

Abstract
In common with several other autoimmune diseases, autoimmune Addison’s disease (AAD) is thought to be caused by a combination of deleterious susceptibility polymorphisms in several genes, together with undefined environmental factors and stochastic events. To date, the strongest genomic association with AAD has been with alleles at the HLA locus, DR3–DQ2 and DR4. The contribution of other genetic variants has been inconsistent. We have studied the association of 16 single-nucleotide polymorphisms (SNPs) within the CD28–CTLA-4–ICOS genomic locus, in a cohort comprising 691 AAD patients of Norwegian and UK origin with matched controls. We have also performed a meta-analysis including 1002 patients from European countries. The G-allele of SNP rs231775 in CTLA-4 is associated with AAD in Norwegian patients (odds ratio (OR)=1.35 (confidence interval (CI) 1.10–1.66), P=0.004), but not in UK patients. The same allele is associated with AAD in the total European population (OR=1.37 (CI 1.13–1.66), P=0.002). A three-marker haplotype, comprising PROMOTER_1661, rs231726 and rs1896286 was found to be associated with AAD in the Norwegian cohort only (OR 2.43 (CI 1.68–3.51), P=0.00013). This study points to the CTLA-4 gene as a susceptibility locus for the development of AAD, and refines its mapping within the wider genomic locus.
Subject terms: Immunogenetics
Introduction
Primary adrenal insufficiency (Addison’s disease, AD) is most often caused by autoimmune destruction of the adrenal cortices, resulting in insufficient production of the steroid hormones, cortisol and aldosterone. Autoimmune diseases, including autoimmune AD (AAD), cluster in families and are believed to share some common etiological factors. The underlying mechanisms resulting in AAD are still obscure, but a contribution from genetic variants is clear.1, 2 The strongest genomic associations have been reported with the human leukocyte antigen (HLA) haplotypes DR3–DQ2 and DR4 (subtype DRB1*0404)-DQ8) in several populations.3, 4, 5, 6 Other associations include variants in genes involved in T-cell signalling and activation, especially the 1858T allele (rs2476601) of the tyrosine-protein phosphatase nonreceptor type 22 (PTPN22) gene.7 The product of this gene, lymphoid tyrosine phosphatase (LYP), acts as an essential mediator of T-cell receptor (TCR) signalling by controlling phosphorylation/dephosphorylation of key signalling pathway kinases.8 Polymorphisms in STAT4, linked to CD4+ cell fate and GATA3, involved in CD8+ cell homeostasis, have also been found to confer susceptibility to AAD.9 In addition, other genes that encode cell-surface receptors that regulate activation responses in T lymphocytes, for example, CD274 (programmed death ligand 1)10 and cytotoxic T-lymphocyte antigen-4, CTLA-4 (CD152)11, 12, 13 have also been linked to AAD susceptibility. CTLA-4, which is encoded by a gene located on chromosome 2q33, competes with the costimulator CD28 for binding to B7 on antigen-presenting cells, and downregulates T-cell responses to antigen-presentation/T-cell receptor engagement. Several studies have implicated CTLA-4 single-nucleotide polymorphisms (SNPs) in autoimmune diseases, including AAD. Recently, Schubert et al.14 described heterozygous mutations in CTLA-4 that resulted in a dominantly inherited complex immune dysregulation syndrome due to disruption of immune cell balance. Importantly, slightly different patterns of genetic association have been seen in different autoimmune conditions, including with alleles of the flanking CD28 and inducible costimulator (ICOS) genes, suggesting that different variants within this extended locus could predispose to different autoimmune conditions.15 Etiological variants at the CTLA-4 locus might result in either a deficient or an overexuberant CTLA-4 response relative to CD28, which interferes with immune homeostasis. Furthermore, CTLA-4 is constitutively expressed on regulatory T cells (Tregs). It has been suggested that this is one of the crucial, innate protective mechanisms against infectious agents and prevents the expansion of pathogenic T-cell clones.16 Blockade of CTLA-4 activity has also been shown to abrogate Treg function in several experimental situations, perhaps promoting an environment permissive to autoreactivity.17, 18
The association between CTLA-4 gene variants and AAD has previously been reported in some but not all studies of different European populations.3, 11, 12, 13, 19, 20, 21, 22 Of particular interest is the common nonsynonymous CTLA-4 polymorphism rs231775, also called exon 1 +49A→G (Ala17Thr). Although this SNP was found to be associated with AAD in Italy,3 this finding was not replicated in studies from Germany,21 Spain22 or Norway.20 In the UK population, results have been conflicting with one study finding an association12 and another failing to replicate this finding.20 Other polymorphisms in CTLA-4 have shown similar inconclusive patterns of association.3, 11, 20
The main limitation of all the previous studies of CTLA-4 gene variation in AAD has been insufficient statistical power due to the small patient cohorts studied, owing to the rarity of AAD. To overcome this, we have now collected DNA from 691 AAD patients from Norway and the UK and explored several polymorphisms in the CD28-CTLA-4-ICOS region within these cohorts using a tag-SNP approach. We also performed a meta-analysis of studies that have examined the CTLA-4 rs231775 variant in European populations.
Results
CD28 and CTLA-4 are associated with AAD in the Norwegian patient cohort, but not in UK AAD patients
Sixteen SNPs were analysed in the extended CTLA-4 gene locus in the UK and Norwegian AAD cohorts and compared with the allele and genotype frequencies of healthy controls of the same ethnic origins (Allele distribution with re-labelling #1–16 shown in Figure 1). Two SNPs did not meet the call rate criteria (that is, combined call rate>95%) from the UK cohort (rs11571308 (#6) and rs231730 (#12)) and from the Norwegian cohort (rs1181389 (#3) and rs5742909 (#7)). These were excluded from further analysis.
Figure 1.
