Neuromodulation of hippocampal synaptic plasticity, learning, and memory by noradrenaline. Cent Nerv Syst Agents Med Chem 7: 17–33. [Google Scholar]
- Gelinas JN, Banko JL, Hou L, Sonenberg N, Weeber EJ, Klann E, Nguyen PV. 2007. ERK and mTOR signaling couple β-adrenergic receptors to translation initiation machinery to gate induction of protein synthesis-dependent long-term potentiation. J Biol Chem 282: 27527–27535. [DOI] [PubMed] [Google Scholar]
- Giovannini MG, Blitzer RD, Wong T, Asoma K, Tsokas P, Morrison JH, Iyengar R, Landau EM. 2001. Mitogen-activated protein kinase regulates early phosphorylation and delayed expression of Ca2+/calmodulin-dependent protein kinase II in long-term potentiation. J Neurosci 21: 7053–7062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granger AJ, Shi Y, Lu W, Cerpas M, Nicoll RA. 2013. LTP requires a reserve pool of glutamate receptors independent of subunit type. Nature 493: 495–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray JA, Shi Y, Usui H, During MJ, Sakimura K, Nicoll RA. 2011. Distinct modes of AMPA receptor suppression at developing synapses by GluN2A and GluN2B: single-cell NMDA receptor subunit deletion in vivo. Neuron 71: 1085–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray EE, Guglietta R, Khakh BS, O'Dell TJ. 2014. Inhibitory interactions between phosphorylation sites in the C terminus of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid-type glutamate receptor GluA1 subunits. J Biol Chem 289: 14600–14611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grudzien A, Shaw P, Weintraub S, Bigio E, Mash DC, Mesulam MM. 2007. Locus coeruleus neurofibrillary degeneration in aging, mild cognitive impairment and early Alzheimer's disease. Neurobiol Aging 28: 327–335. [DOI] [PubMed] [Google Scholar]
- Guo NN, Li BM. 2007. Cellular and subcellular distributions of β1- and β2-adrenoceptors in the CA1 and CA3 regions of the rat hippocampus. Neuroscience 146: 298–305. [DOI] [PubMed] [Google Scholar]
- Haas HL, Konnerth A. 1983. Histamine and noradrenaline decrease calcium-activated potassium conductance in hippocampal pyramidal cells. Nature 302: 432–434. [DOI] [PubMed] [Google Scholar]
- Harley C. 1991. Noradrenergic and locus coeruleus modulation of the perforant path-evoked potential in rat dentate gyrus supports a role for the locus coeruleus in attentional and memorial processes. Prog Brain Res 88: 307–321. [DOI] [PubMed] [Google Scholar]
- Havekes R, Canton DA, Park AJ, Huang T, Nie T, Day JP, Guercio LA, Grimes Q, Luczak V, Gelman IH, et al. 2012. Gravin orchestrates protein kinase A and β2-adrenergic receptor signaling critical for synaptic plasticity and memory. J Neurosci 32: 18137–18149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillman KL, Knudson CA, Carr PA, Doze VA, Porter JE. 2005. Adrenergic receptor characterization of CA1 hippocampal neurons using real time single cell RT-PCR. Brain Res Mol Brain Res 139: 267–276. [DOI] [PubMed] [Google Scholar]
- Hoeffer CA, Cowansage KK, Arnold EC, Banko JL, Moerke NJ, Rodriguez R, Schmidt EK, Klosi E, Chorev M, Lloyd RE, et al. 2011. Inhibition of the interactions between eukaryotic initiation factors 4E and 4G impairs long-term associative memory consolidation but not reconsolidation. Proc Natl Acad Sci 108: 3383–3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeffer CA, Santini E, Ma T, Arnold EC, Whelan AM, Wong H, Pierre P, Pelletier J, Klann E. 2013. Multiple components of eIF4F are required for protein synthesis-dependent hippocampal long-term potentiation. J Neurophysiol 109: 68–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman DA, Johnston D. 1999. Neuromodulation of dendritic action potentials. J Neurophysiol 81: 408–411. [DOI] [PubMed] [Google Scholar]
