

In this study, Chen et al. used Nf1 genetically engineered mice coupled with genetic and pharmacologic inhibitor strategies to investigate the mechanisms of neurofibromin control of NSC proliferation and multilineage differentiation. They demonstrate that neurofibromin differentially controls NSC proliferation and multilineage differentiation through the selective use of the PI3K/AKT and RAF/MEK pathways, providing new insights into the roles for the RAS effector pathways in regulating brain NSC function.
Keywords: neurofibromin, neuroglial progenitor, AKT, MEK, Jagged1, Notch, astrocyte
Abstract
Neurofibromatosis type 1 (NF1) is a common neurodevelopmental disorder caused by impaired function of the neurofibromin RAS regulator. Using a combination of Nf1 genetically engineered mice and pharmacological/genetic inhibition approaches, we report that neurofibromin differentially controls neural stem cell (NSC) proliferation and multilineage differentiation through the selective use of the PI3K/AKT and RAF/MEK pathways. While PI3K/AKT governs neurofibromin-regulated NSC proliferation, multilineage differentiation is MEK-dependent. Moreover, whereas MEK-regulated multilineage differentiation requires Smad3-induced Jagged-1 expression and Notch activation, MEK/Smad3-regulated Hes1 induction is only responsible for astrocyte and neuronal differentiation. Collectively, these findings establish distinct roles for the RAS effector pathways in regulating brain NSC function.
Neurofibromatosis type 1 (NF1) is a common neurogenetic disorder in which individuals manifest numerous CNS abnormalities that reflect impaired neuronal and glial cell lineage function. In this regard, 60%–80% of children with NF1 exhibit impairments in learning, attention, and memory (Diggs-Andrews and Gutmann 2013), and 15%–20% of affected children develop low-grade astrocytomas (gliomas) involving the optic pathway and brain stem (Guillamo et al. 2003). The fact that both neuronal and astroglial lineages are impacted raises the possibility that the NF1 gene product neurofibromin is a critical regulator of neural stem cell (NSC) growth and differentiation. Consistent with this idea, previous reports have revealed that neurofibromin negatively controls NSC proliferation and self-renewal as well as multilineage differentiation (Hegedus et al. 2007; Lee et al. 2010) such that Nf1 inactivation leads to increased numbers of spinal cord neuroglial progenitors (Bennett et al. 2003) and increased telencephalic NSC proliferation (Dasgupta and Gutmann 2005). Similarly, conditional Nf1 gene inactivation in BLBP- or GFAP-expressing neuroglial progenitor cells results in increased NSC proliferation and glial lineage differentiation in vivo (Hegedus et al. 2007; Wang et al. 2012).
Neurofibromin is widely expressed in the developing brain, where it primarily functions as a negative regulator of RAS activity. Previous studies have demonstrated that loss of neurofibromin expression in NSCs results in increased proliferation and glial differentiation in a RAS- and AKT-dependent fashion (Hegedus et al. 2007; Lee et al. 2010). However, other studies have implicated RAS/ERK signaling as the responsible pathway dictating Nf1-deficient neural progenitor cell growth and differentiation in the forebrain (Wang et al. 2012) and cerebellum (Sanchez-Ortiz et al. 2014). Additionally, neurofibromin control of mouse astrocyte and optic glioma growth is mediated by both MEK and AKT effector arms through convergence on the mammalian target of rapamycin (mTOR) complex (Kaul et al. 2015). In contrast, neurofibromin regulation of human and mouse neuronal cyclic AMP homeostasis is RAS-dependent but operates in a MEK/AKT-independent manner through PKCζ (Anastasaki and Gutmann 2014). Together, these observations argue that neurofibromin regulation of nervous system cell biology may be cell type- or cell function-specific.
To mechanistically define the signaling pathways responsible for brain NSC function, we leveraged Nf1 genetically engineered mice and converging inhibition strategies to demonstrate that neurofibromin regulation of NSC proliferation and multilineage differentiation involves the engagement of distinct RAS downstream signaling pathways. Here, we establish that neurofibromin control of NSC proliferation is PI3K/AKT-dependent, while MEK/Smad3/Jagged1/Hes1-dependent signaling is required for neurofibromin-regulated NSC glial and neuronal differentiation in vitro and in vivo.
Results and Discussion
To determine which RAS downstream effectors were hyperactivated following Nf1 inactivation, we focused on third ventricle zone (TVZ) NSCs (Lee et al. 2012). Nf1−/− and wild-type TVZ NSC cultures were generated from postnatal day 1 (P1) Nf1flox/flox pups following Cre or LacZ gene adenovirus infection, respectively. Following neurofibromin loss, increased ERK (3.5-fold; Thr202/Tyr204) and AKT (1.8-fold and threefold; Ser473 and Thr308) phosphorylation was observed (Fig. 1A).
Figure 1.
