

Abstract
Anaerobic metabolism of hydrocarbons proceeds either via addition to fumarate or by hydroxylation in various microorganisms, e.g., sulfate-reducing or denitrifying bacteria, which are specialized in utilizing n-alkanes or alkylbenzenes as growth substrates. General pathways for carbon assimilation and energy gain have been elucidated for a limited number of possible substrates. In this work the metabolic activity of 11 bacterial strains during anaerobic growth with crude oil was investigated and compared with the metabolite patterns appearing during anaerobic growth with more than 40 different hydrocarbons supplied as binary mixtures. We show that the range of co-metabolically formed alkyl- and arylalkyl-succinates is much broader in n-alkane than in alkylbenzene utilizers. The structures and stereochemistry of these products are resolved. Furthermore, we demonstrate that anaerobic hydroxylation of alkylbenzenes does not only occur in denitrifiers but also in sulfate reducers. We propose that these processes play a role in detoxification under conditions of solvent stress. The thermophilic sulfate-reducing strain TD3 is shown to produce n-alkylsuccinates, which are suggested not to derive from terminal activation of n-alkanes, but rather to represent intermediates of a metabolic pathway short-cutting fumarate regeneration by reverse action of succinate synthase. The outcomes of this study provide a basis for geochemically tracing such processes in natural habitats and contribute to an improved understanding of microbial activity in hydrocarbon-rich anoxic environments.
Keywords: anaerobic metabolism, hydrocarbons, activation reactions, n-alkylsuccinates, detoxification
Introduction
Complex mixtures of structurally diverse hydrocarbons occur naturally in subsurface habitats such as marine sediments and petroleum reservoirs (Tissot and Welte, 1984). As hydrocarbons are energy-rich organic compounds it is not surprising that a variety of microorganisms capable of utilizing these substrates have evolved (Widdel et al., 2010). During the Anthropocene the excessive technical use of petroleum and its products has resulted in further proliferation of hydrocarbons into the biosphere creating additional carbon and energy sources for such organisms. These have to cope with unfavorable living conditions including limited availability of electron acceptors and nutrients or stress caused by high concentrations of hydrocarbon solvents.
In many hydrocarbon-rich environments anoxic conditions prevail due to the imbalance of electron donor vs. oxygen availability. For more than 25 years, pure cultures of microorganisms have been known which utilize certain hydrocarbons as sole source of carbon and energy under strictly anoxic conditions (Schink, 1985; Lovley et al., 1989; Aeckersberg et al., 1991; Gilewicz et al., 1991). Instead of oxygen these organisms (mainly proteobacteria) use nitrate, ferric iron, or sulfate as electron acceptors. According to their respective hydrocarbon growth substrates these bacteria can be grouped into alkylbenzene or n-alkane utilizers. Within these subgroups strain-specific substrate ranges exist (for overviews see Rabus, 2005; Widdel et al., 2010). The majority of the known anaerobic n-alkane-utilizing bacteria adds their substrates via carbon atom 2 to fumarate, presumably catalyzed by homologs of the radical enzyme (methylalkyl)succinate synthase (Mas) from “Aromatoleum” sp. strain HxN1 (Rabus et al., 2001; Grundmann et al., 2008). This reaction yields the corresponding (1-methylalkyl)succinates, which are further metabolized to 4-methylalkanoates via CoA-thioester formation, intramolecular rearrangement and decarboxylation (Wilkes et al., 2002). The occurrence of methyl-, ethyl- and propyl-succinates in waters from hot sulfate-reducing environments was reported several times and was linked to the potential activation of the corresponding natural gas hydrocarbons (Duncan et al., 2009; Gieg et al., 2010; Wawrik et al., 2012; Callaghan, 2013). In contrast, alkylsuccinates potentially deriving from higher n-alkane homologs have only very rarely been detected (Duncan et al., 2009; Wawrik et al., 2012; Bian et al., 2015). For activation of toluene, alkylbenzene-utilizing strains employ addition to fumarate by benzylsuccinate synthase (Bss) forming 2-benzylsuccinate (Biegert et al., 1996; Leuthner et al., 1998), but further transformation proceeds via modified β-oxidation (Leutwein and Heider, 1999). In certain denitrifying bacteria alkylbenzenes may alternatively be activated by dehydrogenases forming the corresponding 1-phenylalkan-1-ols (Rabus and Heider, 1998; Kniemeyer and Heider, 2001a); such enzymatic dehydrogenations are so far unknown from sulfate-reducing bacteria.
By now, among the alkylbenzenes only toluene, ethylbenzene, o- and m-xylene, and p-cymene have been reported to be degraded via the corresponding benzylsuccinates in pure cultures of anaerobic bacteria (Biegert et al., 1996; Beller and Spormann, 1997; Krieger et al., 1999; Kniemeyer et al., 2003; Strijkstra et al., 2014). Despite this, Martus and Püttmann (2003) found succinate derivatives originating from C3+ alkylbenzenes in water samples from a jet fuel-contaminated aquifer, thus providing evidence for a broader relevance of this activation mechanism. Other studies have shown that several hydrocarbon-utilizing anaerobic bacteria are capable of activating and transforming hydrocarbons they cannot grow with when supplied as pure substrates. This was shown for strain HxN1 which co-activates cycloalkanes (Wilkes et al., 2003) and toluene (Rabus et al., 2011) while growing with n-hexane. Co-metabolism of toluene was also found in the denitrifying strain OcN1 and the sulfate-reducing strain TD3 (Rabus et al., 2011). In contrast, such kinds of co-activation, especially of alkanes, have not been observed in alkylbenzene-utilizing bacteria. However, a comprehensive survey of co-metabolic capabilities of anaerobic hydrocarbon-degrading bacteria is as yet lacking.
The alkyl-/arylalkyl-succinates formed as initial products of anaerobic metabolism of hydrocarbons contain at least one stereogenic carbon atom. The activation of toluene by strains T and K172 yields exclusively (R)-2-benzylsuccinate proving stereoselective addition to fumarate (Beller and Spormann, 1998; Leutwein and Heider, 1999). During anaerobic growth of strain HxN1 with n-hexane a mixture of (2R,1′R)- and (2S,1′R)-(1-methylpentyl)succinate is formed (Jarling et al., 2012). It is well-known that enzymatic reactions usually lead to enantiopure products (Alworth, 1972). Therefore, it is unlikely that this finding is due to a relaxed stereoselectivity of the activating enzyme Mas, but rather a consequence of a subsequent epimerase activity generating the required stereochemistry for the following rearrangement reaction.
Here, we present a systematic investigation of the formation of alkyl-/arylalkyl-succinates, their stereochemistry and further metabolic activity in hydrocarbon-utilizing anaerobic bacteria. In particular we attempt a detailed comparison of metabolic capabilities of n-alkane- vs. alkylbenzene-utilizing organisms. Moreover, we discuss why enzymes assumed to be Mas-like are potentially polyfunctional.
Materials and methods
General information
Chemicals and solvents were obtained from reputable suppliers. Where necessary, solvents were either dried using standard techniques or purchased as anhydrous. All reference standards used for identification of metabolites were either purchased from reputable suppliers or synthesized in this (see below) or in previous studies (Rabus et al., 2001; Wilkes et al., 2003; Kniemeyer et al., 2007; Jarling et al., 2012; Strijkstra et al., 2014). Glassware was dried in an oven prior to use. Column chromatography was carried out using 40–60 μm mesh silica in glass columns under medium pressure. NMR spectra were recorded at room temperature with a Bruker Avance III 300 or a Bruker Avance II 500 (both Bruker, Karlsruhe, Germany) or a Jeol ECS-400 instrument (JEOL USA, Peabody, Massachusetts, USA) at the radiofrequency given below. Signals were assigned using 1H, 13C, DEPT/APT and H,H-COSY spectra. IR spectra were recorded on a Bio-Rad FTS 3000MX diamond ATR. GC-MS analyses were performed using either a Trace GC-MS system (Thermo Fisher Scientific Inc., Thermo Scientific, Braunschweig, Germany) or an Agilent 6890A gas chromatograph coupled to a MAT95 XL mass spectrometer (Thermo Fisher Scientific Inc., Thermo Scientific, Braunschweig, Germany). The gas chromatograph was equipped with a 5% phenyl polysilphenylene-siloxane fused silica capillary column (BPX-5, SGE; 50 m and 0.22 mm i.d., 0.25 μm film thickness). The column temperature was initially held at 50°C for 1 min, then programmed to 310°C at a rate of 3°C/min with a final hold time of 30 min. Helium was used as the carrier gas. The PTV injector temperature was programmed from 50 to 300°C (10 min isothermal) at a rate of 10°C/sec, and the injection volume was 1 μL in the splitless mode. The mass spectrometer was operated in EI mode (70 eV) at a source temperature of 260°C. Full scan mass spectra were recorded from m/z 50 to 600. For analytical data of the synthesized standards see the Supplementary Material.
Cultivation and extraction
The denitrifying or sulfate-reducing, hydrocarbon-degrading bacteria used in this study are compiled in Table S1 in the Supplementary Material. They have been subcultured in our laboratory since their isolation. General procedures for anaerobic cultivation and preparation of media were as described by Widdel et al. (2010). The anoxic bicarbonate-buffered defined mineral media for sulfate reducers (28 mM sulfate, sulfide-reduced) and denitrifiers (10 mM nitrate, ascorbate-reduced) were composed as described previously (Rabus et al., 1993; Rabus and Widdel, 1995). Anoxic sterile crude oil was prepared and added to the cultures as detailed by Rabus and Widdel (1996). Individual aromatic and/or aliphatic hydrocarbons were provided as dilutions in 2,2,4,4,6,8,8-heptamethylnonane with percentages as listed in the Supplementary Table S2. The individual strains were adapted to anaerobic growth with n-hexane (strain HxN1), n-octane (strain OcN1), n-decane (strain TD3), toluene (strains K172, EbN1, ToN1, T, Tol2), m-xylene (strains mXyN1 and mXyS1), or o-xylene (strain oXyS1) for at least five passages prior to inoculation of the main cultures (400 mL). Main cultures were run in triplicate. Positive controls contained the adaptation substrate only, while negative controls were devoid of electron acceptor or inoculum. All cultures were incubated at 28°C, except for strain TD3 (60°C; Rueter et al., 1994). Harvest of the main cultures for metabolite extraction (see below) was performed when nitrate/nitrite were depleted from the medium and sulfide formation ceased, respectively.