- Hoffman DA, Magee JC, Colbert CM, Johnston D. 1997. K+ channel regulation of signal propagation in dendrites of hippocampal pyramidal neurons. Nature 387: 869–875. [DOI] [PubMed] [Google Scholar]
- Hoffmann R, Baillie GS, MacKenzie SJ, Yarwood SJ, Houslay MD. 1999. The MAP kinase ERK2 inhibits the cyclic AMP-specific phosphodiesterase HSPDE4D3 by phosphorylating it at Ser579. EMBO J 18: 893–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoge EA, Worthington JJ, Nagurney JT, Chang Y, Kay EB, Feterowski CM, Katzman AR, Goetz JM, Rosasco ML, Lasko NB, et al. 2012. Effect of acute posttrauma propranolol on PTSD outcome and physiological responses during script-driven imagery. CNS Neurosci Ther 18: 21–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosokawa T, Mitsushima D, Kaneko R, Hayashi Y. 2015. Stoichiometry and phosphoisotypes of hippocampal AMPA-type glutamate receptor phosphorylation. Neuron 85: 60–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Real E, Takamiya K, Kang MG, Ledoux J, Huganir RL, Malinow R. 2007. Emotion enhances learning via norepinephrine regulation of AMPA-receptor trafficking. Cell 131: 160–173. [DOI] [PubMed] [Google Scholar]
- Huang CC, Tsai JJ, Gean PW. 1993. Enhancement of NMDA receptor-mediated synaptic potential by isoproterenol is blocked by Rp-adenosine 3′,5′-cyclic monophosphothioate. Neurosci Lett 161: 207–210. [DOI] [PubMed] [Google Scholar]
- Izquierdo I, Medina JH, Izquierdo LA, Barros DM, de Souza MM, Mello e Souza T. 1998. Short- and long-term memory are differentially regulated by monoaminergic systems in the rat brain. Neurobiol Learn Mem 69: 219–224. [DOI] [PubMed] [Google Scholar]
- Jenkins MA, Wells G, Bachman J, Snyder JP, Jenkins A, Huganir RL, Oswald RE, Traynelis SF. 2014. Regulation of GluA1 α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor function by protein kinase C at serine-818 and threonine-840. Mol Pharm 85: 618–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji JZ, Wang XM, Li BM. 2003a. Deficit in long-term contextual fear memory induced by blockade of β-adrenoceptors in hippocampal CA1 region. Eur J Neurosci 17: 1947–1952. [DOI] [PubMed] [Google Scholar]
- Ji JZ, Zhang XH, Li BM. 2003b. Deficient spatial memory induced by blockade of β-adrenoceptors in the hippocampal CA1 region. Behav Neurosci 117: 1378–1384. [DOI] [PubMed] [Google Scholar]
- Joiner ML, Lise MF, Yuen EY, Kam AY, Zhang M, Hall DD, Malik ZA, Qian H, Chen Y, Ulrich JD, et al. 2010. Assembly of a β2-adrenergic receptor—GluR1 signalling complex for localized cAMP signalling. EMBO J 29: 482–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandel ER. 2001. The molecular biology of memory storage: a dialogue between genes and synapses. Science 294: 1030–1038. [DOI] [PubMed] [Google Scholar]
- Katsuki H, Izumi Y, Zorumski CF. 1997. Noradrenergic regulation of synaptic plasticity in the hippocampal CA1 region. J Neurophysiol 77: 3013–3020. [DOI] [PubMed] [Google Scholar]
- Kelleher RJ III, Govindarajan A, Tonegawa S. 2004. Translational regulatory mechanisms in persistent forms of synaptic plasticity. Neuron 44: 59–73. [DOI] [PubMed] [Google Scholar]