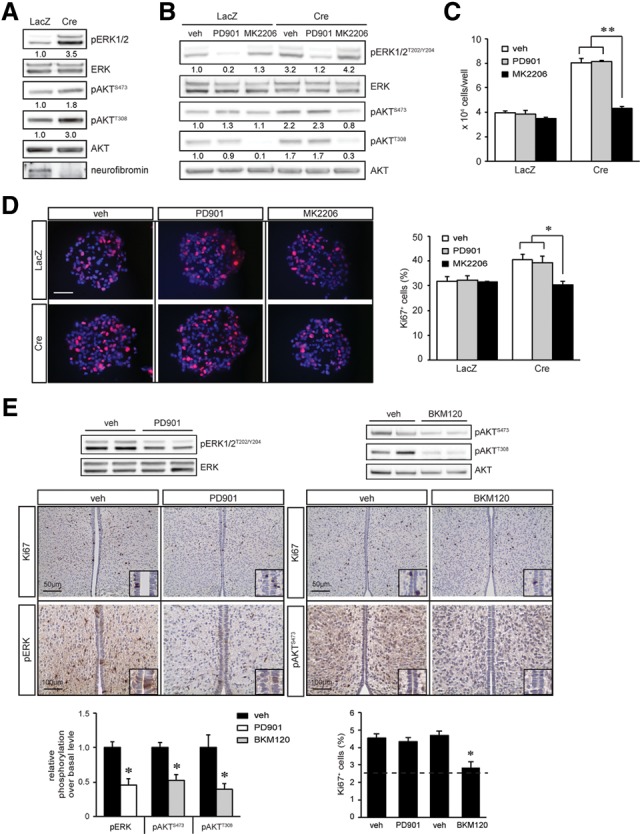
Neurofibromin regulates NSC proliferation in a PI3K/AKT-dependent manner. (A) Nf1 loss in TVZ NSCs resulted in increased ERK and AKT activation (pERKT202/Y204, pAKTS473, and pAKTT308 phosphorylation) relative to wild-type controls. (B) Treatment with the MEK (5 nM PD0325901 [PD901]) or AKT inhibitor (50 nM MK2206) reduced ERK and AKT hyperactivation. Reduced NSC numbers (direct cell counting) (C) and percentage of Ki67+ cells (D) were observed only in Nf1−/− TVZ NSCs treated with MK2206. (E) NVP-BKM120 (BKM120) but not 5 mg/kg PD901 treatment decreased the percentage of Ki67+ cells in the TVZ of P18 Nf1BLBP conditional knockout mice in vivo (n = 4 per group) to nearly wild-type levels (dotted line). ERK and AKT hyperactivation were decreased following PD901 and BKM120 treatment, respectively, relative to wild-type controls. n = 4 per group. (veh) Vehicle. Nuclei were counterstained with DAPI. Error bars denote the mean ± SD. Bar, 100 μm. (*) P < 0.05; (**) P < 0.01.
To identify which RAS effector pathway was responsible for neurofibromin regulation of NSC growth and multilineage differentiation, we used PI3K/AKT and MEK pharmacological inhibitors. While MK2206 treatment inhibited AKT activation (Fig. 1B) and reduced Nf1−/− NSC growth (Fig. 1C [direct cell counting], D [percentage of Ki67+ cells]) to wild-type levels, it had no effect on ERK phosphorylation (Fig. 1B). PD0325901 (PD901) treatment inhibited MEK activation but did not decrease Nf1−/− NSC proliferation (Fig. 1C) or AKT phosphorylation (Fig. 1B). Similar results were obtained with additional PI3K (NVP-BKM120 [BKM120]) and MEK (UO126) inhibitors (Supplemental Fig. S1A,B). The observation that MEK inhibition had no effect on Nf1−/− NSC growth but reduced Nf1−/− astrocyte proliferation (Kaul et al. 2015) further underscores the importance of cell type specificity (Lee et al. 2012).
To determine whether differential RAS pathway control of NSC proliferation was also observed in vivo, we used Nf1BLBP conditional knockout mice to inactivate Nf1 gene expression in BLBP+ NSCs at embryonic day 9.5 (E9.5) (Hegedus et al. 2007). At P0.5, increased ERK phosphorylation (Thr202/Tyr204), AKT phosphorylation (Ser473 and Thr308), and proliferating cells (percentage of Ki67+ cells) were observed in Nf1BLBP conditional knockout mice relative to wild-type controls (Supplemental Fig. S1C,D). Following the treatment of pregnant females with either 5 mg/kg PD901 or 30 mg/kg BKM120 from E15 to E18, the percentage of proliferating Ki67+ cells within the TVZ was quantified at P0.5. ERK hyperactivation in the TVZ of Nf1BLBP conditional knockout pups was reduced by PD901 treatment; however, there was no change in TVZ cell proliferation (Fig. 1E). In contrast, BKM120 treatment inhibited AKT (Ser473 and Thr308 phosphorylation) hyperactivation and reduced TVZ cell proliferation (Fig. 1E). Collectively, these data demonstrate that neurofibromin regulates NSC proliferation in a PI3K/AKT-dependent manner in vitro and in vivo, consistent with previous findings (Lee et al. 2010).
AKT maintenance of NSC growth has been reported in mice and flies (Lee et al. 2010, 2013; Amiri et al. 2012); however, the role of AKT in regulating NSC multilineage differentiation is less clear (Peltier et al. 2007). Following in vitro differentiation, Nf1 loss in NSCs resulted in an increase in the percentage of GFAP+ and O4+ cells (3.4-fold and fourfold, respectively) and a decrease in Tuj1+ cells (2.5-fold) relative to wild-type cells (Fig. 2A). While treatment with the AKT inhibitor (MK2206) had no effect on these Nf1-deficient NSC differentiation defects, MEK inhibition (PD901) restored Nf1−/− NSC astrocyte, oligodendrocyte (Olig2+ and O4+ cells), and neuron differentiation to near wild-type levels (Fig. 2A; Supplemental Fig. S2A). Similar results were observed using other PI3K (BKM120) and MEK (UO126) inhibitors (Supplemental Fig. S2B).
Figure 2.
