

Abstract
Heparan sulphate (HS) sits at the interface of the cell and the extracellular matrix. It is a member of the glycosaminoglycan family of anionic polysaccharides with unique structural features designed for protein interaction and regulation. Its client proteins include soluble effectors (e.g. growth factors, morphogens, chemokines), membrane receptors and cell adhesion proteins such as fibronectin, fibrillin and various types of collagen. The protein-binding properties of HS, together with its strategic positioning in the pericellular domain, are indicative of key roles in mediating the flow of regulatory signals between cells and their microenvironment. The control of transmembrane signalling is a fundamental element in the complex biology of HS. It seems likely that, in some way, HS orchestrates diverse signalling pathways to facilitate information processing inside the cell. A dictionary definition of an orchestra is ‘a large group of musicians who play together on various instruments …’ to paraphrase, the HS orchestra is ‘a large group of proteins that play together on various receptors’. HS conducts this orchestra to ensure that proteins hit the right notes on their receptors but, in the manner of a true conductor, does it also set ‘the musical pulse’ and create rhythm and harmony attractive to the cell? This is too big a question to answer but fun to think about as you read this review.
Keywords: glycosaminoglycan, heparan sulphate, heparan sulphate/heparin
Introduction
Heparan sulphate (HS) belongs to the glycosaminoglycan (GAG) family of linear, anionic polysaccharides in which the basic polymer structure is made up of repeating amino sugar–uronic acid disaccharide units that are commonly modified by sulphation (Sugahara & Kitagawa 2000; Caterson 2012). With the exception of hyaluronic acid, GAGs are normally present in tissues in the form of proteoglycans (PGs), the polymer chains being in covalent linkage to various types of protein core that determine the GAG composition and the cellular/extracellular matrix (ECM) location of the PG. The protein cores also play active roles in many spheres of cell regulation particularly in the key areas of cell growth and cell adhesion (Couchman & Pataki 2012). The principal extracellular heparan sulphate proteoglycans (HSPGs) are perlecan, agrin and collagen XVIII; these PGs possess large modular core proteins that interact extensively with other ECM components and contribute significantly to matrix organization (Whitelock & Melrose 2011). On the cell surface, HS is mainly associated with two core protein families, the transmembrane syndecans and the GPI-anchored glypicans (Figure 1). Neuropilin, betaglycan and CD44 are occasionally modified by HS, although in general they appear on cell surfaces as non-glycanated proteins (Lindahl & Li 2009; Xu & Esko 2014). Cell surface HSPGs act as co-receptors for an extensive and structurally diverse range of extrinsic effector proteins, and it seems that many of the regulatory signals in the microenvironment of cells converge on HSPGs. This review will endeavour to describe the molecular basis of some of the key HS–protein interactions involved in cell regulation and their impact on cell development and disease.
Figure 1.
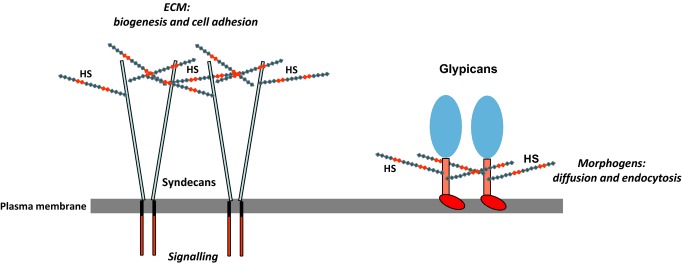
Cell surface heparan sulphate proteoglycans (HSPGs). The major cell surface HSPGs are the transmembrane syndecans and the GPI-anchored glypicans. The syndecans are constitutive dimers and play key roles in matrix biogenesis, cell adhesion to the extracellular matrix (ECM) and transmission of matrix-derived signals to the cell interior. The glypicans regulate morphogen gradients, signalling and the endocytosis of morphogen receptor complexes; these glypican-related specializations may be facilitated by the close proximity of the heparan sulphate (HS) chains to the cell surface. Both HSPG families are probably involved in binding and activating the many growth factors that utilize an HS co-receptor.
Heparan sulphate: structure and biosynthesis
The heparan sulphates are a group of related polymers in which variations in sulphation are imposed on a common structural theme (Casu & Lindahl 2001; Esko & Lindahl 2001; Gallagher 2001). The repeating disaccharide unit in HS consists of an α-/β-1,4-linked N-acetyl or N-sulphoglucosamine (GlcNAc or GlcNS) and uronic acid (glucuronic acid, GlcA, or its C5 epimer iduronic acid, IdoA) with chain lengths ranging in size from about 50 to 200 disaccharide units. The formation of HS begins in the cis-Golgi where an HS co-polymerase complex synthesizes a non-sulphated N-acetylated (NA) polymer named heparan composed of repeating units of -4-β-GlcA 1–4 α-GlcNAc 1-.
This precursor is assembled on core proteins primed by the common GAG linkage sequence GlcA-Gal-Gal-Xyl-Ser. As it transits the Golgi, the heparan precursor is enzymatically modified in a sequential and stepwise manner by a series of N- and O-HS-sulphotransferases and an HS epimerase to produce the mature HS chain [Figure 2; for reviews, see Lindahl et al. (1989); Kreuger and Kjellen (2012); Rudd and Yates (2012)].
Figure 2.
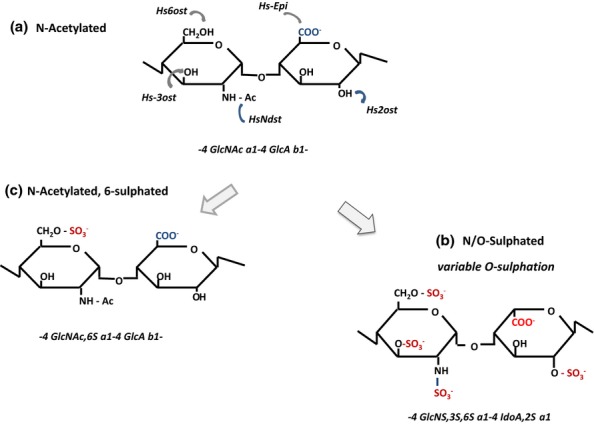
Enzymatic modifications in the biosynthesis of heparan sulphate (HS). The N-acetylated repeat disaccharide unit (a) in the HS precursor, heparan, is converted to HS by a series of modification enzymes (HS-MEs) that act in the following order: NDST, N-deactylase/N-sulphotransferase; C-5 epimerase (converts GlcA to IdoA); 2OST, 2-O-sulphotransferase; 6OST, 6-O-sulphotransferase; and 3OST, 3-O-sulphotransferase. The sequential actions of these enzymes produce a fully modified disaccharide (b) that contains IdoA and sulphate groups at all potential sites of modification. However, the modifications are incomplete at each stage, generally clustered in domains, and give rise to considerable variability in the structure of HS. Extensive regions of the heparan chain remain unmodified. S domains are formed by repeat GlcNS-IdoA, 2S units modified to varying degrees by sulphation at C6 and occasionally at C3. GlcNAc residues may be a target for 6OSTs when positioned next to an N-sulphated unit. As a consequence of this restriction, GlcNAc,6S (c) is found only in (NA)/NS regions of HS.
The controlled actions of the HS-modifying enzymes (HSMEs) lead to the formation of an ordered polymeric structure distinguished by a unique domain organization in which IdoA-rich, N-/O-sulphated regions, the S-domains, from two to nine disaccharides in length, are distributed in a fairly regular manner along the GAG chain. The S-domains are separated by unmodified (i.e. non-sulphated) NA regions deficient in N- and O-sulphate groups (Figure 3) (Turnbull & Gallagher 1990, 1991). The predominant S-domain sequence is
Figure 3.
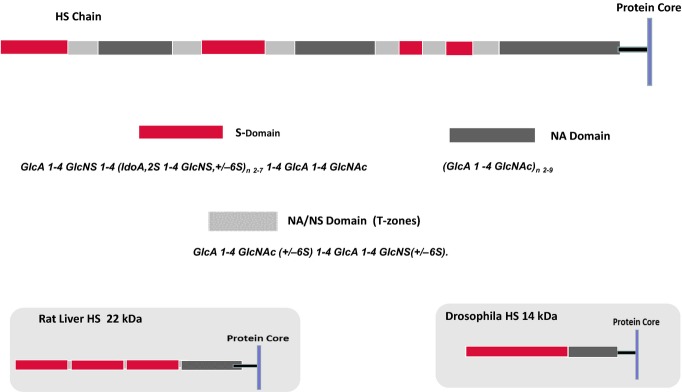
Domain structure of heparan sulphate (HS). The models illustrate a typical HS species from mammalian cells, rat liver and Drosophila. Mammalian HS is an ordered structure composed of an alternating arrangement of hypervariable sulphated regions [S- and N-acetylated (NA)/NS domains] and non-sulphated regions (NA domains) spaced in a fairly regular manner along the polymer; chain lengths vary from about 50 to 200 disaccharide units. An internal NA domain of approximately 10 disaccharides is contiguous with the glycosaminoglycan–protein linkage sequence. An S domain, often highly sulphated, is common at the distal, non-reducing end of the chain. Rat liver HS is a notable exception to the general design of mammalian HS species; it is an asymmetric structure with three, closely spaced S domains arranged towards the chain periphery but with retention of the internal, non-sulphated NA domain. HS synthesized by Drosophila is a relatively short, two-domain polymer in which a core NA sequence is connected to a longer, heparin-like distal region (Kusche-Gullberg et al. 2012). HS thus appears to have acquired a more complex structure during the course of evolution with an extension of chain length accompanied by the emergence of internal sulphated regions but with retention of the core NA domain.
with variable O-sulphation at C6 (and occasionally C3) of the amino sugars (Merry et al. 1999). The proximal region of HS close to the protein core is an extended non-sulphated NA-domain about 10 disaccharides in length (Lyon et al. 1987), whereas an S-domain, often highly sulphated, is common at the distal, non-reducing end of the chain (Staples et al. 2010; Naimy et al. 2011). Regions of intermediate sulphation called transition (T-) zones (or NA-/NS-domains), composed of alternate N-acetylated and N-sulphated disaccharides, are situated between the NA- and S-domains (Murphy et al. 2004). In these regions, the glucosamine residues are frequently sulphated at C6, but C2 sulphation is uncommon. Despite the lack of any known biosynthetic template, the fine structure of HS appears to be tightly regulated at the cellular level with variations in sulphation being characteristic of the cell or tissue of origin (Gallagher & Walker 1985; Ledin et al. 2004; Shi & Zaia 2009). An extreme example of this variability is the rat liver HS that differs in the overall design from the majority of mammalian HS species. It is a relatively short chain (approximately 60 disaccharides in length) with a highly asymmetric structure (Figure 3) in which an unmodified, core NA-domain is connected to three highly sulphated S-domains clustered towards the distal end of the chain (Lyon et al. 1994). The composition of the rat liver S-domains is similar to heparin, a highly sulphated chemical analogue of HS (Gallagher & Walker 1985). From an evolutionary perspective, it is interesting that the Drosophila HS is a much simpler structure than the mammalian counterpart (Figure 3); the chain length is quite short (approximately 30 disaccharides) with a core NA region and a single, distal S-domain (Kusche-Gullberg et al. 2012).
