

Abstract
The capacity of mosquitoes to resist insecticides threatens the control of diseases such as dengue and malaria. Until alternative control tools are implemented, characterizing resistance mechanisms is crucial for managing resistance in natural populations. Insecticide biodegradation by detoxification enzymes is a common resistance mechanism; however, the genomic changes underlying this mechanism have rarely been identified, precluding individual resistance genotyping. In particular, the role of copy number variations (CNVs) and polymorphisms of detoxification enzymes have never been investigated at the genome level, although they can represent robust markers of metabolic resistance. In this context, we combined target enrichment with high-throughput sequencing for conducting the first comprehensive screening of gene amplifications and polymorphisms associated with insecticide resistance in mosquitoes. More than 760 candidate genes were captured and deep sequenced in several populations of the dengue mosquito Ae. aegypti displaying distinct genetic backgrounds and contrasted resistance levels to the insecticide deltamethrin. CNV analysis identified 41 gene amplifications associated with resistance, most affecting cytochrome P450s overtranscribed in resistant populations. Polymorphism analysis detected more than 30,000 variants and strong selection footprints in specific genomic regions. Combining Bayesian and allele frequency filtering approaches identified 55 nonsynonymous variants strongly associated with resistance. Both CNVs and polymorphisms were conserved within regions but differed across continents, confirming that genomic changes underlying metabolic resistance to insecticides are not universal. By identifying novel DNA markers of insecticide resistance, this study opens the way for tracking down metabolic changes developed by mosquitoes to resist insecticides within and among populations.
Mosquitoes are vectors of numerous human diseases, representing a major threat for public health worldwide (Lounibos 2002). Dengue and Chikungunya viruses are both transmitted by the mosquito Aedes aegypti and represent a burden in more than 100 countries putting more than 2.5 billion people at risk (WHO 2009, 2014). Since the Second World War, chemical insecticides have been massively used for controlling vector populations and reducing disease transmission, but their efficacy is now threatened by resistance mechanisms developed by mosquitoes. Insecticide resistance is widespread in Ae. aegypti and affects most insecticides used for vector control (Ranson et al. 2010). Resistance to pyrethroid insecticides, the primary insecticide family used against adult mosquitoes, is particularly worrying in the context of the re-emergence of dengue and other arboviruses worldwide (Bhatt et al. 2013). Although attempts are made to develop new insecticides or alternative mosquito control strategies (Scholte et al. 2004; Lacey 2007; Hoffmann et al. 2011; Walker et al. 2011; Harris et al. 2012), their large-scale implementation in tropical regions will require at least a decade. Until this, characterizing molecular mechanisms underlying resistance is crucial for tracking down resistance alleles and improving resistance management strategies (Corbel et al. 2013).
Resistance to pyrethroids can be the consequence of various mechanisms, such as nonsynonymous mutations affecting the voltage-gated sodium channel targeted by these insecticides, i.e., knockdown resistance (kdr) mutations, a lower insecticide penetration, its sequestration, or its biodegradation (metabolic resistance) (Hemingway et al. 2004; Li et al. 2007). Kdr mutations and metabolic resistance are known as the two primary resistance mechanisms in mosquitoes. Several kdr mutations have been identified in Ae. aegypti, and the association between the V1016G/I and the F1534C mutations and pyrethroid resistance has been confirmed (Brengues et al. 2003; Saavedra-Rodriguez et al. 2007; Yanola et al. 2011). Monitoring the frequency of these mutations in field populations is possible through simple DNA-based diagnostic assays and provides key data for resistance management strategies.
Metabolic resistance is far less understood, although this type of resistance is frequent and often accounts for a significant part of the resistance phenotype (Li et al. 2007; Nkya et al. 2013). Such a resistance mechanism is caused by an increased activity of detoxification enzymes. These detoxification enzymes include cytochrome P450 monooxygenases (P450s or CYPs for genes), carboxy/cholinesterases (CCEs), glutathione S-transferases (GSTs), and UDP-glycosyl-transferases (UDPGTs), although other families can be involved (Hemingway et al. 2004; David et al. 2013). Their high diversity (roughly 300 genes in Ae. aegypti) and the complexity of insecticide biodegradation pathways make the identification of those conferring resistance challenging. Theoretically, metabolic resistance can be the consequence of an increased expression of one or multiple detoxification enzymes capable of metabolizing the insecticide and/or the selection of variants showing a higher insecticide metabolism rate due to conformational modifications. Because overexpression is frequently associated with overtranscription, most candidate genes were identified based on their differential transcription in resistant populations compared to susceptible counterparts using transcriptomics (for review, see David et al. 2013). Although subjected to inherent difficulties associated with gene expression studies (e.g., RNA handling and degradation, uncontrolled variation across time, tissues, and populations), these approaches identified several detoxification enzymes overtranscribed in resistant populations, some of them being later validated as pyrethroid metabolizers (for review, see Vontas et al. 2012; David et al. 2013). Although these approaches proved their value for identifying metabolic resistance genes, they failed to pinpoint the genomic changes associated to their overexpression. In addition, because a limited quantity of mRNA can be extracted from a single mosquito, these studies were conducted at the population level without estimating the frequency of resistant alleles within populations. A constitutive overtranscription can be the consequence of an up-regulation controlled by cis/trans genomic regulatory elements or an increased gene copy number (Li et al. 2007). Although gene amplifications have been associated with the overtranscription of a few pyrethroid resistance genes in mosquitoes (Wondji et al. 2009; Itokawa et al. 2010, 2011; Bariami et al. 2012), no comprehensive screening of gene amplifications linked to insecticide resistance has ever been conducted. Considering the central role of copy number variation (CNV) in adaptations associated with a “gene dosage” effect (for review, see Kondrashov 2012) and the richness of the Ae. aegypti genome in transposable elements known to favor duplication events (Nene et al. 2007), conducting a comprehensive screening of gene amplifications associated with insecticide resistance may allow identifying reliable DNA markers of metabolic resistance. Besides, most genes linked to metabolic resistance were identified based on their differential expression while the importance of nonsynonymous changes affecting detoxification enzymes have clearly been neglected by molecular screenings. As for gene amplifications, these polymorphisms are of high interest for monitoring the dynamics of insecticide resistance mechanisms among and within mosquito populations.
In this context, the present study aimed at combining the target enrichment technology with high-throughput sequencing for identifying gene amplifications and polymorphisms linked to pyrethroid resistance in the dengue vector, Ae. aegypti. More than 760 genes, including those encoding for detoxification enzymes, cuticle proteins, ATP-binding cassette (ABC) transporters, neurotransmitter receptors, and voltage-gated channels, were captured and deep sequenced in several susceptible and pyrethroid-resistant populations from three geographical regions (Asia, South America, and laboratory populations). After phenotyping populations for resistance to the pyrethroid deltamethrin and segregating most resistant individuals, gene amplifications and polymorphisms associated with deltamethrin resistance were identified by deep targeted sequencing of large pools of individuals. Gene amplifications were validated on individual mosquitoes, and their impact on transcription levels were validated. Polymorphism data were first used to identify genomic regions under selection through a hierarchical Bayesian approach taking into account population structure. Then, nonsynonymous variants strongly linked to deltamethrin resistance were identified by combining those identified by the Bayesian approach and those displaying allele frequencies mostly associated with resistance. Quantitative PCR assays were used for cross validating polymorphism data and studying the association between gene amplifications and known target-site mutations. Results are discussed in regard to known insecticide resistance mechanisms and the identification of novel DNA markers of insecticide resistance.
Results
Deltamethrin resistance levels
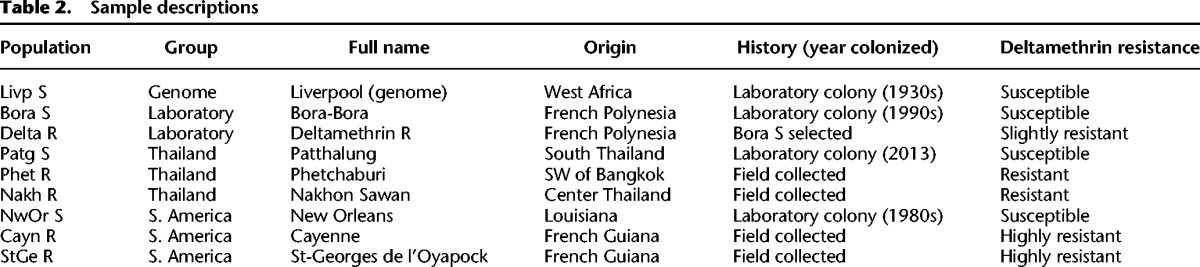
Bioassays on adult females revealed a broad range of resistance to deltamethrin across populations (Table 1). Exposing mosquitoes for 1 h to various doses of insecticide showed that the lethal dose killing 50% of individuals (LD50) varied up to 750-fold between susceptible populations and the most resistant populations from French Guiana. Resistant populations from Thailand showed an intermediate resistance level (about 250-fold), and the laboratory selected population showed a slight resistance (approximately fivefold). These different resistance levels were confirmed by WHO diagnostic assays, in which the time necessary to knock down 50% of individuals (KDT50) varied from 11 min for susceptible populations to >8 h for the most resistant ones.