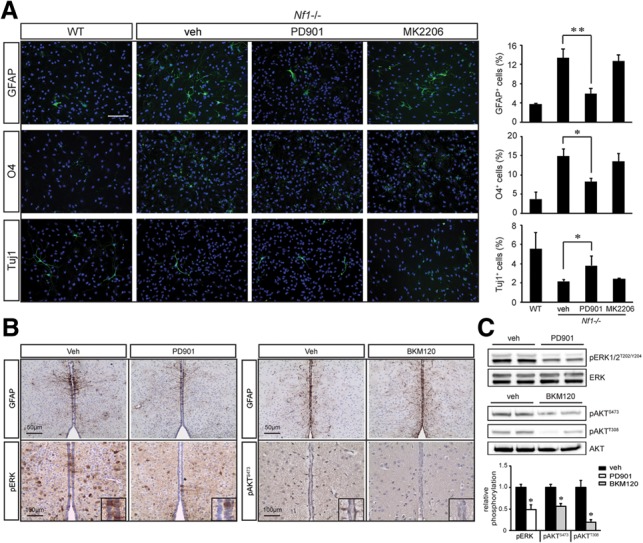
Neurofibromin regulation of NSC multilineage differentiation is MEK-dependent. (A) The astrocyte, oligodendrocyte, and neuronal differentiation defects observed in Nf1−/− TVZ NSCs were restored to wild-type (WT) levels following PD901, but not MK2206, treatment. (B) PD901-treated Nf1BLBP conditional knockout mice exhibited reduced astrocyte numbers compared with vehicle-treated controls. n = 4 per group. No change in astrocyte numbers were observed following BKM120 treatment. n = 4 per group. (C) Densitometric analyses of pERKT202/Y204, pAKTS473, and pAKTT308 immunoblots relative to total ERK and AKT levels. n = 4 per group. Error bars denote the mean ± SD. (veh) Vehicle. Bar, 100 μm. (*) P < 0.05; (**) P < 0.01.
To determine whether MEK activation was responsible for these multilineage defects in vivo, Nf1BLBP conditional knockout pups were treated from P0.5 to P18 (astrocytes and oligodendrocytes) or from E15 to E18 (neurons and Olig2+ progenitors) with PD901 or BKM120. While Nf1 inactivation in BLBP+ neural progenitor cells led to increased numbers of GFAP+ astrocytes and APC+ oligodendrocytes at P18 (Supplemental Fig. S2C), decreased numbers of NeuN+ neurons and increased numbers of Olig2+ cells were observed at P0.5 (Supplemental Fig. S2E). As observed in vitro, MEK, but not PI3K/AKT, inhibition restored astrocyte (Fig. 2B,C) and oligodendrocyte numbers in Nf1BLBP conditional knockout mice to wild-type levels at P18 (Supplemental Fig. S2D) as well as ameliorated the increase in Olig2+ cells and decrease in NeuN+ cells at P0.5 (Supplemental Fig. S2E). Together with the in vitro results, these data reveal that AKT and MEK independently regulate NSC proliferation and multilineage differentiation, respectively.
The observation that MEK is a central driver of gliogenesis is consistent with prior reports demonstrating that Mek1/2-deficient mice exhibit impaired glial cell specification (Li et al. 2012) and that neonatal MEK inhibition rescues the developmental defects in Nf1-deficient brains by restoring normal neuron–glial specification (Wang et al. 2012). However, the mechanism responsible for neurofibromin/MEK-driven multilineage differentiation has not been elucidated. Two transcription factors, Erm and Ascl1, can regulate gliogenesis in response to elevated RAS/ERK signaling (Li et al. 2012; Breunig et al. 2015). While Nf1−/− NSCs exhibit increased Erm expression by quantitative RT–PCR (qRT–PCR) and Western blotting, this was not attenuated following MEK inhibition (PD901) (Supplemental Fig. S3A,B). In addition, no change in Asc1 protein levels was observed after Nf1 loss, and nearly 100% of wild-type and Nf1−/− NSCs were Ascl1+ (Supplemental Fig. S3B,C). Since Erm and Ascl1 function can also be regulated by phosphorylation (Li et al. 2014), these proteins could still play a role in Nf1−/− NSC gliogenesis.
Based on increased Jagged-1 expression in Nf1-deficient mouse astrocytes (Banerjee et al. 2011) and numerous studies highlighting the critical role of Notch1 signaling in specifying neural cell fate during development (Lutolf et al. 2002; Stump et al. 2002), we examined Jagged1/Notch pathway activation. Following neurofibromin loss in NSCs, there was increased Jagged1 expression and Notch activation (Notch intracellular domain [NICD] expression) in vitro (Fig. 3A) and in vivo (Fig. 3B). However, in contrast to Nf1-deficient astrocytes (Banerjee et al. 2011), Jagged1 was not regulated by mTOR activation. While there was a 2.1-fold and 3.6-fold increase in S6 Ser240/244 and Ser235/236 phosphorylation, respectively, mTOR inhibition with rapamycin did not affect Jagged1 or NICD expression in Nf1−/− NSCs (Supplemental Fig. S3D). Consistent with the hypothesis that neurofibromin control of Jagged1 in NSCs is AKT/mTOR-independent, MK2206 treatment of Nf1−/− NSCs did not reduce Jagged1 or NICD expression (Fig. 3C) or attenuate ERK hyperphosphorylation (Supplemental Fig. S3E). Instead, MEK inhibition (PD901) restored Jagged1 and NICD expression in Nf1−/− NSCs to wild-type levels (Fig. 3D) without any change in AKT phosphorylation (Supplemental Fig. S3F). Similar results were observed with additional MEK (UO126) and PI3K (BKM120) inhibitors (Supplemental Fig. S3G). Moreover, Jagged1 and NICD expression in the TVZ of Nf1BLBP conditional knockout mice was reduced following PD901 treatment (Fig. 3E) but not by PI3K/AKT (BKM120) inhibition. Collectively, these results establish that neurofibromin regulation of Jagged1/Notch activation is mediated by MEK/ERK signaling in vitro and in vivo.
Figure 3.