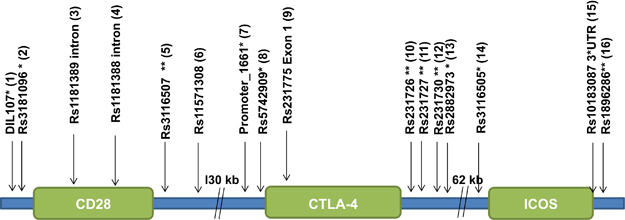
An overview of the SNPs that were analysed in this study. For simplicity, the SNPs are also labelled 1–16. *5′ near gene; **3′ near gene.
Genotypes and allele frequencies of one SNP, rs3181096 (#2), located close to the start of the CD28 gene (Figure 1), were associated with AAD in the combined cohort (all UK and Norwegian patients), and in the Norwegian cohort alone (Table 1). When we further separated the Norwegian patients into isolated AAD (iAAD) or APS II, or those with 21-hydroxylase (21-OH) antibodies, all subgroups remained significantly associated. However, there was no association in the UK patients when analysed alone (Table 1).
Table 1.
The distribution of alleles of 16 SNPs in the extended CTLA-4 gene locus encompassing CD28, CTLA-4 and ICOS in the Norwegian and UK patients and controls cohorts
| DIL107 | AAD G | AAD T | Ctrls G | Ctrls T | Call rate | OR | Z | P | Q hetero | P hetero | Weight | iAAD | APS II | 21-OH+ N |
| UK | 489 | 129 | 500 | 150 | 0.985 | 1.14 | 0.95 | 0.344 | 48.24 | P=0.52 | P=0.39 | |||
| Norway | 575 | 177 | 591 | 155 | 0.98 | 0.85 | −1.29 | 0.199 | 51.76 | P=0.99 | P=0.57 | P=0.22 | ||
| Meta random | 0.98 | −0.14 | 0.885 | 2.45 | 0.12 | P=0.65 | P=0.06 | |||||||
| rs3181096 | AAD C | AAD T | Ctrls C | Ctrls T | Call rate | OR | Z | P | Q hetero | P hetero | Weight | iAAD | APS II | 21-OH+ N |
| UK | 412 | 198 | 412 | 218 | 0.965 | 1.1 | 0.8 | 0.424 | 45.6 | P=0.31 | P=0.73 | |||
| Norway | 504 | 236 | 464 | 284 | 0.975 | 1.31 | 2.46 | 0.014 | 54.4 | P=0.054 | P=0.041 | P=0.006 | ||
| Meta random | 1.21 | 2.22 | 0.027 | 1.12 | 0.29 | P=0.03 | P=0.13 | |||||||
| rs1181389 | AAD T | AAD C | Ctrl T | Ctrl C | Call rate | OR | Z | P | Q hetero | P hetero | Weight | iAAD | APS II | 21-OH+ N |
| UK | 476 | 104 | 583 | 102 | 0.95 | 0.8 | −1.46 | 0.145 | 45.98 | P=0.43 | P=0.46 | |||
| rs1181388 | AAD G | AAD A | Ctrl G | Ctrl A | Call rate | OR | Z | P | Q hetero | P hetero | Weight | iAAD | APS II | 21-OH+ N |
| UK | 515 | 81 | 551 | 105 | 0.97 | 1.21 | 1.2 | 0.231 | 47.55 | P=0.29 | P=0.15 | |||
| Norway | 636 | 114 | 631 | 103 | 0.975 | 0.91 | −0.64 | 0.525 | 52.45 | P=0.50 | P=0.69 | P=0.52 | ||
| Meta random | 1.04 | 0.3 | 0.767 | 1.72 | 0.19 | P=0.84 | P=0.77 | |||||||
| rs3116507 | AAD A | AAD G | Ctrl A | Ctrl G | Call rate | OR | Z | P | Q hetero | P hetero | Weight | iAAD | APS II | 21-OH+ N |
| UK | 475 | 141 | 496 | 144 | 0.98 | 0.98 | −0.16 | 0.869 | 45.38 | 0.40 | P=0.57 | |||
| Norway | 565 | 183 | 570 | 164 | 0.975 | 0.89 | −0.96 | 0.335 | 54.62 | 0.54 | P=0.35 | P=0.21 | ||
| Meta random | 0.93 | −0.82 | 0.41 | 0.28 | 0.6 | P=0.92 | P=0.18 | |||||||
| rs11571308 | AAD C | AAD T | Ctrl C | Ctrl T | Call rate | OR | Z | P | Q hetero | P hetero | Weight | iAAD | APS II | 21-OH+ N |
| Norway | 646 | 92 | 651 | 87 | 0.97 | 0.94 | −0.4 | 0.69 | 59.04 | P=0.47 | P=1.0 | P=0.90 | ||
| rs5742909 | AAD C | AAD T | Ctrl C | Ctrl T | Call rate | OR | Z | P | Q hetero | P hetero | Weight | iAAD | APS II | 21-OH+ N |
| UK | 560 | 44 | 588 | 36 | 0.955 | 0.78 | −1.07 | 0.283 | 37.72 | P=0.57 | P=0.11 | |||
| Promoter_1661 | AAD A | AAD G | Ctrl A | Ctrl G | Call rate | OR | Z | P | Q hetero | P hetero | Weight | iAAD | APS II | 21-OH+ N |
| UK | 486 | 94 | 556 | 94 | 0.956 | 0.87 | −0.85 | 0.396 | 42.94 | 1.00 | P=0.92 | |||
| Norway | 611 | 135 | 598 | 122 | 0.965 | 0.92 | −0.58 | 0.562 | 57.06 | 0.53 | P=0.73 | P=0.61 | ||
| Meta random | 0.9 | −0.99 | 0.32 | 0.07 | 0.79 | P=0.63 | P=0.27 | |||||||
| rs231775 | AAD A | AAD G | Ctrl A | Ctrl G | Call rate | OR | Z | P | Q hetero | P hetero | Weight | iAAD | APS II | 21-OH+ N |
| UK | 361 | 257 | 400 | 260 | 0.995 | 0.91 | −0.8 | 0.425 | 47.52 | 0.19 | P=0.013 | |||
| Norway | 382 | 370 | 437 | 313 | 0.985 | 0.74 | −2.9 | 0.004 | 52.48 | 0.10 | P=0.003 | P=0.003 | ||
| Meta random | 0.82 | −1.91 | 0.055 | 1.87 | 0.17 | P=0.04 | P=0.27 | |||||||