Table 1.
Deltamethrin resistance levels
Sequencing metrics and coverage
The use of target enrichment technology followed by multiplexed Illumina sequencing allowed obtaining an average of 4.35 million 75-bp reads per sample (Supplemental Table 1). Filtering them according to read pairing, sequence quality, and mapping quality allowed successful mapping of 68% of reads on the Ae. aegypti genome. The majority of mapped reads consisted in perfect matches (46.6%), whereas reads matching with one and two or more mismatches represented 12.6% and 9.2% of sequenced reads, respectively. The mean coverage was homogenous across samples with a total mean coverage of 83× (Supplemental Fig. 1).
Gene amplifications linked to deltamethrin resistance
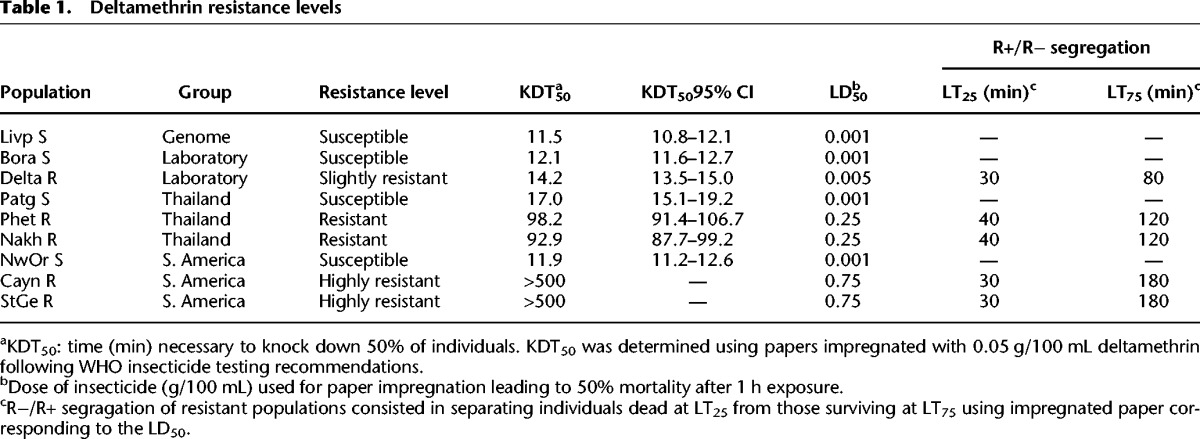
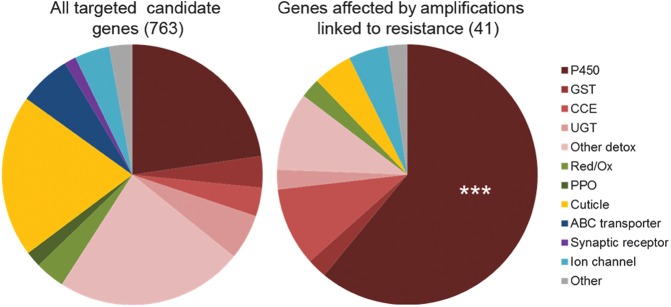
Of the 763 targeted candidate genes, 727 showed a mean coverage greater than 90 reads per kilobase per million reads (RPKM) and were considered for further analysis. Among them, 41 genes showed an increased coverage >1.5-fold in any resistant population compared to the mean coverage of all susceptible ones, together with an increased coverage >1.0-fold in LT75 survivors (R+ phenotype) compared to individuals dead at LT25 (R− phenotype). These genes were considered amplified in association with deltamethrin resistance (Supplemental Table 2). These genes mostly encoded detoxification enzymes with a significant enrichment in P450s (Fig. 1). Most gene amplifications occurred in gene clusters located within a few genomic supercontigs spread across all chromosomes (Fig. 2). Gene amplification profiles were highly conserved within each region but showed marked differences between continents. Thai resistant populations (Nakh R and Phet R) showed a strong amplification of four CCEs located in supercontig 1.142 and another one in supercontig 1.1678. Two CYP6s located in supercontig 1.702 and two sulfotransferases in supercontig 1.975 were also amplified in Thai resistant populations. In contrast, resistant populations from South America (Cayn R and StGe R) showed a marked amplification of four CYP6s located in supercontig 1.1327. Resistant populations from both regions also shared the amplifications of several CYP9Js in supercontigs 1.1188 and 1.221. As expected, the less resistant laboratory population (Delta R) exhibited fewer gene amplifications associated with resistance. The amplification profile of the Delta R population shared particular gene amplifications with field resistant populations. This included a chloride channel also detected in Thailand together with two CYP4Js and a glycosyltransferase also detected in South America.
Figure 1.
Gene families affected by gene amplifications associated with deltamethrin resistance. (Left) Frequency of each gene family among the 763 captured genes. (Right) Frequency of each gene family among the 41 genes affected by genic amplifications associated with deltamethrin resistance. Proportions of each gene family between all captured genes and genes affected by genic amplifications were compared using a one-sided Fisher's exact test: (***) P < 0.001.
Figure 2.

Gene amplifications associated with deltamethrin resistance. (Left) CNV profiles of genes affected by gene amplifications associated with deltamethrin resistance. Color scale shows (R+)/(meanS) CNV for each resistant population, and overimposed “+” marks show (R+)/(R−) CNV. (Right) Location of gene amplifications on genomic supercontigs. Amplified genes are shown in red. Nonamplified genes are shown in maroon. Genes not included in the capture design are shown in gray. Chromosomal locations are shown as described in Timoshevskiy et al. (2014) and Juneja et al. (2014).
Validation of gene amplifications on individual mosquitoes
Nine gene amplifications identified by deep sequencing belonging to two P450 clusters and one CCE cluster heavily impacted by gene amplifications were cross-validated by qPCR on individual mosquitoes. The presence of gene amplifications was confirmed for all targeted genes. A good correlation was observed between CNVs obtained by deep sequencing and qPCR (Supplemental Fig. 2). Individual qPCR data reveal high copy number polymorphism within resistant populations (Supplemental Fig. 3). Estimating the frequency of gene amplifications within each population revealed that amplifications are frequent in resistant populations and sometimes also present at low frequencies in susceptible populations (Supplemental Fig. 4). Comparing the amplification profile of each gene across all individuals revealed CYP6 and CCE clusters are likely affected by single amplification events (Supplemental Fig. 5). In contrast, different copy numbers were frequently observed for genes belonging to the CYP9J cluster on supercontig 1.1188, suggesting that distinct duplication events affect this large P450 cluster.
Impact of gene amplifications on transcription levels
Comparing gene amplification levels obtained by DNA-sequencing with transcription levels obtained by RT-qPCR for the nine candidate genes across all resistant populations confirmed that gene amplifications are often associated with increased transcription (Supplemental Fig. 6). High and significant positive correlations were observed for most genes except for two CYP9Js in supercontig 1.1118, where positive correlations were not significant.
Association of gene amplifications with target-site mutations
Among the nine gene amplifications genotyped by qPCR, only one was found associated with kdr mutations (Supplemental Table 3). This association occurred in the Nakh population from Thailand, where a positive correlation between the presence of CYP9J28 amplification and the presence of the V1016G mutation was found (r = 0.61; P = 0.025). As a consequence of the total segregation of V1016G and F1534C mutations in Thailand, the F1534C mutation was negatively correlated to CYP9J28 amplification in this population (r = −058; P = 0.036).
Polymorphisms identified by deep targeted sequencing
A total of 41,469 polymorphisms supported by an important coverage were called against the reference genome. Among them, 30,400 were polymorphic among populations. A principal component analysis (PCA) based on allele frequency variation across all populations confirmed the overall population structure and the different genetic background of Thai, South-American, and laboratory (French Polynesia) populations (Supplemental Fig. 7). The reliability of polymorphism data was confirmed by comparing the frequencies of V1016I, V1016G, and F1534C kdr mutations estimated by deep targeted sequencing and those obtained by qPCR on individual mosquitoes (Supplemental Fig. 8). The presence of distinct kdr mutations at position 1016 in Thailand and South America was confirmed, and the F1534C mutation was also detected in most populations.