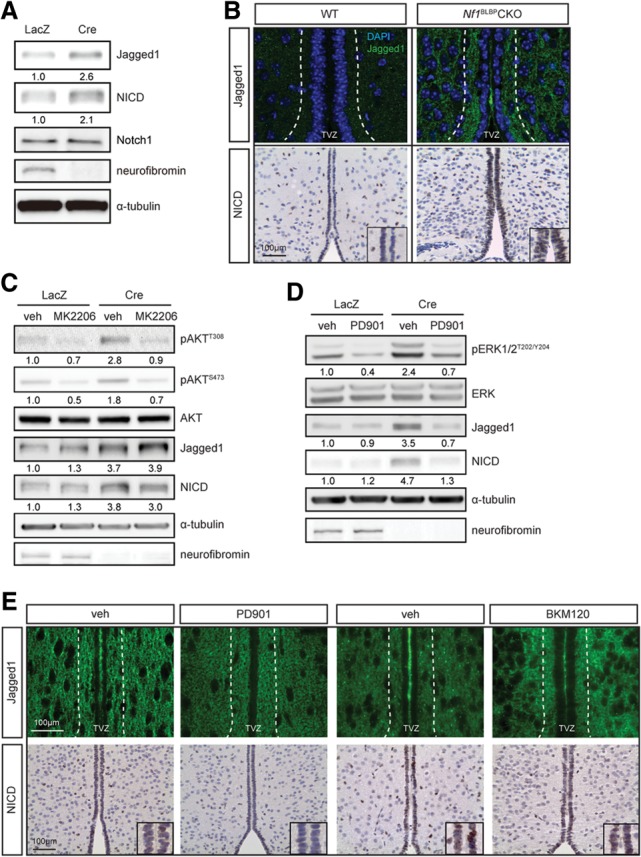
Neurofibromin loss in NSCs results in MEK-dependent Jagged1/Notch activation. Neurofibromin loss resulted in increased Jagged1 and cleaved Notch1 (NICD) expression in vitro (A) and in vivo (B) relative to controls. While MK2206 treatment did not reduce Jagged1 and NICD expression (C), PD901 treatment reduced Nf1-deficient NSC Jagged1 and NICD expression to wild-type (WT) levels (D). (E) Increased Jagged1 and NICD expression was observed in the TVZ of Nf1BLBP conditional knockout (CKO) mice, which was ameliorated by PD901, but not BKM120, treatment. n = 4 per group. (veh) Vehicle.
The importance of Jagged1 to gliogenesis is further supported by studies using conditional Jagged1 deletion in cerebellar neuroepithelial cells (Weller et al. 2006) as well as reports demonstrating that Jagged1-mediated Notch pathway activation promotes astrogliogenesis in vivo (Hu et al. 2013) and inhibits neurogenesis in vitro (Wilhelmsson et al. 2012). The ability of activated Notch1 to dictate multilineage differentiation in neural progenitor cells typically involves the Hes1 and Hes5 transcription factors (Furukawa et al. 2000; Hojo et al. 2000). Following Nf1 loss in NSCs, there was increased Hes1 and Hes5 expression (Fig. 4A), which was reduced by PD901 treatment (Fig. 4B) in vitro. Moreover, ectopic expression of an activated MEK (caMEK), but not an activated AKT (myrAKT), molecule in NSCs increased Jagged1, NICD, Hes1, and Hes5 levels (Supplemental Fig. S4A). Finally, MEK inhibition restored Hes1 and Hes5 expression to wild-type levels in the TVZ of Nf1BLBP conditional knockout mice in vivo (Fig. 4C). Together, these findings demonstrate that the Notch1 signaling pathway is activated following neurofibromin loss in a MEK-dependent manner.
Figure 4.
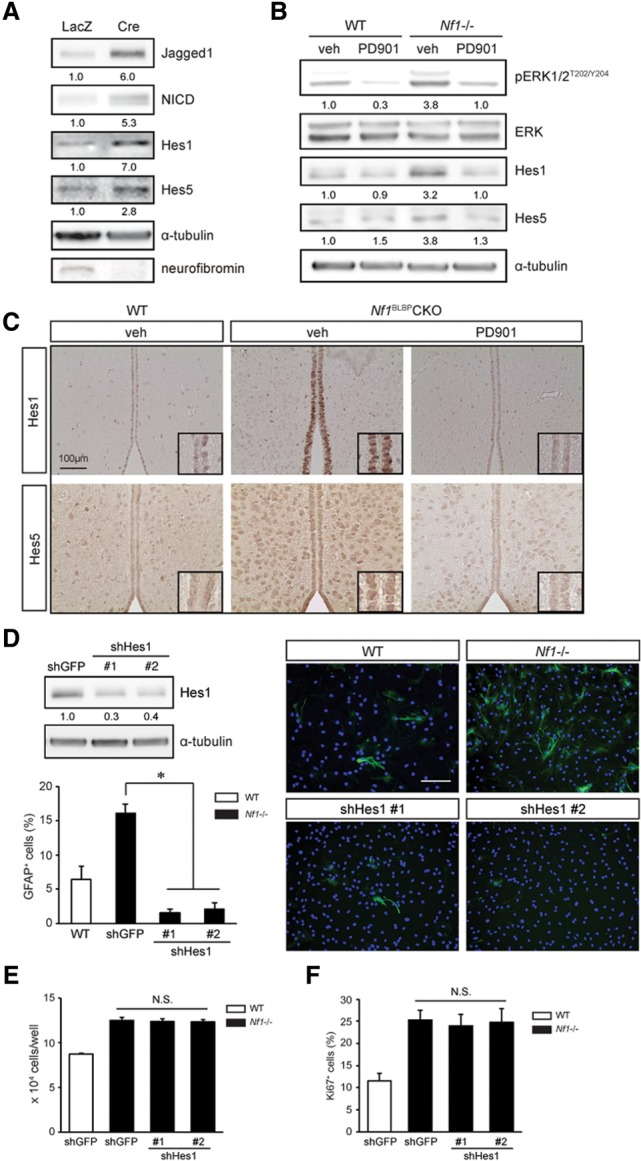
Neurofibromin regulation of NSC astrocyte differentiation is Hes1-dependent. Nf1 loss results in increased expression of the Hes1 and Hes5 Notch downstream effectors (A), which was restored to wild-type (WT) levels following 5 nM PD901 treatment (B). (C) Nf1BLBP conditional knockout (CKO) mice treated with 5 mg/kg PD901 (P0.5–P18) have reduced Hes1 and Hes5 expression relative to vehicle-treated mice. n = 4 per group. (D) Hes1 shRNA knockdown reduced the percentage GFAP+ astrocytes following Nf1−/− TVZ NSC differentiation. Hes1 knockdown (shHes1) did not reduce Nf1−/− TVZ NSC growth (direct cell counting) (E) or proliferation (percentage of Ki67+ cells) (F). (veh) Vehicle. Error bars denote mean ± SD. Nuclei were counterstained with DAPI. Bar, 100 μm. (*) P < 0.01; (N.S.) not significant.