| rs231726 | AAD C | AAD T | Ctrl C | Ctrl T | Call rate | OR | Z | P | Q hetero | P hetero | Weight | iAAD | APS II | 21-OH+ N |
| UK | 387 | 211 | 410 | 222 | 0.955 | 0.99 | −0.06 | 0.954 | 48.49 | P=0.67 | P=0.008 | |||
| Norway | 411 | 325 | 467 | 269 | 0.965 | 0.73 | −2.97 | 0.003 | 51.51 | P=0.10 | P=0.002 | P=0.002 | ||
| Meta random | 0.85 | −1.08 | 0.282 | 3.75 | 0.05 | P=0.16 | P=0.40 | |||||||
| rs231727 | AAD G | AAD A | Ctrl G | Ctrl A | Call rate | OR | Z | P | Q hetero | P hetero | Weight | iAAD | APS II | 21-OH+ N |
| UK | 381 | 207 | 433 | 225 | 0.965 | 0.96 | −0.37 | 0.709 | 47.47 | P=0.41 | P=0.023 | |||
| Norway | 420 | 328 | 457 | 269 | 0.97 | 0.75 | −2.66 | 0.008 | 52.53 | P=0.09 | P=0.007 | P=0.007 | ||
| Meta Random | 0.84 | −1.43 | 0.154 | 2.22 | 0.14 | P=0.10 | P=0.36 | |||||||
| rs231730 | AAD T | AAD A | Ctrl T | Ctrl A | Call rate | OR | Z | P | Q hetero | P hetero | Weight | iAAD | APS II | 21-OH+ N |
| Norway | 586 | 150 | 579 | 141 | 0.955 | 0.95 | −0.38 | 0.704 | 56.97 | P=0.35 | P=0.85 | P=0.85 | ||
| rs2882973 | AAD T | AAD C | Ctrl T | Ctrl C | Call rate | OR | Z | P | Q hetero | P hetero | Weight | iAAD | APS II | 21-OH+ N |
| UK | 401 | 215 | 431 | 225 | 0.985 | 0.97 | −0.23 | 0.821 | 46.53 | P=0.56 | P=0.13 | |||
| Norway | 453 | 301 | 490 | 262 | 0.99 | 0.8 | −2.04 | 0.042 | 53.47 | P=0.20 | P=0.048 | P=0.02 | ||
| Meta random | 0.88 | −1.35 | 0.176 | 1.44 | 0.23 | P=0.18 | P=0.37 | |||||||
| rs3116505 | AAD C | AAD T | Ctrl C | Ctrl T | Call rate | OR | Z | P | Q hetero | P hetero | Weight | iAAD | APS II | 21-OH+ N |
| UK | 395 | 211 | 411 | 217 | 0.96 | 0.99 | -0.1 | 0.922 | 46.71 | P=0.68 | P=0.10 | |||
| Norway | 428 | 286 | 477 | 259 | 0.95 | 0.81 | -1.91 | 0.056 | 53.29 | P=0.30 | P=0.045 | P=0.03 | ||
| Meta random | 0.89 | -1.19 | 0.235 | 1.47 | 0.23 | P=0.26 | P=0.46 | |||||||
| rs10183087 | AAD A | AAD C | Ctrl A | Ctrl C | Call rate | OR | Z | P | Q hetero | P hetero | Weight | iAAD | APS II | 21-OH+ N |
| UK | 444 | 158 | 456 | 172 | 0.955 | 1.06 | 0.45 | 0.651 | 50.91 | P=0.89 | P=0.66 | |||
| Norway | 594 | 146 | 595 | 143 | 0.97 | 0.98 | −0.17 | 0.864 | 49.09 | P=0.45 | P=0.39 | P=0.94 | ||
| Meta random | 1.02 | 0.2 | 0.839 | 0.19 | 0.66 | P=0.54 | P=0.83 | |||||||
| rs1896286 | AAD T | AAD G | Ctrl T | Ctrl G | Call rate | OR | Z | P | Q hetero | P hetero | Weight | iAAD | APS II | 21-OH+ N |
| UK | 372 | 238 | 382 | 274 | 0.985 | 1.12 | 1 | 0.319 | 46.82 | P=0.67 | P=0.42 | |||
| Norway | 445 | 275 | 444 | 290 | 0.955 | 1.06 | 0.51 | 0.607 | 53.18 | P=0.38 | P=0.96 | P=0.46 | ||
| Meta random | 1.09 | 1.06 | 0.29 | 0.14 | 0.71 | P=0.35 | P=0.42 |
Abbreviations: AAD, autoimmune Addison’s disease; APS II, autoimmune polyendocrinopathy type II; APS II, P-value for analysis of only APS II; Ctrl, control subjects; OR, odds ratio; Z, Z-value for meta-analysis; P, P-value for the different cohorts and for the meta-analysis using the random model for calculations; Q hetero, Q-value for heterogeneity; P hetero, P-value for heterogeneity; Weight, relative weight using the random model; iAAD, P-value for analysis of only isolated AAD; 21-OH+ N, P-value for analysis of Norwegian patients with positive anti-21-OH autoantibodies.
The distribution of alleles is given in numbers for the cohort of patients with AAD (cases) and for control subjects (Ctrl). The call rates given in the table are the combined call rate for controls and patients. All SNPs with a combined call rate of <95% have been rejected. Analyses have been executed by the program ‘Comprehensive meta-analysis version 2.0’ (www.meta-analysis.com) using a random effects model. P-values which mark a significant association (P<0.05) are marked in bold text. Norwegian cohort: Total AAD cases: N=382 (iAAD N=162; APS II N=220). Total controls: N=380. UK cohort: Total AAD cases: N=309 (iAAD N=135; APS II N=174). Total controls: N=335.