Polymorphisms associated with deltamethrin resistance
Searching for selection footprints using a Bayesian approach taking into account population structure (Foll et al. 2014) identified 156 outlier loci significantly associated with deltamethrin resistance. These outlier loci included 31 nonsynonymous variants and affected multiple supercontigs spread over all chromosomes. Selection footprints varied between geographical regions with outlier loci densities ranging from 0 to 4.1 per kb of captured sequence (Fig. 3). In Thailand, supercontigs 1.142 and 1.4629 showed high outlier loci densities with three CCE and one ABC transporter being affected. Six other supercontigs also displayed selection footprints impacting two P450s, two other detox enzymes, one PPO, and one cuticle protein. High outlier loci densities were identified in laboratory populations in supercontigs 1.371, 1.47, and 1.51 affecting 17 P450s and two cuticle proteins. Selection footprints were less pronounced in South America but showed overlap with laboratory and Thai populations.
Figure 3.
Supercontigs showing selection footprint. For each region, loci displaying a significant BayeScan3 Fsc Q-value <0.05 and both f(R+)-f(S) and f(R+)-f(R−) allele frequency variation occurring in the same direction were considered as outliers. For each supercontig, outlier loci densities were obtained by dividing the number of outlier loci by the total length of all captured genes (outlier loci per kb of captured region). Outlier loci densities are shown as a blue color scale. Gray stands for an absence of outlier loci. For each supercontig, horizontal bars show the number of captured genes from each gene family (upper bar) compared to those affected by outlier loci (lower bar). Supercontigs are clustered according to their putative chromosomal location as described in Timoshevskiy et al. (2014) and Juneja et al. (2014).
Of the 30,400 polymorphic loci, 3054 loci showed an allele frequency variation between R+ phenotypes and their respective susceptible population ≥40% together with an allele frequency variation between R+ and R− phenotypes ≥5%. These loci included 465 nonsynonymous variants (Supplemental Table 4). Comparing the Bayesian and the frequency filtering approaches revealed that the Bayesian approach was overall more stringent (Supplemental Fig. 9). However, the stringency of the Bayesian approach was high in South America, medium in Thailand, and low in laboratory populations following the decreasing diversity of these groups. The frequency filtering approach showed an opposite stringency with more variants identified in South America than Thailand and even less in laboratory populations. Frequency filtering identified 16 nonsynonymous variants shared by distinct geographic regions, whereas the Bayesian approach only identified variants being specific to each region.
Combining the best candidates identified by each approach identified 55 nonsynonymous variants strongly associated with deltamethrin resistance (Fig. 4; Supplemental Table 4). These variants affected 39 distinct genes mainly belonging to P450s, UGTs, CCEs, and cuticle proteins. As for gene amplifications, allele frequency patterns were conserved within regions but often differed among continents. Most nonsynonymous variants affecting P450s were identified from South American or laboratory populations. These P450s mostly belong to CYP6, CYP9, and CYP12 families. Conversely, most variants affecting CCEs were identified from Thai populations. Two were both affected by gene amplifications and nonsynonymous variants with CCEAE4A bearing height distinct nonsynonymous variants associated with resistance. Several UGTs were also affected by nonsynonymous variants, with three showing an increased frequency across multiple regions. ABC transporters and cuticle proteins were most frequently affected in South America. Several other enzymes potentially involved in insecticide detoxification were also affected by nonsynonymous variants, including two aldehyde oxidases, one alcohol dehydrogenase, and one sulfotransferase. Finally, the nicotinic acetylcholine receptor (nAchR) AAEL004935 targeted by neonicotinoid insecticides and the voltage-gated sodium channel (VSGC) AAEL006019 targeted by pyrethroid insecticides were also affected. Variants detected in the VGSC corresponded to the S989P and F1534C kdr mutations previously described in this species. None of the V1016I/G kdr mutations previously associated with pyrethroid resistance was retained as best candidates. Neither of them was retained by the Bayesian approach, and the V1016G mutation only passed the allele frequency filtering in Thailand (Supplemental Table 4).
Figure 4.

Best nonsynonymous polymorphisms associated with deltamethrin resistance. Only the 55 best differential nonsynonymous variants identified from frequency-based filtering and the Bayesian approach are shown (see Methods). For each region, allele frequency variation between each resistant population (R+ phenotypes) and their susceptible counterpart (S) are shown as a blue-yellow color scale. Blue indicates an enrichment in the reference allele, whereas yellow indicates an enrichment in the variant allele. Variants identified by the Bayesian approach are indicated by “B” marks. For variants passing frequency-based filtering or BayeScan3 filtering, allele frequency variation between R+ and R− phenotypes are shown as overimposed “+” marks. Variants are grouped by gene families and are described by the following annotations: chromosomal location (according to Juneja et al. 2014; Timoshevskiy et al. 2014), supercontig position, nucleotide change, amino acid position, amino acid change, gene accession number, and gene description. (*) Genes also found affected by CNVs linked to deltamethrin resistance. (**) The sodium channel S729P and F1249C variants correspond to the S989P and F1534C kdr mutations described in the literature due to changes in AAEL006019-RD transcript annotation.
Discussion
Deep targeted sequencing for studying insecticide resistance
Screening for genomic changes associated with insecticide resistance remained challenging until the sequencing of mosquito genomes (Holt et al. 2002; Nene et al. 2007; Arensburger et al. 2010). Afterward, the development of mosquito DNA microarrays allowed screening for resistance genes based on their differential transcription, leading to the identification of detoxification genes conferring resistance (David et al. 2005, 2013; Strode et al. 2008; Vontas et al. 2010; Edi et al. 2014). However, these screenings focused on differential transcription while genomic changes such as CNVs and polymorphisms were neglected. Then, the development of high-throughput DNA sequencing allowed screening for genomic variants across whole genomes. However, such an approach required a huge amount of sequences to reach the minimum genome coverage necessary for accurately quantifying CNV and allele frequencies, making it unaffordable when several samples have to be compared. Although RNA-seq is more affordable and can generate gene expression and variant data concomitantly, this approach is not suitable for identifying CNVs and does not provide reliable polymorphism data from poorly expressed genes. In addition, allele frequencies inferred from RNA-seq are to be taken with caution because they can be altered by allele-specific expression events (Gaur et al. 2013).
Recently, the development of target enrichment techniques allowed conducting DNA-seq focused on genomic regions of interest, hence, drastically increasing the sequencing depth while maintaining costs at a reasonable level (Altmüller et al. 2014). By targeting more than 760 genes from all protein families potentially involved in insecticide resistance in the dengue mosquito, our study screened for both CNVs and polymorphisms across several samples. Our experimental design allowed reaching a very deep coverage (>80×) across multiple resistant and susceptible populations originating from different geographical areas, providing the first comprehensive screening of CNVs and polymorphisms associated with insecticide resistance. Targeted DNA-seq data were consistent with qPCR data obtained from individual mosquitoes validating the robustness of the approach. Altogether, this study demonstrates that target enrichment coupled with high-throughput sequencing is a powerful tool for pinpointing genomic changes associated with adaptive traits from which candidate gene families are known.
Gene amplifications associated with pyrethroid resistance
Only a few CNVs were previously associated with insecticide resistance, and no comprehensive screening has ever been conducted in mosquitoes (for review, see Bass and Field 2011). The present study identified 41 genes affected by gene amplifications linked to deltamethrin resistance. Most were previously found significantly overtranscribed in pyrethroid resistant Ae. aegypti populations according to data publicly available in the VectorBase expression browser (https://vectorbase.org, data sets from Strode et al. 2008; Marcombe et al. 2009; Poupardin et al. 2012; David et al. 2014). The majority of CNVs observed between resistant and susceptible populations were <2.5-fold, suggesting a predominance of duplications. However several genes displayed CNV >fivefold in particular resistant populations, suggesting that copy number may vary within continents. This was confirmed by qPCR on individual mosquitoes showing an important copy number polymorphism within and across resistant populations. Such polymorphism suggests that most amplifications are not fixed in resistant populations. The strong relationship between gene amplification and overtranscription was confirmed by RT-qPCR. Although, overexpression can also be triggered by cis/trans regulatory elements or post-translational events, the present study suggests that gene amplification is a common adaptive mechanism allowing mosquitoes to overexpress detoxification genes conferring resistance to insecticides. The importance of such mechanism may even be greater in species highly infected by transposable elements like Ae. aegypti because their presence is known to favor duplication events (Nene et al. 2007).
The present study revealed distinct gene amplification patterns among resistant populations according to their geographical origin. This supports that the selection of gene amplifications conferring resistance depends on the genetic background of populations, their population dynamics, and on the selection pressures they undergo. Most amplified genes encoded detoxification enzymes, suggesting that gene amplification is rather linked to metabolic resistance than resistance conferred by an altered transport/penetration of the insecticide.