Metabolite extraction was essentially performed as described (Rabus et al., 2001). After heat inactivation, the culture broth was separated from the carrier phase (in the case of crude oil experiments, the nonpolar fraction was removed by successive extraction with n-pentane), acidified with HCl and extracted 3 times with 50 mL diethyl ether. The combined ether extracts were dried over anhydrous sodium sulfate and stored at 4°C in glass bottles sealed with Teflon-coated screw caps. The extracts were divided into two aliquots, each reduced to about 1 mL using a TurboVap® 500 (Biotage, Uppsala, Sweden). One aliquot was subsequently methylated with diazomethane as described elsewhere (Rabus et al., 2001). The other aliquot was used for stereochemical investigation as described below.
Syntheses of succinate derivatives potentially deriving from butyl- and pentyl-benzene
With one exception (see below), a procedure was adapted from Müller et al. (1999) and Concellon et al. (2004), for the condensation after Stobbe (1899) and the reduction, respectively. The Stobbe condensation of diethyl succinate and suitable ketones yields the 1-ethyl esters of the corresponding itaconates. Taking this into account we started with dimethyl succinate to obtain the corresponding 1-methyl esters of the desired products, which after methylation with diazomethane served as standard compounds for comparison with metabolites detected in methylated extracts of bacterial cultures.
About 1 g (44 mmol) sodium was completely dissolved in 8 mL methanol under reflux. After cooling to room temperature, 1.5 mL (1.7 g, 11.6 mmol) dimethyl succinate and subsequently about 6.5 mmol of the respective ketone were added. The mixture was refluxed for 2 h and left overnight at room temperature. After addition of 25 mL water the mixture was extracted three times with 20 mL chloroform. The organic extracts were discarded and the aqueous phase was acidified to pH < 1 using concentrated hydrochloric acid and extracted three times with 20 mL chloroform and once with 30 mL ethyl acetate. The combined organic extracts were dried over sodium sulfate and evaporated under reduced pressure. The yellow-brown crude product was pre-purified by medium pressure chromatography on silica by gradient elution with methanol in chloroform. The product mixture obtained was pure enough for the next step.
About 50–100 mg of the product from the first step were dissolved in 2 mL tetrahydrofuran and 2 mL water, de-aerated und kept under nitrogen. To this solution 12.5 mL 0.1 M blue samarium diiodide solution in tetrahydrofuran was added and the mixture was stirred under nitrogen for about 90 min until the pinkish color diminished. After addition of 5 mL 0.1 M hydrochloric acid the solution was extracted three times with ethyl acetate. The combined extracts were dried over sodium sulfate and evaporated under reduced pressure. The desired products were isolated from the crude product mixtures using semi-preparative HPLC (Prominence system, Shimadzu, Duisburg, Germany) equipped with an Acclaim® OA column (5 μm, 120 Å, 4.0 × 250 mm; Thermo Fisher Scientific Inc., Thermo Scientific Dionex, Dreieich, Germany) applying gradients of acetonitrile in water (both acidified with 0.1% formic acid).
Syntheses of dimethyl 2-(1-phenylethyl)succinate
(1-Iodoethyl)benzene (1.00 g, 4.31 mmol), tris(trimethylsilyl)silane (1.34 g, 5.39 mmol), dimethyl fumarate (1.86 g, 12.9 mmol), and azobis-isobutyronitrile (0.5 eq) in α,α,α-trifluorotoluene (40 mL) was heated under reflux for 12 h. The mixture was cooled and concentrated to an oil that was fractionated by medium pressure chromatography, eluting with petrol—ethyl acetate (8: 2, v/v) to afford dimethyl 2-(1-phenylethyl)succinate (0.54 g, 50%), as a colorless oil. Rf 0.40 (petrol—ethyl acetate, 8: 2, v/v). 1H and 13C NMR indicated a 1:1 mixture of two diastereoisomers.
Syntheses of 2-(1-phenylethyl)succinic acid
To dimethyl 2-(1-phenylethyl)succinate (0.70 g, 2.79 mmol) in tetrahydrofuran (55 mL) was added, dropwise with stirring, lithium hydroxide (0.27 g, 11 mmol) in water (20 mL). After stirring overnight at room temperature, the tetrahydrofuran was removed under reduced pressure and the remaining aqueous phase was acidified with 6 M hydrochloric acid. The mixture was extracted with ethyl acetate (3 × 20 mL). The combined organic extracts were dried (Na2SO4) and the solvent was removed to give the crude 2-alkylsuccinic acid. Crystallization of the crude product from ethyl acetate-petrol by the vapor diffusion technique gave 2-(1-phenylethyl)succinic acid (0.433 g, 70%) as white crystals. 1H NMR indicated that the product was a 1:1 mixture of diastereoisomers.
Succinimide formation from diacid derivatives
The method used here was derived from a method published earlier (Jarling et al., 2012). The culture extract or about 100 μg of the standard compound was introduced into a 10 mL pear-shaped flask. 20 μL (R)-1-phenylethanamine in diethyl ether (7% v/v) was added and the mixture was dried under a stream of nitrogen. 1 mL toluene and 20 μL pyridine were added und the mixture was refluxed for 1 h. After cooling, 1 mL 3M hydrochloric acid was added and the organic phase was separated. The remaining acidified aqueous solution was extracted twice with 1 mL diethyl ether. The combined organic phases were dried under reduced pressure at 40°C and re-dissolved in 1 mL dichloromethane for GC-MS measurement.
Results
Hydrocarbon substrates for In vivo formation of succinate derivatives
The metabolic potential and activity of alkyl-/arylalkyl-succinate formation was investigated with 11 obligately or facultatively anaerobic bacterial strains (Tables 1, 2, Supplementary Table S1). The analyses revealed a high diversity in succinate derivatives formed, especially in case of n-alkane-utilizing strains (Figure 1, Supplementary Table S3). To assign unequivocally the succinate derivatives detected in experiments with crude oil to their respective hydrocarbon-precursors, the same bacterial strains were cultivated with defined binary mixtures of a strain-specific hydrocarbon substrate and a second (mostly non growth supporting) hydrocarbon and comparatively analyzed for metabolites. Succinate derivatives can be identified as dimethyl esters in mass chromatograms due to their specific fragmentation pattern, e.g., m/z 114, 146, [M-73] as well as [M-31] and m/z 131, 145, [M-145], [M-60] as well as [M] for alkyl- and benzyl-succinates, respectively (for further details see Supplementary Material).
Table 1.
Number of gas chromatographically separable alkyl-/arylalkyl-succinates formed from different hydrocarbons by n-alkane-utilizing bacteria under anoxic conditions.a
| Substrate | Strain | |||||
|---|---|---|---|---|---|---|
| Electron acceptor | ||||||
| Cultivation conditionb | ||||||
| HxN1c | OcN1c | TD3d | ||||
| B | O | B | O | B | O | |
| Methane | 0 | 0 | 0 | 0 | 0 | 0 |
| Ethane | 0 | 0 | 0 | 0 | 0 | 0 |
| Propane | 1 | 1 | 2 | 1 | 2 | 2 |
| n-Butane | 2 | 2 | 2 | 2 | 2 | 2 |
| n-Pentane | 3 | 3 | 3 | 3 | 3 | 3 |
| n-Hexane | 3 | 3 | 3 | 3 | 3 | 3 |
| n-Heptane | 3 | 3 | 3 | 3 | 3 | 3 |
| n-Octane | 3 | 3 | 3 | 3 | 3 | 3 |
| n-Nonane | 3 | 3 | nt | 3 | 3 | 3 |
| n-Decane | 0 | 0 | 2 | 2 | 2 | 2 |
| n-Undecane | nt | 0 | 2 | 2 | 2 | 2 |
| n-Dodecane | nt | 0 | 2 | 2 | 2 | 2 |
| n-Tridecane | nt | 0 | 2 | 1 | 2 | 1 |
| n-Tetradecane | nt | 0 | nt | 0 | nt | 1 |
| n-Hexadecane | nt | 0 | 0 | 0 | 1 | 0 |
| 2-Methylpentane | 1 | 1 | 1 | 1 | 1 | 1 |
| 3-Methylpentane | 0 | 0 | 0 | 0 | 0 | 0 |
| 2,2-Dimethylbutane | 0 | 0 | 0 | 0 | 0 | 0 |
| 2,3-Dimethylbutane | 0 | 0 | 0 | 0 | 0 | 0 |
| Cyclopentane | 1 | 1 | 1 | 1 | 1 | 1 |
| Methylcyclopentane | 1 | 1 | 1 | 1 | 1 | 1 |
| Ethylcyclopentane | nt | 4 | 4 | 4 | 4 | 4 |
| Cyclohexane | 1 | 0 | nt | 0 | nt | 0 |
| Toluene | 1 | 1 | 1 | 1 | 1 | 1 |
| Ethylbenzene | 2 | 2 | 2 | 2 | 2 | 2 |
| Propylbenzene | 3 | 0 | 1 | 0 | 0 | 0 |
| Butylbenzene | 1 | 0 | 1 | 0 | 1 | 0 |
| Pentylbenzene | 2 | 0 | 2 | 0 | 0 | 0 |
| Hexylbenzene | 2 | 0 | 2 | 0 | 2 | 0 |
| Heptylbenzene | 0 | 0 | 2 | 0 | 2 | 0 |
| Octylbenzene | 0 | 0 | 2 | 0 | 2 | 0 |
| Nonylbenzene | nt | 0 | 2 | 0 | 2 | 0 |
| Decylbenzene | nt | 0 | 2 | 0 | 2 | 0 |
| o-Xylene | 1 | 0 | 1 | 0 | 1 | 0 |
| o-Ethyltoluene | nt | 0 | 0 | 0 | 0 | 0 |
| o-Isopropyltoluene | nt | 0 | 0 | 0 | 0 | 0 |
| m-Xylene | 1 | 0 | 1 | 0 | 1 | 1 |
| m-Ethyltoluene | nt | 0 | 3 | 0 | 3 | 3 |
| m-Isopropyltoluene | nt | 0 | 0 | 0 | 1 | 0 |
| p-Xylene | 1 | 1 | 1 | 1 | 1 | 1 |
| p-Ethyltoluene | nt | 3 | 3 | 3 | 3 | 3 |
| p-Propyltoluene | nt | 0 | nt | 1 | nt | 1 |
| p-Isopropyltoluene | nt | 0 | 1 | 1 | 1 | 1 |
| p-Butyltoluene | nt | 0 | nt | 1 | nt | 1 |
| p-tert-Butyltoluene | nt | 0 | 1 | 0 | 1 | 0 |
| p-Pentyltoluene | nt | 0 | nt | 0 | nt | 1 |
| p-Diethylbenzene | nt | 2 | nt | 1 | nt | 2 |
| p-Ethylpropylbenzene | nt | 0 | nt | 2 | nt | 2 |
| p-Ethylisopropylbenzene | nt | 0 | nt | 1 | nt | 2 |
| p-Butylethylbenzene | nt | 0 | nt | 2 | nt | 2 |
| 1,2,3-Trimethylbenzene | nt | 0 | 0 | 0 | 0 | 0 |
| 1,2,4-Trimethylbenzene | nt | 0 | 0 | 0 | 1 | 1 |
| 1,3,5-Trimethylbenzene | nt | 0 | 0 | 0 | 0 | 0 |
| 1,2,3,5-Tetramethylbenzene | nt | 0 | 0 | 0 | 0 | 0 |
| 1,2,4,5-Tetramethylbenzene | nt | 0 | 0 | 0 | 0 | 0 |
| Pentamethylbenzene | nt | 0 | 0 | 0 | 0 | 0 |
| Hexamethylbenzene | nt | 0 | 0 | 0 | 0 | 0 |
| 1-Methylnaphthalene | nt | 0 | nt | 0 | nt | 1 |
| Growth substrate | Co-substrate | No substrate | ||||
The numbers qualitatively indicate the count of succinates (as dimethyl esters) formed from the respective substrate detected as chromatographically separable peaks during analyses with GC-MS.