For most GAGs, the sulphation is established at the time of biosynthesis. However, this is not the case for HS; on cell surfaces, HS is prone to partial C6 desulphation by two endo-6-sulphatases or Sulfs (Dhoot et al. 2001; Ai et al., 2006, Frese et al. 2009). These enzymes are quite specific in their actions, targeting mainly GlcNS,6S residues in the S-domains rather than the T zones (Viviano et al. 2004; Seffouh et al. 2013).
There is little information on the three-dimensional structure of HS, but it is probable that the S- and NA-domains have distinctive conformational features. The S-domains are assumed to adopt a relatively rigid twofold helical symmetry similar to that of heparin in which the trisulphated disaccharide: IdoA,2S - GlcNS,6S is the main repeat unit (Mulloy & Forster 2000). The rotation of the heparin helix positions clusters of three sulphate groups on opposite faces of the helical axis, enabling proteins to bind to both sides of the saccharide chain. The plasticity of the iduronate ring, which oscillates mainly between two equi-energetic 1C4 and 2So conformers, alters the spatial disposition of the carboxyl and 2-O-sulphate groups with little apparent effect on the geometry of the glycosidic linkages (Mulloy 2012, for review). In principle then, S-domains in HS have a well-defined, heparin-like helical shape in which variations in density and disposition of sulphate groups and the flexible character of the iduronate ring offer a range of protein recognition motifs with variable affinities and specificities.
The regularly spaced NA-domains in HS appear to be considerably less constrained in structure than the S-domains (Figure 4). These flexible regions are predicted to expand the interaction range of HS and to support chain reactivity by conferring considerable orientational freedom on the S-domains (Mobli et al. 2008). The biological importance of HS chain flexibility may explain the strict conservation of the long NA-domain at the point of attachment of HS to the PG core protein.
Figure 4.
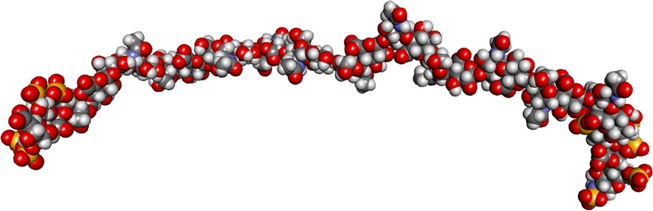
Molecular model of a flexible N-acetylated (NA) region of heparin sulphate flanked by short S domains. The model representing a long NA region in heparan sulphate (HS) was made on the basis of one of the models in the ensemble 4KHL.pdb, currently available as supplementary material to Khan et al. J. Biol. Chem. 2013, 288:27737–27751. This was a 24-mer of the heparan GlcA-GlcNAc sequence, consistent with X-ray scattering results. The two short S domains added at each end are made up of trisaccharides from the NMR structure of heparin, HPN1.pdb. This representation is an illustration, not the results of a simulation exercise. The model was kindly prepared by Professor Barbara Mulloy.
Heparan sulphate/heparin–protein interactions
Heparin is often used as an alternative to HS in protein interaction studies; it is a useful substitute for the S-domains of HS but lacks the organizational features that define the HS family (Skidmore et al. 2008). Nevertheless, all proteins known to interact with HS also bind efficiently to heparin and its commercial availability has led to its widespread use in the detection and characterization of potential HS-binding sites in proteins. Using the geometry of heparin as a guide, it has been possible to apply molecular modelling to accurately predict the location of several HS-binding domains in proteins (Mulloy & Forster 2000; Forster & Mulloy 2006).
Heparan sulphate/heparin–protein interactions are largely electrostatic mediated mainly by ion pairing between positively charged lysine, arginine and occasionally histidine residues exposed on protein surfaces and sulphate and carboxyl groups in the GAG chain; additional binding energy is often derived from hydrogen bonding and van der Waals' contacts (Capila & Linhardt 2002; Raman et al. 2005; Kreuger et al. 2006). The molecular architecture and overall flexibility of the HS chain appear to be designed to accommodate a variety of binding modes to meet the demands of many client proteins in the ‘heparanome’ (Ori et al. 2008); there is no single protein motif or fold that defines an HS (or heparin)-binding site (Mulloy & Linhardt 2001; Xu & Esko 2014) although some common conformational and sequence characteristics can be discerned. In general, HS-/heparin-binding regions in proteins are found in secondary structural elements, conformational sites or relatively unstructured regions.
In the first detailed study of heparin interaction sites in several proteins, Cardin and Weintraub (1989) identified two important consensus sequences for heparin binding: X-B-B-X-B-X and X-B-B-B-X-X-B-X, where B is a basic arginine (R) or lysine (K) residue, and X is a hydrophobic amino acid. These ‘CW motifs’ were located in regions of amphipathic secondary structural elements (beta-strands or α-helices) with the solvent-exposed basic residues projecting away from the protein surface and available for heparin binding. Although it is now clear that CW motifs are not a universal feature in heparin-/HS-binding proteins, they are perhaps more widespread than is generally appreciated. The Cardin/Weintraub paper was influential in drawing attention to the fact that GAG-binding sites in proteins are not simply defined by regions of positive charge and emphasized the importance of specific arrangements of basic residues for electrostatic compatibility with the charge distribution patterns in heparin and HS.
In many proteins such as the fibroblast growth factors (FGFs) and antithrombin, the HS-/heparin-binding region is a conformational site in which peptide loops (regions of connectivity between secondary structural elements) with one or more lys/arg residues converge in the folded protein, often forming a shallow binding pocket or cleft on the protein surface (Capila & Linhardt 2002). These regions sometimes contain a CW motif as a component of an otherwise discontinuous HS-/heparin-binding site (Hileman et al. 1998). In a careful analysis of several co-crystals of protein–heparin complexes, Sasisekharan et al. (Raman et al. 2005) detected local distortions, or kinks in areas of the heparin helical structure that interacted directly with protein surfaces. These deviations, which spanned a trisaccharide sequence of GlcNS,6S–IdoA,2S–GlcNS,6S with the iduronate in the 1C4 conformer, enabled close matching of the surface geometries of heparin and protein approaching an optimal fit for ionic and H bonds and van der Waals' contacts. Iduronate ring plasticity was deemed essential for ‘relaxing’ the helical architecture of heparin.
Relatively unstructured HS-/heparin-binding regions are present in a number of proteins including interferon (IFN)-gamma and the vascular endothelial growth factor (VEGF)/platelet-derived growth factor (PDGF) family. These binding regions are functionally significant. For example in the dimeric IFN, two linear HS-/heparin-binding sites (125KTGKRKR131 and 137RGRR141) in the unfolded C-terminal region of each monomer act in concert to enable the protein to accumulate on the cell surface HS (Lortat-Jacob et al. 1995). This serves two functions: it localizes IFN-gamma in the vicinity of its receptor and shields the receptor binding site from proteolytic attack.
Heparan sulphate in cell growth and development
Heparan sulphate is involved in many aspects of cell regulation during embryonic and post-natal development (for reviews, see Lin 2004; Bulow & Hobert 2006; Matsuo & Kimura-Yoshida 2014). Some selected examples of the various means through which HS–protein interactions regulate the growth, diffusion, migration and differentiation of cells are described below.
Soluble effectors: growth factors, morphogens, migration factors
Three HS-mediated regulatory mechanisms of soluble effectors can be discerned:
Co-receptor function
Localization and guidance
Diffusion effects
Co-receptor function
Heparan sulphate interacts with an extensive range of growth factors, morphogenic proteins and other soluble effectors that have recruited heparan sulphates to fulfil the role of ‘low-affinity’ cell surface co-receptors that operate in dual receptor systems to facilitate ligand binding to other higher affinity receptors that transduce signals into the cell (Lindahl & Li 2009). Examples of proteins that rely upon an HS co-receptor are the family of FGFs, hepatocyte growth factor/scatter factor (HGF/SF), VEGF and various neuroactive proteins including midkine, pleiotropin and glial-derived neurotrophic factor (GDNF). With these examples alone, it is clear that HS reaches into all areas of cell development and function.
In fulfilling its role as a co-receptor, HS is usually considered to be on the same cell as the signalling receptor, but it should be kept in mind that HS also acts in a trans co-receptor mode in which an HS/growth factor complex on a ‘presenting cell’ delivers signals to receptors on a nearby ‘receiving cell’. By separating the signal from the cell, trans co-reception is an attractive mechanism for defining cell migration tracts and for setting the boundaries of signalling activity in the stem cell niche (Kramer & Yost 2002; Dejima et al. 2011).