The majority of detoxification enzymes affected by gene amplifications encoded P450s belonging to CYP9J and CYP6 families that were frequently associated with insecticide resistance in mosquitoes (Vontas et al. 2012; David et al. 2013). The overrepresentation of P450s was expected because their overexpression is known to play a key role in pyrethroid resistance, and they are frequently affected by copy number variants (Feyereisen 2006, 2011). Several amplified CYP9Js were previously found overtranscribed in resistant populations and some of them (CYP9J24, CYP9J28, CYP9J32) have been validated as able to metabolize pyrethroids (Stevenson et al. 2012), supporting the importance of their amplification in resistance. Among the CYP6s amplified, most were previously found overtranscribed in resistant populations, and CYP6BB2 was recently involved in pyrethroid metabolism (Kasai et al. 2014). In addition, some of their close orthologs in Anopheles (CYP6P3, CYP6P7, CYP6P9, CYP6M2, and CYP6AA5) are also capable of degrading pyrethroids (Boonsuepsakul et al. 2008; Müller et al. 2008; Duangkaew et al. 2011; Stevenson et al. 2011; Riveron et al. 2013). Altogether, these results confirm the key role of P450 in the resistance of mosquitoes to pyrethroids and their propensity to increase their expression level through gene amplification when undergoing a strong selection pressure from insecticides.
Three CCEs (CCEAE3A, CCEAE4A, and CCEAE6A) belonging to a single genomic cluster were highly amplified in Thai resistant populations leading to their strong overtranscription. Highest amplification levels were found in the Nakh R population which is also resistant to organophosphates (Poupardin et al. 2014). In this previous study, the overtranscription of CCEAE3A and CCEAE6A through gene amplification was associated with temephos resistance at the larval stage, although resistance to pyrethroids was also observed. Other studies performed on South American and Caribbean populations confirmed the association of some of these CCEs with temephos resistance (Marcombe et al. 2009, 2012; Saavedra-Rodriguez et al. 2014). Our data revealed higher CCE copy number in mosquitoes surviving deltamethrin exposure (R+ phenotype), supporting their role in pyrethroid resistance. Although the association between pyrethroid resistance and these CCE genes in Thailand might be due to multiresistant individuals carrying both pyrethroid- and organophosphate-resistance genes, the role of CCEs in pyrethroid metabolism has been demonstrated in mammals (Hodgson 2003; Nakamura et al. 2007) and mosquitoes (Somwang et al. 2011; Chandor-Proust et al. 2013). However, no individual mosquito CCE has yet been validated as a pyrethroid metabolizer, and insecticide sequestration may also confer resistance. Further functional studies are required for validating the precise role of these amplified CCEs in pyrethroid resistance.
Two sulfotransferases were amplified in Thai resistant populations. The overtranscription of these enzymes has been frequently described in insecticide resistant mosquitoes and deserve further attention (Poupardin et al. 2012; Marcombe et al. 2013; Nkya et al. 2013). Indeed, their involvement in insecticide degradation pathways has been validated in mammals (Lee et al. 2007).
Among GSTs, only GSTE2 was found amplified in Thai resistant populations. This GST is known for its ability to metabolize DDT in various mosquito species (Ortelli et al. 2003; Lumjuan et al. 2005), and recent studies confirmed its role in pyrethroid resistance although pyrethroid metabolism has not been observed (Lumjuan et al. 2011; Riveron et al. 2014b).
A recent study suggested that the duplication of the gene encoding the Ae. aegypti voltage-gated sodium channel (VGSC) targeted by pyrethroids contributed to resistance by maintaining together wild-type allele and kdr mutations and reducing mutant allele deleterious effects (Martins et al. 2013). A similar role was suggested for the duplication of the acetylcholinesterase gene (carrying the Ace1 mutation) in the resistance of An. gambiae to carbamates (Edi et al. 2014). Although such a mechanism is likely contributing to resistance in mosquitoes, no amplification of genes encoding proteins targeted by insecticides were detected in our study. In addition, a poor association was observed between the presence of kdr mutations and gene amplifications affecting detoxification enzymes in individual mosquitoes. Indeed, recombination events are probably countering such association, especially as the VGSC gene and amplified detoxification genes are located on distant genomic regions. In addition, fitness costs associated with both kdr mutations and gene amplifications have been identified (Kondrashov 2012; Brito et al. 2013; Katju and Bergthorsson 2013; Rinkevich et al. 2013).Therefore, although kdr mutations and gene amplifications of detoxification enzymes co-occur in resistant populations, additive fitness costs may contribute to segregate them at the individual level.
Polymorphisms associated with pyrethroid resistance
Apart from mutations affecting the proteins targeted by insecticides, very few nonsynonymous variants have been associated with insecticide resistance. In this context, the present study represents the first DNA-seq screening of variants associated with pyrethroid resistance in mosquitoes. Searching for selection footprints identified multiple genomic regions under selection, suggesting that pyrethroid resistance is polygenic. Several nonsynonymous variants were associated with deltamethrin resistance, but few were found in multiple geographic regions, confirming the strong influence of genetic background and population history on the selection of resistant alleles.
Although several nonsynonymous variants affecting P450s were identified from each region, those mostly associated with resistance were mainly detected in South American and laboratory populations. Some were previously identified in a permethrin-resistant strain by RNA-seq (David et al. 2014). In Drosophila melanogaster, functional studies demonstrated that nonsynonymous mutations in the P450 CYP6A2 identified in the RDDTR resistant strain have a prominent role in resistance by enhancing DDT metabolism (Amichot et al. 2004). In the mosquito, Anopheles funestus, directional selection footprints were found in the coding sequence of two CYP6Ps capable of metabolizing pyrethroids (Riveron et al. 2014a). Most nonsynonymous variants impacting CCEs were identified in Thailand with CCEAE4A and CCEAE3A impacted by multiple variants strongly associated with deltamethrin resistance. Elevated copy numbers of these genes were also identified in Thailand, suggesting that their amplification may have been followed by their neofunctionalization under insecticide selection pressure (Katju and Bergthorsson 2013). Interestingly, the best orthologous gene of CCEAE4A in the sheep blowfly, Lucilia cuprina (LcαE7), encodes an esterase associated with organophosphate resistance through a single point mutation enhancing its activity toward these chemicals (Newcomb et al. 1997). Later, it was shown that another mutation of this gene allowed this enzyme to metabolize synthetic pyrethroids (Heidari et al. 2005; Devonshire et al. 2007). More recently, multiple randomly generated nonsynonymous mutations of this gene were shown to enhance its activity toward various pyrethroids including deltamethrin (Coppin et al. 2012). Although additional work is necessary for validating the role of these nonsynonymous variants, our data support the selection of detoxification enzyme variants as being a key process in the adaptation of mosquitoes to chemical insecticides.
Finally, multiple nonsynonymous variants were detected in the gene encoding the voltage-gated sodium channel targeted by pyrethroids (kdr mutations). Their frequencies were consistent with those previously reported in Thailand and South America (Stenhouse et al. 2013; Linss et al. 2014). However, neither of the two mutations extensively used as resistance markers (V1016I/G) were retained as best candidates. Although the V1016I mutation showed high frequencies in South American resistant populations, it showed a poor association with deltamethrin survival phenotype. The V1016G mutation was associated with resistance in one Thai population but was not retained among the best candidates according to our criteria. Instead, two other less-known mutations (S989P and F1534C) showed a stronger association with deltamethrin resistance. Such a result supports the substantial contribution of other mechanisms in the resistance phenotype.
New tools for monitoring resistance genes in mosquito populations
Overall, the present study confirms that gene amplifications affecting detoxification enzymes are good markers of metabolic resistance in mosquitoes. Novel nonsynonymous variants potentially enhancing their ability to detoxify insecticides were also identified. Despite significant research efforts, developing molecular tests to track the overexpression of these enzymes in field populations has proved challenging in terms of affordability, throughput, and reliability; and current vector population monitoring tools do not include metabolic resistance markers (Bass et al. 2010). Detecting their increased copy number or the presence of specific variants conferring resistance through simple PCR-based DNA assays opens up new perspectives for managing insecticide resistance. Indeed, such novel resistance markers will be accessible to developing countries in terms of technology and affordability and will allow tracking down both target-site and metabolic-resistance alleles in individual mosquitoes and monitoring their frequencies through time and space among and within natural populations.
Methods
Mosquitoes
Nine Ae. aegypti populations of distinct genetic backgrounds were used. These included four populations susceptible to insecticides and five populations showing elevated resistance to deltamethrin. Two distant geographic areas where pyrethroid resistance is threatening mosquito control were studied: South East Asia (Thailand) and South America (French Guiana). Two resistant and one susceptible population having a close genetic background were studied in each area (see Table 2 for populations’ information). In addition, one laboratory population selected with deltamethrin for five generations was used together in comparison to its susceptible “parental” population. The laboratory Liverpool population used for public genome sequencing (Liverpool population) (Nene et al. 2007) was used as an additional susceptible population. Resistant populations from Thailand and French Guiana were colonized for two generations in the laboratory without insecticide selection pressure before assessing resistance levels and sample collection.
Table 2.