Anaerobic growth with binary substrate mixtures (B) or crude oil (O); nt, not tested. In the case of the binary substrate mixtures, the hydrocarbons listed in the left column served as co-substrate, with n-hexane (HxN1), n-octane (OcN1), or n-decane (TD3) as the strain-specific main substrates.
Ehrenreich et al., 2000.
Rueter et al., 1994.
Table 2.
Number of gas chromatographically separable arylalkyl-succinates formed from different hydrocarbons by alkylbenzene-utilizing bacteria under anoxic conditions.a
| Substrate | Strain | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Electron acceptor | ||||||||||||||||
| Cultivation conditionb | ||||||||||||||||
| K172c | EbN1d | ToN1d | mXyN1d | Te | Tol2f | oXyS1g | mXyS1g | |||||||||
| B | O | B | O | B | O | B | O | B | O | B | O | B | O | B | O | |
| n-Hexane | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Cyclohexane | 0 | 0 | nt | 0 | nt | 0 | nt | 0 | nt | 0 | nt | 0 | nt | 0 | nt | 0 |
| Methylcyclohexane | nt | 0 | nt | 0 | nt | 0 | nt | 0 | 0 | 0 | nt | 0 | nt | 0 | nt | 0 |
| Toluene | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 0 | 1 | 1 |
| Ethylbenzene | nt | 0 | 0 | 0 | nt | 0 | nt | 0 | nt | 0 | nt | 0 | nt | 0 | nt | 0 |
| Butylbenzene | nt | 0 | nt | 0 | 0 | 0 | nt | 0 | nt | 0 | 0 | 0 | nt | 0 | nt | 0 |
| Pentylbenzene | nt | 0 | nt | 0 | 0 | 0 | nt | 0 | nt | 0 | 0 | 0 | nt | 0 | nt | 0 |
| o-Xylene | nt | 1 | 1 | 1 | nt | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 0 | 1 | 1 |
| o-Ethyltoluene | nt | 1 | 0 | 1 | nt | 1 | nt | 1 | nt | 1 | nt | 0 | 1 | 0 | nt | 0 |
| m-Xylene | nt | 1 | 1 | 1 | nt | 0 | 1 | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 1 | 1 |
| m-Ethyltoluene | nt | 0 | 0 | 0 | nt | 0 | nt | 0 | nt | 0 | nt | 0 | nt | 0 | 1 | 1 |
| m-Propyltoluene | nt | 0 | nt | 0 | nt | 0 | nt | 0 | nt | 0 | nt | 0 | nt | 0 | nt | 1 |
| m-Isopropyltoluene | nt | 0 | nt | 0 | nt | 0 | nt | 0 | nt | 0 | nt | 0 | nt | 0 | nt | 1 |
| p-Xylene | nt | 0 | 0 | 0 | nt | 1 | 1 | 1 | 1 | 1 | 1 | 0 | nt | 0 | 1 | 1 |
| p-Ethyltoluene | nt | 0 | 0 | 0 | nt | 0 | nt | 0 | nt | 0 | nt | 0 | nt | 0 | nt | 0 |
| 1,2,3-Trimethylbenzene | nt | 0 | nt | 0 | nt | 0 | nt | 1 | nt | 1 | nt | 0 | nt | 1 | nt | 1 |
| 1,2,4-Trimethylbenzene | nt | 1 | nt | 1 | nt | 1 | nt | 3 | nt | 3 | nt | 0 | 1 | 1 | 3 | 3 |
| 1,3,5-Trimethylbenzene | nt | 0 | nt | 0 | nt | 0 | nt | 1 | nt | 1 | nt | 0 | nt | 0 | 1 | 1 |
| 1-Methylnaphthalene | nt | 0 | nt | 0 | nt | 0 | nt | 0 | nt | 1 | nt | 0 | nt | 0 | nt | 0 |
| 2-Methylnaphthalene | nt | 0 | nt | 0 | nt | 0 | nt | 1 | nt | 1 | nt | 0 | nt | 0 | nt | 1 |
| Growth substrate | Co-substrate | No substrate | ||||||||||||||
The numbers qualitatively indicate the count of succinates (as dimethyl esters) formed from the respective substrate detected as chromatographically separable peaks during analyses with GC-MS.
Anaerobic growth with binary substrate mixtures (B) or crude oil (O); nt, not tested. In the case of the binary substrate mixtures, the hydrocarbons listed in the left column served as co-substrate, with toluene (K172, EbN1, ToN1, T, Tol2), m-xylene (mXyN1, mXyS1), or o-xylene (oXyS1) as the strain-specific main-substrates.
Anders et al., 1995.
Rabus and Widdel, 1995.
Dolfing et al., 1990.
Rabus et al., 1993.
Harms et al., 1999.
Figure 1.

Partial ion chromatograms (upper trace, m/z 114; lower trace, m/z 226+236 + 250+264+278+292+306) of a methylated extract of strain TD3 after anaerobic growth with crude oil. Annotated peaks represent alkyl-/arylalkyl-succinates formed from respective hydrocarbon precursors. (A) Homologous series of activation products of n-alkanes in blue, branched alkanes in orange and cycloalkanes in green. The red series indicates n-alkylsuccinates not directly derived from n-alkanes (Annotation code: n-Cx, n-alkanes; 2-MC5, 2-methylpentane; brCx, branched alkanes; Cy5, cyclopentane; MCy5, methylcyclopentane; ECy5, ethylcyclopentane). (B) Succinates derived from aromatic hydrocarbons as indicated by their structures. Their respective activation positions are highlighted by black dots. (green, homologous series derived from p-n-alkyltoluenes; blue, homologous series derived from p-n-alkylethylbenzenes; magenta, succinates derived from m-dialkylbenzenes; orange, succinates derived from p-dialkylbenzenes with one branched side chain).
The hydrocarbons found to be transformed to succinate derivatives by each strain are compiled in Tables 1, 2. It is evident that the range of hydrocarbons transformable by n-alkane-utilizing strains markedly exceeds that of the hydrocarbons supporting growth. In contrast, the putative benzylsuccinate synthases in all tested alkylbenzene-utilizing strains are restricted to a rather limited number of alkylbenzenes containing at least one benzylic methyl group. Most notably, these strains are apparently incapable of activating n-alkanes by addition to fumarate. In several cases formation of the corresponding succinate derivative was only observed, if the respective hydrocarbon was supplied in high (but sub-lethal) concentration in a binary substrate mixture experiment, while it could not be detected upon growth of the respective strain with crude oil, which contained the same hydrocarbon, however, in significantly lower concentration (Tables 1, 2). In general, the n-alkane-utilizing strains also transform n-alkanes with chain lengths somewhat longer or shorter than the growth-supporting n-alkanes, as well as a restricted range of branched alkanes apparently with low degree of alkyl substitution, certain monocyclic alkanes and various alkylated aromatic hydrocarbons (Table 1, Figure 1). The broadest range of transformable hydrocarbons is observed with strain TD3.
Significant amounts of succinates deriving from activation of cyclopentane and its methyl and ethyl homologs are detected in cultures of n-alkane degraders (Figure 1A, light green). In contrast, products of cyclohexane and methylcyclohexane transformation are near detection limit if present at all, even though the cyclohexanes are similar in concentration or even more abundant in the crude oil compared to the cyclopentanes (see for example Dahlgren et al., 1998). Succinate derivatives of branched alkanes appear at similar relative concentrations as those of methyl- and ethyl-cyclopentane (Figure 1A, orange). From the four branched C6-alkanes, only 2-methylpentane is activated by all three n-alkane-utilizing strains incubated with binary mixtures or crude oil.
Noteworthy, all three n-alkane-utilizing strains also transform monoalkylbenzenes with unbranched alkyl chains of different length as well as certain o-, m-, and p-dialkylbenzenes (Table 1). Upon anaerobic growth of these three strains with crude oil containing similar amounts of the xylene isomers, the succinate derivative of the p-isomer is always strongly dominating. Similarly, higher abundance of succinates deriving from other p-dialkylbenzenes, i.e., p-alkylated toluenes and ethylbenzenes, is observed as depicted in Figure 1B. This preferential accumulation of succinates formed from p-dialkylbenzenes is further supported by the finding that during anaerobic growth of strain TD3 with a binary mixture of n-decane and m-cymene (0.5% of the p-isomer present as impurity) equal levels of succinates from both cymene isomers are produced.