Five SNPs, all located within, or very close to, CTLA-4, were associated with AAD in the Norwegian cohort: rs231775 (#9), rs231726 (10), rs231727 (#11), rs2882973 (#13) and rs3116505 (#14) (Table 1). In the combined cohort, the G-allele of rs231775 (#1), was associated. This association was also seen in both the Norwegian and UK APS II subgroups, and in the 21-OH positive Norwegian subgroup. At rs231726 (#10), rs231727 (#11), rs2882973 (#13) and rs3116505 (#14), the association was specific for Norwegian APS II patients, rather than for iAAD. SNPs in ICOS were not associated with AAD in the either cohort.
The haplotype ‘PROMOTER_1661, rs231726 and rs1896286’ is associated with AAD in the Norwegian cohort
A three-marker haplotype, comprising PROMOTER_1661 (#7), rs231726 (#10) and rs1896286 (#16) was found to be associated with AAD in the Norwegian cohort only. The major haplotype (A–C–T) was found with a frequency of 34.9% in the Norwegian control cohort. Association was found with the A–T–T haplotype, found in 25.2% of Norwegian cases compared with only 15.8% of Norwegian controls (odds ratio (OR) 2.43 (confidence interval (CI) 1.68–3.51)). The stepwise inclusion of additional markers did not improve the model, and when the data set was conditioned for SNPs within the CTLA-4 gene, markers in the CD28 gene were not found to be exerting an independent effect.
CTLA-4 (rs231775) is strongly associated with AAD in the European population; a meta-analysis from studies including patients from Norway, UK, Germany, Spain and Italy
The SNP rs231775 (#9) has previously been studied in other AAD populations and found to be associated with disease. We confirm a strong correlation between this SNP and development of AAD in a random effects meta-analysis comprising 1002 patients and 1614 controls (Table 2), of which ~60% came from our current study. Overall, the OR for the association of the G-allele with AAD was 1.37 (CI 1.13–1.66), P=0.002.
Table 2.
Meta-analysis on the effect of the CTLA-4 SNP rs231775 for AAD
| Study name | A AAD | G AAD | A Ctrls | G Ctrls | Odds ratio for G (95% CI) | Z-value | P-v alue | Relative weight | |
|---|---|---|---|---|---|---|---|---|---|
| UK Euradrenal |
AAD N=309; Ctrls N=335 |
361 | 257 | 400 | 260 | 1.10 (0.88–1.37) | −0.80 | 0.425025 | 28.13 |
| Norway Euradrenal |
AAD, N=382; Ctrls, N=380 |
382 | 370 | 437 | 313 | 1.35 (1.10–1.66) | −2.90 | 0.003688 | 33.87 |
| Brozzetti et al.3 |
AAD, N=180; Ctrls, N=394 |
214 | 146 | 578 | 210 | 1.88 (1.44–2.44) | −4.69 | 0.000003 | 20.31 |
| Donner et al.21 |
AAD, N=76 Ctrls; N=394 |
85 | 67 | 581 | 358 | 1.28 (0.91–1.81) | −1.39 | 0.163298 | 11.73 |
| Perez de Nanclares et al.22 |
AAD, N=57; Ctrls, N=111 |
75 | 39 | 160 | 62 | 1.34 (0.83-2.18) | −1.19 | 0.235072 | 5.96 |
| TOTAL random modela |
AAD, N=1002; Ctrls, N=1614 |
1121 | 885 | 2136 | 1186 | 1.37 (1.13–1.66) | −3.15 | 0.002 |
Meta-analysis was performed using the program ‘Comprehensive meta-analysis version 2.0’ (www.meta-analysis.com), random effects model. Significant associations are marked with bold text in the P-value column.
aBeslow-Day test of heterogeneity: P=0.05; Hedges’s g test of heterogeneity: P=0.05.
Discussion
Variants in the CTLA-4 gene have been found to have a role in susceptibility to several autoimmune diseases in different populations, including autoimmune endocrinopathies such as Graves’ disease, autoimmune hypothyroidism and type 1 diabetes, (reviewed by Kavvoura et al.23, Kristiansen et al.24 and Ueda et al.25). The significance of CTLA-4 polymorphisms in AAD susceptibility remains controversial, as different studies have reported conflicting findings.3, 11, 12, 13, 19, 20, 21, 22 We have studied the contribution of SNPs in and around the CTLA-4 gene to AAD development in a large population comprising almost 700 patients from Norway and the UK. We conclude that several of the SNPs located in close proximity to the CD28 gene and to CTLA-4, including the G-allele of the common polymorphism rs231775 (#9), were significantly associated with AAD in the Norwegian population, but not in the UK patient cohort. This finding is in agreement with those of a previous study by Vaidya from 2000 which included 90 patients,12 but conflicts with the findings reported by Blomhoff from 2004 on 134 UK patients.20 However, these previous studies were conducted on much smaller cohorts, and hence are significantly less powerful than the current study. Other studies have indicated that CTLA-4, and not the neighbouring genes encoding CD28 and ICOS, are involved in susceptibility to autoimmune diseases.25, 26, 27 This study shows an association with CTLA-4 and AAD but no independent association between CD28 or ICOS and AAD, the latter of which had been previously suggested. We further show that for some of the SNPs in CTLA-4, the association with AAD is only true when AAD presents in combination with type 1 diabetes or thyroid disease (that is, APS II). It is possible that the positive association with these variants is due to both or one of these accompanying endocrine autoimmune conditions rather than to AAD in particular.