Sample descriptions
Deltamethrin resistance levels
For each population, the exposure time necessary to knock down 50% of individuals (KDT50) was determined on 2- to 4-d-old females using test tubes equipped with Whatman filter paper impregnated with 0.05% deltamethrin in silicone oil according to standard WHO procedure (WHO 2006). Then, a collection of Whatman filter papers impregnated with different concentrations of deltamethrin (from 0.005% to 0.75%) was used for evaluating the lethal dose for 50% of individuals (LD50) of each population. Bioassays were conducted in duplicates of 40 2- to 4-d-old females, and mortality was recorded after 1 h exposure to deltamethrin and 24 h recovery without insecticide. For each resistant population, the impregnated paper killing 40%–60% mosquitoes after 1 h exposure was then used to segregate the most susceptible individuals (dead at LT25: R− phenotype) from the most resistant ones (survivors at LT75: R+ phenotype) by adjusting the exposure time from 10 min to 3 h.
Sample preparation
All sampled mosquitoes consisted of 2- to 4-d-old non-blood-fed Ae. aegypti females grown in standardized laboratory conditions. For each population/phenotype, 200 individuals were collected and stored individually in silica gel. For each population, three pools of 30 females not exposed to insecticide were also collected and stored in RNAlater (Life Technologies) for gene expression analyses. Gene amplification screening by exon capture and mass sequencing was performed on gDNA extracted from two pools of 65 individuals per population/phenotype. Genomic DNA was extracted using the PureGene Core Kit A (Qiagen) following the manufacturer's instructions. For each population/phenotype, gDNA from each pool were then combined in equal quantity in order to be representative of a total of 130 individuals. Quantitative PCR validation of gene amplifications and genotyping of kdr mutations was performed on gDNA extracted from 12 single adult females per population/phenotype using the method described in Collins et al. (1987) and resuspended in 20 µL nuclease-free water. Gene expression analyses were performed on RNA extracted from three pools of 30 adult females per population (R and S populations) using the RNAqueous4PCR kit (Life Technologies) according to the manufacturer's instructions and resuspended in 50 µL nuclease-free water.
Capture of target regions and sequencing
Gene capture was performed by Hybrigenics-Helixio (Clermont-Ferrand, France) using the SureSelect target enrichment system (Agilent). Capture library consisted in 51,073 overlapping RNA probes of 120 bp (baits) targeting 3458 exons belonging to 789 genes. The mean coverage of target regions was 4×. These genes were chosen according to their putative role in insecticide resistance. These genes include all detoxification enzymes sensu lato, all cuticle proteins, all ABC transporters together with several ion channels, redox enzymes, and synaptic proteins (list of captured genes in Supplemental Table 5). Capture of target genes was performed according to “SureSelect Target Enrichment System for Illumina Paired-end Sequencing Library version 1.5” protocol. Briefly, 3 µg gDNA were fragmented using a Bioruptor (Diagenode), ligated to adaptors, and amplified by PCR using Herculase II DNA polymerase (Agilent). After QC analysis, libraries were hybridized to biotinylated baits and purified using Dynabeads MyOne Streptavidin T1 beads (Life Technologies). Captured DNA fragments were amplified, purified, and multiplexed before sequencing. Paired-end sequencing was performed on a Genome Analyzer IIx (Illumina) producing 76 bp reads. An average sequencing coverage of greater than 60× was expected for each sample.
Reads filtering and mapping
Sequenced reads were assigned to each sample (unplexing), and adaptors were removed. Overall read quality was checked for each sample using FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc). Reads were then filtered based on their length, pairing, and quality using Trimmomatic (Bolger et al. 2014). Parameters were set as follows: read length ≥76 bp; mean Phred quality score greater than 30; and Phred quality score greater than 30 across all reads (10-bp sliding window). Only paired reads were kept for mapping. Reads were mapped to the Ae. Aegypti genome (AaegL2 assembly) using BWA (Li and Durbin 2009) with default parameters implemented into a Galaxy pipeline (http://galaxyproject.org). Mapped reads were loaded into Genespring NGS version 12.5 (Agilent) and further filtered according to their mapping quality (alignment score of 95 or above). In case of multiple mapped reads, only primary hits were conserved.
Copy number variation analysis
Coverage of target genes was quantified for each sample and normalized according to the total number of filtered reads per library. Only genes showing more than 90 reads per kilobase exon model per million sequenced reads (RPKM) in all samples were considered for further analysis. Detection of copy number variation (CNV) was based on differential coverage between samples obtained from resistant and susceptible populations. Fold changes (FC) between resistant samples (R+ and R− phenotypes) and all susceptible populations (meanS) were computed for each gene. Genes satisfying the following criteria in any resistant population were considered affected by gene amplifications in association with deltamethrin resistance: [(R+)/(meanS) FC ≥ 1.5] and [(R+)/(R−) FC] > 1 (i.e., 1.5-fold more copies in resistant samples and more copies in LD75 survivors compared to LD25 dead).
Validation of gene amplifications on individual mosquitoes
Gene amplifications detected by previous CNV analysis were further studied by qPCR on 156 individual mosquitoes (12 from each population/phenotype). Nine candidate genes located in three different genomic clusters affected by gene amplifications were selected: Supercontig 1.1188: AAEL014614 (CYP9J?), AAEL014615 (CYP9J23), and AAEL014617 (CYP9J28); Supercontig 1.1327: AAEL014890 (CYP6CC1), AAEL014891 (CYP6P?), and AAEL014893 (CYP6BB2); Supercontig 1.142: AAEL05112 (CCEAE3A), AAEL005101(CCEAE4A), and AAEL005122 (CCEAE6A). PCR primers targeting exonic regions were designed using NCBI Primer BLAST and checked for specificity against the whole Ae. aegypti genome (Supplemental Table 6). Two target genes showing a constant copy number across all samples from sequencing data (see above) were used for normalization (AAEL005950 and AAEL007808). Real-time quantitative PCR was performed on an iQ5 cycler (Bio-Rad). PCR reactions consisted of 3 µL gDNA template (see above), 3.6 µL nuclease free water, 0.45 µL of each primer (10 mM), and 7.5 µL of SYBR Green Supermix 2× (Bio-Rad). A dilution scale made from a pool of all gDNA samples was used for assessing PCR efficiency and quantification. All samples were amplified in triplicates (14 populations × 12 individuals × 3 replicates). After normalization, CNVs were expressed as mean relative gDNA quantity compared to one individual of the Livp S population.
Impact of gene amplifications on transcription levels
The link between gene amplifications and increased transcription levels was investigated on nine genes (see above) by RT-qPCR. Total RNA samples from each population were treated with DNase I (Invitrogen) to remove genomic DNA following the manufacturer's instructions. Reverse transcription and qPCR reactions were performed as described in Nkya et al. (2014). Data analysis was performed according to the ΔΔCt method taking into account PCR efficiency (Pfaffl 2001) and using the housekeeping genes encoding the ribosomal proteins L8 (AGAP005802) and S7 (AGAP010592) for normalization. Three technical replicates were performed per population, and results were expressed as mean transcription ratio in each resistant population ± SD relative to the mean transcription ratio of all susceptible populations.
Polymorphism calling and population structure
Polymorphisms (SNPs, multiple nucleotide polymorphisms [MNPs], and indels) were called against the reference genome using filtered reads by using Genespring NGS version 12.5 (Agilent). Calling parameters were as follows for each sample: calling score of 50 or above (P-value < 10−5); ignore homopolymer 10 or higher and surrounding positions; locus coverage of 30 or more. Variants identified from each sample were further filtered based on their coverage and strand bias across all samples (coverage 30 or more; strand bias 140 or less). For each variant, allele frequencies were estimated for each population based on the number of reads supporting each allele, and genic effects were computed against the reference genome. A principal component analysis (PCA) based on allele frequencies of all polymorphic variants across all samples was used for inferring the genetic structure of the studied populations and confirming the relevance of the geographical groups.
Searching for selection footprint using a hierarchical Bayesian approach
BayeScan3 was used for identifying loci under natural selection through a hierarchical Bayesian model, taking into account population structure and usable on pools of individuals (Foll et al. 2014). This approach estimates the probability that each locus is subject to selection by discriminating population-specific and locus-specific components of the fixation index using a logistic regression. The posterior probability of a given locus being under selection is assessed by defining two alternative models, one including the locus-specific effect and the other excluding it. Departure from neutrality is supposed when the locus-specific component is necessary to explain the observed pattern of diversity using a reversible jump Markov chain Monte Carlo (MCMC). Because the high genetic proximity between R+ and R− phenotypes can bias the estimation of the neutral model, only S populations and R+ phenotypes from resistant populations were used. For the same reasons, loci showing allele frequency <5% or >95% in all samples were not considered. BayeScan3 was run with default parameters and the following hierarchical structure: Thailand (Patg S, Nakh R+, and Phet R+), South America (NwOr S, Cayn R+, StGe R+), and Laboratory (Bora S, Delta R+). Loci showing a Fsc Q-value ≤ 0.05 in any geographical group were considered under selection (i.e., outlier locus). These loci were further filtered based on their association with the resistance phenotype by retaining only those showing both [f(R+)-f(S)] and [f(R+)-f(R−)] metrics being positive (enrichment of variant allele) or negative (enrichment of reference allele). Because the Ae. aegypti genome is not assembled, selection footprints were looked for at the supercontigs scale. For each supercontig, the density of outlier loci was computed by dividing the number of outlier loci by the total length of captured regions within the supercontig. In order to avoid inferring selection footprint from limited genomic coverage, only supercontigs represented by not more than four genes or from which the captured region represented >10% of supercontig length were considered.