Based on conflicting reports regarding Hes5 regulation of gliogenesis (Hojo et al. 2000; Wu et al. 2003) and the nearly exclusive expression of Hes1 within the TVZ, we chose to focus on Hes1. Using two independently generated Hes1 shRNA constructs to decrease Hes1 expression in NSCs (60% reduction), the increased astrocyte differentiation observed following neurofibromin loss was reduced to near wild-type levels (Fig. 4D). Importantly, Hes1 reduction had no effect on Nf1−/− NSC growth (direct cell counting) (Fig. 4E) or proliferation (percentage of Ki67+ cells) (Fig. 4F). Moreover, Hes1 knockdown ameliorated the decrease in neuronal differentiation in Nf1-deficient NSCs but surprisingly had no effect on oligodendrocyte differentiation (percentage of O4+ cells) (Supplemental Fig. S4B). These findings demonstrate that neurofibromin regulation of astrocyte and neuronal differentiation is mediated by Hes1 in a reciprocally coordinated fashion, whereas other mechanisms underlie MEK-dependent oligodendrocyte differentiation. In this regard, Hes1 knockdown did not reduce the number of Olig2+ cells (Supplemental Fig. S4C). While RAF/MEK signaling is an important determinant of oligodendrocyte differentiation in mice (Galabova-Kovacs et al. 2008) and zebrafish (Shin et al. 2012), other neurofibromin-regulated MEK downstream pathways are likely responsible for governing oligodendrocyte differentiation.
To determine how neurofibromin controls Jagged1 expression, we examined Jagged1 mRNA expression using real-time qRT–PCR (Fig. 5A). Jagged-1 mRNA expression was regulated in Nf1−/− NSCs on the transcriptional level through MEK, since PD901 treatment restored Jagged1 mRNA to wild-type levels. Several potential regulators of Jagged1 expression have been identified, including β-catenin, YAP, and TGFβ/Smad3 (Chen et al. 2010; Zhang et al. 2010; Tschaharganeh et al. 2013). While we observed no changes in β-catenin activation or YAP expression following neurofibromin loss, the increased Smad3 expression observed in Nf1-deficient NSCs was reduced to wild-type levels following PD901 treatment (Fig. 5B,C).
Figure 5.
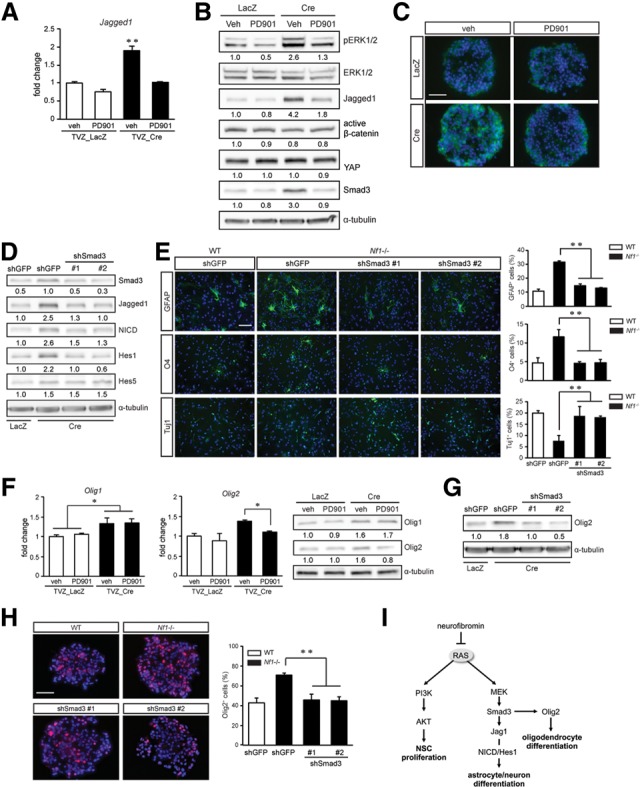
Neurofibromin/Jagged1 regulation of astrocyte differentiation requires MEK-mediated Smad3 expression. Increased Jagged1 transcription (A) and protein levels (B) in Nf1−/− NSCs were reduced to wild-type levels following 5 nM PD901 treatment. PD901 treatment of Nf1−/− NSCs had no effect on β-catenin activity or YAP expression but reduced Smad3 expression by Western blotting (B) and immunocytochemistry (C). (D) Smad3 knockdown restored Jagged1, NICD, and Hes1, but not Hes5, expression to wild-type levels. (E) The astrocyte, oligodendrocyte, and neuronal differentiation defects observed in Nf1−/− TVZ NSCs were restored to near wild-type levels following Smad3 knockdown. (F) MEK inhibition (5 nM PD901) reduced Olig2, but not Olig1, expression by qRT–PCR and Western blotting. Smad3 knockdown reduced Olig2 expression by Western blotting (G) as well as the percentage of Olig2+ cells within the Nf1−/− neurospheres (H). (I) Proposed model of neurofibromin/RAS regulation of NSC growth and multilineage differentiation. (veh) Vehicle. Error bars denote mean ± SD. Bar, 100 μm. (*) P < 0.05; (**) P < 0.01.