Structure elucidation of alkyl-/arylalkyl-succinates and regiochemistry of hydrocarbon activation
In case of n-alkanes it is generally accepted that these molecules are activated at carbon atom 2, leading to a prominent homologous series of (1-methylalkyl)succinate diastereoisomers, when the respective bacteria anaerobically grow with crude oil (Figure 1A, dark blue; see also Wilkes et al., 2003). Only in a few cases activations at the carbon atom 3 (n-hexane, strain HxN1) or the terminal methyl group (propane, strain BuS5) have been documented to occur as side reactions (Rabus et al., 2001; Kniemeyer et al., 2007). As confirmed by experiments with single substrates or binary substrate mixtures, all tested n-alkane-utilizing strains form succinate derivatives eluting in front of the (1-methylalkyl)succinates (Figure 1A, light blue). These by-products have identical mass spectrometric fragmentation patterns and originate from less favorable transformations of the n-alkanes. The respective product from n-hexane was identified by comparison with a reference standard as (1-ethylbutyl)succinate in strains OcN1 and TD3 (this study) and HxN1 (Rabus et al., 2001), while tentative assignment of its homologs is based on relative retention times and mass spectrometric fragmentation patterns. Moreover, strains OcN1 and TD3 form isopropyl- as well as n-propylsuccinate in cultures with a mixture of their growth substrates (n-octane and n-decane, respectively) and propane, which were identified by comparisons with reference standards. However, these two strains significantly differ with respect to the ratio of isopropyl- to n-propylsuccinate (231.6 in OcN1 vs. 6.5 in TD3). In addition, strain TD3 unexpectedly also formed C1–C7 n-alkylsuccinates during anaerobic growth with pure n-alkanes (> C6), binary substrate mixtures containing at least one such n-alkane or crude oil (Figure 1A, red peaks). Methyl-, n-propyl-, and n-butylsuccinate were identified by comparison with reference standards, while assignment of the other n-alkylsuccinates is based on relative GC retention times and mass spectra. Notably, C-even n-alkylsuccinates were predominant upon growth with C-odd n-alkanes and vice versa (Figure 2A).
Figure 2.
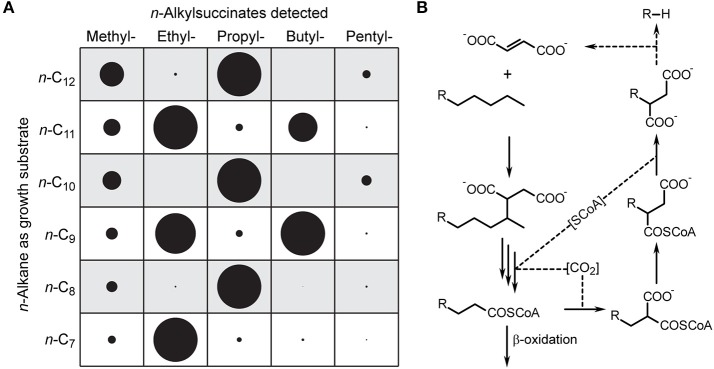
Formation of short chain n-alkylsuccinates from n-alkanes with chain length > C6 in strain TD3. (A) Relative amounts of n-alkylsuccinates (as dimethyl esters) found in cultures of strain TD3 grown with the mentioned n-alkane as sole source of carbon and energy. Relative amounts were determined by integration of respective summed mass traces from GC-MS analyses and normalization to 100% (circle diameter represents the relative amount). (B) Potential pathway for formation and further utilization of short chain n-alkylsuccinates.
The succinates deriving from activation of branched alkanes have not unambiguously been identified due to the lack of standards. We would like to emphasize that only 2-methylpentane, but none of the three other branched C6-alkanes, is activated. Compared to the other branched C6-alkanes the unique structural feature of 2-methylpentane is an unsubstituted propyl moiety. Thus, we propose that the product of 2-methylpentane should be (1,3-dimethylbutyl)succinate. The regiochemistry of activation of methyl- and ethyl-cyclopentane remains unclear.
To investigate the activation of alkylbenzenes in the three n-alkane-utilizing strains, the latter were grown with binary mixtures of a C1–10 n-alkylbenzene and a growth-supporting n-alkane. All of these monoalkylbenzenes were found to be activated, but exclusively the succinate derivatives of toluene and ethylbenzene were detected in cultures grown anaerobically with crude oil, too. By comparison with standards they were identified as benzylsuccinate and the two diastereoisomers of (1-phenylethyl)succinate, respectively, confirming the results of Rabus et al. (2011) for strain HxN1. In case of n-propylbenzene strain HxN1 forms 3 activation products. The two metabolites eluting later share the same mass spectrometric fragmentation pattern, i.e., base peak at m/z 119 while the one eluting first has a base peak at m/z 91. These products were tentatively assigned as the two diastereoisomers of (1-methyl-2-phenylethyl)succinate and (1-phenylpropyl)succinate, respectively. Furthermore, the activation products of n-butyl- and n-pentyl-benzene from all three n-alkane utilizers were identified by comparison with synthetic standards as (1-methyl-3-phenylpropyl)- and (1-methyl-4-phenylbutyl)succinate, respectively (Supplementary Figure S1). The mass spectra of the dimethyl esters of these succinates display m/z 91 and [M-64] as most intense signals of typically similar height. In comparison, the methylated (1-phenylalkyl)succinates (potential benzyl position activation products) show a clear predominating base peak at m/z 91 (Supplementary Figure S1) and elute significantly earlier. The respective main activation products of hexyl- to decyl-benzene were tentatively identified as (1-methyl-omega-phenylalkyl)succinates based on their relative retention times and homologous fragmentation patterns. In addition to these activation products a second series was detected in extracts from cultures containing pentyl- to decyl-benzene, which always elute shortly before the main products. In analogy to the activation of n-alkanes and taking into account their homologous mass spectrometric fragmentation patterns these were tentatively assigned as the (1-ethyl-omega-phenylalkyl)succinates.
It is noteworthy that presumptive Mas-like enzymes apparently activate the methyl or ethyl group in alkyltoluenes and alkylethylbenzenes. The resulting alkyl-substituted benzyl- or (1-phenylethyl)succinates (as dimethyl esters) show distinct mass spectrometric fragmentation patterns. While m/z [M-60] is the base peak of the former, the latter exhibit m/z [M-145] as the base peak. Moreover, due to the formation of an additional stereogenic center, the products from alkylethylbenzenes always occur as two chromatographically separable diastereoisomers. Taking also into account the above mentioned preferential accumulation of p-dialkylbenzene activation products by n-alkane-utilizing strains we assign the two homologous series of arylsuccinates formed during anaerobic growth with crude oil as (p-n-alkylbenzyl)succinates (Figure 1B, green) and [1-(p-n-alkylphenyl)ethyl]succinates (Figure 1B, blue). This is corroborated by results obtained from experiments with binary mixtures of selected dialkylbenzenes and a growth-supporting n-alkane (Table 1). Particularly, (p-isopropylbenzyl)succinate the activation product of p-cymene was identified by standard comparison. It thus is reasonable to assume that [1-(p-isopropylphenyl)ethyl]succinate is formed from p-ethylisopropylbenzene (Figure 1B, orange). For p-cymene no activation at the isopropyl group was observed.
We suppose that the same is true for activation of o- and m-dialkylbenzenes, as succinate derivatives formed from them show mass spectrometric fragmentation patterns (as dimethyl esters) very similar to those of the respective para-compounds. However, in case of ortho-substituted derivatives additional peaks at m/z [M-18] and [M-64] were observed, probably due to an ortho-effect (Schwarz, 1978). This kind of ortho-effect can be used to distinguish the three succinates derived from 1,2,4-trimethylbenzene (Supplementary Figure S2). Among the alkylbenzene utilizers, strain mXyS1 forms all three products, whereas strain oXyS1 produces only the second, and n-alkane-utilizing strain TD3, exclusively the third eluting one. The latter (as dimethyl ester) in comparison to the others does not exhibit fragments m/z 200 [M-64] and 246 [M-18] in its mass spectrum. Thus, we assign the last eluting isomer as (3,4-dimethylbenzyl)succinate containing no substituent in ortho-position to the activated carbon atom. The mass spectra of the other two isomers are rather similar; however, their GC retention times differ significantly. In all other cases of arylalkylsuccinates (as dimethyl esters) the para-isomer elutes at a later retention time than the meta-isomer. It is therefore very probable, that the earlier eluting one is (2,5-dimethylbenzyl)succinate and the second is (2,4-dimethylbenzyl)succinate.
Stereochemistry of alkyl-/arylalkyl-succinates
The alkyl-/arylalkyl-succinates produced contain at least one stereocenter. For chromatographic separation of the stereoisomers the succinates were transformed to succinimides by reaction with (R)-1-phenylethanamine (Figure 3) using a modification of the method described (Jarling et al., 2012). The scope of this derivatisation was evaluated by applying it to a broad range of diacids, including succinic acids with polar and nonpolar substituents as well as malonic and glutaric acids (Supplementary Table S4). Separation efficiency was determined for two achiral GC columns of different polarity. All tested succinic acids with alkyl and (alkyl)benzyl substituents could be separated on both columns under appropriate GC conditions. On the other hand, butanedioic acids with hydroxyl or amino groups (e.g., malic or aspartic acid), as well as malonic or glutaric acids, either gave no imide or the succinimide formed was not separable. Unfortunately, employment of too basic conditions leads to formation of by-products from aryl- and arylalkyl-succinates which might influence the determination of diastereoisomer ratios. The optimized dervatization was then applied to the extracts from the cultures with crude oil, which were analyzed by GC-MS. The succinimides show very characteristic fragmentation patterns allowing assessment of the stereochemistry even in complex mixtures (Supplementary Figure S3, Table S5).
Figure 3.
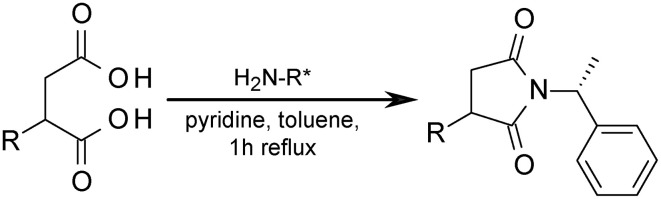
Formation of diastereoisomeric succinimides from succinate derivatives for stereochemical investigation. H2N-R* = (R)-1-phenylethanamine.