Haplotype analysis revealed that the haplotype PROMOTER_1661(A)-rs231726 (T)-rs1896286 (T) (#7#10#16) was significantly associated with AAD in the Norwegian cohort, strengthening the hypothesis of a role for CTLA-4 in AAD susceptibility. Markers in the CD28 gene were not found to exert an independent effect; hence, we speculate that the CD28 association with AAD is the result of linkage disequilibrium between CTLA-4 and CD28 alleles. A recent study suggested that the conflicting results of CD28, CTLA-4 and ICOS candidate gene association studies is due to population stratification. That is, the presence of systematic differences in allele frequencies between covert subpopulations within a population.28 This phenomenon is likely to explain the differing results in the UK and Norwegian cohorts studied here, even though the formal tests for genetic heterogeneity between the two populations were not overtly discrepant. It is also plausible that the real percentage of patients with Addison’s disease in whom the disease is in fact caused by pathological autoimmunity differ. However, we have tested the Norwegian cohort in this study and in a pilot subgroup of British AAD patients for autoantibodies against 21-OH, a positive marker for an ongoing autoimmune condition against the adrenals, and found similar results (data not shown). Deviations of results in different populations have also been reported for CTLA-4 associations with, for example, type 1 diabetes and autoimmune thyroid diseases (reviewed in Hou et al.29 and Wang et al.30). Notably, however, when the data indicate an association, it is most often with the same allele (for example, the G-allele concerning rs231775). As most autoimmune diseases are thought to be caused by a combination of unfavourable genetic factors (in addition to environmental triggers), a possibility exist that different genes have different impact on the association risk based on underlying genetics specific for each populations and ethnic groups. Hence, CTLA polymorphisms might be important for autoimmunity susceptibility in some populations, but overruled by other factors in other populations.
This study also confirms that the rs231775 (#9) SNP in CTLA-4 is significantly associated with AAD in a European meta-analysis. Interestingly, a previous report has demonstrated that this polymorphism exerts an effect on AAD susceptibility independent of prevalent HLA haplotypes.3 CD28 and CTLA-4 are important for T-cell function and signalling, and their regulation is therefore important for maintenance of normal lymphocyte biology. A polymorphism that diminishes CTLA-4 function or which increases CD28 function would be predicted to result in down-regulation of T-cell responses, favouring autoreactivity and therefore the development of autoimmune conditions. One important mechanism for controlling CTLA-4 and CD28 activity is by altering their cell-surface expression.31 In 2003, Ueda et al.25 demonstrated that a 3′-UTR haplotype of CTLA-4, including the CT60 SNP (rs30807243), was associated with altered levels of full-length (transmembrane) and soluble (s)CTLA-4 isoforms, the susceptibility haplotype being associated with reduced sCTLA-4 mRNA levels in man. Paradoxically, several studies have found increased levels of sCTLA-4 in the sera of individuals with autoimmune diseases, although newer data have challenged this hypothesis and the effect of certain genotypes on sCTLA-4 concentration remain controversial.32, 33, 34, 35, 36, 37, 38 People over 70 years of age have also been reported to have an increased serum level of sCTLA-4,39 which could be a contributing factor to the high prevalence of autoimmune diseases amongst the elderly. In accordance with our results, the G-allele of rs231775 (#9) has been shown to confer higher T-cell proliferation than the A-allele, indicating a lower degree of inhibition of T-cell activity, which may promote autoimmunity.40 However, this SNP, which is located on the peptide leader sequence of CTLA-4, may potentially also interfere with mRNA stability.
The impact of heterogeneity in our meta-analysis was borderline significant (P=0.05). We therefore used a random effects model which does not make the assumption that all studies included had a uniform direction of effect. The varying inclusion criteria between different studies and the differing frequency of HLA haplotypes between different European subpopulations are factors which might explain the genetic heterogeneity between the included studies. Nevertheless, all studies included in the meta-analysis indicate the same trend; that the G-allele of CTLA-4 SNP rs231775 (#9) contributes to AAD susceptibility.
In conclusion, this is the largest genetic study that has ever been conducted in AAD in relation to the CTLA-4 gene and we strengthen the evidence of an association between the CTLA-4 gene and AAD. In particular, the rs231775 (#9) polymorphism in the CTLA-4 signal peptide is associated with AAD in the European population, which supports a role for CTLA-4 in the pathogenesis of AAD.
Patients and methods
Patient material
AAD patients were recruited from the UK (N=309: iAAD N=135, autoimmune polyendocrinopathy type II (APS II) N=174) and Norway (N=382: iAAD N=162, APS II N=220). For the Norwegian cohort, 290 patients (76%) were 21-OH autoantibody positive, measured by radioimmunoassay as described elsewhere.41 In each AAD subject, the clinical diagnosis was confirmed by either a low basal cortisol with a high ACTH level or a subnormal response to the ACTH1–24 stimulation test (short synacthen test using 250 μg parenteral tetracosactide). Patients with primary adrenal failure owing to infiltrative or infective causes or secondary adrenal failure were excluded. In addition, patients with APS1, on the grounds of mucocutaneous candidiasis, hypoparathyroidism and ectodermal dystrophy, were also excluded and to our knowledge, all included individuals were nonconsanguineous. Patients with type 1 diabetes and/or autoimmune thyroid disease in addition to AAD were classified as having APS II. As controls, blood samples were collected from 335 and 380 healthy people from the UK and Norway, respectively. All individuals participating in this study gave informed, written consent. The study was approved by the Regional Ethical Committees (Leeds East in the UK; Western Norway in Norway).
Genotyping
DNA was extracted from peripheral blood. Sixteen SNPs within the extended CTLA-4 gene locus (encompassing CD28, CTLA-4 and ICOS) were chosen (Figure 1). Fourteen tag-SNPs were selected from the HapMap database, and an additional two SNPs (DIL107, promoter_1661) were chosen for their association with the autoimmune condition systemic lupus erythematosus.15 The DNA sequence flanking each SNP was found in the Ensembl database (~300 bp in length) and this sequence data was then used to generate a Sequenom iPlex assay, using the MassARRAY Designer software (Sequenom, San Diego, CA, USA). Compatibility of the multiplex assays was checked in silico. SNPs were genotyped using the Sequenom MassARRAY technology (Sequenom). The Iplex assay was followed according to manufacturer’s instructions (www.sequenom.com) using 30 ng of genomic DNA template.