The FGF family of growth factors and morphogens
General features
The FGFs are a family of 23 heparin-binding growth factors with essential functions in embryogenesis and post-natal growth. They are relatively small, compact, globular proteins with similar tertiary structures of 12 antiparallel beta-strands arranged in a threefold internal symmetry like that of IL-1-α and IL-1-β. This type of common fold favours the co-existence of two or more non-overlapping binding sites on the protein surface. Fibroblast growth factors can be grouped into the majority paracrine or the minor endocrine subfamilies; the latter, which have weak heparin affinities, comprise FGFs 19, 21 and 23 and are involved in the control of carbohydrate and lipid metabolisms (Goetz & Mohammadi 2013). Their actions are largely independent of HS.
The paracrine FGFs operate through HS co-receptors to transmit signals to cells through four genetically distinct, tyrosine kinase receptors (FGFRs 1–4), three of which (FGFR1–FGFR3) are alternatively spliced giving rise to b- and c-isoforms that differ in ligand binding and biological functions (Mohammadi et al. 2005; Beenken & Mohammadi 2009). X-ray diffraction analyses of several co-crystals of FGFs in complexes with heparin have identified the primary GAG-binding sites as conformational sites composed of three peptide loops brought into close proximity in the native proteins. In several of the paracrine FGFs with solved crystal structures (FGF1, FGF2, FGF4, FGF7, FGF9, FGF10), the topologies of the loop regions are similar but with small differences in peptide sequence that probably explains their preferences for different sulphation patterns in heparin and HS (Ashikari-Hada et al. 2004; Raman et al. 2005; Xu et al. 2012a). In addition to their primary sites, secondary low-affinity HS-binding sites have been identified in the majority of the FGFs. These are not directly implicated in effector functions but may be important for FGF stability, resistance to proteolysis and diffusion in the ECM (Xu et al. 2012a).
Heparan sulphate-/heparin-binding properties of the FGFs
The FGFs discriminate between different potential binding sites in HS and heparin by recognition of sulphation patterns, domain length and conformation. To date, the most detailed investigations of the FGF interactions with HS and heparin have been carried out with FGF1 and FGF2, the founding members of the FGF family that provided the earliest in vitro experimental evidence for growth factors that depend on an HS co-receptor (Rapraeger et al. 1991; Yayon et al. 1991).
The HS-/heparin-binding domains in FGF1 and FGF2 are similar in shape and composition accommodating a minimum of five sugar residues in each case, but minor variations in protein structure lead to subtle but important differences in their interactions with HS/heparin (Faham et al. 1996; DiGabriele et al. 1998). FGF2 binds to a minimal, N-sulphated dp5 sequence (Figures5 and 6) that contains a key IdoA,2S residue (Maccarana et al. 1993; Faham et al. 1996); 6-sulphates are not required for the FGF2 interaction, nor can they substitute for the 2-sulphate groups (Habuchi et al. 1992; Turnbull et al. 1992), but 6-sulphates are required for FGF1 (Kreuger et al. 2001; Guerrini et al. 2002; Ashikari-Hada et al. 2004). A 2, 6, 2 O-sulphation triad IdoA,2S–GlcNS,6S–IdoA,2S was identified as a consistent motif in the FGF1-binding sites in HS (Kreuger et al. 2001). Co-crystals of FGF1 and a dp14 heparin demonstrate that a single FGF1 monomer binds to one side of the heparin helix; sulphates on the other side bind to a second monomer with opposite polarity, (DiGabriele et al. 1998) but the binding of individual monomers is to only one side. Monomeric FGF2 also binds to one side of the heparin chain (Faham et al. 1996). In the heparin polymer, clusters of sulphation, with one NS, one 2S and one 6S from a sequence of three sugar residues, are positioned along both sides of the helical axis, separated by approximately 17 Å. This arrangement is depicted in the simple layout recommended by Mulloy (2005) (Figure 7). It is clear from these structure-based models that the distribution of sulphates along the helical axis is a key issue when relating the FGF-binding properties of heparin or HS to charge density (Pellegrini 2001). Sulphates on the ‘wrong side’ will be largely inaccessible to FGF monomers. With this consideration in mind, Mulloy notes that hidden specificities may be discerned from the arrangement of sulphate groups in protein-binding sequences in heparin and HS (Mulloy 2005).Thus, it may be inferred that FGF1 and FGF2 recognize specific patterns of sulphate modification as distinct from binding affinities being mainly dependent on sulphate density (Figure 7).
Figure 5.
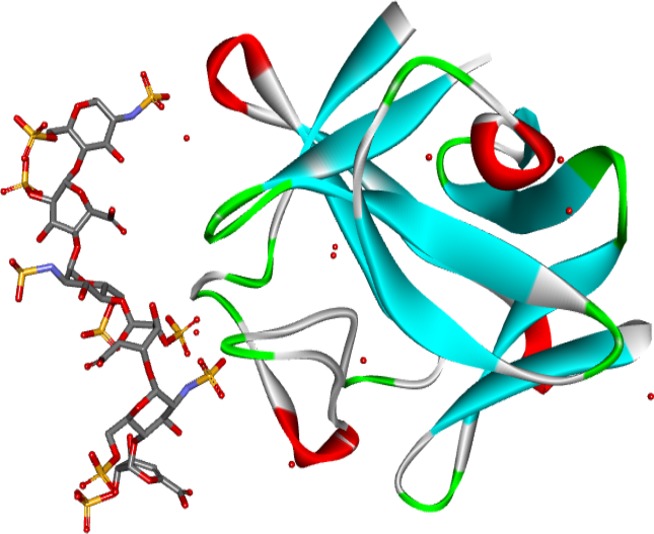
Crystal structure of a heparin dp6/FGF2 complex (Faham et al. 1996). The model shows the structure of FGF2 and a heparin hexasaccharide from the pdb file 1BFC.pdb. The protein is shown as a solid ribbon coloured by secondary structure: blue for beta-strands, red for helices, green for turns and white otherwise. Water molecules are red circles. FGF, fibroblast growth factors.
Figure 6.
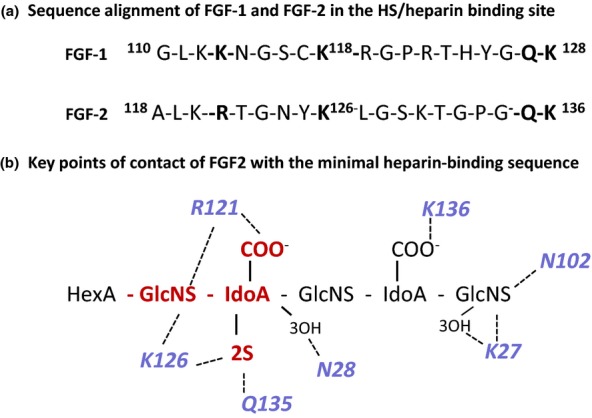
The interaction of FGF2 and FGF1 with heparin. (a) Sequence alignment of FGF2 and FGF1 in the main heparin/heparan sulphate binding sequence; conserved residues involved in heparin binding are in bold text. (b) Schematic diagram of the main FGF2 heparin contacts in the co-crystal FGF2 heparin complex in Figure 4. The GlcNS-IdoA,2S sequence (red) interacts with a high affinity subsite in FGF2; R121 are K126 are critical residues in this site. The predominant interactions are electrostatic, but Asn (N28 and N102) and Gln (Q131) participate in important H-bonds with the bound heparin. In the crystal structure, the IdoA,2S residue in the high-affinity site is in 1C4 chair conformation and the non-sulphated IdoA is in the 2SO skew boat conformer. For simplicity, non-interacting 6-O-sulphate groups on the amino sugars are not shown. FGF, fibroblast growth factors.
Figure 7.
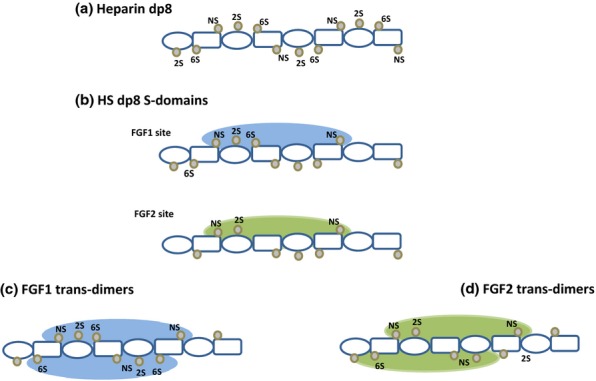
Sulphation clusters in heparin and HS: FGF1- and FGF2-binding sites. The diagrams follow the proposals of Pellegrini (2001) and Mulloy (2005) for illustrating the disposition of sulphate groups in the heparin helix. In the dp8 fragments, disaccharide repeats of iduronate (ovals) and glucosamine (rectangles) are inverted to show the clusters of three sulphates (NS, 2S, 6S) in sequences of three residues on either side of the molecule. Heparin dp8 is shown as a fully sulphated molecule. The HS dp8 fragments have a lower degree of sulphation than heparin. The proposed minimal binding sites for FGF1 and FGF2 extend over a similar sugar sequence of five monosaccharides but differ in the required degree of sulphation. FGF monomers bind to only one side of the saccharide chain. In the asymmetric model of a proposed mitogenically active configuration of FGF as shown in Figure 8, the growth factor assembles on HS in a trans-dimer arrangement. FGF1 dimers form on HS sequences with two trisulphation clusters as shown. Although monomeric binding of FGF2 is not dependent on 6-sulphates, at least one 6S group is required for activation. This key 6S may be positioned towards the end of a bioactive sequence where it could interact with an FGF2 monomer that binds with opposite polarity to that shown for FGF2 in the primary binding site (see text for details). FGF, fibroblast growth factors; HS, heparan sulphate.