All investigated alkylbenzene-utilizing strains formed exclusively (R)-2-benzylsuccinate from toluene, which was confirmed by comparison with a standard. All other succinate derivatives detected in these cultures consisted of exclusively one stereoisomer. As in all cases the later eluting isomer was found, we propose that always the (R)-stereoisomer of the succinate is formed (Figure 4). In contrast, the alkyl-/arylalkyl-succinates formed by n-alkane utilizers were generally present as two stereoisomers (Figure 4). In case of n-hexane-derived (1-methylpentyl)succinate, strains HxN1, OcN1, and TD3 formed the (2R,1′R)- as well as the (2S,1′R)-isomer, as confirmed by standard comparison. These are the first and the last eluting of the four possible isomers (Jarling et al., 2012). Likewise, in the cases of (1-phenylethyl)succinate (ethylbenzene-derived) and (1-methyl-4-phenylbutyl)succinate (n-pentylbenzene-derived), only the first and last eluting isomers were formed. From equidistant GC retention times we infer that the same is true for all other succinate derivatives with two stereocenters and suggest that they were also present as the epimers at carbon atom 2. Succinate derivatives with only a single stereocenter located in the succinate moiety were always found as mixtures of both enantiomers.
Figure 4.
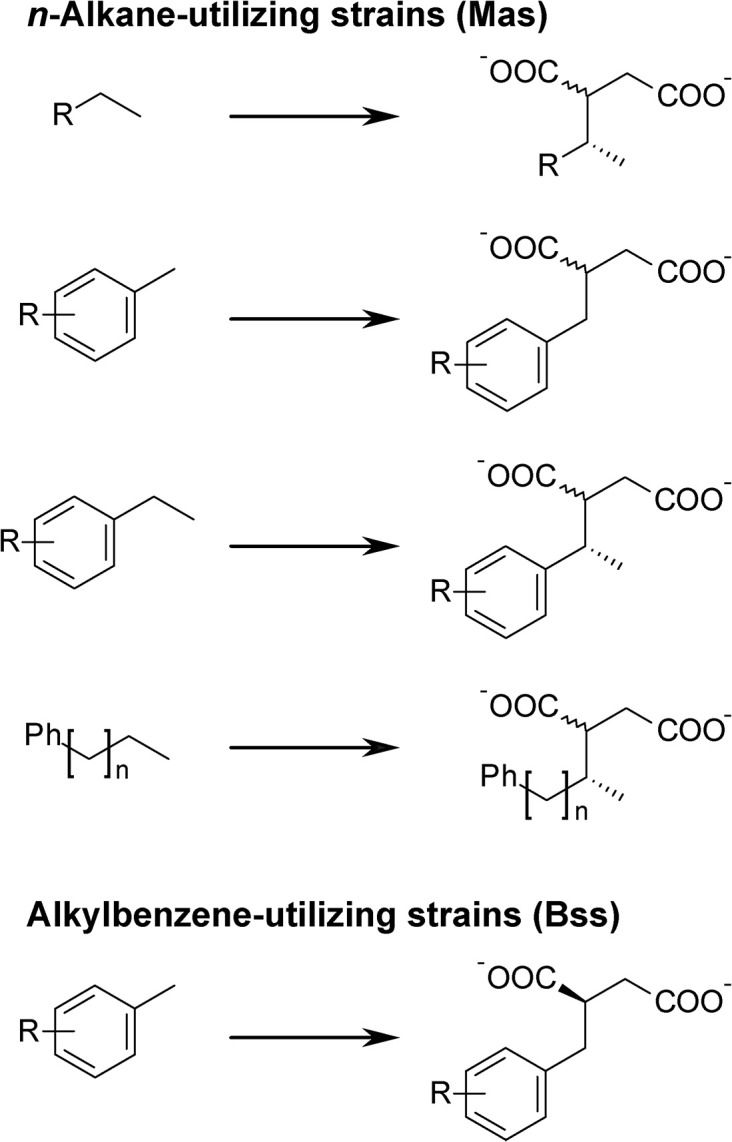
Stereochemistry of alkyl-/arylalkyl-succinates formed by different strains. Mas, methylalkylsuccinate synthase; Bss, benzylsuccinate synthase; R = H, alkyl; n = 1…8.
Further potential activation products from alkylbenzenes
Unexpectedly, a number of strains produced the corresponding benzyl alcohols and/or 1-phenylalkan-1-ols during anaerobic growth in the presence of certain alkylbenzenes. Furthermore, several strains formed benzaldehydes or 1-phenylalkanones as well as benzoates representing the expected oxidation products of the alcohols (Figure 5, Table 3, Supplementary Table S9). These transformation processes were observed in n-alkane- and alkylbenzene-utilizing bacteria and, more importantly, not only in denitrifying but also in sulfate-reducing bacteria. For n-alkane-utilizing strains in general, hydroxy- and oxo-derivatives occurred in comparably higher relative abundances in extracts of cultures with ortho-di-, tri-, tetra-, penta-, and hexa-alkylbenzenes in comparison with mono- and other dialkylbenzenes. Notably, these appear to be alkylbenzenes that are not or only poorly transformed to succinate derivatives. However, in certain cases succinate derivatives and hydroxylation products originating from the same alkylbenzene were present in the extracts. Respective benzyl alcohols and benzaldehydes were not detected for alkylated toluenes carrying exclusively meta-substituents, i.e., m-xylene, m-ethyltoluene, and 1,3,5-trimethylbenzene. The only exception is tentatively identified m-isopropylbenzyl alcohol formed from m-cymene in strain TD3.
Figure 5.
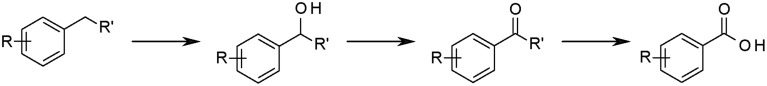
Anaerobic hydroxylation of aromatic hydrocarbons and further transformations. R = H, alkyl; R′ = H, methyl, ethyl, propyl.
Table 3.
Anaerobic transformation of hydrocarbons by selected bacteria via succinate formation or hydroxylation.a
| Hydrocarbon | Strain | |||||||
|---|---|---|---|---|---|---|---|---|
| Electron acceptor | ||||||||
| Substrate activationb | ||||||||
| EbN1 | mXyS1 | OcN1 | TD3 | |||||
| AS | H | AS | H | AS | H | AS | H | |
| Toluene | + | + | + | − | + | + | + | + |
| Ethylbenzene | − | + | nt | + | + | + | + | |
| Propylbenzene | nt | nt | − | + | − | + | ||
| Butylbenzene | nt | nt | + | + | + | − | ||
| o-Xylene | + | + | + | − | + | + | + | + |
| o-Ethyltoluene | + | + | nt | − | + | − | + | |
| o-Isopropyltoluene | nt | nt | − | + | − | + | ||
| m-Xylene | + | − | + | − | + | − | + | − |
| m-Ethyltoluene | − | + | + | − | + | + | + | + |
| m-Isopropyltoluene | nt | nt | − | − | + | + | ||
| p-Xylene | − | + | + | + | + | + | + | + |
| p-Ethyltoluene | − | + | nt | + | + | + | + | |
| p-Isopropylbenzene | nt | nt | + | − | + | − | ||
| p-tert-Butylbenzene | nt | nt | + | − | + | − | ||
| 1,2,3-Trimethylbenzene | nt | nt | − | + | − | + | ||
| 1,2,4-Trimethylbenzene | nt | + | + | − | + | + | + | |
| 1,3,5-Trimethylbenzene | nt | + | − | − | − | − | − | |
| 1,2,3,5-Tetramethylbenzene | nt | nt | − | + | − | + | ||
| 1,2,4,5-Tetramethylbenzene | nt | nt | − | + | − | + | ||
| Pentamethylbenzene | nt | nt | − | + | − | + | ||
| Hexamethylbenzene | nt | nt | − | + | − | + | ||
| Hydroxylation | Both activations | Succinate formation | ||||||
Transformation and further oxidation of the respective hydrocarbon during anaerobic growth of the used bacteria in binary substrate mixture experiments (+, detected; −, not detected; nt, not tested), as revealed by identification of the respective metabolites. For detailed characterization of product formation see Supplementary Table S9.
Discussion
Hydrocarbon range, regiochemistry, and stereochemistry
The experiments with crude oil as a complex mixture of hydrocarbons are particularly useful to examine possible preferences for certain hydrocarbon substrates, although it has to be considered that the relative concentrations of metabolites are controlled both by their generation as well as further transformation rates. In case of non-growth supporting co-substrates which are not completely oxidized, the suite of different metabolites formed from each co-substrate represents the actual susceptibility to the enzymes involved. Thus, presumptive Mas-like enzymes seem to prefer as substrates cyclopentane over cyclohexane derivatives, normal over branched alkanes, and para- over ortho- and meta-substituted alkylbenzenes. In contrast, presumptive Bss-like enzymes are restricted to activation of benzylic methyl groups and seem to prefer substrates with low degree of substitution in the aromatic ring. Moreover, all tested alkylbenzene-utilizing strains can activate toluene and o-xylene, but further substrates of the respective Bss-like enzymes are specific for each strain (e.g., presumptive Bss from strain oXyS1 requires ortho-substituted alkylbenzenes). As these restrictions are likely to be the result of structural differences in the Bss-like enzymes involved, i.e., their amino acid sequences, a phylogenetic analysis would be expected to plot together those enzymes with similar (co-)activation pattern. Indeed, this is the case for BssAs from strains K172 and EbN1, having an identical co-substrate range (Table 2) and giving a distinct branch in the phylogenetic analysis (Strijkstra et al., 2014). In comparison, BssAs from strains T and Tol2, which differ in their (co-)substrate ranges from each other and the former, plot in different, more distant branches. Unfortunately, only very few Bss amino acid sequences are known preventing a detailed comparison of their structure-activity relationship.
In the case of n-alkane-utilizing strains the experiments with branched alkanes provide evidence for the necessity of a terminal n-propyl group in acyclic aliphatic substrates to enable successful addition to fumarate. In cyclopentane derivatives a ring-CH2 group may also act as reactive position, if it is not sterically hindered by neighboring substituents. Rios-Hernandez et al. (2003) provided evidence that carbon atom 3 of ethylcyclopentane is added to fumarate in a sulfate-reducing enrichment culture. Even though the steric constraints at benzylic positions are higher, presumptive Mas-like enzymes seem to be able to activate these positions in toluene, ethylbenzene and their derivatives due to the lower C-H-bond dissociation energy of the benzylic methylene group. In n-alkylbenzenes with side chain lengths greater than three, exclusively omega-2 and to a lesser extent omega-3 activation products are formed. Obviously, these substrates are recognized as substituted n-alkanes by Mas-like enzymes thus exhibiting the same incomplete regioselectivity as observed for n-alkanes (see above). The steric hindrance at the benzylic positions prevents activation at that site, despite the lesser activation energy needed. Steric hindrance might also be the reason why no omega-3 activation by-product of butylbenzene was detected. Propylbenzene is an intermediate case as here the benzylic methylene group is also the omega-3-position; therefore, both activation products were found.