Single-locus tests in Norwegian and English AAD cohorts
The UK and Norwegian cohort data were analysed separately and then analysed in combination by meta-analysis. For each SNP a count was made of each genotype (AA, AB and BB) in cases and controls. Genotype frequencies were used to calculate call rates for each SNP (AA+AB+BB/sample size × 100). Genotypes at all markers were checked for Hardy–Weinberg Equilibrium (threshold P<0.01). SNPs with a combined call rate (healthy controls and patients) of <95% were excluded from the analysis. The program ‘Comprehensive meta-analysis version 2.0’ (www.meta-analysis.com) using a random effects model was used in order to calculate ORs, z-scores, P-values. A subgroup analysis was then performed, whereby each cohort was divided into iAAD and APS II, (with and without 21-OH antibodies in the Norwegian cohort only).
Haplotype analysis
Haploview42 was used to calculate nonrandom association of alleles (linkage disequilibrium expressed as r2 in the UK and Norwegian control cohorts (Figure 2)). The UNPHASED program43 was used to estimate haplotype frequencies and association in the UK and Norwegian cases and controls.
Figure 2.
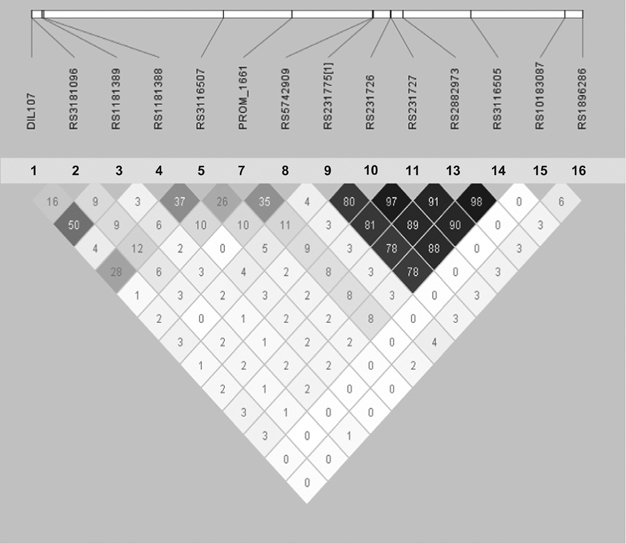
Haploview plots of linkage disequilibrium (LD; r2) between the SNPs for the UK control cohort analysed in this study. The plot for the Norwegian controls looked very similar. Some of the SNPs for each control cohort were excluded due to low call rate and these were not included in the Figure. To clarify which SNPs that are included in the graph, the SNPs have also been relabelled 1–16, which corresponds to the numbers given in Figure 1.
1.2.5 Meta-analysis for rs231775
‘CTLA-4’ and ‘autoimmune Addison’s disease’ were used as search terms in Pubmed aiming to find all studies examining this gene in AAD patients. Studies comprising over 50 patients which investigated rs231775, and which showed no obvious overlap with the AAD cohorts described here, were included in the meta-analysis. The program ‘Comprehensive meta-analysis version 2.0’ (www.meta-analysis.com) using a random effects model was used in order to calculate ORs, z-scores and P-values. The impact of heterogeneity in the material was calculated by Hegdes’s G’ test and by Breslow–Day test of heterogeneity.
Acknowledgements
We sincerely thank all patients who have participated in this study and the medical doctors who have recruited patients to the project. This study has been funded by EU FP7, Grant number 201167, Euradrenal, the Western Norway Health Authorities and University of Bergen. ASBW is funded by Bergen Medical Research Foundation. ALM is funded by the Medical Research Council, UK (grant number G0900390).
Competing interests
The authors declare no conflict of interest.
Footnotes
A S B Wolff and A L Mitchell: These authors contributed equally to this work.
References
- 1.Falorni A, Brozzetti A, Torre DL, Tortoioli C, Gambelunghe G. Association of genetic polymorphisms and autoimmune Addison's disease. Expert Rev Clin Immunol. 2008;4:441–456. doi: 10.1586/1744666X.4.4.441. [DOI] [PubMed] [Google Scholar]
- 2.Mitchell AL, Pearce SH. Autoimmune Addison disease: pathophysiology and genetic complexity. Nature reviews. Endocrinology. 2012;8:306–316. doi: 10.1038/nrendo.2011.245. [DOI] [PubMed] [Google Scholar]
- 3.Brozzetti A, Marzotti S, Tortoioli C, Bini V, Giordano R, Dotta F. Cytotoxic T lymphocyte antigen-4 Ala17 polymorphism is a genetic marker of autoimmune adrenal insufficiency: Italian association study and meta-analysis of European studies. Eur J Endocrinol. 2010;162:361–369. doi: 10.1530/EJE-09-0618. [DOI] [PubMed] [Google Scholar]
- 4.Erichsen MM, Lovas K, Skinningsrud B, Wolff AB, Undlien DE, Svartberg J. Clinical, immunological, and genetic features of autoimmune primary adrenal insufficiency: observations from a Norwegian registry. J Clin Endocrinol Metab. 2009;94:4882–4890. doi: 10.1210/jc.2009-1368. [DOI] [PubMed] [Google Scholar]
- 5.Maclaren NK, Riley WJ. Inherited susceptibility to autoimmune Addison's disease is linked to human leukocyte antigens-DR3 and/or DR4, except when associated with type I autoimmune polyglandular syndrome. J Clin Endocrinol Metab. 1986;62:455–459. doi: 10.1210/jcem-62-3-455. [DOI] [PubMed] [Google Scholar]
- 6.Ross IL, Babu S, Armstrong T, Zhang L, Schatz D, Pugliese A. HLA similarities indicate shared genetic risk in 21-hydroxylase autoantibody positive South African and United States Addison's disease. Tissue Antigens. 2014;84:361–369. doi: 10.1111/tan.12407. [DOI] [PubMed] [Google Scholar]
- 7.Skinningsrud B, Husebye ES, Gervin K, Lovas K, Blomhoff A, Wolff AB. Mutation screening of PTPN22: association of the 1858T-allele with Addison's disease. Eur J Hum Genet. 2008;16:977–982. doi: 10.1038/ejhg.2008.33. [DOI] [PubMed] [Google Scholar]