- Kim J, Hoffman DA. 2008. Potassium channels: newly found players in synaptic plasticity. Neuroscientist 14: 276–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Jung SC, Clemens AM, Petralia RS, Hoffman DA. 2007. Regulation of dendritic excitability by activity-dependent trafficking of the A-type K+ channel subunit Kv4.2 in hippocampal neurons. Neuron 54: 933–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kindt M, Soeter M, Vervliet B. 2009. Beyond extinction: erasing human fear responses and preventing the return of fear. Nat Neurosci 12: 256–258. [DOI] [PubMed] [Google Scholar]
- Klann E, Antion MD, Banko JL, Hou L. 2004. Synaptic plasticity and translation initiation. Learn Mem 11: 365–372. [DOI] [PubMed] [Google Scholar]
- Krupp JJ, Vissel B, Thomas CG, Heinemann SF, Westbrook GL. 2002. Calcineurin acts via the C-terminus of NR2A to modulate desensitization of NMDA receptors. Neuropharmacology 42: 593–602. [DOI] [PubMed] [Google Scholar]
- Laggerbauer B, Ostareck D, Keidel EM, Ostareck-Lederer A, Fischer U. 2001. Evidence that fragile X mental retardation protein is a negative regulator of translation. Human Mol Gen 10: 329–338. [DOI] [PubMed] [Google Scholar]
- Leonard AS, Hell JW. 1997. Cyclic AMP-dependent protein kinase and protein kinase C phosphorylate N-methyl-D-aspartate receptors at different sites. J Biol Chem 272: 12107–12115. [DOI] [PubMed] [Google Scholar]
- Li B, Devidze N, Barengolts D, Prostak N, Sphicas E, Apicella AJ, Malinow R, Emamian ES. 2009. NMDA receptor phosphorylation at a site affected in schizophrenia controls synaptic and behavioral plasticity. J Neurosci 29: 11965–11972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Jin M, Zhang D, Yang T, Koeglsperger T, Fu H, Selkoe DJ. 2013. Environmental novelty activates β2-adrenergic signaling to prevent the impairment of hippocampal LTP by Aβ oligomers. Neuron 77: 929–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YW, Min MY, Chiu TH, Yang HW. 2003. Enhancement of associative long-term potentiation by activation of β-adrenergic receptors at CA1 synapses in rat hippocampal slices. J Neurosci 23: 4173–4181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MT, Lujan R, Watanabe M, Adelman JP, Maylie J. 2008. SK2 channel plasticity contributes to LTP at Schaffer collateral-CA1 synapses. Nat Neurosci 11: 170–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Ma L, Li HH, Huang B, Li YX, Tao YZ, Ma L. 2015. β-Arrestin-biased signaling mediates memory reconsolidation. Proc Natl Acad Sci 112: 4483–4488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonergan MH, Olivera-Figueroa LA, Pitman RK, Brunet A. 2013. Propranolol's effects on the consolidation and reconsolidation of long-term emotional memory in healthy participants: a meta-analysis. J Psychiatry Neurosci 38: 222–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Roche KW. 2012. Posttranslational regulation of AMPA receptor trafficking and function. Curr Opin Neurobiol 22: 470–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Shi Y, Jackson AC, Bjorgan K, During MJ, Sprengel R, Seeburg PH, Nicoll RA. 2009. Subunit composition of synaptic AMPA receptors revealed by a single-cell genetic approach. Neuron 62: 254–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luttrell LM, Ferguson SSG, Daaka Y, Miller WE, Maudsley S, Della Rocca GJ, Lin F-T, Kawakatsu H, Owada K, Luttrell DK, et al. 1999. β-arrestin-dependent formation of β2 adrenergic receptor-Src protein kinase complexes. Science 283: 655–661. [DOI] [PubMed] [Google Scholar]
- Ma T, Tzavaras N, Tsokas P, Landau EM, Blitzer RD. 2011. Synaptic stimulation of mTOR is mediated by Wnt signaling and regulation of glycogen synthetase kinase-3. J Neurosci 31: 17537–17546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madison DV, Nicoll RA. 1982. Noradrenaline blocks accommodation of pyramidal cell discharge in the hippocampus. Nature 299: 636–638. [DOI] [PubMed] [Google Scholar]