Identifying polymorphisms mostly associated with deltamethrin resistance
Allele frequency-based filtering was combined with the Bayesian approach for identifying polymorphisms most strongly associated with deltamethrin resistance. Frequency-based filtering consisted in retaining alleles satisfying the following conditions in any resistant/susceptible population pair from each geographical group: [f(R+)-f(S) ≥ 40%] and [f(R+)-f(R−) ≥ 5%] and both metrics being positive or negative. Alleles passing both the frequency-based filtering and the Bayesian approach were retained. In addition, the best candidates obtained by each approach were also retained. For the Bayesian approach, this included loci showing Fsc Q-values within the best 10th percentile from each region. For frequency-based filtering, this includes alleles passing frequency-based filtering and showing the five highest Abs{[f(R+)-f(S)]+[f(R+)-f(R−)]} metric from each region. Variants most associated with deltamethrin resistance were then filtered according to their genic effects and those being nonsynonymous (i.e., affecting protein sequence) were retained.
Validation of polymorphism data and association between kdr mutations and gene amplifications
Allele frequencies obtained by high-throughput sequencing were validated on kdr mutations (V1016I, V1016G, and F1534C) on 156 individuals (12 individual mosquitoes from each population/phenotype). Genomic DNA was extracted as described above, and kdr mutations were detected by qPCR following the method described in Saavedra-Rodriguez et al. (2007). Associations between the presence of kdr mutations and gene amplifications were tested across the five resistance populations for the three kdr mutations and the nine genotyped gene amplifications by testing linear correlations between the kdr gentoype (0: homozygote wildtype, 0.5: heterozygote, 1: homozygote resistant) and gene amplification levels.
Data access
The sequence data from this study have been submitted to the European Nucleotide Archive (ENA; http://www.ebi.ac.uk/ena) under accession number PRJEB7976.
Supplementary Material
Acknowledgments
This project was primarily funded by the Centre National de la Recherche Scientifique (CNRS, INEE department) (Grant programme APEGE 2013, project Mosqui-Target). Additional funding was received from the French Institut de Microbiologie et Maladies Infectieuses (grant IMMI 109764). We also thank the Laboratoire d'Ecologie Alpine of Grenoble for additional funding. F.F. was supported by a PhD fellowship from the Grenoble-Alpes University. We acknowledge support from the federative structure Environmental and Systems Biology (BEeSy) of Grenoble-Alpes University. This work was also supported by the French-Thai cooperation programme-PHC Siam project (RESA 2013–2014) funded by the French embassy and the Office of the Higher Education Commission of Thailand. We also thank the Thailand International Development Cooperation Agency (TICA) through the STOP-VEC programme. This work was also supported by the Center for Advanced Studies for Agriculture and Food, Institute for Advanced Studies, Kasetsart University under the Higher Education Research Promotion and National Research University Project of Thailand, Office of the Higher Education Commission, Ministry of Education, Thailand. W.J. was supported by the Thailand Research Fund (senior research scholarship RTA 5558002 and grant for new researchers MRG 5380102) and Kasetsart University Research and Development Institute (KURDI). We also acknowledge the UMR 5558 and the Pôle Rhône-Alpin de Bioinformatique (PRABI) for providing access to their computational cluster. Finally, we thank Dr. A. Bonin, Dr. M. Weill, and the anonymous reviewers for their constructive comments on the manuscript.
Author contributions: F.F. was involved in all aspects of the study and helped draft the manuscript. I.D. contributed to field sampling and molecular analyses. T.G. provided technical support for mosquito rearing and contributed to sample preparation. V.N. and F.B. contributed to data analyses and helped draft the manuscript. F.C., W.J., T.C., R.G., I.D., and V.C. contributed to study design and helped draft the manuscript. P.S., K.T., and R.P. contributed to field sampling. S.R. contributed to sample preparation and helped analyze data and draft the manuscript. J.P.D. conceived the study, contributed to field sampling and sample preparation, analyzed data, and wrote the manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.189225.115.
Freely available online through the Genome Research Open Access option.
References
- Altmüller J, Budde BS, Nürnberg P. 2014. Enrichment of target sequences for next-generation sequencing applications in research and diagnostics. Biol Chem 395: 231–237. [DOI] [PubMed] [Google Scholar]
- Amichot M, Tarés S, Brun-Barale A, Arthaud L, Bride JM, Bergè JB. 2004. Point mutations associated with insecticide resistance in the Drosophila cytochrome P450 Cyp6a2 enable DDT metabolism. Eur J Biochem 1: 1250–1257. [DOI] [PubMed] [Google Scholar]
- Arensburger P, Megy K, Waterhouse RM, Abrudan J, Amedeo P, Antelo B, Bartholomay L, Bidwell S, Caler E, Camara F, et al. 2010. Sequencing of Culex quinquefasciatus establishes a platform for mosquito comparative genomics. Science 330: 86–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bariami V, Jones CM, Poupardin R, Vontas J, Ranson H. 2012. Gene amplification, ABC transporters and cytochrome P450s: unraveling the molecular basis of pyrethroid resistance in the dengue vector, Aedes aegypti. PLoS Negl Trop Dis 6: e1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass C, Field LM. 2011. Gene amplification and insecticide resistance. Pest Manag Sci 67: 886–890. [DOI] [PubMed] [Google Scholar]
- Bass C, Nikou D, Vontas J, Donnelly MJ, Williamson MS, Field LM. 2010. The Vector Population Monitoring Tool (VPMT): high-throughput DNA-based diagnostics for the monitoring of mosquito vector populations. Malar Res Treat 2010: 190434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatt S, Gething PW, Brady OJ, Messina JP, Farlow AW, Moyes CL, Drake JM, Brownstein JS, Hoen AG, Sankoh O, et al. 2013. The global distribution and burden of dengue. Nature 496: 504–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boonsuepsakul S, Luepromchai E, Rongnoparut P. 2008. Characterization of Anopheles minimus CYP6AA3 expressed in a recombinant baculovirus system. Arch Insect Biochem Physiol 69: 13–21. [DOI] [PubMed] [Google Scholar]
- Brengues C, Hawkes NJ, Chandre F, McCarroll L, Duchon S, Guillet P, Manguin S, Morgan JC, Hemingway J. 2003. Pyrethroid and DDT cross-resistance in Aedes aegypti is correlated with novel mutations in the voltage-gated sodium channel gene. Med Vet Entomol 17: 87–94. [DOI] [PubMed] [Google Scholar]
- Brito LP, Linss JG, Lima-Camara TN, Belinato TA, Peixoto AA, Lima JB, Valle D, Martins AJ. 2013. Assessing the effects of Aedes aegypti kdr mutations on pyrethroid resistance and its fitness cost. PLoS One 8: e60878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandor-Proust A, Bibby J, Regent-Kloeckner M, Roux J, Guittard-Crilat E, Poupardin R, Riaz MA, Paine M, Dauphin-Villemant C, Reynaud S, et al. 2013. The central role of mosquito cytochrome P450 CYP6Zs in insecticide detoxification revealed by functional expression and structural modelling. Biochem J 455: 75–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins FS, Drumm ML, Cole JL, Lockwood WK, Vandewoude GF, Iannuzzi MC. 1987. Construction of a general human-chromosome jumping library, with application to cystic-fibrosis. Science 235: 1046–1049. [DOI] [PubMed] [Google Scholar]
- Coppin CW, Jackson CJ, Sutherland T, Hart PJ, Devonshire AL, Russell RJ, Oakeshott JG. 2012. Testing the evolvability of an insect carboxylesterase for the detoxification of synthetic pyrethroid insecticides. Insect Biochem Mol Biol 42: 343–352. [DOI] [PubMed] [Google Scholar]
- Corbel V, Nosten F, Thanispong K, Luxemburger C, Kongmee M, Chareonviriyaphap T. 2013. Challenges and prospects for dengue and malaria control in Thailand, Southeast Asia. Trends Parasitol 29: 623–633. [DOI] [PubMed] [Google Scholar]