- 8.Zhang J, Zahir N, Jiang Q, Miliotis H, Heyraud S, Meng X. The autoimmune disease-associated PTPN22 variant promotes calpain-mediated Lyp/Pep degradation associated with lymphocyte and dendritic cell hyperresponsiveness. Nat Genet. 2011;43:902–907. doi: 10.1038/ng.904. [DOI] [PubMed] [Google Scholar]
- 9.Mitchell AL, Macarthur KD, Gan EH, Baggott LE, Wolff AS, Skinningsrud B. Association of autoimmune Addison's disease with alleles of STAT4 and GATA3 in European cohorts. PLoS ONE. 2014;9:e88991. doi: 10.1371/journal.pone.0088991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mitchell AL, Cordell HJ, Soemedi R, Owen K, Skinningsrud B, Wolff AB. Programmed death ligand 1 (PD-L1) gene variants contribute to autoimmune Addison's disease and Graves' disease susceptibility. J Clin Endocrinol Metab. 2009;94:5139–5145. doi: 10.1210/jc.2009-1404. [DOI] [PubMed] [Google Scholar]
- 11.Kemp EH, Ajjan RA, Husebye ES, Peterson P, Uibo R, Imrie H. A cytotoxic T lymphocyte antigen-4 (CTLA-4) gene polymorphism is associated with autoimmune Addison's disease in English patients. Clin Endocrinol (Oxf) 1998;49:609–613. doi: 10.1046/j.1365-2265.1998.00579.x. [DOI] [PubMed] [Google Scholar]
- 12.Vaidya B, Imrie H, Geatch DR, Perros P, Ball SG, Baylis PH. Association analysis of the cytotoxic T lymphocyte antigen-4 (CTLA-4) and autoimmune regulator-1 (AIRE-1) genes in sporadic autoimmune Addison's disease. J Clin Endocrinol Metab. 2000;85:688–691. doi: 10.1210/jcem.85.2.6369. [DOI] [PubMed] [Google Scholar]
- 13.Vaidya B, Pearce S. The emerging role of the CTLA-4 gene in autoimmune endocrinopathies. Eur J Endocrinol. 2004;150:619–626. doi: 10.1530/eje.0.1500619. [DOI] [PubMed] [Google Scholar]
- 14.Schubert D, Bode C, Kenefeck R, Hou TZ, Wing JB, Kennedy A. Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations. Nat Med. 2014;20:1410–1416. doi: 10.1038/nm.3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cunninghame Graham DS, Wong AK, McHugh NJ, Whittaker JC, Vyse TJ. Evidence for unique association signals in SLE at the CD28-CTLA4-ICOS locus in a family-based study. Hum Mol Genet. 2006;15:3195–3205. doi: 10.1093/hmg/ddl395. [DOI] [PubMed] [Google Scholar]
- 16.Takahashi T, Tagami T, Yamazaki S, Uede T, Shimizu J, Sakaguchi N. Immunologic self-tolerance maintained by CD25(+)CD4(+) regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J Exp Med. 2000;192:303–310. doi: 10.1084/jem.192.2.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kolar P, Knieke K, Hegel JK, Quandt D, Burmester GR, Hoff H. CTLA-4 (CD152) controls homeostasis and suppressive capacity of regulatory T cells in mice. Arthritis Rheum. 2009;60:123–132. doi: 10.1002/art.24181. [DOI] [PubMed] [Google Scholar]
- 18.Sojka DK, Hughson A, Fowell DJ. CTLA-4 is required by CD4+CD25+ Treg to control CD4+ T-cell lymphopenia-induced proliferation. Eur J Immunol. 2009;39:1544–1551. doi: 10.1002/eji.200838603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Blomhoff A, Kemp EH, Gawkrodger DJ, Weetman AP, Husebye ES, Akselsen HE. CTLA4 polymorphisms are associated with vitiligo, in patients with concomitant autoimmune diseases. Pigment Cell Res. 2005;18:55–58. doi: 10.1111/j.1600-0749.2004.00196.x. [DOI] [PubMed] [Google Scholar]
- 20.Blomhoff A, Lie BA, Kemp EH, Weetman AP, Akselsen HE, Myhre AG. Polymorphisms in the CTLA4 gene region confer susceptibility to Addison's disease. J Clin Endocrinol Metab. 2004;89:3474–3476. doi: 10.1210/jc.2003-031854. [DOI] [PubMed] [Google Scholar]
- 21.Donner H, Rau H, Walfish PG, Braun J, Siegmund T, Finke R. CTLA4 alanine-17 confers genetic susceptibility to Graves' disease and to type 1 diabetes mellitus. J Clin Endocrinol Metab. 1997;82:143–146. doi: 10.1210/jcem.82.1.3699. [DOI] [PubMed] [Google Scholar]
- 22.Perez de Nanclares G, Martin-Pagola A, Ramon Bilbao J, Vazquez F, Castano L. No evidence of association of CTLA4 polymorphisms with Addison's disease. Autoimmunity. 2004;37:453–456. doi: 10.1080/08916930400001917. [DOI] [PubMed] [Google Scholar]
- 23.Kavvoura FK, Ioannidis JP. CTLA-4 gene polymorphisms and susceptibility to type 1 diabetes mellitus: a HuGE Review and meta-analysis. Am J Epidemiol. 2005;162:3–16. doi: 10.1093/aje/kwi165. [DOI] [PubMed] [Google Scholar]
- 24.Kristiansen OP, Larsen ZM, Pociot F. CTLA-4 in autoimmune diseases—a general susceptibility gene to autoimmunity? Genes Immun. 2000;1:170–184. doi: 10.1038/sj.gene.6363655. [DOI] [PubMed] [Google Scholar]
- 25.Ueda H, Howson JM, Esposito L, Heward J, Snook H, Chamberlain G. Association of the T-cell regulatory gene CTLA4 with susceptibility to autoimmune disease. Nature. 2003;423:506–511. doi: 10.1038/nature01621. [DOI] [PubMed] [Google Scholar]