We next used genetic and pharmacologic approaches to reduce Smad3 function. Following Smad3 knockdown (using two different shRNA constructs), Jagged1, NICD, and Hes1 expression in Nf1−/− NSCs was restored to wild-type levels (Fig. 5D). Similar to PD901 treatment, Smad3 knockdown of Nf1−/− NSCs restored astrocyte, oligodendrocyte, and neuronal differentiation to wild-type levels (Fig. 5E). It should be noted that Smad3 knockdown did not change Hes5 expression (Fig. 5D), arguing against Hes5 as a mediator of neurofibromin-controlled multilineage differentiation. Similar results were observed in Nf1-deficient NSCs treated with the SIS3 Smad3 pharmacological inhibitor (Supplemental Fig. S5A,B; Jinnin et al. 2006). Collectively, these results support a mechanism by which neurofibromin/MEK control of astrocyte and neuron differentiation operates in a Smad3/Jagged1/Hes1-dependent manner.
Neurofibromin regulation of Smad3 function could operate at the level of transcription, protein degradation, or phosphorylation (Massague et al. 2005). While neurofibromin loss results in increased Smad3 levels, subcellular fractionation revealed an enrichment of Smad3 in the nucleus of Nf1−/− NSCs (34%) relative to wild-type NSCs (8%) (Supplemental Fig. S5C). However, the mechanism underlying this increase in Smad3 expression was not the result of increased Smad3 RNA levels (qRT–PCR) (Supplemental Fig. S5D) or degradation mediated by increased SCF/ROC1 and GSK3-β binding (Fukuchi et al. 2001; Guo et al. 2008). Whereas Smad3 physically interacted with GSK3-β, but not with SCF/ROC1 (Supplemental Fig. S5E), neurofibromin loss or MEK inhibition (PD901 treatment) had no effect on Smad3 and GSK3-β binding, as assessed by immunoprecipitation. Finally, MEK-dependent Smad3 regulation was not dependent on TGFβ-induced phosphorylation, as Smad3-Ser423/425 phosphorylation was similar in wild-type and Nf1−/− NSCs (Supplemental Fig. S5F) and did not involve phosphorylation at the best-characterized ERK phosphorylation site (Ser208) (Supplemental Fig. S5F). Future studies will be required to identify the mechanism responsible for neurofibromin regulation of Smad3 levels.
Taken together, our findings establish that neurofibromin control of NSC function involves the selective use of distinct RAS effector pathways. In this regard, RAS activation is critical for both neurofibromin-regulated NSC proliferation and multilineage differentiation such that inhibition using the nonselective RAS inhibitor (lovastatin) (Li et al. 2005) restored both Nf1-deficient NSC growth and multilineage differentiation to wild-type levels (Supplemental Fig. S5G–I). However, whereas neurofibromin control of neuron and astrocyte differentiation requires Smad3 regulation of Hes1, the mechanism underlying Smad3-mediated oligodendrocyte differentiation likely involves other transcription factors. In this manner, Olig-1 and Olig-2 have been identified as essential factors for specifying oligodendrogliogenesis (Lu et al. 2000; Takebayashi et al. 2002). Consistent with recent findings demonstrating that Olig1 is not essential for oligodendrocyte development in mice (Paes de Faria et al. 2014), MEK inhibition (PD901) reduced the increased Olig2, but not Olig1, mRNA and protein expression in Nf1-deficient NSCs (Fig. 5F). Moreover, the elevated Olig2 expression in Nf1-deficient NSCs was decreased following Smad3 genetic (Fig. 5G,H) or pharmacologic (Supplemental Fig. S5J) inhibition. These findings suggest a model in which neurofibromin control of NSC multilineage differentiation involves distinct transcriptional programs: Neuron and astrocyte differentiation requires MEK/Smad3-dependent Hes1 induction, whereas MEK/Smad3-dependent oligodendrocyte differentiation involves Olig2 function (Fig. 5I).
Coupled with observations that other RAS downstream pathways have cell type-specific functions (neurons vs. astrocytes) relevant to brain cell function (Hegedus et al. 2007; Anastasaki and Gutmann 2014), the observations reported here establish that differential use of distinct RAS effector signaling pathways can govern separable cellular functions even within the same cell type, further underscoring the importance of cellular context in interpreting the impact of genetic mutations on brain function.
Materials and methods
Mice
BLBP-Cre; Nf1flox/wt transgenic mice were crossed with Nf1flox/flox mice to generate BLBP-Cre; Nf1flox/flox (conditional knockout) mice. Nf1flox/flox mice were used as wild-type controls. All strains were maintained on a C57BL/6 background and used in accordance with an approved animal studies protocol at Washington University.
Primary NSC analysis
TVZ NSCs were established from the TVZ of P1 Nf1flox/flox mouse pups and analyzed as previously described (Lee et al. 2010). Retroviral and lentivirus (Supplemental Table 1) transduction was performed for overexpression and knockdown studies, respectively. All experiments were performed at least three times using primary NSCs generated from independent litters.
Western blotting
Western blotting was performed as previously reported (Lee et al. 2010) using primary antibodies (Supplemental Table 2) and was quantified by densitometry using a chemiluminescence imaging system (UVP). Each experiment was performed at least three times, and representative blots are presented.
Immunostaining
Paraffin or frozen sections were processed (Dasgupta and Gutmann 2005) prior to staining with the appropriate antibodies. The percentage of Ki67+ cells lining the TVZ was quantified as previously reported (Lee et al. 2012).
Pharmacologic inhibition studies
Neurospheres were trypsinized into single cells, and 5 × 105 cells per well were plated onto ultralow-binding 60-mm plates. Cells were treated with specific inhibitors for 4–5 d. PD901 (5 mg/kg/day; Selleck), BKM120 (30 mg/kg; Selleck), or matched vehicle (0.5% hydroxypropyl methycellulose with 0.2% Tween 80 [Sigma-Aldrich] or 10/90 [v/v] N-methyl-2-pyrrolidone [NMO]/PEG300, respectively) was injected intraperitoneally into pregnant females from E15 to E18. Postnatal PD901 or NVP-BKM120 administration to lactating females (P0.5–P18) was achieved by oral gavage, and the mice were perfused at P18.