- Madison DV, Nicoll RA. 1986. Cyclic adenosine 3′,5′-monophosphate mediates β-receptor actions of noradrenaline in rat hippocampal pyramidal cells. J Physiol 372: 245–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maki BA, Cole R, Popescu GK. 2013. Two serine residues on GluN2A C-terminal tails control NMDA receptor current decay times. Channels 7: 126–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makino Y, Johnson RC, Yu Y, Takamiya K, Huganir RL. 2011. Enhanced synaptic plasticity in mice with phosphomimetic mutation of the GluA1 AMPA receptor. Proc Natl Acad Sci 108: 8450–8455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man HY, Sekine-Aizawa Y, Huganir RL. 2007. Regulation of {α}-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor trafficking through PKA phosphorylation of the Glu receptor 1 subunit. Proc Natl Acad Sci 104: 3579–3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer ML, Westbrook GL, Guthrie PB. 1984. Voltage-dependent block by Mg2+ of NMDA responses in spinal cord neurones. Nature 309: 261–263. [DOI] [PubMed] [Google Scholar]
- McFall ME, Murburg MM, Ko GN, Veith RC. 1990. Autonomic responses to stress in Vietnam combat veterans with posttraumatic stress disorder. Biol Psychiatry 27: 1165–1175. [DOI] [PubMed] [Google Scholar]
- McGaugh JL. 2013. Making lasting memories: remembering the significant. Proc Natl Acad Sci 110Suppl 2: 10402–10407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGhee LL, Maani CV, Garza TH, Desocio PA, Gaylord KM, Black IH. 2009. The effect of propranolol on posttraumatic stress disorder in burned service members. J Burn Care Res 30: 92–97. [DOI] [PubMed] [Google Scholar]
- Moncada D, Viola H. 2007. Induction of long-term memory by exposure to novelty requires protein synthesis: evidence for a behavioral tagging. J Neurosci 27: 7476–7481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monyer H, Burnashev N, Laurie DJ, Sakmann B, Seeburg PH. 1994. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron 12: 529–540. [DOI] [PubMed] [Google Scholar]
- Moody TD, Thomas MJ, Makhinson M, O'Dell TJ. 1998. 5-Hz stimulation of CA3 pyramidal cell axons induces a β-adrenergic modulated potentiation at synapses on CA1, but not CA3, pyramidal cells. Brain Res 794: 75–79. [DOI] [PubMed] [Google Scholar]
- Moody TD, Watabe AM, Indersmitten T, Komiyama NH, Grant SG, O'Dell TJ. 2011. β-adrenergic receptor activation rescues θ frequency stimulation-induced LTP deficits in mice expressing C-terminally truncated NMDA receptor GluN2A subunits. Learn Mem 18: 118–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muravieva EV, Alberini CM. 2010. Limited efficacy of propranolol on the reconsolidation of fear memories. Learn Mem 17: 306–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murchison CF, Zhang XY, Zhang WP, Ouyang M, Lee A, Thomas SA. 2004. A distinct role for norepinephrine in memory retrieval. Cell 117: 131–143. [DOI] [PubMed] [Google Scholar]
- Murphy JA, Stein IS, Lau CG, Peixoto RT, Aman TK, Kaneko N, Aromolaran K, Saulnier JL, Popescu GK, Sabatini BL, et al. 2014. Phosphorylation of Ser1166 on GluN2B by PKA is critical to synaptic NMDA receptor function and Ca2+ signaling in spines. J Neurosci 34: 869–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nader K, Schafe GE, Le Doux JE. 2000. Fear memories require protein synthesis in the amygdala for reconsolidation after retrieval. Nature 406: 722–726. [DOI] [PubMed] [Google Scholar]