Fibroblast growth factors in vivo
Heparan sulphate synthesized by different tissues provides additional evidence for specific interactions within the FGF signalling system. For example, the HS component of perlecan synthesized by human tracheal chondrocytes contains distinct structural features for binding FGF18 and signalling through FGFR3 (Chuang et al. 2010). By using a novel, in situ binding technique (LACE; ligand and carbohydrate engagement assay), Rapraeger et al. (Allen & Rapraeger 2003; Allen et al. 2003) showed that spatial and temporal changes in HS structure in mouse embryos are linked to the expression of distinct binding sites for several FGFs (FGFs 1, 2, 4 and 8) and for supporting their interactions with cognate FGFRs. Epithelial branching and morphogenesis in murine salivary and lacrimal glands are dependent on FGF7 and its close relative, FGF10. These FGFs bind to the same receptor subtype FGFR2b. They are synthesized in the embryonic mesenchyme and diffuse to epithelial buds where FGF10 induces bud elongation and FGF7 epithelial branching. Genetic and biochemical evidence indicates that the diffusion range and morphogenic actions of these FGFs are defined by their recognition of ligand-specific patterns of sulphate modification in HS rather than being determined solely by charge density (Patel et al. 2008; Makarenkova et al. 2009; Qu et al. 2011). FGF7 binds with high affinity to novel, low-sulphated sequences in HS that contain the rare 3-O-S group (Luo et al. 2006). These motifs may be expressed on responsive epithelial cells with lower affinity HS species directing FGF7 diffusion through the ECM.
Mechanisms of FGF activation
Minimal binding pentasaccharide sequences are unable to promote the mitogenic action of FGF1 and FGF2. An active site sequence in HS (i.e. one that has equivalent potency to the parent HS) appears to comprise a core of IdoA, 2S–GlcNS repeats (three of these units in a decasaccharide, dp10, S-domain seem to be the minimum requirement) substituted with one or more 6-sulphates (Walker et al. 1994). FGF1 activation is favoured if at least two of the core disaccharides are 6-sulphated, whereas only one seems to be needed for FGF2 (Pye et al. 1998; Sugaya et al. 2008). Recent interesting findings from studies using hsulf-2 to progressively remove 6-OS groups from heparin saccharides (Seffouh et al. 2013) also strongly suggest that FGF1 and FGF2 recognize particular patterns of 6-sulphation rather than simply charge density.
Various models have been put forward to explain FGF activation by HS. Yayon et al. (1991) proposed an allosteric mechanism in which HS induced a conformational change in FGF required for the efficient engagement of its receptor. An alternative idea was that active sites in HS contain two subsites: one for the FGFs and the other for FGFRs (Guimond et al. 1993). In this model, HS saccharides serve as templates, bringing ligands and receptors into close alignment for efficient binding. The requirement for longer chains than minimal binding sequences lends credence to this idea. The 6-sulphate group needed for FGF2 activation could be a key modification in the subsite for the FGFRs. Heparin and HS are known to stabilize the tertiary structures of the FGFs, especially FGF1, which is said to exist in solution in a partially unfolded, ‘molten globule’ state and to require HS to act as a molecular chaperone to maintain an active conformation (Uniewicz et al. 2010).
The publication of two crystal structures of ternary complexes of FGF/FGFR/heparin decasaccharides (dp10) raised the possibility of two distinct but related signalling architectures that were anticipated to assemble at different sites on HS chains (Figure 8). In the symmetrical model, a 2:2:2 FGF:FGFR:heparin complex contains two heparin saccharides that terminate with their non-reducing ends at the centre of the structure (Schlessinger et al. 2000). On cell surfaces, this species would assemble only at the periphery of two proximal polymer chains. As noted above, S-domains are often found at the ends of HS chains and they are strong activators of FGF2 (Naimy et al. 2011; see also Sterner et al. 2014).
Figure 8.
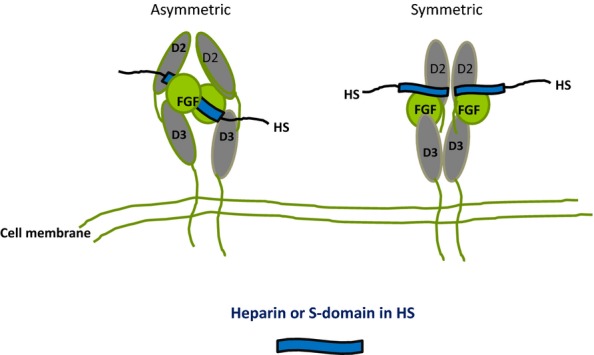
Diagrammatic models of the crystal structures of FGF/FGFR/(D2 and D3 domains)/heparin complexes. In the asymmetric model (Pellegrini et al. 2000), two FGFs bind on opposite sides of a heparin dp10 saccharide and recruit two FGFRs in a stable 2:2:1 complex with minimal protein:protein contacts. In the symmetric model (Schlessinger et al. 2000), two half complexes (1:1:1 FGF:FGFR:dp10) assemble at the non-reducing ends of two dp10 heparin saccharides and these then combine, primarily by means of extensive FGFR interactions, to form the symmetric complex. In the diagrams, the heparin saccharides in the crystal structures are imagined as S domains in HS positioned internally in the asymmetric model or at the periphery of the HS chain in the symmetric version. FGF, fibroblast growth factors; HS, heparan sulphate.
In contrast, in the so-called asymmetric 2:2:1 model (Pellegrini et al. 2000), two FGF: FGFR complexes are positioned around a central heparin fragment and could form on an internal S-domain sequence in HS. The FGF1 and FGF2 interactions with bioactive sequences in heparin are strongly cooperative with the first bound FGF monomer inducing a higher affinity site in heparin for a second FGF (Robinson et al. 2005; Brown et al. 2013). A discernible kink seen in the heparin helix in the region where the FGFs bind may enhance the affinity of the FGF interaction by creating more favourable binding orientations of sulphate and COO− groups. Both the symmetric and asymmetric architectures contain a common substructure in which heparin binds to a cationic cleft that extends along the FGF–FGFR interface. Analysis of complexes by mass spectrometry, analytical ultracentrifugation and gel filtration indicate that both the symmetric and asymmetric complexes exist in solution and may reflect alternative states for FGF signalling in vivo (Harmer et al., 2004, Goodger et al. 2008).
Hepatocyte growth factor/scatter factor: HGF/SF
Hepatocyte growth factor/scatter factor was discovered independently by two groups: one investigating ‘scattering’ of cell monolayers (SF) and the other, hepatocyte proliferation (HGF). Hepatocyte growth factor/scatter factor is now recognized as an important paracrine growth and motility protein synthesized by mesenchymal cells and active on epithelial and endothelial cells, neural cells and progenitor cells in the haemopoietic lineage. It is an essential morphogenetic factor during embryogenesis and has critical functions in the regeneration and repair of adult tissues. Its key role in organ development is probably due to an ability to support epithelial branching and stimulate ‘invasive growth’. HGF/SF is involved in tumour–stroma interactions, and its aberrant expression in the stromal environment is a major factor in tumour angiogenesis and metastasis (for review, see Birchmeier et al. 2003). In human myeloma cells, it is an autocrine, rather than a paracrine, factor and its effector functions are mediated by syndecan-1 that is overexpressed in this disease (Ramani et al. 2011 and references therein).
HGF/SF is a plasminogen-related protein and acts on a single tyrosine kinase receptor, the c-Met proto-oncogene. It is released from cells as a 90-kDa pro-protein, which is then activated by protease scission to form a two-chain heterodimer composed of 60-kDa A- and 30-kDa B-subunits. The A-subunit is a modular element with an N-terminal hairpin loop (N-domain) and four kringle domains (K1–K4), whilst the B-subunit is closely related to the inactive serine protease domain of plasminogen (Figure 9a). Two naturally occurring truncated variants, NK1 and NK2, with weak agonist activity, arise by alternative splicing of the hgf/sf gene (Figure 9b). The main GAG-binding region in HGF/SF is in the hairpin loop of the N-domain with secondary interactions in the K2 region and the B-subunit (Holmes et al. 2007). NMR analysis, crystallography and deletion mutations have identified the main binding site in the N-domain as a shallow groove formed by two spatially close, small clusters of basic residues, K60, K62, K63 and R73, R76, K78; an additional minor contribution to heparin binding is derived from a more remote third hairpin cluster of K91, R93, K94 (Hartmann et al. 1998; Zhou et al. 1998; Lietha et al. 2001).
Figure 9.
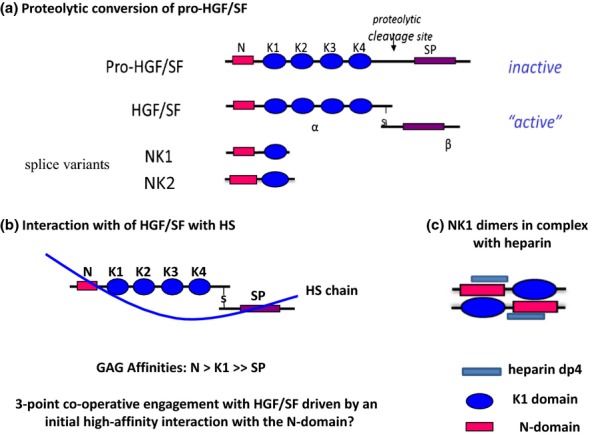
Hepatocyte growth factor/scatter factor (HGF/SF), its splice variants and interactions with heparan sulphate (HS). (a) HGF/SF is a disulphide-linked heterodimer with an N-terminal hairpin loop (N), four kringle domains (K1–K4) and an inactive serine protease (SP) domain. The primary HS-binding site is in the hairpin loop, with accessory sites in the K1 and SP regions. NK1 and NK2 are splice variants of the hgf/sf gene. (b) HS S domains of length dp12-dp14 are the optimum size for high-affinity binding to HGF/SF. In principle, HS fragments of this length are sufficient to engage in a three-point attachment to HGF/SF that may stabilize an active conformation of the modular elements in the native protein. (c) NK1 has an absolute requirement for HS or heparin to bind the Met receptor and for signalling activity in cultured cells. In crystal structures, NK1 forms dimers in the presence of heparin and four sulphated monosaccharides make contact with the binding site in the N-domain. Heparin (or HS) may stabilize the dimer and/or expose the dimerization surfaces in the N and K1 regions. The tendency for NK1 domains to form stable interactions in the presence of heparin suggests a mechanism for dimerization and activation of native HGF/SF.