- David JP, Strode C, Vontas J, Nikou D, Vaughan A, Pignatelli PM, Louis C, Hemingway J, Ranson H. 2005. The Anopheles gambiae detoxification chip: a highly specific microarray to study metabolic-based insecticide resistance in malaria vectors. Proc Natl Acad Sci 102: 4080–4084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David JP, Ismail HM, Chandor-Proust A, Paine MJI. 2013. Role of cytochrome P450s in insecticide resistance: impact on the control of mosquito-borne diseases and use of insecticides on Earth. Philos Trans R Soc Lond B Biol Sci 368: 20120429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David JP, Faucon F, Chandor-Proust A, Poupardin R, Riaz MA, Bonin A, Navratil V, Reynaud S. 2014. Comparative analysis of response to selection with three insecticides in the dengue mosquito Aedes aegypti using mRNA sequencing. BMC Genomics 15: 174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devonshire AL, Heidari R, Huang HZ, Hammock BD, Russell RJ, Oakeshott JG. 2007. Hydrolysis of individual isomers of fluorogenic pyrethroid analogs by mutant carboxylesterases from Lucilia cuprina. Insect Biochem Mol Biol 37: 891–902. [DOI] [PubMed] [Google Scholar]
- Duangkaew P, Pethuan S, Kaewpa D, Boonsuepsakul S, Sarapusit S, Rongnoparut P. 2011. Characterization of mosquito CYP6P7 and CYP6AA3: differences in substrate preference and kinetic properties. Arch Insect Biochem Physiol 76: 236–248. [DOI] [PubMed] [Google Scholar]
- Edi CV, Djogbénou L, Jenkins AM, Regna K, Muskavitch MA, Poupardin R, Jones CM, Essandoh J, Kétoh GK, Paine MJ, et al. 2014. CYP6 P450 enzymes and ACE-1 duplication produce extreme and multiple insecticide resistance in the malaria mosquito Anopheles gambiae. PLoS Genet 10: e1004236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feyereisen R. 2006. Evolution of insect P450. Biochem Soc Trans 34(Pt 6): 1252–1255. [DOI] [PubMed] [Google Scholar]
- Feyereisen R. 2011. Arthropod CYPomes illustrate the tempo and mode in P450 evolution. Biochim Biophys Acta 1814: 19–28. [DOI] [PubMed] [Google Scholar]
- Foll M, Gaggiotti OE, Daub JT, Vatsiou A, Excoffier L. 2014. Widespread signals of convergent adaptation to high altitude in Asia and America. Am J Hum Genet 95: 394–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaur U, Li K, Mei S, Liu G. 2013. Research progress in allele-specific expression and its regulatory mechanisms. J Appl Genet 54: 271–283. [DOI] [PubMed] [Google Scholar]
- Harris AF, McKemey AR, Nimmo D, Curtis Z, Black I, Morgan SA, Oviedo MN, Lacroix R, Naish N, Morrison NI, et al. 2012. Successful suppression of a field mosquito population by sustained release of engineered male mosquitoes. Nat Biotechnol 30: 828–830. [DOI] [PubMed] [Google Scholar]
- Heidari R, Devonshire AL, Campbell BE, Dorrian SJ, Oakeshott JG, Russell RJ. 2005. Hydrolysis of pyrethroids by carboxylesterases from Lucilia cuprina and Drosophila melanogaster with active sites modified by in vitro mutagenesis. Insect Biochem Mol Biol 35: 597–609. [DOI] [PubMed] [Google Scholar]
- Hemingway J, Hawkes NJ, McCarroll L, Ranson H. 2004. The molecular basis of insecticide resistance in mosquitoes. Insect Biochem Mol Biol 34: 653–665. [DOI] [PubMed] [Google Scholar]
- Hodgson E. 2003. In vitro human phase I metabolism of xenobiotics I: pesticides and related compounds used in agriculture and public health, May 2003. J Biochem Mol Toxicol 17: 201–206. [DOI] [PubMed] [Google Scholar]
- Hoffmann AA, Montgomery BL, Popovici J, Iturbe-Ormaetxe I, Johnson PH, Muzzi F, Greenfield M, Durkan M, Leong YS, Dong Y, et al. 2011. Successful establishment of Wolbachia in Aedes populations to suppress dengue transmission. Nature 476: 454–457. [DOI] [PubMed] [Google Scholar]
- Holt RA, Subramanian GM, Halpern A, Sutton GG, Charlab R, Nusskern DR, Wincker P, Clark AG, Ribeiro JM, Wides R, et al. 2002. The genome sequence of the malaria mosquito Anopheles gambiae. Science 298: 129–149. [DOI] [PubMed] [Google Scholar]
- Itokawa K, Komagata O, Kasai S, Okamura Y, Masada M, Tomita T. 2010. Genomic structures of Cyp9m10 in pyrethroid resistant and susceptible strains of Culex quinquefasciatus. Insect Biochem Mol Biol 40: 631–640. [DOI] [PubMed] [Google Scholar]
- Itokawa K, Komagata O, Kasai S, Masada M, Tomita T. 2011. Cis-acting mutation and duplication: history of molecular evolution in a P450 haplotype responsible for insecticide resistance in Culex quinquefasciatus. Insect Biochem Mol Biol 41: 503–512. [DOI] [PubMed] [Google Scholar]
- Juneja P, Osei-Poku J, Ho YS, Ariani CV, Palmer WJ, Pain A, Jiggins FM. 2014. Assembly of the genome of the disease vector Aedes aegypti onto a genetic linkage map allows mapping of genes affecting disease transmission. PLoS Negl Trop Dis 8: e2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasai S, Komagata O, Itokawa K, Shono T, Ng LC, Kobayashi M, Tomita T. 2014. Mechanisms of pyrethroid resistance in the dengue mosquito vector, Aedes aegypti: target site insensitivity, penetration, and metabolism. PLoS Negl Trop Dis 8: e2948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katju V, Bergthorsson U. 2013. Copy-number changes in evolution: rates, fitness effects and adaptive significance. Front Genet 4: 273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondrashov FA. 2012. Gene duplication as a mechanism of genomic adaptation to a changing environment. Proc Biol Sci 279: 5048–5057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacey LA. 2007. Bacillus thuringiensis serovariety israelensis and Bacillus sphaericus for mosquito control. J Am Mosq Control Assoc 232 Suppl: 133–163. [DOI] [PubMed] [Google Scholar]
- Lee CH, Kamijima M, Li C, Taneda S, Suzuki AK, Nakajima T. 2007. 3-Methyl-4-nitrophenol metabolism by uridine diphosphate glucuronosyltransferase and sulfotransferase in liver microsomes of mice, rats, and Japanese quail (Coturnix japonica). Environ Toxicol Chem 26: 1873–1878. [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Schuler MA, Berenbaum MR. 2007. Molecular mechanisms of metabolic resistance to synthetic and natural xenobiotics. Annu Rev Entomol 52: 231–253. [DOI] [PubMed] [Google Scholar]
- Linss JG, Brito LP, Garcia GA, Araki AS, Bruno RV, Lima JB, Valle D, Martins AJ. 2014. Distribution and dissemination of the Val1016Ile and Phe1534Cys Kdr mutations in Aedes aegypti Brazilian natural populations. Parasit Vectors 7: 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lounibos LP. 2002. Invasions by insect vectors of human disease. Annu Rev Entomol 47: 233–266. [DOI] [PubMed] [Google Scholar]
- Lumjuan N, McCarroll L, Prapanthadara LA, Hemingway J, Ranson H. 2005. Elevated activity of an Epsilon class glutathione transferase confers DDT resistance in the dengue vector, Aedes aegypti. Insect Biochem Mol Biol 35: 861–871. [DOI] [PubMed] [Google Scholar]
- Lumjuan N, Rajatileka S, Changsom D, Wicheer J, Leelapat P, Prapanthadara LA, Somboon P, Lycett G, Ranson H. 2011. The role of the Aedes aegypti Epsilon glutathione transferases in conferring resistance to DDT and pyrethroid insecticides. Insect Biochem Mol Biol 41: 203–209. [DOI] [PubMed] [Google Scholar]
- Marcombe S, Poupardin R, Darriet F, Reynaud S, Bonnet J, Strode C, Brengues C, Yébakima A, Ranson H, Corbel V, et al. 2009. Exploring the molecular basis of insecticide resistance in the dengue vector Aedes aegypti: a case study in Martinique Island (French West Indies). BMC Genomics 10: 494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcombe S, Mathieu RB, Pocquet N, Riaz MA, Poupardin R, Sélior S, Darriet F, Reynaud S, Yébakima A, Corbel V, et al. 2012. Insecticide resistance in the dengue vector Aedes aegypti from Martinique: distribution, mechanisms and relations with environmental factors. PLoS One 7: e30989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcombe S, Paris M, Paupy C, Bringuier C, Yebakima A, Chandre F, David JP, Corbel V, Despres L. 2013. Insecticide-driven patterns of genetic variation in the dengue vector Aedes aegypti in Martinique Island. PLoS One 8: e77857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins AJ, Brito LP, Linss JG, Rivas GB, Machado R, Bruno RV, Lima JB, Valle D, Peixoto AA. 2013. Evidence for gene duplication in the voltage-gated sodium channel gene of Aedes aegypti. Evol Med Public Health 2013: 148–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller P, Warr E, Stevenson BJ, Pignatelli PM, Morgan JC, Steven A, Yawson AE, Mitchell SN, Ranson H, Hemingway J, et al. 2008. Field-caught permethrin-resistant Anopheles gambiae overexpress CYP6P3, a P450 that metabolises pyrethroids. PLoS Genet 4: e1000286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura Y, Sugihara K, Sone T, Isobe M, Ohta S, Kitamura S. 2007. The in vitro metabolism of a pyrethroid insecticide, permethrin, and its hydrolysis products in rats. Toxicology 235: 176–184. [DOI] [PubMed] [Google Scholar]