- Navakkode S, Sajikumar S, Sacktor TC, Frey JU. 2010. Protein kinase Mζ is essential for the induction and maintenance of dopamine-induced long-term potentiation in apical CA1 dendrites. Learn Mem 17: 605–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngo-Anh TJ, Bloodgood BL, Lin M, Sabatini BL, Maylie J, Adelman JP. 2005. SK channels and NMDA receptors form a Ca2+-mediated feedback loop in dendritic spines. Nat Neurosci 8: 642–649. [DOI] [PubMed] [Google Scholar]
- Nguyen PV, Woo NH. 2003. Regulation of hippocampal synaptic plasticity by cyclic AMP-dependent protein kinases. Prog Neurobiol 71: 401–437. [DOI] [PubMed] [Google Scholar]
- Nguyen PV, Abel T, Kandel ER. 1994. Requirement of a critical period of transcription for induction of a late phase of LTP. Science 265: 1104–1107. [DOI] [PubMed] [Google Scholar]
- Nicholas AP, Pieribone VA, Hokfelt T. 1993. Cellular localization of messenger RNA for β-1 and β-2 adrenergic receptors in rat brain: an in situ hybridization study. Neuroscience 56: 1023–1039. [DOI] [PubMed] [Google Scholar]
- Nowak L, Bregestovski P, Ascher P, Herbet A, Prochiantz A. 1984. Magnesium gates glutamate-activated channels in mouse central neurones. Nature 307: 462–465. [DOI] [PubMed] [Google Scholar]
- Nusser Z, Lujan R, Laube G, Roberts JD, Molnar E, Somogyi P. 1998. Cell type and pathway dependence of synaptic AMPA receptor number and variability in the hippocampus. Neuron 21: 545–559. [DOI] [PubMed] [Google Scholar]
- O'Dell TJ, Connor SA, Gelinas JN, Nguyen PV. 2010. Viagra for your synapses: enhancement of hippocampal long-term potentiation by activation of β-adrenergic receptors. Cell Signal 22: 728–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh MC, Derkach VA, Guire ES, Soderling TR. 2006. Extrasynaptic membrane trafficking regulated by GluR1 serine 845 phosphorylation primes AMPA receptors for long-term potentiation. J Biol Chem 281: 752–758. [DOI] [PubMed] [Google Scholar]
- Papay R, Gaivin R, Jha A, McCune DF, McGrath JC, Rodrigo MC, Simpson PC, Doze VA, Perez DM. 2006. Localization of the mouse α1A-adrenergic receptor (AR) in the brain: α1AAR is expressed in neurons, GABAergic interneurons, and NG2 oligodendrocyte progenitors. J Comp Neurol 497: 209–222. [DOI] [PubMed] [Google Scholar]
- Przybyslawski J, Roullet P, Sara SJ. 1999. Attenuation of emotional and nonemotional memories after their reactivation: role of β adrenergic receptors. J Neurosci 19: 6623–6628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi XL, Zhu B, Zhang X-H, Li B-M. 2008. Are β-adrenergic receptors in the hippocampal CA1 region required for retrieval of contextual fear memory? Biochem Biophys Res Commun 368: 186–191. [DOI] [PubMed] [Google Scholar]
- Qian H, Matt L, Zhang M, Nguyen M, Patriarchi T, Koval OM, Anderson ME, He K, Lee HK, Hell JW. 2012. β2-Adrenergic receptor supports prolonged θ tetanus-induced LTP. J Neurophysiol 107: 2703–2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman IM, Tong G, Jahr CE. 1996. β-adrenergic regulation of synaptic NMDA receptors by cAMP-dependent protein kinase. Neuron 16: 415–421. [DOI] [PubMed] [Google Scholar]
- Redondo RL, Morris RG. 2011. Making memories last: the synaptic tagging and capture hypothesis. Nat Rev Neurosci 12: 17–30. [DOI] [PubMed] [Google Scholar]