Native HGF/SF binds to heparin and to the S-domains of HS with high affinity (Kd of 0.2–0.3 nM) but low sequence specificity accommodating a range of sulphation patterns and densities that enable it to support cell activation via the c-Met receptor (Lyon et al. 1994, 2002). Although the strength of binding and bioactivity both correlate with saccharide length and degree of sulphation (Ashikari-Hada et al. 2004), a variety of low sulphated sequences will interact with HGF/SF and there appears to be no preference for any specific position of sulphation (Catlow et al. 2008). The low specificity of the GAG-binding site in the N-domain is reflected in its ability to bind di-O-sulphated sequences in dermatan sulphate (DS) with comparable affinity to HS (Lyon et al. 2004; Deakin et al. 2008). Dermatan sulphate binds to the same site as HS and is an efficient activator of HGF/SF.
The mechanism of HS-mediated activation of native HGF/SF is unclear. Saccharides containing three or four sugar units will bind and elicit a very weak signalling response from full-length HGF/SF (Deakin et al. 2008; Li et al. 2010), but its highest affinity and activity are associated with HS S-domains of 10–12 monosaccharides in length (Lyon et al. 1994; Delehedde et al. 2002). The dimensions of HGF/SF are such that bioactive dp12/dp14 HS S-domains could in principle contact the three GAG-binding regions (i.e. N- and K2-domains and the B-subunit; Figure 9b) in a multipoint interaction that favours not only HGF/SF dimerization but also stabilizes a receptor-compatible conformation of the other modular elements in the native protein.
Heparin induces HGF/SF to form dimers and oligomers (Zioncheck et al. 1995), and HGF/SF dimerization is likely to be a prerequisite for the efficient stimulation of the Met receptor. The mechanism of dimerization is probably determined by interactions in the NK1 region. In co-crystal structures with heparin, NK1 is a homodimer with a span of four monosaccharides in contact with each NK1 molecule; the dimer interface is formed by direct protein–protein contacts between the N- and K1-domains of the two monomers (Kemp et al. 2006; Figure 9c). It is thus possible that cell surface HS can induce conformational changes in the NK1 region of HGF/SF that drives dimer formation leading to the presentation of HGF/SF dimers to c-Met receptors.
Neurotrophic and angiogenic factors
Midkine- and heparin-binding growth-associated molecule, HB-GAM (pleiotropin)
Midkine and HB-GAM comprise a small family of dimeric, neuroactive growth and differentiation factors with fully conserved disulphide bonds and an exceptionally high content of lysine residues (Rauvala 1989; Muramatsu, 1994; Kaneda et al. 1996). Midkine synthesis is enhanced during the mid-gestation period of embryogenesis, and it has angiogenic as well as neurotrophic actions; its expression level in neuronal tumours correlates with poor progonosis (Kadomatsu et al. 2013).
The midkine monomer (13 kDa) is composed of N- and C-terminal modules of broadly similar size and overall conformation with two principal HS-binding sites located in the C-terminal region (Figure 10). NMR analysis of the solution structure of midkine/heparin complexes together with mutagenesis studies implicated two short, basic clusters on one side of each monomer (Iwasaki et al. 1997). The key sequence was a CW-type motif of 85XKKXRX90 found in a flexible hairpin loop region that slopes towards a second cluster of K79, R81 and R102 positioned on an adjacent beta-sheet (Muramatsu et al. 1994). The midkine dimer is formed by non-covalent association of monomers that combine in a symmetrical head-to-head fashion. An extended high-affinity GAG-binding site is located at the elongated dimer interface by the close proximity of the CW motifs (Iwasaki et al. 1997). The midkine monomer binds efficiently to a heparin sequence of 6 disaccharide units (dp12), but an oligosaccharide of 10 disaccharides is needed to fully occupy the composite binding site in the dimer. The use of selectively desulphated heparins indicate that N-, 2- and 6-sulphate groups participate in the interaction. Midkine interacts strongly with a highly sulphated species of HS isolated by affinity chromatography from organ cultures of 13-day embryonic brains, but interestingly, it also binds a brain-derived chondroitin sulphate enriched in type E disulphated units (i.e. GalNAc, 4S,6S–GlcA; Li et al. 2010).
Figure 10.
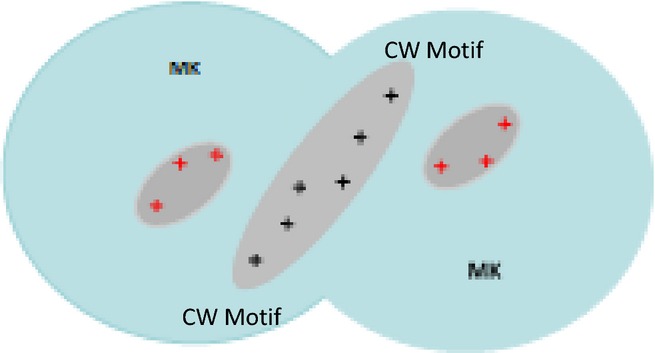
Heparan sulphate-binding sites in the midkine (MK) dimer. In the head-to-head midkine dimer, two CW motifs (+++) form an extended heparan sulphate (HS)-binding site at the dimer interface; in each monomer, the CW motif slopes towards three additional basic residues that further enhance HS affinity. Cell surface HS stabilizes the midkine dimer and is essential for midkine signalling.
The high level of sequence identity between midkine and HB-GAM and conservation of all the basic residues bar one (R89) involved in heparin binding is reflected in their similar tertiary structures and HS (and CS)-binding characteristics with perhaps minor differences in affinity due to the missing arg residue (Kilpeläinen et al. 2000). Midkine and HB-GAM are examples of the significance of the basic peptide environment in GAG recognition; the proteins are rich in lysines, and yet GAG interactions are determined by a minor fraction of basic residues with the required orientation and spacing of their charged side chains.
In neuronal cell cultures, the neurite outgrowth-promoting activity of midkine is impaired when cells are enzymatically depleted of HS and the activity is suppressed by the addition of dp20 heparin saccharides that presumably bind and occlude the HS interaction site at the interface of the dimer (Asai et al. 1997; Zou et al. 2003). It appears that midkine requires HS to be present on the cell surface and unlike the FGFs and HGF/SF, it cannot be activated by soluble HS saccharides. This suggests that HSPGs may directly participate in the signalling mechanism perhaps in conjunction with another receptor that transduces the signal.
Similar to midkine, HB-GAM also forms inert complexes with heparin in solution and its activity is compromised in neuronal cells treated with heparinase III. HB-GAM binds to syndecan-3 (N-syndecan) isolated from rat brain and syndecan-3 may be the membrane receptor for HB-GAM-stimulated neurite outgrowth, acting independently of more conventional receptors (Raulo et al. 1994; Kinnunen et al. 1996). The cytoplasmic domain of syndecan-3 binds several cytosolic proteins including src kinases and src kinase substrates such as cortactin and b-tubulin; binding of HB-GAM to syndecan-3 may transmit signals for cortactin polymerization via the phosphorylation and activation of c-src (Kinnunen et al. 1998).
The CS-binding properties of midkine and HB-GAM are also biologically relevant. A CS-bearing receptor protein tyrosine phosphatase has been identified as a receptor for midkine- and HB-GAM-stimulated migration of embryonic neurones and osteoblasts (Maeda & Noda 1998). Thus, depending on the cell type, both HS and CS, probably with high levels of sulphation, directly participate in midkine and HB-GAM signalling.
Glial cell line neurotrophic factor
The GDNF family ligands (GFLs) comprise four related growth factors that include neurturin, artemin and persephin, and all except persephin are heparin-/HS-binding proteins. They fall within the larger family of TGF-beta-related cytokines with the cystine-knot motif as a major element in their tertiary structures (Baloh et al. 2000). The GFLs are essential for the growth, development and maintenance of the nervous system. They prolong the viability of dopaminergic and motor neurones and have considerable potential for the treatment of neurological conditions such as Parkinson's disease and amyotrophic lateral sclerosis.
Glial-derived neurotrophic factor was the first member of the GFL family to be identified and is the most extensively investigated. Its actions extend beyond the nervous system to include key roles in spermatogenesis and in the development of the embryonic kidney. Glial-derived neurotrophic factor is a disulphide-linked homodimer composed of approximately 20-kDa N-glycosylated monomeric units bound in an antiparallel fashion with the cystine knot positioned close to the dimer interface. A relatively long (45 amino acids), apparently unstructured N-terminal region extends from the core of the monomer units. Although this region contains two sections rich in basic amino acids, the main heparin-/HS-binding site is located in the one more distant from the N-terminus (NT) with seven semi-contiguous arg/lys residues in the sequence R-G-K-G-R-R-G-Q-R-G-K-N-R (Alfano et al. 2007). The heparin-/HS-binding region in GDNF accommodates a dp12/dp14 saccharide that may bridge the two binding sites in the native dimer (Rickard et al. 2003). All the main sites of sulphation in heparin contribute to the interaction with GDNF but with a significant degree of dependence on 2-O-sulphation suggesting some specificity in GDNF–heparin/HS recognition (Davies et al. 2003; Rickard et al. 2003). The requirement for C2-sulphation is interesting because it may explain the renal agenesis in mice deficient in the HS-2-O-sulphotransferase gene (Hs2ost−/−) (Bullock et al. 1998; Merry et al. 2001). Glial-derived neurotrophic factor in the meta-nephric mesenchyme is a chemoattractant for the ureteric bud, and HS deficient in 2-O-sulphation may not support the formation of a stable GDNF gradient at the bud tip. Recent studies in vivo have confirmed that the expression of the Hs2ost gene is essential for mesenchyme induction but suggest that the defect may be due more to disruption in Wnt signalling than in GDNF (Shah et al. 2010). GDNF gradients are important in other developmental processes. For example, an HS-dependent chemoattractant gradient of GDNF directs the migration of neural progenitors during oesophageal innervation, but in this setting the activity is dependent on the regulated expression of Sulf enzymes that bring about a controlled release of GDNF from the oesophageal ECM (Ai et al. 2007).