Real-time qRT–PCR
Real-time qRT–PCR was performed as previously described (Yeh et al. 2009) with specific primers (Supplemental Table 3), and ΔΔCT values were calculated using H3f3a as an internal control.
Statistical analysis
Each experiment was performed with samples from at least three independent groups. Statistical significance was set at P < 0.05 using the Student's t-test.
Supplementary Material
Acknowledgments
We thank the Broad Institute RNAi Consortium (TRC), the Children's Discovery Institute (CDI), and The Genome Institute (TGI) at Washington University. We also thank Dr. Raphael Kopan for helpful discussions during the execution of these studies. Y.-H.C. is a recipient of the fellowship from the American Brain Tumor Association supported by the Emily Dorfman Foundation for Children in memory of Emily Ann Dorfman. This work was partially supported by a grant from the National Institutes of Health (R01-NS065547-01) to D.H.G.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.261677.115.
References
- Amiri A, Cho W, Zhou J, Birnbaum SG, Sinton CM, McKay RM, Parada LF. 2012. Pten deletion in adult hippocampal neural stem/progenitor cells causes cellular abnormalities and alters neurogenesis. J Neurosci 32: 5880–5890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anastasaki C, Gutmann DH. 2014. Neuronal NF1/RAS regulation of cyclic AMP requires atypical PKC activation. Hum Mol Genet 23: 6712–6721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee S, Gianino SM, Gao F, Christians U, Gutmann DH. 2011. Interpreting mammalian target of rapamycin and cell growth inhibition in a genetically engineered mouse model of Nf1-deficient astrocytes. Mol Cancer Ther 10: 279–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett MR, Rizvi TA, Karyala S, McKinnon RD, Ratner N. 2003. Aberrant growth and differentiation of oligodendrocyte progenitors in neurofibromatosis type 1 mutants. J Neurosci 23: 7207–7217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breunig JJ, Levy R, Antonuk CD, Molina J, Dutra-Clarke M, Park H, Akhtar AA, Kim GB, Hu X, Bannykh SI, et al. 2015. Ets factors regulate neural stem cell depletion and gliogenesis in Ras pathway glioma. Cell Rep 12: 258–271. [DOI] [PubMed] [Google Scholar]
- Chen X, Stoeck A, Lee SJ, Shih Ie M, Wang MM, Wang TL. 2010. Jagged1 expression regulated by Notch3 and Wnt/β-catenin signaling pathways in ovarian cancer. Oncotarget 1: 210–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasgupta B, Gutmann DH. 2005. Neurofibromin regulates neural stem cell proliferation, survival, and astroglial differentiation in vitro and in vivo. J Neurosci 25: 5584–5594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diggs-Andrews KA, Gutmann DH. 2013. Modeling cognitive dysfunction in neurofibromatosis-1. Trends Neurosci 36: 237–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuchi M, Imamura T, Chiba T, Ebisawa T, Kawabata M, Tanaka K, Miyazono K. 2001. Ligand-dependent degradation of Smad3 by a ubiquitin ligase complex of ROC1 and associated proteins. Mol Biol Cell 12: 1431–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa T, Mukherjee S, Bao ZZ, Morrow EM, Cepko CL. 2000. rax, Hes1, and notch1 promote the formation of Muller glia by postnatal retinal progenitor cells. Neuron 26: 383–394. [DOI] [PubMed] [Google Scholar]
- Galabova-Kovacs G, Catalanotti F, Matzen D, Reyes GX, Zezula J, Herbst R, Silva A, Walter I, Baccarini M. 2008. Essential role of B-Raf in oligodendrocyte maturation and myelination during postnatal central nervous system development. J Cell Biol 180: 947–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillamo JS, Creange A, Kalifa C, Grill J, Rodriguez D, Doz F, Barbarot S, Zerah M, Sanson M, Bastuji-Garin S, et al. 2003. Prognostic factors of CNS tumours in Neurofibromatosis 1 (NF1): a retrospective study of 104 patients. Brain 126: 152–160. [DOI] [PubMed] [Google Scholar]
- Guo X, Ramirez A, Waddell DS, Li Z, Liu X, Wang XF. 2008. Axin and GSK3-β control Smad3 protein stability and modulate TGF-β signaling. Genes Dev 22: 106–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegedus B, Dasgupta B, Shin JE, Emnett RJ, Hart-Mahon EK, Elghazi L, Bernal-Mizrachi E, Gutmann DH. 2007. Neurofibromatosis-1 regulates neuronal and glial cell differentiation from neuroglial progenitors in vivo by both cAMP- and Ras-dependent mechanisms. Cell Stem Cell 1: 443–457. [DOI] [PubMed] [Google Scholar]
- Hojo M, Ohtsuka T, Hashimoto N, Gradwohl G, Guillemot F, Kageyama R. 2000. Glial cell fate specification modulated by the bHLH gene Hes5 in mouse retina. Development 127: 2515–2522. [DOI] [PubMed] [Google Scholar]
- Hu X, He W, Luo X, Tsubota KE, Yan R. 2013. BACE1 regulates hippocampal astrogenesis via the Jagged1–Notch pathway. Cell Rep 4: 40–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinnin M, Ihn H, Tamaki K. 2006. Characterization of SIS3, a novel specific inhibitor of Smad3, and its effect on transforming growth factor-β1-induced extracellular matrix expression. Mol Pharm 69: 597–607. [DOI] [PubMed] [Google Scholar]