- Roberson ED, English JD, Adams JP, Selcher JC, Kondratick C, Sweatt JD. 1999. The mitogen-activated protein kinase cascade couples PKA and PKC to cAMP response element binding protein phosphorylation in area CA1 of hippocampus. J Neurosci 19: 4337–4348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche KW, O'Brien RJ, Mammen AL, Bernhardt J, Huganir RL. 1996. Characterization of multiple phosphorylation sites on the AMPA receptor GluR1 subunit. Neuron 16: 1179–1188. [DOI] [PubMed] [Google Scholar]
- Sara SJ. 2009. The locus coeruleus and noradrenergic modulation of cognition. Nat Rev Neurosci 10: 211–223. [DOI] [PubMed] [Google Scholar]
- Sara SJ, Roullet P, Przybyslawski J. 1999. Consolidation of memory for odor-reward association: β-adrenergic receptor involvement in the late phase. Learn Mem 6: 88–96. [PMC free article] [PubMed] [Google Scholar]
- Schafe GE, Nadel NV, Sullivan GM, Harris A, LeDoux JE. 1999. Memory consolidation for contextual and auditory fear conditioning is dependent on protein synthesis, PKA, and MAP kinase. Learn Mem 6: 97–110. [PMC free article] [PubMed] [Google Scholar]
- Scharf MT, Woo NH, Lattal KM, Young JZ, Nguyen PV, Abel T. 2002. Protein synthesis is required for the enhancement of long-term potentiation and long-term memory by spaced training. J Neurophysiol 87: 2770–2777. [DOI] [PubMed] [Google Scholar]
- Schmitt JM, Stork PJ. 2000. β2-adrenergic receptor activates extracellular signal-regulated kinases (ERKs) via the small G protein rap1 and the serine/threonine kinase B-Raf. J Biol Chem 275: 25342–25350. [DOI] [PubMed] [Google Scholar]
- Schwabe L, Nader K, Wolf OT, Beaudry T, Pruessner JC. 2012. Neural signature of reconsolidation impairments by propranolol in humans. Biol Psychiatry 71: 380–386. [DOI] [PubMed] [Google Scholar]
- Seol GH, Ziburkus J, Huang S, Song L, Kim IT, Takamiya K, Huganir RL, Lee HK, Kirkwood A. 2007. Neuromodulators control the polarity of spike-timing-dependent synaptic plasticity. Neuron 55: 919–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng M, Hoogenraad CC. 2007. The postsynaptic architecture of excitatory synapses: a more quantitative view. Ann Rev Biochem 76: 823–847. [DOI] [PubMed] [Google Scholar]
- Shukla AK, Xiao K, Lefkowitz RJ. 2011. Emerging paradigms of β-arrestin-dependent seven transmembrane receptor signaling. Trends Biochem Sci 36: 457–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skeberdis VA, Chevaleyre V, Lau CG, Goldberg JH, Pettit DL, Suadicani SO, Lin Y, Bennett MV, Yuste R, Castillo PE, et al. 2006. Protein kinase A regulates calcium permeability of NMDA receptors. Nat Neurosci 9: 501–510. [DOI] [PubMed] [Google Scholar]
- Song RS, Massenburg B, Wenderski W, Jayaraman V, Thompson L, Neves SR. 2013. ERK regulation of phosphodiesterase 4 enhances dopamine-stimulated AMPA receptor membrane insertion. Proc Natl Acad Sci 110: 15437–15442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanton PK, Sarvey JM. 1984. Blockade of long-term potentiation in rat hippocampal CA1 region by inhibitors of protein synthesis. J Neurosci 4: 3080–3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steckler T, Risbrough V. 2012. Pharmacological treatment of PTSD—established and new approaches. Neuropharmacology 62: 617–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein MB, Kerridge C, Dimsdale JE, Hoyt DB. 2007. Pharmacotherapy to prevent PTSD: results from a randomized controlled proof-of-concept trial in physically injured patients. J Trauma Stress 20: 923–932. [DOI] [PubMed] [Google Scholar]
- Strange BA, Dolan RJ. 2004. β-adrenergic modulation of emotional memory-evoked human amygdala and hippocampal responses. Proc Natl Acad Sci 101: 11454–11458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strawn JR, Geracioti TD Jr. 2008. Noradrenergic dysfunction and the psychopharmacology of posttraumatic stress disorder. Depress Anxiety 25: 260–271. [DOI] [PubMed] [Google Scholar]