Cell surface HS is essential for efficient GDNF-mediated cell signalling; following the treatment of cells with heparinase III to remove cell surface HS, GDNF cannot stimulate axon growth in either cultured neurones or PC12 cells nor can it induce a scattering response in MDCK epithelial cells (Barnett et al. 2002). The addition of heparin is unable to restore the activity in cells lacking HS, and low concentrations of heparin (1 μg/ml) inhibit GDNF signalling in cells with normal levels of cell surface HS. These findings indicate that unlike the FGFs but in common with midkine and HB-GAM, GDNF activity is dependent on the presence of HS at the cell surface, and soluble GAG is not an adequate means of stimulation.
Glial-derived neurotrophic factor signals to cells through a membrane receptor system composed of a 2:2 complex of a GPI-linked co-receptor GFRa1 and RET, a tyrosine kinase signalling protein (Baloh et al. 2000). The critical requirement for cell surface HS suggests that it may interact in a very specific manner with the GDNF-receptors. Cell surface HS concentrates GDNF on the cell surface (Kd 0.22 nM), bringing it into intimate contact with its receptor system. The GRLa1 co-receptor contains two positively charged regions, and one of these, arranged along a surface-exposed α-helix, conforms to a CW motif; co-crystals of GRLa with sucrose octasulphate (a mimic for HS and heparin) and molecular modelling suggest that this motif is a binding site for HS (Parkash et al. 2008). In the assembly of a signalling complex, GDNF initially binds to GRLa1 and this interaction may be facilitated if the GFRa1 is already held in a favourable orientation for dimerization by its association with the S domains of HS. HS is unlikely to form a stable complex with GFRLa1 because its binding site partially overlaps with the RET-binding site on GRLa1. HS may thus act as a ‘catalyst of encounter’ (Lander 1998) on the cell surface by transient association with GDNF and GFRa1, accelerating the rate at which the 1:2 GDNF-GRLa1 complex forms and then rapidly dissociating as RET engages the complex and initiates signalling.
An alternative signalling mechanism for GDNF operates when it is adsorbed to culture surfaces. Surface-immobilized GDNF is believed to mimic the mode of action of GDNF in tissues where it is often present in the ECM. The adsorbed GDNF stimulates cells in a similar manner to HB-GAM by binding to syndecan-3 (Bespalov et al. 2011). Response is critically dependent on the HS chains of syndecan-3 and leads to a rapid and extensive neurite outgrowth in rat embryonic hippocampal neurones and cell spreading in neuroblastoma cells. The mechanism of signal transfer by syndecan-3 is unclear, but perhaps GDNF occupancy of specific S-domains in HS elicits a conformational change in the cytoplasmic region of the protein that stimulates src activation. Parallel signalling may occur through RET if syndecan-3 can substitute for GRLa1 as a RET co-receptor. Syndecan-3 (N-syndecan) was first isolated from neonatal rat Schwann cells and appears to have a unique signalling role in the nervous system (Carey 1996).
Vascular endothelial growth factor
Vascular endothelial growth factor belongs to the PDGF family of covalently linked dimeric growth and morphogenic factors that play critical roles in all stages of embryonic angiogenesis from the derivation of endothelial progenitors to the establishment of a mature, highly branched vascular network. In adults, the vasculature is relatively quiescent but is induced by VEGF and other factors during the female reproductive cycle and in wound healing and tissue repair. Various forms of pathological angiogenesis seen in malignancy, arthritis and retinal disease (e.g. diabetic retinopathy, macular degeneration) are in part driven by VEGF, and the VEGF signalling system is a recognized and valuable therapeutic target in these diseases (Ferrara 2004).
Alternate mRNA splicing of a single VEGF gene gives rise to several VEGF isoforms with identical N-terminal receptor-binding domains but with variations in the composition of the C-terminal, heparin-/HS-binding region due to the presence or absence of sequences encoded by exons 6, 7 and 8. The main VEGF isoforms are the freely soluble VEGF121 that lacks the heparin binding domain, HBD, VEGF189 that is tightly associated with the ECM and VEGF165 (VEGF-A), the most common and widely studied of the VEGFs. VEGF165 (or VEGF164 in mice) contains the 44-residue exon 7 sequence essential for binding to heparin and HS. It is functionally versatile, being able to fully support embryonic angiogenesis in the absence of other variants (Krilleke et al. 2009). It is the main VEGF isoform induced in areas of retinal ischaemia, and the expression of VEGF165 in hypoxic regions of tumours is correlated with the transition from localized to disseminated disease (Ng et al. 2006).
VEGF165 signals to endothelial cells by activating two related receptor tyrosine kinases VEGFR1 and VEGFR2 (Grünewald et al. 2010). It binds with higher affinity to VEGFR1, but VEGFR2 is the main receptor for transmitting angiogeneic signals. Cell culture studies and experiments in vivo have shown that HS is an important co-receptor for VEGF165; cultured endothelial cells depleted of HS respond poorly to VEGF165 and the activity is only partially restored by the addition of heparin or HS, suggesting that optimum activity requires HS to be retained on the cell membrane (Ashikari-Hada et al. 2005; Robinson & Stringer 2006; Robinson et al. 2006; Fuster et al. 2007; Zhao et al. 2012). Heparan sulphate is essential for the activity of VEGF in the in vitro haemopoietic differentiation of mouse ES cells (Holley et al. 2011). Particular patterns and densities of N- and 6-O-sulphation may be necessary for VEGF binding and signalling to endothelial cells, but 2-O-sulphates appear to be less important (Robinson et al. 2006). The reduced expression of Hs6ost-1 or Hs6ost-2 significantly impairs the activity of VEGF165 in assays of sprouting and tube formation in cultured endothelial cells (Ferreras et al. 2012). The pivotal role of the HBD in VEGF was shown in VEGF120/120 mice (equivalent to human VEGF 121) generated by the deletion of exons 6 and 7 of the vegf gene and devoid of heparin-binding isoforms. In these embryos, VEGF chemoattractant gradients and differentiation signals were defective, leading to gross abnormalities in vessel sprouting and vascular patterning (Ruhrberg et al. 2002). Heparan sulphate also binds to the VEGF receptors and may promote receptor ligation by a proximity-based mechanism (Xu et al. 2011).
Modelling of the VEGF165 HBD in complex with heparin (Figure 11a,b) indicated a conformational binding site formed by a shallow groove orthogonal to the main axis of the protein (Robinson et al. 2006). The groove was lined by eight basic residues and accommodated a heparin dp7 saccharide with six of the monosaccharides in close contact with the protein surface. Mutagenesis identified Arg 124, 125 and 149 in a loop region of the binding groove of HBD as a critical subsite for heparin recognition (Krilleke et al. 2007). The interaction of HS with VEGF dimers is complex and determined by the spacing of S-domains along the GAG chain. Vascular endothelial growth factor dimers associate in a side-by-side antiparallel arrangement stabilized by disulphide binds. Binding data using long HS fragments derived by K5 lyase scission (specific for the non-sulphated NA regions of HS) were compatible with a model in which the two HBDs in the VEGF dimer bind simultaneously with two S-domains connected by a short transition zone sequence (Figure 11c; Robinson et al. 2006). The bidentate nature of this interaction is likely to be very stable and may represent the mode of interaction of VEGF with HS in establishing migration tracts in the embryonic matrix.
Figure 11.
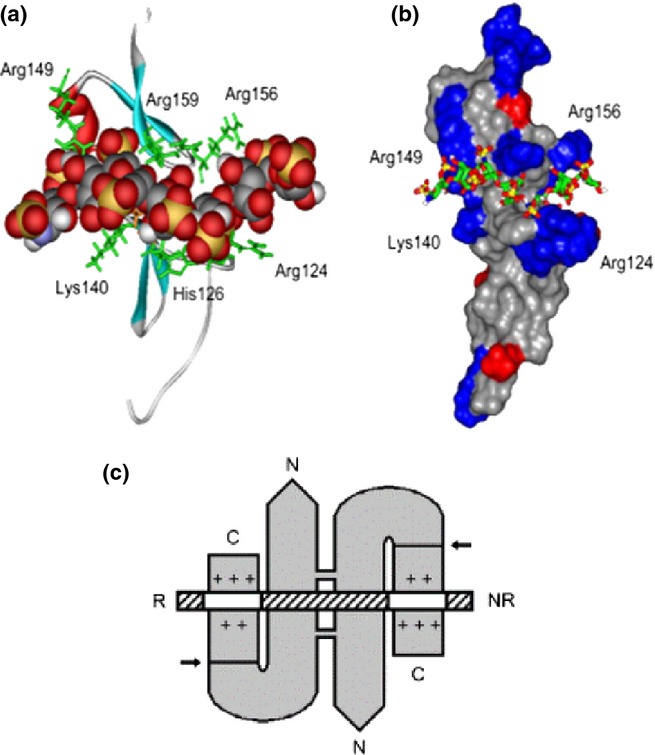
Heparin-/heparan sulphate (HS)-binding sites in the VEGF165 heparin-binding domain (HBD) and in the VEGF165 dimer. (a) Ribbon diagram of the VEGF165 HBD (residues 111–165; Protein Data Bank code 2VGH) with a docked heparin dp7 [space-filling representation: carbon (grey), oxygen (red), sulphur (yellow), nitrogen (blue) and hydrogen (white)]. Basic residues lining the shallow binding groove are shown in a stick representation (green). (b) The same complex with 2VGH depicted as a protein surface. Arg and Lys residues are shown in blue, and Glu and Asp residues are shown in red. The heparin dp7 saccharide is a stick representation. The atomic coloration is as in (a), except that carbons are shown in green. (c) A K5 lyase-resistant HS fragment (white and hatched boxes) is bound to the HBDs of VEGF165 homodimer (grey). The N- and C-termini of VEGF165 and the reducing (R) and nonreducing (NR) ends of the HS chain are indicated. The VEGF165 subunits are held by disulfide bonds in an antiparallel ‘side-by-side’ orientation. Arrows indicate plasmin cleavage at the sites that release the HBDs. Basic residues in each heparin-binding cleft are shown (+). The two clefts are occupied by separate S domains (white boxes) in the same HS chain. The S domains are at least dp6 in length and are 6-O-sulphated. This research was originally published in Journal of Biological Chemistry. Robinson et al. 2006 © the American Society for Biochemistry and Molecular Biology.” VEGF, vascular endothelial growth factor.