- Nene V, Wortman JR, Lawson D, Haas B, Kodira C, Tu ZJ, Loftus B, Xi ZY, Megy K, Grabherr M, et al. 2007. Genome sequence of Aedes aegypti, a major arbovirus vector. Science 316: 1718–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newcomb RD, Campbell PM, Ollis DL, Cheah E, Russell RJ, Oakeshott JG. 1997. A single amino acid substitution converts a carboxylesterase to an organophosphorus hydrolase and confers insecticide resistance on a blowfly. Proc Natl Acad Sci 94: 7464–7468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nkya TE, Akhouayri I, Kisinza W, David JP. 2013. Impact of environment on mosquito response to pyrethroid insecticides: facts, evidences and prospects. Insect Biochem Mol Biol 43: 407–416. [DOI] [PubMed] [Google Scholar]
- Nkya T, Akhouayri I, Poupardin R, Batengana B, Mosha F, Magesa S, Kisinza W, David JP. 2014. Insecticide resistance mechanisms associated with different environments in the malaria vector Anopheles gambiae: a case study in Tanzania. Malar J 13: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortelli F, Rossiter LC, Vontas J, Ranson H, Hemingway J. 2003. Heterologous expression of four glutathione transferase genes genetically linked to a major insecticide-resistance locus from the malaria vector Anopheles gambiae. Biochem J 373(Pt 3): 957–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffl MW. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poupardin R, Riaz MA, Jones CM, Chandor-Proust A, Reynaud S, David JP. 2012. Do pollutants affect insecticide-driven gene selection in mosquitoes? Experimental evidence from transcriptomics. Aquat Toxicol 114–115: 49–57. [DOI] [PubMed] [Google Scholar]
- Poupardin R, Srisukontarat W, Yunta C, Ranson H. 2014. Identification of carboxylesterase genes implicated in temephos resistance in the dengue vector Aedes aegypti. PLoS Negl Trop Dis 8: e2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranson H, Burhani J, Lumjuan N, Black WC. 2010. Insecticide resistance in dengue vectors. TropIKA.net 1: 1–12. [Google Scholar]
- Rinkevich FD, Du Y, Dong K. 2013. Diversity and convergence of sodium channel mutations involved in resistance to pyrethroids. Pestic Biochem Physiol 106: 93–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riveron JM, Irving H, Ndula M, Barnes KG, Ibrahim SS, Paine MJ, Wondji CS. 2013. Directionally selected cytochrome P450 alleles are driving the spread of pyrethroid resistance in the major malaria vector Anopheles funestus. Proc Natl Acad Sci 110: 252–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riveron JM, Ibrahim SS, Chanda E, Mzilahowa T, Cuamba N, Irving H, Barnes KG, Ndula M, Wondji CS. 2014a. The highly polymorphic CYP6M7 cytochrome P450 gene partners with the directionally selected CYP6P9a and CYP6P9b genes to expand the pyrethroid resistance front in the malaria vector Anopheles funestus in Africa. BMC Genomics 15: 817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riveron JM, Yunta C, Ibrahim SS, Djouaka R, Irving H, Menze BD, Ismail HM, Hemingway J, Ranson H, Albert A, et al. 2014b. A single mutation in the GSTe2 gene allows tracking of metabolically based insecticide resistance in a major malaria vector. Genome Biol 15: R27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saavedra-Rodriguez K, Urdaneta-Marquez L, Rajatileka S, Moulton M, Flores AE, Fernandez-Salas I, Bisset J, Rodriguez M, McCall PJ, Donnelly MJ, et al. 2007. A mutation in the voltage-gated sodium channel gene associated with pyrethroid resistance in Latin American Aedes aegypti. Insect Mol Biol 16: 785–798. [DOI] [PubMed] [Google Scholar]
- Saavedra-Rodriguez K, Strode C, Flores AE, Garcia-Luna S, Reyes-Solis G, Ranson H, Hemingway J, Black WC IV. 2014. Differential transcription profiles in Aedes aegypti detoxification genes after temephos selection. Insect Mol Biol 23: 199–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholte EJ, Knols BG, Samson RA, Takken W. 2004. Entomopathogenic fungi for mosquito control: a review. J Insect Sci 4: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somwang P, Yanola J, Suwan W, Walton C, Lumjuan N, Prapanthadara LA, Somboon P. 2011. Enzymes-based resistant mechanism in pyrethroid resistant and susceptible Aedes aegypti strains from northern Thailand. Parasitol Res 109: 531–537. [DOI] [PubMed] [Google Scholar]
- Stenhouse SA, Plernsub S, Yanola J, Lumjuan N, Dantrakool A, Choochote W, Somboon P. 2013. Detection of the V1016G mutation in the voltage-gated sodium channel gene of Aedes aegypti (Diptera: Culicidae) by allele-specific PCR assay, and its distribution and effect on deltamethrin resistance in Thailand. Parasit Vectors 6: 253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevenson BJ, Bibby J, Pignatelli P, Muangnoicharoen S, O'Neill PM, Lian LY, Muller P, Nikou D, Steven A, Hemingway J, et al. 2011. Cytochrome P450 6M2 from the malaria vector Anopheles gambiae metabolizes pyrethroids: sequential metabolism of deltamethrin revealed. Insect Biochem Mol Biol 41: 492–502. [DOI] [PubMed] [Google Scholar]
- Stevenson BJ, Pignatelli P, Nikou D, Paine MJ. 2012. Pinpointing P450s associated with pyrethroid metabolism in the dengue vector, Aedes aegypti: developing new tools to combat insecticide resistance. PLoS Negl Trop Dis 6: e1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strode C, Wondji CS, David JP, Hawkes NJ, Lumjuan N, Nelson DR, Drane DR, Karunaratne SH, Hemingway J, Black WC IV, et al. 2008. Genomic analysis of detoxification genes in the mosquito Aedes aegypti. Insect Biochem Mol Biol 38: 113–123. [DOI] [PubMed] [Google Scholar]
- Timoshevskiy VA, Kinney NA, deBruyn BS, Mao C, Tu Z, Severson DW, Sharakhov IV, Sharakhova MV. 2014. Genomic composition and evolution of Aedes aegypti chromosomes revealed by the analysis of physically mapped supercontigs. BMC Biol 12: 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vontas J, Ranson H, Alphey L. 2010. Transcriptomics and disease vector control. BMC Biol 8: 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vontas J, Kioulos E, Pavlidi N, Morou E, Torre AD, Ranson H. 2012. Insecticide resistance in the major dengue vectors Aedes albopictus and Aedes aegypti. Pestic Biochem Physiol 104: 126–131. [Google Scholar]
- Walker T, Johnson PH, Moreira LA, Iturbe-Ormaetxe I, Frentiu FD, McMeniman CJ, Leong YS, Dong Y, Axford J, Kriesner P, et al. 2011. The wMel Wolbachia strain blocks dengue and invades caged Aedes aegypti populations. Nature 476: 450–453. [DOI] [PubMed] [Google Scholar]
- WHO. 2006. Guidelines for testing mosquito adulticides for indoor residual spraying and treatment of mosquito nets. Document WHO/CDS/NTD/WHOPES/GCDPP/3 World Health Organization, Geneva, Switzerland. [Google Scholar]
- WHO. 2009. Dengue and dengue hemorrhagic fever. World Health Organization Factsheet no117 revised March 2009 World Health Organization, Geneva, Switzerland. [Google Scholar]
- WHO. 2014. Chikungunya. World Health Organization Fact Sheet N° 327 World Health Organization, Geneva, Switzerland. [Google Scholar]
- Wondji CS, Irving H, Morgan J, Lobo NF, Collins FH, Hunt RH, Coetzee M, Hemingway J, Ranson H. 2009. Two duplicated P450 genes are associated with pyrethroid resistance in Anopheles funestus, a major malaria vector. Genome Res 19: 452–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanola J, Somboon P, Walton C, Nachaiwieng W, Somwang P, Prapanthadara LA. 2011. High-throughput assays for detection of the F1534C mutation in the voltage-gated sodium channel gene in permethrin-resistant Aedes aegypti and the distribution of this mutation throughout Thailand. Trop Med Int Health 16: 501–509. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.