- Sutton MA, Schuman EM. 2006. Dendritic protein synthesis, synaptic plasticity, and memory. Cell 127: 49–58. [DOI] [PubMed] [Google Scholar]
- Swanson-Park JL, Coussens CM, Mason-Parker SE, Raymond CR, Hargreaves EL, Dragunow M, Cohen AS, Abraham WC. 1999. A double dissociation within the hippocampus of dopamine D1/D5 receptor and β-adrenergic receptor contributions to the persistence of long-term potentiation. Neuroscience 92: 485–497. [DOI] [PubMed] [Google Scholar]
- Sweatt JD. 2004. Mitogen-activated protein kinases in synaptic plasticity and memory. Curr Opin Neurobiol 14: 311–317. [DOI] [PubMed] [Google Scholar]
- Szot P, White SS, Greenup JL, Leverenz JB, Peskind ER, Raskind MA. 2006. Compensatory changes in the noradrenergic nervous system in the locus ceruleus and hippocampus of postmortem subjects with Alzheimer's disease and dementia with Lewy bodies. J Neurosci 26: 467–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi J, Shibata T, Sasaki M, Kudo M, Yanezawa H, Obara S, Kudo K, Ito K, Yamashita F, Terayama Y. 2015. Detection of changes in the locus coeruleus in patients with mild cognitive impairment and Alzheimer's disease: high-resolution fast spin-echo T1-weighted imaging. Geriatr Gerontol Int 15: 334–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenorio G, Connor SA, Guevremont D, Abraham WC, Williams J, O'Dell TJ, Nguyen PV. 2010. ‘Silent’ priming of translation-dependent LTP by β-adrenergic receptors involves phosphorylation and recruitment of AMPA receptors. Learn Mem 17: 627–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas MJ, Moody TD, Makhinson M, O'Dell TJ. 1996. Activity-dependent β-adrenergic modulation of low frequency stimulation induced LTP in the hippocampal CA1 region. Neuron 17: 475–482. [DOI] [PubMed] [Google Scholar]
- Thomas MJ, Watabe AM, Moody TD, Makhinson M, O'Dell TJ. 1998. Postsynaptic complex spike bursting enables the induction of LTP by θ frequency synaptic stimulation. J Neurosci 18: 7118–7126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tingley WG, Ehlers MD, Kameyama K, Doherty C, Ptak JB, Riley CT, Huganir RL. 1997. Characterization of protein kinase A and protein kinase C phosphorylation of the N-methyl-D-aspartate receptor NR1 subunit using phosphorylation site-specific antibodies. J Biol Chem 272: 5157–5166. [DOI] [PubMed] [Google Scholar]
- Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, Hansen KB, Yuan H, Myers SJ, Dingledine R. 2010. Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev 62: 405–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tronson NC, Taylor JR. 2007. Molecular mechanisms of memory reconsolidation. Nat Rev Neurosci 8: 262–275. [DOI] [PubMed] [Google Scholar]
- Tsokas P, Ma T, Iyengar R, Landau EM, Blitzer RD. 2007. Mitogen-activated protein kinase upregulates the dendritic translation machinery in long-term potentiation by controlling the mammalian target of rapamycin pathway. J Neurosci 27: 5885–5894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tully K, Bolshakov VY. 2010. Emotional enhancement of memory: how norepinephrine enables synaptic plasticity. Mol Brain 3: 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaiva G, Ducrocq F, Jezequel K, Averland B, Lestavel P, Brunet A, Marmar CR. 2003. Immediate treatment with propranolol decreases posttraumatic stress disorder two months after trauma. Biol Psychiatry 54: 947–949. [DOI] [PubMed] [Google Scholar]