The neuropilin co-receptor for VEGF
Neuropilin-1 (Nrp-1) is a co-receptor for VEGF165, essential for VEGF binding to VEGFR-2 and the induction of angiogenesis (Grünewald et al. 2010). The ternary complex formed by VEGF165 with Nrp-1 and HS is believed to display the two N-terminal receptor-binding regions of the VEGF dimer in a favourable orientation for interaction with VEGFR2.
VEGF165 binds to Nrp-1 via its C-terminal HBD region, and protein–protein and protein–HS interactions are required. Nrp-1 itself is a dimeric heparin-binding transmembrane protein and consists of three domains in its extracellular region. The heparin-/HS-binding site is in the central b-domain, mainly in the b2-subdomain (Mamluk et al. 2002) where a BBXB CW motif (513RKFK516) on a beta-strand is the main interaction site; residues Arg 359 and Lys 373 form a second minor site in the b1-subdomain, and the two sites combine to form a stretch of electropositivity that can be occupied by six disaccharides (Vander Kooi et al. 2007). In high-resolution structures, the heparin- and HS-binding domains of Nrp-1 and VEGF are aligned to form a composite HBD of length equivalent to about 10 disaccharides. These results fit well with the requirement for heparin saccharides of this size to strongly potentiate the interaction of a recombinant b1b2-domain and VEGF165 in co-immunoprecipitation experiments (Mamluk et al. 2002).
It is interesting that Nrp-1 in muscle and nerve cells is glycanated by a single HS chain and in these cells, HS glycanation of Nrp-1 is necessary for its action as a VEGFR2 co-receptor (Shintani et al. 2006). Perhaps the Nrp-1 HS expresses distinctive sulphation motifs, not found on other nerve/muscle HSPGs, essential for binding VEGF.
Chemokines
The major class of proteins that depend on HS for strategic localization on the vascular endothelium are the chemokines, a large family of chemoattractant and migration factors that direct the trans-endothelial movement of circulating leucocytes from blood to tissues. As such, they have essential functions in immune surveillance and in directing the infiltration of neutrophils at sites of infection and injury. Chemokines induce the migration of mesenchymal cells during wound healing, and aberrant chemokine activity is a key factor in inflammation, autoimmunity, transplant rejection and the invasive properties of malignant tumours (Handel et al. 2005; Lortat-Jacob 2009; Dai et al. 2010).
Chemokines are small monomeric proteins (8–12 kDa), and the majority contain four conserved cystines that form internal disulphide bonds essential for the 3-D structure of the folded proteins. There are over 50 members of the chemokine family classified into four groups, CCL, CXCL, CX3CL and CL, according to the number and spacing of cystine residues in a conserved N-terminal fold. Within this classification, by far, the major groups are the CC and CXC chemokines, in which two cystine pairs are either contiguous or separated by a single amino acid. Chemokines share the same tertiary structure but differ in the mode of dimerization, receptor binding and HS/heparin recognition (Lortat-Jacob et al. 2002). In cell cultures, chemokine monomers are biologically active and able to elicit cell signals via G-protein-coupled receptors, but in physiological conditions, in the presence of HS, the majority associate as dimers or tetramers and higher-order structures may form in areas of high HS chain density (Proudfoot et al. 2003).
Heparan sulphate is essential for the attachment of chemokines to the endothelial surface and for the formation of chemokine gradients in the subendothelial matrix. Heparan sulphate may also play a more active role through conformational effects that enhance the stability of the chemokine receptor complex (Goger et al. 2001). In this regard, the CC chemokine MCP-1 (CCL-2) forms complexes with heparin and HS as a monomer, dimer or tetramer with the aggregation state predicted to be a means of regulating its bioactivity and restricting receptor cross-reactivity (Lau et al. 2004; Yu et al. 2005).
The HS-binding sites in chemokine monomers are positively charged areas with the majority of contacts formed by ionic interactions between arg and lys residues and N- and O-sulphate groups in HS. Molecular modelling data predict up to four binding modes largely determined by variable patterns of association of basic amino acids and their location on the chemokine surface (Lortat-Jacob et al. 2002). These observations suggest considerable selectivity in the chemokine recognition of structural motifs in HS. In principle, the regulation of fine structure of HS on the vascular endothelium will define specific chemokine interactions and direct the tissue-specific emigration of circulating leucocyte subpopulations.
Platelet factor 4, PF4 (CXCL4) and IL-8 (CXCL8)
IL-8, also known as neutrophil chemotaxis factor, and platelet factor 4, PF-4, a chemokine released from a-granules of activated platelets, are CXCL chemokines, and both have been investigated in some detail with regard to their HS-/heparin-binding characteristics. The tertiary structures of their monomeric units are typical of the chemokine family in which a C-terminal α-helix lies across a core of three-stranded antiparallel beta-sheets (Clore et al. 1990; Shute 2012). In PF4, the side chains of two pairs of lysines in the sequence 61KKIIKK66 project from the exposed face of the α-helix; the corresponding sequence in IL-8 (54KENWVQRVVEKFLKR68) reveals a more dispersed arrangement of basic lys/arg residues in which the presence of an acidic glutamate (E) may have some bearing on HS sequence recognition. In both proteins, the monomers bind at their N-terminal beta-sheets to form flat antiparallel dimers, although in PF4, two dimers then stack to give an asymmetric tetramer. In IL-8, the two basic clusters are on the same dimer surface, but the stacking arrangement in PF4 positions two pairs of basic clusters on opposite sides of the tetramer (Gallagher & Lyon 2000). Chain flexibility is important for HS reactivity with PF4 and IL8; both bind along extended regions of HS with the interaction sites incorporating two or more S-domains connected by flexible NA segments.
The tetrameric PF4 binds with high affinity to a 9-kDa saccharide in HS (approximately 19–20 disaccharides in length); the saccharide was identified by a ‘footprinting’ method (Lortat-Jacob et al. 1995) in which the ligand (PF4) was used to protect its HS-binding region from degradation by heparinase enzymes. Based on the structural analysis of the protected fragment (PPD) and its predicted complementarity to the lys clusters in PF4, a model was proposed in which a long HS saccharide tracks a ring of positive charge that lies over the surface of the tetramer with two short, closely spaced S-domains positioned at both ends of the PPD (Stringer & Gallagher 1997). In the model structure, the two peripheral S-domains, which contain IdoA 2-sulphates shown to be essential for binding, are correctly spaced to associate with lysine clusters in the antiparallel α-helices on opposite faces of the tetramer. Other important cationic residues, such as arginines 20, 22, and 46, which encircle PF4, are assumed to interact with the GlcA in the in the central NA region of PPD (Figure 12a). High-resolution studies and mutagenesis of specific basic residues (Zhang et al. 1994; Mayo et al. 1995) support the idea that saccharides encircle the tetramer by electrostatic interactions along a ring of positive charges with the S-domains perpendicular to the lysine-containing α-helices. The ring of charge on PF4 is conserved in several other CXC chemokines, emphasizing its likely physiological importance for chemokine function.
Figure 12.
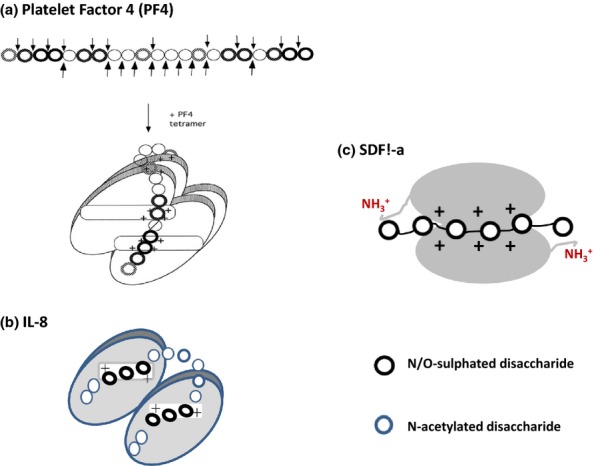
Models of CXCL chemokines in complexes with heparan sulphate (HS) and heparin. The models proposed for HS in complexes with PF4 and IL-8 are based on the structures of chemokine-binding domains in HS protected from degradation by heparinase enzymes (see text for details). In PF4, the S domains run perpendicular to the alpha-helices but adopt a parallel orientation to the alpha-helices in IL-8. The alpha-helices in SDF1-α are not involved in HS–heparin binding. Molecular docking reveals that heparin (dp12) binds along a positively charged ‘crevasse'at the interface of the SDF1-a dimer and then extends to the N-terminal lysines in each monomer (Sadir et al. 2001). Ref. PF4 model in (a): This research was originally published in Journal of Biological Chemistry. Authors: Sally E. Stringer and John T. Gallagher Title: Specific Binding of the Chemokine Platelet Factor 4 to Heparan Sulfate. J. Biol. Chem. (1997) 272, 20508–20514 © the American Society for Biochemistry and Molecular Biology.”