- Kaul A, Toonen JA, Cimino PJ, Gianino SM, Gutmann DH. 2015. Akt- or MEK-mediated mTOR inhibition suppresses Nf1 optic glioma growth. Neuro Oncol 17: 843–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DY, Yeh TH, Emnett RJ, White CR, Gutmann DH. 2010. Neurofibromatosis-1 regulates neuroglial progenitor proliferation and glial differentiation in a brain region-specific manner. Genes Dev 24: 2317–2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DY, Gianino SM, Gutmann DH. 2012. Innate neural stem cell heterogeneity determines the patterning of glioma formation in children. Cancer Cell 22: 131–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KS, Wu Z, Song Y, Mitra SS, Feroze AH, Cheshier SH, Lu B. 2013. Roles of PINK1, mTORC2, and mitochondria in preserving brain tumor-forming stem cells in a noncanonical Notch signaling pathway. Genes Dev 27: 2642–2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Cui Y, Kushner SA, Brown RA, Jentsch JD, Frankland PW, Cannon TD, Silva AJ. 2005. The HMG-CoA reductase inhibitor lovastatin reverses the learning and attention deficits in a mouse model of neurofibromatosis type 1. Curr Biol 15: 1961–1967. [DOI] [PubMed] [Google Scholar]
- Li X, Newbern JM, Wu Y, Morgan-Smith M, Zhong J, Charron J, Snider WD. 2012. MEK is a key regulator of gliogenesis in the developing brain. Neuron 75: 1035–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Mattar P, Dixit R, Lawn SO, Wilkinson G, Kinch C, Eisenstat D, Kurrasch DM, Chan JA, Schuurmans C. 2014. RAS/ERK signaling controls proneural genetic programs in cortical development and gliomagenesis. J Neurosci 34: 2169–2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu QR, Yuk D, Alberta JA, Zhu Z, Pawlitzky I, Chan J, McMahon AP, Stiles CD, Rowitch DH. 2000. Sonic hedgehog-regulated oligodendrocyte lineage genes encoding bHLH proteins in the mammalian central nervous system. Neuron 25: 317–329. [DOI] [PubMed] [Google Scholar]
- Lutolf S, Radtke F, Aguet M, Suter U, Taylor V. 2002. Notch1 is required for neuronal and glial differentiation in the cerebellum. Development 129: 373–385. [DOI] [PubMed] [Google Scholar]
- Massague J, Seoane J, Wotton D. 2005. Smad transcription factors. Genes Dev 19: 2783–2810. [DOI] [PubMed] [Google Scholar]
- Paes de Faria J, Kessaris N, Andrew P, Richardson WD, Li H. 2014. New Olig1 null mice confirm a non-essential role for Olig1 in oligodendrocyte development. BMC Neurosci 15: 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peltier J, O'Neill A, Schaffer DV. 2007. PI3K/Akt and CREB regulate adult neural hippocampal progenitor proliferation and differentiation. Dev Neurobiol 67: 1348–1361. [DOI] [PubMed] [Google Scholar]
- Sanchez-Ortiz E, Cho W, Nazarenko I, Mo W, Chen J, Parada LF. 2014. NF1 regulation of RAS/ERK signaling is required for appropriate granule neuron progenitor expansion and migration in cerebellar development. Genes Dev 28: 2407–2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin J, Padmanabhan A, de Groh ED, Lee JS, Haidar S, Dahlberg S, Guo F, He S, Wolman MA, Granato M, et al. 2012. Zebrafish neurofibromatosis type 1 genes have redundant functions in tumorigenesis and embryonic development. Dis Model Mech 5: 881–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stump G, Durrer A, Klein AL, Lutolf S, Suter U, Taylor V. 2002. Notch1 and its ligands Delta-like and Jagged are expressed and active in distinct cell populations in the postnatal mouse brain. Mech Dev 114: 153–159. [DOI] [PubMed] [Google Scholar]
- Takebayashi H, Nabeshima Y, Yoshida S, Chisaka O, Ikenaka K, Nabeshima Y. 2002. The basic helix-loop-helix factor olig2 is essential for the development of motoneuron and oligodendrocyte lineages. Curr Biol 12: 1157–1163. [DOI] [PubMed] [Google Scholar]
- Tschaharganeh DF, Chen X, Latzko P, Malz M, Gaida MM, Felix K, Ladu S, Singer S, Pinna F, Gretz N, et al. 2013. Yes-associated protein up-regulates Jagged-1 and activates the Notch pathway in human hepatocellular carcinoma. Gastroenterology 144: 1530–1542.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Kim E, Wang X, Novitch BG, Yoshikawa K, Chang LS, Zhu Y. 2012. ERK inhibition rescues defects in fate specification of Nf1-deficient neural progenitors and brain abnormalities. Cell 150: 816–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weller M, Krautler N, Mantei N, Suter U, Taylor V. 2006. Jagged1 ablation results in cerebellar granule cell migration defects and depletion of Bergmann glia. Dev Neurosci 28: 70–80. [DOI] [PubMed] [Google Scholar]
- Wilhelmsson U, Faiz M, de Pablo Y, Sjoqvist M, Andersson D, Widestrand A, Potokar M, Stenovec M, Smith PL, Shinjyo N, et al. 2012. Astrocytes negatively regulate neurogenesis through the Jagged1-mediated Notch pathway. Stem Cells 30: 2320–2329. [DOI] [PubMed] [Google Scholar]
- Wu Y, Liu Y, Levine EM, Rao MS. 2003. Hes1 but not Hes5 regulates an astrocyte versus oligodendrocyte fate choice in glial restricted precursors. Dev Dyn 226: 675–689. [DOI] [PubMed] [Google Scholar]
- Yeh TH, Lee DY, Gianino SM, Gutmann DH. 2009. Microarray analyses reveal regional astrocyte heterogeneity with implications for neurofibromatosis type 1 (NF1)-regulated glial proliferation. Glia 57: 1239–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Zhang J, Navrazhina K, Argaw AT, Zameer A, Gurfein BT, Brosnan CF, John GR. 2010. TGFβ1 induces Jagged1 expression in astrocytes via ALK5 and Smad3 and regulates the balance between oligodendrocyte progenitor proliferation and differentiation. Glia 58: 964–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.