- 26.Ahmed S, Ihara K, Kanemitsu S, Nakashima H, Otsuka T, Tsuzaka K. Association of CTLA-4 but not CD28 gene polymorphisms with systemic lupus erythematosus in the Japanese population. Rheumatology. 2001;40:662–667. doi: 10.1093/rheumatology/40.6.662. [DOI] [PubMed] [Google Scholar]
- 27.Ihara K, Ahmed S, Nakao F, Kinukawa N, Kuromaru R, Matsuura N. Association studies of CTLA-4, CD28, and ICOS gene polymorphisms with type 1 diabetes in the Japanese population. Immunogenetics. 2001;53:447–454. doi: 10.1007/s002510100351. [DOI] [PubMed] [Google Scholar]
- 28.Butty V, Roy M, Sabeti P, Besse W, Benoist C, Mathis D. Signatures of strong population differentiation shape extended haplotypes across the human CD28, CTLA4, and ICOS costimulatory genes. Proc Natl Acad Sci USA. 2007;104:570–575. doi: 10.1073/pnas.0610124104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hou HF, Jin X, Sun T, Li C, Jiang BF, Li QW. Cytotoxic T lymphocyte-associated antigen 4 gene polymorphisms and autoimmune thyroid diseases: an updated systematic review and cumulative meta-analysis. Int J Endocrinol. 2015;2015:747816. doi: 10.1155/2015/747816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang J, Liu L, Ma J, Sun F, Zhao Z, Gu M. Common variants on cytotoxic T lymphocyte antigen-4 polymorphisms contributes to type 1 diabetes susceptibility: evidence based on 58 studies. PLoS ONE. 2014;9:e85982. doi: 10.1371/journal.pone.0085982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Valk E, Rudd CE, Schneider H. CTLA-4 trafficking and surface expression. Trends Immunol. 2008;29:272–279. doi: 10.1016/j.it.2008.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Anjos SM, Shao W, Marchand L, Polychronakos C. Allelic effects on gene regulation at the autoimmunity-predisposing CTLA4 locus: a re-evaluation of the 3' +6230G>A polymorphism. Genes Immun. 2005;6:305–311. doi: 10.1038/sj.gene.6364211. [DOI] [PubMed] [Google Scholar]
- 33.Daroszewski J, Pawlak E, Karabon L, Frydecka I, Jonkisz A, Slowik M. Soluble CTLA-4 receptor an immunological marker of Graves' disease and severity of ophthalmopathy is associated with CTLA-4 Jo31 and CT60 gene polymorphisms. Eur J Endocrinol. 2009;161:787–793. doi: 10.1530/EJE-09-0600. [DOI] [PubMed] [Google Scholar]
- 34.Esposito L, Hunter KM, Clark J, Rainbow DB, Stevens H, Denesha J. Investigation of soluble and transmembrane CTLA-4 isoforms in serum and microvesicles. J Immunol. 2014;193:889–900. doi: 10.4049/jimmunol.1303389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maier LM, Anderson DE, De Jager PL, Wicker LS, Hafler DA. Allelic variant in CTLA4 alters T cell phosphorylation patterns. Proc Natl Acad Sci USA. 2007;104:18607–18612. doi: 10.1073/pnas.0706409104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mayans S, Lackovic K, Nyholm C, Lindgren P, Ruikka K, Eliasson M. CT60 genotype does not affect CTLA-4 isoform expression despite association to T1D and AITD in northern Sweden. BMC Med Genet. 2007;8:3. doi: 10.1186/1471-2350-8-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Purohit S, Podolsky R, Collins C, Zheng W, Schatz D, Muir A. Lack of correlation between the levels of soluble cytotoxic T-lymphocyte associated antigen-4 (CTLA-4) and the CT-60 genotypes. J Autoimmune Dis. 2005;2:8. doi: 10.1186/1740-2557-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Repnik K, Potocnik U. CTLA4 CT60 single-nucleotide polymorphism is associated with Slovenian inflammatory bowel disease patients and regulates expression of CTLA4 isoforms. DNA Cell Biol. 2010;29:603–610. doi: 10.1089/dna.2010.1021. [DOI] [PubMed] [Google Scholar]
- 39.Sakthivel P, Wermeling F, Elmgren A, Hulthe J, Kakoulidou M, Lefvert AK. Circulating soluble CTLA-4 is related to inflammatory markers in the 70 year old population. Scand J Clin Lab Invest. 2010;70:237–243. doi: 10.3109/00365511003695608. [DOI] [PubMed] [Google Scholar]
- 40.Ban Y, Davies TF, Greenberg DA, Kissin A, Marder B, Murphy B. Analysis of the CTLA-4, CD28, and inducible costimulator (ICOS) genes in autoimmune thyroid disease. Genes Immun. 2003;4:586–593. doi: 10.1038/sj.gene.6364018. [DOI] [PubMed] [Google Scholar]
- 41.Ekwall O, Hedstrand H, Grimelius L, Haavik J, Perheentupa J, Gustafsson J. Identification of tryptophan hydroxylase as an intestinal autoantigen. Lancet. 1998;352:279–283. doi: 10.1016/S0140-6736(97)11050-9. [DOI] [PubMed] [Google Scholar]
- 42.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 43.Dudbridge F. Likelihood-based association analysis for nuclear families and unrelated subjects with missing genotype data. Hum Hered. 2008;66:87–98. doi: 10.1159/000119108. [DOI] [PMC free article] [PubMed] [Google Scholar]