- Vanhoose AM, Winder DG. 2003. NMDA and β1-adrenergic receptors differentially signal phosphorylation of glutamate receptor type 1 in area CA1 of hippocampus. J Neurosci 23: 5827–5834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhoose AM, Clements JM, Winder DG. 2006. Novel blockade of protein kinase A-mediated phosphorylation of AMPA receptors. J Neurosci 26: 1138–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang QW, Rowan MJ, Anwyl R. 2009. Inhibition of LTP by β-amyloid is prevented by activation of β2 adrenoceptors and stimulation of the cAMP/PKA signalling pathway. Neurobiol Aging 30: 1608–1613. [DOI] [PubMed] [Google Scholar]
- Wang SH, Redondo RL, Morris RG. 2010. Relevance of synaptic tagging and capture to the persistence of long-term potentiation and everyday spatial memory. Proc Natl Acad Sci 107: 19537–19542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Yuen EY, Zhou Y, Yan Z, Xiang YK. 2011. Amyloid β peptide-(1–42) induces internalization and degradation of β2 adrenergic receptors in prefrontal cortical neurons. J Biol Chem 286: 31852–31863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Lin MT, Adelman JP, Maylie J. 2014. Distinct Ca2+ sources in dendritic spines of hippocampal CA1 neurons couple to SK and Kv4 channels. Neuron 81: 379–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe S, Hoffman DA, Migliore M, Johnston D. 2002. Dendritic K+ channels contribute to spike-timing dependent long-term potentiation in hippocampal pyramidal neurons. Proc Natl Acad Sci 99: 8366–8371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenthold RJ, Petralia RS, Blahos J II, Niedzielski AS. 1996. Evidence for multiple AMPA receptor complexes in hippocampal CA1/CA2 neurons. J Neurosci 16: 1982–1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westphal RS, Tavalin SJ, Lin JW, Alto NM, Fraser ID, Langeberg LK, Sheng M, Scott JD. 1999. Regulation of NMDA receptors by an associated phosphatase-kinase signaling complex. Science 285: 93–96. [DOI] [PubMed] [Google Scholar]
- Winder DG, Martin KC, Muzzio IA, Rohrer D, Chruscinski A, Kobilka B, Kandel ER. 1999. ERK plays a regulatory role in induction of LTP by θ frequency stimulation and its modulation by β-adrenergic receptors. Neuron 24: 715–726. [DOI] [PubMed] [Google Scholar]
- Yehuda R, Southwick S, Giller EL, Ma X, Mason JW. 1992. Urinary catecholamine excretion and severity of PTSD symptoms in Vietnam combat veterans. J Nerv Ment Dis 180: 321–325. [DOI] [PubMed] [Google Scholar]
- Young JZ, Nguyen PV. 2005. Homosynaptic and heterosynaptic inhibition of synaptic tagging and capture of long-term potentiation by previous synaptic activity. J Neurosci 25: 7221–7231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JZ, Isiegas C, Abel T, Nguyen PV. 2006. Metaplasticity of the late-phase of long-term potentiation: a critical role for protein kinase A in synaptic tagging. Eur J Neurosci 23: 1784–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan LL, Adams JP, Swank M, Sweatt JD, Johnston D. 2002. Protein kinase modulation of dendritic K+ channels in hippocampus involves a mitogen-activated protein kinase pathway. J Neurosci 22: 4860–4868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Patriarchi T, Stein IS, Qian H, Matt L, Nguyen M, Xiang YK, Hell JW. 2013. Adenylyl cyclase anchoring by a kinase anchor protein AKAP5 (AKAP79/150) is important for postsynaptic β-adrenergic signaling. J Biol Chem 288: 17918–17931. [DOI] [PMC free article] [PubMed] [Google Scholar]