In general the findings from the stereochemical investigations lead to the assumption, that alkyl- as well as arylalkylsuccinates initially formed by n-alkane-utilizing strains were further converted, because epimerization should follow CoA-thioester formation. This is further corroborated by the identification of potential downstream products of these succinate derivatives in the respective cultures (see the Supplementary Tables S7, S8 for selected cases).
n-alkylsuccinates in strain TD3 and the possibility of alkanogenesis
The distribution patterns of n-alkylsuccinates formed by strain TD3 (i.e., the predominance of C-odd or C-even n-alkylsuccinates upon growth with C-even or C-odd n-alkanes; Figure 2A, Supplementary Table S6) cannot be explained by assuming a terminal activation of n-alkanes as a side-reaction. We rather propose that their formation may be linked to relaxed specificity of the enzymes involved in regeneration of fumarate. Wilkes et al. (2002) had suggested that during anaerobic degradation of n-hexane in strain HxN1 propionyl-CoA formed during β-oxidation is used to regenerate fumarate via transcarboxylation and intramolecular rearrangement. The degradation of n-alkanes leads to n-alkanoyl-CoAs, e.g., octanoyl-, hexanoyl-, and butanoyl-CoA are intermediates of n-decane degradation. If these n-alkanoyl-CoAs instead of propionyl-CoA are transformed by the fumarate-regenerating enzymes, the corresponding n-alkylsuccinates will be formed with the observed predominance of odd or even side chains depending on the chain length of the n-alkane provided as growth substrate (Figure 2B).
We assume that these metabolites do not reflect “erroneous” side reactions but rather occur in the course of processes that have a specific physiological function. Thus, n-alkylsuccinates formed from alkanoyl-CoAs other than propionyl-CoA may serve for alternative fumarate regeneration by reverse action of Mas, which at the same time would result in alkanogenesis. Especially, in case of methylsuccinate (formed from n-butanoyl-CoA) methane would be produced. It has been calculated that the overall formation of methylsuccinate from methane and fumarate by Mas is exothermic by 9–11 kcal/mol at room temperature (Beasley and Nanny, 2012). However, we consider that the reverse reaction may take place, if certain thermodynamic conditions favor it: (i) elevated temperature, (ii) very low intracellular concentrations of fumarate, and (iii) removal of methane from the chemical equilibrium out of the cell. Thermophilic strain TD3 compared to mesophilic strains HxN1 and OcN1 (Ehrenreich et al., 2000) grows well at 60°C (Rueter et al., 1994) and solvent stress may lead to fumarate deficiency (see chapter below). It was already shown, that Bss is reversible (Li and Marsh, 2006). Therefore, reversibility of Mas is very likely as well, possibly helping the bacterium to sustain certain combined temperature and solvent stress conditions by avoiding the involvement of the ethylmalonyl-CoA pathway (Erb et al., 2007; Thauer and Shima, 2008) and thus short-cutting the supply of fumarate. Finally, such a pathway may even contribute to biogenic formation of ethane and propane (Hinrichs et al., 2006; Xie et al., 2013) in sediments and possibly also in oil reservoirs.
Further potential activation products from alkylbenzenes
Based on our observations mentioned above we propose that the respective bacteria listed in Table 3 and Supplementary Table S9 (Supplementary Material) possess enzymes that enable direct oxidation of alkylbenzenes to alcohols. Such activation reactions have previously been reported for anaerobic degradation of ethylbenzene by strain EbN1, propylbenzene by strain PbN1 and p-ethyltoluene and p-cymene by strain pCyN1 (Rabus and Widdel, 1995; Ball et al., 1996; Johnson and Spormann, 1999; Spormann and Widdel, 2000; Strijkstra et al., 2014). Available evidence suggests that these reactions are catalyzed by dehydrogenases employing molybdenum-containing cofactors and proceed via benzylic carbenium ions (Kniemeyer and Heider, 2001a,b; Kloer et al., 2006; Knack et al., 2012). Remarkably, in the case of p-ethyltoluene and p-cymene activation in strain pCyN1 benzyl alcohols are formed by activation of the methyl groups, which is energetically favorable for p- and putatively o-substituted dialkylbenzenes due to the hyperconjugative effects of the alkyl substituents stabilizing the intermediary carbenium ion (Strijkstra et al., 2014). This might explain why oxidation products of meta-substituted alkylbenzenes were generally not detectable. Interestingly, while such oxidation reactions have so far exclusively been reported from denitrifiers, we here provide evidence that they occur in sulfate-reducing bacteria, too.
Formation of alkyl-/arylalkyl-succinates and their further transformation as a potential mechanism of hydrocarbon detoxification
Compared with alkylbenzene utilizers, the range of hydrocarbons activated by n-alkane utilizers by far exceeds the range of the growth substrates, including the activation of alkylbenzenes, which apparently do not promote growth. In addition, these products of regioselective substrate activation are metabolically further transformed, indicating promiscuity along the downstream degradation steps as well. Webner (2012) showed that a wide range of hydrocarbons, including n-alkanes, cyclic alkanes, and alkylbenzenes, induce expression of the mas gene in strain HxN1 in vivo, and suggested that this may reflect an expanded functionality of this enzyme.
In agreement with this we propose that certain bacteria may benefit from these broad range transformations during detoxification of hydrocarbons. The strong toxic effects of hydrocarbons and other nonpolar solvents on microorganisms and the mechanisms involved in solvent tolerance of bacteria have been investigated intensively (for further information see Sikkema et al., 1995; Kobayashi et al., 2000; Trautwein et al., 2008; Zink and Rabus, 2010; Stancu and Grifoll, 2011; Baumgarten et al., 2012; Segura et al., 2012; Alvarez-Ortega et al., 2013).
We suggest that the alkyl-/arylalkyl-succinate synthase-catalyzed transformation of hydrocarbons to diacids, which under physiological conditions are unable to penetrate the membrane due to their two negative charges, immediately reduces their toxic effects. The further metabolism of these succinates, if formed from substrates not supporting growth may be required for carbon and energy recycling to compensate for the fumarate molecule consumed during the activation. The products of these transformations are monoacids, e.g., benzoate from toluene (Rabus et al., 2011), which may accumulate at the surface of the inner membrane from where they may be extruded. This may explain the typical very low alkyl-/arylalkyl-succinate concentrations in natural environments, in which anaerobic biodegradation of hydrocarbons occurs. Alternatively, if the Mas-mediated reaction is reversible, the intracellular alkyl-/arylalkyl-succinates may be decomposed again, after surrounding concentrations of hydrocarbons have lowered due to external influences. In this case formation of alkyl-/arylalkyl-succinates would work as a fast responding system buffering high intracellular hydrocarbon concentrations and releasing the required fumarate later. If under certain solvent stress conditions intracellular fumarate concentrations are abrogated and potential alternative pathways for regeneration of fumarate are already exploited, the bacteria will still be able to lower their intracellular hydrocarbon concentrations by utilization of the proposed, obviously fumarate-independent oxidation of alkyltoluenes to benzoic acids.
Indeed, a continuous co-transformation of toxic hydrocarbons to their respective succinates and further to monoacids would be expected to result in a substantial accumulation of these compounds in the culture broth which was not observed within the timescales of the cultivation experiments performed in this study. The occurrence of aromatic monoacids in waters from natural hydrocarbon-bearing environments has been reported (e.g., Martus and Püttmann, 2003; Barman Skaare et al., 2007). Here, a massive accumulation might be prevented by the activity of other microorganisms able to further degrade these metabolites and/or under suitable hydrological conditions by water transport processes removing potential dead-end products from the sampling locations. Close to an oil-water contact these amphiphilic acids may also contribute to emulsification of the hydrophobic phase, thus improving the accessibility of the substrates.
Conclusions
In this study we demonstrate a significantly higher metabolic versatility of n-alkane-utilizing anaerobic bacteria with respect to the range of transformable hydrocarbons as compared to known alkylbenzene utilizers. It is thus very likely that an even broader range of nonpolar compounds, which still has to be explored, may represent potential substrates of (methylalkyl)succinate synthases. The requirement for detoxification of hydrocarbons that compromise membranes and do not support growth may offer an explanation for this promiscuity. Conceptually, this would aid especially n-alkane-utilizing anaerobic bacteria to survive in environments with relatively high concentrations of complex hydrocarbon mixtures, e.g., oil contaminated aquifers or petroleum reservoirs. The preferential early degradation of n-alkanes in oil reservoirs may thus be explained by the more pronounced solvent resistance of n-alkane-utilizing anaerobic bacteria which may enable them to survive even in close vicinity to the oil water-contact where their growth substrates are sufficiently bioavailable. Alkylbenzene-utilizing bacteria on the other hand would benefit from the lower oil-water-partition coefficients of their growth substrates (Harms et al., 2010) allowing them to thrive at a greater distance from the oil-water contact, i.e., at lower solvent concentration. The detection of the metabolites identified in this study in natural habitats may be useful for tracing the underlying biochemical processes. However, according to our present results the occurrence of methylsuccinate, particularly if accompanied by other n-alkylsuccinates (Gieg et al., 2010; Agrawal and Gieg, 2013), would not necessarily represent an indication of addition of methane to fumarate. Consistently, detection of arylalkylsuccinates together with their respective downstream metabolites (e.g., alkylbenzoic acids) does not necessarily confirm the presence of alkylbenzene-degrading bacteria in the investigated environment (see also Rabus et al., 2011). We point out that the assessment of succinate stereochemistry will help to distinguish the groups of bacteria involved. Furthermore, it may be envisaged that transformation products extruded for the purpose of detoxification may serve as substrates for other members of the microbial community. If this were the case, n-alkane-utilizing bacteria may have an elevated role in the subsurface carbon cycle by providing access to inert carbon sources to other kinds of bacteria. In the subsurface microbial community they may take over the part of pioneer organisms by reworking hydrocarbon-infested areas and make them accessible for other community members. Finally, the enormous overall versatility of anaerobic bacteria initiating hydrocarbon metabolism via C-H-activation as documented in this study should prompt further investigations on detailed reaction mechanisms, which in the long term may even be relevant with respect to the sustainable use of hydrocarbon feedstocks for the chemical industry.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Kristin Günther, Anke Kaminski, Conny Karger, Anne Köhler, Hardy Weißhoff, and Anika Zabel for technical assistance. We are indebted to Fritz Widdel for his trailblazing contributions to the field of anaerobic degradation of hydrocarbons and his constant support. This project was funded by the Deutsche Forschungsgemeinschaft (program SPP 1319; HW, RR, and BG), COST Action CM0603 (BG), the University of Oldenburg (RR), and the Helmholtz Gemeinschaft (HW).
Supplementary material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2015.00880
References
- Aeckersberg F., Bak F., Widdel F. (1991). Anaerobic oxidation of saturated hydrocarbons to CO2 by a new type of sulfate-reducing bacterium. Arch. Microbiol. 156, 5–14. 10.1007/BF00418180 [DOI] [Google Scholar]
- Agrawal A., Gieg L. M. (2013). In situ detection of anaerobic alkane metabolites in subsurface environments. Front. Microbiol. 4:140. 10.3389/fmicb.2013.00140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Ortega C., Olivares J., Martínez J. L. (2013). RND multidrug efflux pumps: what are they good for? Front.Microbiol. 4:7. 10.3389/fmicb.2013.00007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alworth W. L. (1972). Stereochemistry and its Application in Biochemistry: The Relation between Substrate Symmetry and Biological Stereospecificity. New York, NY:Wiley-Interscience. [Google Scholar]
- Anders H. J., Kaetzke A., Kämpfer P., Ludwig W., Fuchs G. (1995). Taxonomic position of aromatic-degrading denitrifying pseudomonad strains K 172 and KB 740 and their description as new members of the genera Thauera, as Thauera aromatica sp. nov., and Azoarcus, as Azoarcus evansii sp. nov., respectively, members of the beta subclass of the Proteobacteria. Int. J. Syst. Bacteriol. 45, 327–333. 10.1099/00207713-45-2-327 [DOI] [PubMed] [Google Scholar]
- Ball H. A., Johnson H. A., Reinhard M., Spormann A. M. (1996). Initial reactions in anaerobic ethylbenzene oxidation by a denitrifying bacterium, strain EB1. J. Bacteriol. 178, 5755–5761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barman Skaare B., Wilkes H., Vieth A., Rein E., Barth T. (2007). Alteration of crude oils from the Troll area by biodegradation: analysis of oil and water samples. Org. Geochem. 38, 1865–1883. 10.1016/j.orggeochem.2007.07.007 [DOI] [Google Scholar]
- Baumgarten T., Sperling S., Seifert J., von Bergen M., Steiniger F., Wick L. Y., et al. (2012). Membrane vesicle formation as a multiple-stress response mechanism enhances Pseudomonas putida DOT-T1E cell surface hydrophobicity and biofilm formation. Appl. Environ. Microbiol. 78, 6217–6224. 10.1128/AEM.01525-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beasley K. K., Nanny M. A. (2012). Potential energy surface for anaerobic oxidation of methane via fumarate addition. Environ. Sci. Technol. 46, 8244–8252. 10.1021/es3009503 [DOI] [PubMed] [Google Scholar]
- Beller H. R., Spormann A. M. (1997). Anaerobic activation of toluene and o-Xylene by addition to fumarate in denitrifying strain T. J. Bacteriol. 179, 670–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beller H. R., Spormann A. M. (1998). Analysis of the novel benzylsuccinate synthase reaction for anaerobic toluene activation based on structural studies of the product. J. Bacteriol. 180, 5454–5457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bian X.-Y., Mbadinga S. M., Liu Y.-F., Yang S.-Z., Liu J.-F., Ye R.-Q., et al. (2015). Insights into the anaerobic biodegradation pathway of n-alkanes in oil reservoirs by detection of signature metabolites. Sci. Rep. 5:9801. 10.1038/srep09801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biegert T., Fuchs G., Heider J. (1996). Evidence that anaerobic oxidation of toluene in the denitrifying bacterium Thauera aromatica is initiated by formation of benzylsuccinate from toluene and fumarate. Eur. J. Biochem. 238, 661–668. 10.1111/j.1432-1033.1996.0661w.x [DOI] [PubMed] [Google Scholar]
- Callaghan A. V. (2013). Metabolomic investigations of anaerobic hydrocarbon-impacted environments. Curr. Opin. Biotechnol. 24, 506–515. 10.1016/j.copbio.2012.08.012 [DOI] [PubMed] [Google Scholar]
- Concellon J. M., Rodriguez-Solla H., Concellon C. (2004). Deuteration of α,β-acetylenic esters, amides, or carboxylic acids without using deuterium gas: synthesis of 2,2,3,3-tetradeuterioesters, amides, or acids. Tetrahedron Lett. 45, 2129–2131. 10.1016/j.tetlet.2004.01.05126325618 [DOI] [Google Scholar]
- Dahlgren S., Hanesand T., Mills N., Patience R., Brekke T., Sinding-Larsen R. (1998). Norwegian Geochemical Standard samples: North Sea Oil-1 (NGS NSO-1). Norwegian Geochemical Standards Newsletter, Vol. 3. Norwegian Petroleum Directorate: Stavanger.
- Dolfing J., Zeyer J., Binder-Eicher P., Schwarzenbach R. P. (1990). Isolation and characterization of a bacterium that mineralizes toluene in the absence of molecular oxygen. Arch. Microbiol. 154, 336–341. 10.1007/BF00276528 [DOI] [PubMed] [Google Scholar]
- Duncan K. E., Gieg L. M., Parisi V. A., Tanner R. S., Tringe S. G., Bristow J., et al. (2009). Biocorrosive thermophilic microbial communities in Alaskan North Slope oil facilities. Environ. Sci. Technol. 43, 7977–7084. 10.1021/es9013932 [DOI] [PubMed] [Google Scholar]
- Ehrenreich P., Behrends A., Harder J., Widdel F. (2000). Anaerobic oxidation of alkanes by newly isolated denitrifying bacteria. Arch. Microbiol. 173, 58–64. 10.1007/s002030050008 [DOI] [PubMed] [Google Scholar]
- Erb T. J., Berg I. A., Brecht V., Müller M., Fuchs G., Alber B. E. (2007). Synthesis of C5-dicarboxylic acids from C2-units involving crotonyl-CoA carboxylase/reductase: The ethylmalonyl-CoA pathway. Proc. Natl. Acad. Sci. U.S.A. 104, 10631–10636. 10.1073/pnas.0702791104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gieg L. M., Davidova I. A., Duncan K. E., Suflita J. M. (2010). Methanogenesis, sulfate reduction and crude oil biodegradation in hot Alaskan oilfields. Environ. Microbiol. 12, 3074–3086. 10.1111/j.1462-2920.2010.02282.x [DOI] [PubMed] [Google Scholar]
- Gilewicz M., Monpert G., Acquaviva M., Mille G., Bertrand J.-C. (1991). Anaerobic oxidation of 1-heptadecene by a marine denitrifying bacterium. Appl. Microbiol. Biotechnol. 36, 252–256. 10.1007/BF00164430 [DOI] [Google Scholar]
- Grundmann O., Behrends A., Rabus R., Amann J., Halder T., Heider J., et al. (2008). Genes encoding the candidate enzyme for anaerobic activation of n-alkanes in the denitrifying bacterium, strain HxN1. Environ. Microbiol. 10, 376–385. 10.1111/j.1462-2920.2007.01458.x [DOI] [PubMed] [Google Scholar]
- Harms G., Zengler K., Rabus R., Aeckersberg F., Minz D., Rosselló-Mora R., et al. (1999). Anaerobic oxidation of o-xylene, m-xylene, and homologous alkylbenzenes by new types of sulfate-reducing bacteria. Appl. Environ. Microbiol. 65, 999–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harms H., Smith K. E. C., Wick L. Y. (2010). Introduction: problems of hydrophobicity/bioavailability, in Handbook of Hydrocarbon and Lipid Microbiology, ed Timmis K. N. (Heidelberg: Springer; ), 1439–1450. [Google Scholar]
- Hinrichs K.-U., Hayes J. M., Bach W., Spivack A. J., Hmelo L. R., Holm N. G., et al. (2006). Biological formation of ethane and propane in the deep marine subsurface. Proc. Natl. Acad. Sci. U.S.A. 103, 14684–14689. 10.1073/pnas.0606535103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarling R., Sadeghi M., Drozdowska M., Lahme S., Buckel W., Rabus R., et al. (2012). Stereochemical investigations reveal the mechanism of the bacterial activation of n-Alkanes without oxygen. Angew. Chem. Int. Ed. 51, 1334–1338. 10.1002/anie.201106055 [DOI] [PubMed] [Google Scholar]
- Johnson H. A., Spormann A. M. (1999). In Vitro studies on the initial reactions of anaerobic ethylbenzene mineralization. J. Bacteriol. 181, 5662–5668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloer D. P., Hagel C., Heider J., Schulz G. E. (2006). Crystal structure of ethylbenzene dehydrogenase from Aromatoleum aromaticum. Structure 14, 1377–1388. 10.1016/j.str.2006.07.001 [DOI] [PubMed] [Google Scholar]
- Knack D., Hagel C., Szaleniec M., Dudzik A., Salwinski A., Heider J. (2012). Substrate and inhibitor spectra of ethylbenzene dehydrogenase: perspectives on application potential and catalytic mechanism. Appl. Environ. Microbiol. 78, 6475–6482. 10.1128/AEM.01551-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kniemeyer O., Fischer T., Wilkes H., Glöckner F. O., Widdel F. (2003). Anaerobic degradation of ethylbenzene by a new type of marine sulfate-reducing bacterium. Appl. Environ. Microbiol. 69, 760–768. 10.1128/AEM.69.2.760-768.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kniemeyer O., Heider J. (2001a). Ethylbenzene dehydrogenase, a novel hydrocarbon-oxidizing molybdenum/iron-sulfur/heme enzyme. J. Biol. Chem. 276, 21381–21386. 10.1074/jbc.M101679200 [DOI] [PubMed] [Google Scholar]
- Kniemeyer O., Heider J. (2001b). (S)-1-Phenylethanol dehydrogenase of Azoarcus sp. strain EbN1, an enzyme of anaerobic ethylbenzene catabolism. Arch. Microbiol. 176, 129–135. 10.1007/s002030100303 [DOI] [PubMed] [Google Scholar]
- Kniemeyer O., Musat F., Sievert S. M., Knittel K., Wilkes H., Blumenberg M., et al. (2007). Anaerobic oxidation of short-chain hydrocarbons by marine sulphate-reducing bacteria. Nature 449, 898–901. 10.1038/nature06200 [DOI] [PubMed] [Google Scholar]
- Kobayashi H., Uematsu K., Hirayama H., Horikoshi K. (2000). Novel toluene elimination system in a toluene-tolerant microorganism. J. Bacteriol. 182, 6451–6455. 10.1128/JB.182.22.6451-6455.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieger C. J., Beller H. R., Reinhard M., Spormann A. M. (1999). Initial reactions in anaerobic oxidation of m-Xylene by the denitrifying bacterium Azoarcus sp. strain T. J. Bacteriol. 181, 6403–6410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuthner B., Leutwein C., Schulz H., Hörth P., Haehnel W., Schiltz E., et al. (1998). Biochemical and genetic characterization of benzylsuccinate synthase from Thauera aromatica: a new glycyl radical enzyme catalysing the first step in anaerobic toluene metabolism. Mol. Microbiol. 28, 615–628. 10.1046/j.1365-2958.1998.00826.x [DOI] [PubMed] [Google Scholar]
- Leutwein C., Heider J. (1999). Anaerobic toluene-catabolic pathway in denitrifying Thauera aromatica: activation and β-oxidation of the first intermediate, (R)-(+)-benzylsuccinate. Microbiology 145, 3265–3271. [DOI] [PubMed] [Google Scholar]
- Li L., Marsh E. N. G. (2006). Mechanism of benzylsuccinate synthase probed by substrate and isotope exchange. J. Am. Chem. Soc. 128, 16056–16057. 10.1021/ja067329q [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovley D. R., Baedecker M. J., Lonergan D. J., Cozzarelli I. M., Phillips E. J. P., Siegel D. I. (1989). Oxidation of aromatic contaminants coupled to microbial iron reduction. Nature 339, 297–300. 10.1038/339297a018075087 [DOI] [Google Scholar]
- Martus P., Püttmann W. (2003). Formation of alkylated aromatic acids in groundwater by anaerobic degradation of alkylbenzenes. Sci. Total. Environ. 307, 19–33. 10.1016/S0048-9697(02)00512-0 [DOI] [PubMed] [Google Scholar]
- Müller J. A., Galushko A. S., Kappler A., Schink B. (1999). Anaerobic degradation of m-cresol by Desulfobacterium cetonicum is initiated by formation of 3-hydroxybenzylsuccinate. Arch. Microbiol. 172, 287–294. 10.1007/s002030050782 [DOI] [PubMed] [Google Scholar]
- Rabus R. (2005). Functional genomics of an anaerobic aromatic-degrading denitrifying bacterium, strain EbN1. Appl. Microbiol. Biotechnol. 68, 580–587. 10.1007/s00253-005-0030-x [DOI] [PubMed] [Google Scholar]
- Rabus R., Heider J. (1998). Initial reactions of anaerobic metabolism of alkylbenzenes in denitrifying and sulfate-reducing bacteria. Arch. Microbiol. 170, 377–384. 10.1007/s002030050656 [DOI] [Google Scholar]
- Rabus R., Jarling R., Lahme S., Kühner S., Heider J., Widdel F., et al. (2011). Co-metabolic conversion of toluene in anaerobic n-alkane-degrading bacteria. Environ. Microbiol. 13, 2576–2586. 10.1111/j.1462-2920.2011.02529.x [DOI] [PubMed] [Google Scholar]
- Rabus R., Nordhaus R., Ludwig W., Widdel F. (1993). Complete oxidation of toluene under strictly anoxic conditions by a new sulfate-reducing bacterium. Appl. Environ. Microbiol. 59, 1444–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabus R., Widdel F. (1995). Anaerobic degradation of ethylbenzene and other aromatic hydrocarbons by new denitrifying bacteria. Arch. Microbiol. 163, 96–103. 10.1007/BF00381782 [DOI] [PubMed] [Google Scholar]
- Rabus R., Widdel F. (1996). Utilization of alkylbenzenes during anaerobic growth of pure cultures of denitrifying bacteria on crude oil. Appl. Environ. Microbiol. 62, 1238–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabus R., Wilkes H., Behrends A., Armstroff A., Fischer T., Pierik A. J., et al. (2001). Anaerobic initial reaction of n-Alkanes in a denitrifying bacterium: evidence for (1-Methylpentyl)succinate as initial product and for involvement of an organic radical in n-Hexane metabolism. J. Bacteriol. 183, 1707–1715. 10.1128/JB.183.5.1707-1715.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rios-Hernandez L. A., Gieg L. M., Suflita J. M. (2003). Biodegradation of an alicyclic hydrocarbon by a sulfate-reducing enrichment from a gas condensate-contaminated aquifer. Appl. Environ. Microbiol. 69, 434–443. 10.1128/AEM.69.1.434-443.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rueter P., Rabus R., Wilkes H., Aeckersberg F., Rainey F. A., Jannasch H. W., et al. (1994). Anaerobic oxidation of hydrocarbons in crude oil by new types of sulphate-reducing bacteria. Nature 372, 455–458. 10.1038/372455a0 [DOI] [PubMed] [Google Scholar]
- Schink B. (1985). Fermentation of acetylene by an obligate anaerobe, Pelobacter acetylenicus sp. nov. Arch. Microbiol. 142, 295–301. 10.1007/BF00693407 [DOI] [Google Scholar]
- Schwarz H. (1978). Some newer aspects of mass spectrometric Ortho effects, in Topics in Current Chemistry, Vol. 73, ed Boschke F. L. (Heidelberg: Springer; ), 231–263. [Google Scholar]
- Segura A., Molina L., Fillet S., Krell T., Bernal P., Muñoz-Rojas J., et al. (2012). Solvent tolerance in Gram-negative bacteria. Curr. Opin. Biotechnol. 23, 415–421. 10.1016/j.copbio.2011.11.015 [DOI] [PubMed] [Google Scholar]
- Sikkema J., de Bont J. A., Poolman B. (1995). Mechanisms of membrane toxicity of hydrocarbons. Microbiol. Rev. 59, 201–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spormann A. M., Widdel F. (2000). Metabolism of alkylbenzenes, alkanes, and other hydrocarbons in anaerobic bacteria. Biodegradation 11, 85–105. 10.1023/A:1011122631799 [DOI] [PubMed] [Google Scholar]
- Stancu M. M., Grifoll M. J. (2011). Multidrug resistance in hydrocarbon-tolerant Gram-positive and Gram-negative bacteria. J. Gen. Appl. Microbiol. 57, 1–18. 10.2323/jgam.57.1 [DOI] [PubMed] [Google Scholar]
- Stobbe H. (1899). Condensation des Benzophenons mit Bernsteinsäureester. Liebigs Ann. Chem. 308, 89–115. 10.1002/jlac.18993080106 [DOI] [Google Scholar]
- Strijkstra A., Trautwein K., Jarling R., Wöhlbrand L., Dörries M., Reinhardt R., et al. (2014). Anaerobic activation of p-Cymene in denitrifying betaproteobacteria: methyl group hydroxylation versus addition to fumarate. Appl. Environ. Microbiol. 80, 7592–7603. 10.1128/AEM.02385-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thauer R. K., Shima S. (2008). Methane as fuel for anaerobic microorganisms. Ann. N. Y. Acad. Sci. 1125, 158–170. 10.1196/annals.1419.000 [DOI] [PubMed] [Google Scholar]
- Tissot B. P., Welte D. H. (1984). Petroleum Formation and Occurrence. Heidelberg: Springer; 10.1007/978-3-642-87813-8 [DOI] [Google Scholar]
- Trautwein K., Kühner S., Wöhlbrand L., Halder T., Kuchta K., Steinbüchel A., et al. (2008). Solvent Stress Response of the Denitrifying Bacterium “Aromatoleum aromaticum” Strain EbN1. Appl. Environ. Microbiol. 74, 2267–2274. 10.1128/AEM.02381-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wawrik B., Mendivelso M., Parisi V. A., Suflita J. M., Davidova I. A., Marks C. R., et al. (2012). Field and laboratory studies on the bioconversion of coal to methane in the San Juan Basin. FEMS Microbiol. Ecol. 81, 26–42. 10.1111/j.1574-6941.2011.01272.x [DOI] [PubMed] [Google Scholar]
- Webner K. (2012). Die Gene der (1-Methylalkyl)succinat-Synthase im anaeroben n-Alkanabbau des Betaproteobakteriums Stamm HxN1. Dissertation, University of Bremen. [Google Scholar]
- Widdel F., Knittel K., Galushko A. (2010). Anaerobic hydrocarbon-degrading microorganisms: an overview, in Handbook of Hydrocarbon and Lipid Microbiology, ed Timmis K. N. (Heidelberg: Springer; ), 1997–2021. 10.1007/978-3-540-77587-4_146 [DOI] [Google Scholar]
- Wilkes H., Kühner S., Bolm C., Fischer T., Classen A., Widdel F., et al. (2003). Formation of n-alkane- and cycloalkane-derived organic acids during anaerobic growth of a denitrifying bacterium with crude oil. Org. Geochem. 34, 1313–1323. 10.1016/S0146-6380(03)00099-8 [DOI] [Google Scholar]
- Wilkes H., Rabus R., Fischer T., Armstroff A., Behrends A., Widdel F. (2002). Anaerobic degradation of n-hexane in a denitrifying bacterium: further degradation of the initial intermediate (1-methylpentyl)succinate via C-skeleton rearrangement. Arch. Microbiol. 177, 235–243. 10.1007/s00203-001-0381-3 [DOI] [PubMed] [Google Scholar]
- Xie S., Lazar C. S., Lin Y.-S., Teske A., Hinrichs K.-U. (2013). Ethane- and propane-producing potential and molecular characterization of an ethanogenic enrichment in an anoxic estuarine sediment. Org. Geochem. 59, 37–48. 10.1016/j.orggeochem.2013.03.001 [DOI] [Google Scholar]
- Zink K. G., Rabus R. (2010). Stress-induced changes of phospholipids in betaproteobacterium Aromatoleum aromaticum strain EbN1 due to alkylbenzene growth substrates. J. Mol. Microbiol. Biotechnol. 18, 92–101. 10.1159/000287988 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.