The IL-8-binding domain in HS (also identified by the footprinting method) is shorter than the comparable PF4-binding region but similar in the overall structure being composed of about 12 disaccharides with two short dp6 S-domains connected by a predominantly NA region (Spillmann et al. 1998). In common with the model for PF4, this latter region is assumed to loop over and around IL-8, maintaining close contact with the protein surface (Figure 12b); however, the two S-domains in the IL-8 dimer lie almost parallel to the basic clusters on the a-helices in contrast to the perpendicular trajectory of the S-domains in PF4. The overall symmetry of the IL-8/HS model is attractive because it allows the S-domains to bind the IL-8 monomers with the same polarity. An alternative model to that based on footprinting is the one derived from molecular docking that shows a single heparin sequence of approximately 10 monosaccharides lying perpendicular to the C-terminal a-helices of IL-8 but extending to positively charged residues in a loop region towards the N-terminal strand (Lortat-Jacob 2009). A similar binding mode has been proposed for CXCL11, a T-cell-derived chemokine (Severin et al. 2010). Perhaps both the perpendicular and parallel modes of HS binding to IL-8 are physiologically relevant with the structure of the GAG–chemokine complex being determined by the length, sulphation and spacing of the S-domains in HS.
MIP1-α and other CCL chemokines
MIP1a (CCL3) is in the CC chemokine subfamily, and these use a different basic amino acid cluster from the CXCL subgroup to bind HS (Koopmann & Krangel 1997). The C-terminal a-helix is not involved in GAG binding. The interaction site contains a short basic CW motif of 44XKRXRX49 on a solvent-exposed beta-turn arranged in close proximity to arginine 18 (R18); three critical arg residues (R46, R48 and R18) are strictly conserved across the CC subfamily. There are some additional but non-conserved basic residues in the vicinity of this motif that may lead to variations in HS affinity or sequence preference. In CCL2 (monocyte chemoattractant protein, MCP-1) for example, the additional basic residues K19 and R24 are strong enhancers of HS affinity (Lau et al. 2004).
In the dimeric MIP1a, the only CC chemokine investigated for HS reactivity by the footprinting method, the protected region in HS was quite distinctive being composed of two long highly sulphated S-domains (dp12-dp14) placed at the ends of a short NA region of four to five disaccharide units (Stringer et al. 2002). The protected fragment was approximately 140 Å in length, sufficient to wrap around MIP1α in a horseshoe shape. Given the relatively low frequency of long dp12-dp14 S-domains in HS, the MIP1α-binding site is likely to be quite specific and potentially very stable. The difference in the overall structure of the MIP1a-protected site compared to that protected by IL-8 and PF4 is quite striking and reflects the potential for highly selective chemokine–HS interactions on cell surfaces.
MIP1α is a major regulatory component in the bone marrow where it functions as a reversible inhibitor of haemopoietic stem cell (HPC) proliferation and thus helps to maintain stem cells in a quiescent state. Heparan sulphate significantly augments its action in supporting the long-term maintenance of HPCs in culture and may also determine its location in the bone marrow stem cell niche (Stringer et al. 2003).
The CCL chemokine RANTES, a T-cell-derived chemotactic factor, shares some binding characteristics with MIP1a. It is monomeric in solution but forms dimers in the presence of heparin oligosaccharides of size dp16/dp18 (Vives et al. 2002); synthetic oligomers composed of two dp8 heparin-like sequences connected by a flexible linker that mimics the spacing of S-domains in HS bind and dimerize RANTES with high efficiency. This interesting observation supports the general proposition that HS chain flexibility will enable two spaced S-domains to engage simultaneously with two positively charged areas on the surface of chemokine dimers.
SDF1-α and SDF1-gamma: novel interactions with HS
SDF1-α (CXCL12) is a widely expressed CXCL chemokine that acts as a migration factor for a variety of circulating blood cells including monocytes and T cells. In the bone marrow endothelium, it is one of the main attractants for homing of circulating HPCs and for successful engraftment of the transplanted bone marrow (Netelenbos et al. 2003). The HS-binding domain in SDF1-α is a novel conformational site situated along the interface of the monomer units and then extending to two N-terminal lysines at the periphery of the protein (Sadir et al. 2001; Figure 12c). In the SDF1-α monomer, a crestlike ‘half-site’ is formed by a CW motif in the first beta-strand and Arg 41/Lys 43 in the second beta-strand; at the junction of the antiparallel dimer, these half-sites converge to form a positively charged ‘crevasse-like’ region that accommodates a dp6 heparin sequence. But for the full occupancy of the binding site, contact with the two terminal lysines is also needed, and this is attained by dp12 to dp14 saccharides (Sadir et al. 2001).
SDF1-α-binding affinity is dominated by the two lysines (K24 and K27) in the CW motif and on the GAG side by N- and 2-O-sulphates, with 6-O-sulphates playing only a minor role. The HS-binding site in SDF1-α is clearly different from PF-4 and IL-8 where the interactions are mainly focused on the α-helices. The extension of the SDF1-α site to the NT is significant; this region contains key elements for receptor binding, and HS efficiently protects it from cleavage by serine proteases (Sadir et al. 2004).
SDF1-gamma – a chemokine with repetitive CW motifs
Several isoforms of SDF1 are generated by alternative splicing of the sdf1 gene including a distinctive splice variant named SDF1-gamma composed of an identical core to SDF1-α but with the addition of an unstructured, positively charged C-terminal region with four CW-type XBBXBX motifs (Laguri et al. 2007; Rueda et al. 2008). This region acts together with the basic residues in the core of SDF1-gamma to form a very high-affinity region for HS (Kd 0.9 mM) that leads to a tight and prolonged association with cell surfaces close to the site of release. Despite some attenuation of agonist function by comparison with SDF1-α, the stable binding of SDF1-gamma to HS was instrumental in its superior potency in assays of intraperitoneal leucocyte migration and in in vitro angiogenesis models (Rueda et al. 2012). SDF1-gamma is an important chemoattractant for the neovascularization of ischaemic muscle tissue and a key migration factor in embryogenesis. Its tight and persistent association with HS may enable it to form durable concentration gradients that serve as guidance paths for the homing of specific progenitor cells in the early stages of organ development (Rueda et al. 2012).
Diffusion effects
Embryogenesis is dependent on a complex interplay of diffusible signalling proteins (morphogens) that emit from ‘organizing centres’ and specify distinct cell fates across fields of cells in a concentration-dependent manner. Genetic screens in Drosophila have elucidated important roles for HS polymerases, HSMEs and HSPG core proteins in various differentiation pathways controlled by the spatial distribution of the major morphogenic proteins, hedgehog (Hh), wingless (W) and decapentaplegic (DPP); these are Drosophila orthologues of the vertebrate hedgehog (Hh), Wnt and bone morphogenetic protein (BMP) families of morphogens and growth factors, and all are heparin-/HS-binding proteins (Yan & Lin 2009).
In Drosophila, morphogen diffusion is mainly regulated by the GPI-anchored Dally (division abnormally delayed) and Dally-like (Dlp) cell surface HSPG core proteins, the counterparts of lon1/lon2 in Caenorhabditis elegans and glypicans in mammals (Lin & Perrimon 2000; Selleck 2001; Lin 2004). In these PGs, the HS chains are positioned close to the cell surface where they may form organized polymer networks that define (and restrict) diffusion paths whilst maintaining intimate contact between morphogens and cognate signalling receptors (Hufnagel et al. 2006). Heparan sulphate also affects morphogen stability and conformation, and there may be an active component in the diffusion mechanism because morphogens tend not to move across cell clones deficient in HS. Wg/Wnt and Hh are lipid-modified proteins, and HS is important for maintaining them in a soluble form in the gradient field (Yan & Lin 2009). The HS/glypican regulation of Wg/Wnt proteins is complex and involves negative as well as positive features. The developmentally regulated enzyme Notum was recently shown to impair the signalling activity of Wnt by specifically removing its lipid component (Kakugawa et al. 2015); in this important study, Notum was also identified as an HS-binding enzyme that would bring it into close proximity with its Wnt substrate.
Local variations in HS structure along the gradient may influence morphogen signalling. In quail embryos, Sulf-mediated modification of the 6-O-sulphation patterns in HS mediates the transfer of Wnt from HS to its signalling receptor frizzled (Ai et al. 2003). Sulfs may be strategically positioned in Wnt gradients to elicit key events in cell specification and patterning. Although endocytosis appears to play only a minor role in gradient formation, it is essential for morphogen signalling and HSPGs, in close contact with membrane receptors, may have a significant influence on the internalization of signalling complexes. The mechanisms for establishing morphogen gradients are not fully understood, and in some developmental settings, there may be a significant contribution from free diffusion (Lander et al. 2002) with HSPGs employed in gradient sensing and transmission of the morphogenic signal. An elegant new technique developed by Fernig et al. for tracking single proteins in the pericellular matrix of cultured cells (Duchesne et al. 2012) offers a novel approach for the analysis of morphogen diffusion and the mechanisms involved in shaping morphogen gradients.
Hedgehog (Hh) proteins
Hh was first identified in Drosophila as a segment polarity gene essential for establishing the basic body plan. In mammals, there are three genetically distinct Hh proteins: sonic, Shh; Indian, Ihh; and Desert, Dhh (see McMahon et al. 2003 for review). During embryogenesis, many of the pleiotropic actions of Hh proteins that include cell patterning, differentiation and progenitor cell proliferation result in part from eliciting specific modifications in gene expression as a function of Hh concentration. In adults, Hh signalling is largely concerned with tissue maintenance, repair and supporting the integrity of stem cell niches in the brain. Shh is fundamental to the proliferation of cerebellar granule cells during central nervous system development. Its action is dependent on HS and correlates with increased expression of ext1/ext2 HS co-polymerases (Rubin et al. 2002). The HSPG that mediates this effect is glypican-5. The Hh pathway is commonly reactivated in human tumours, often in a paracrine pathway in which Hh released by tumour cells brings about changes in cellular composition of the surrounding stroma (Filmus & Capurro 2014). Hh signalling is of particular importance in pancreatic cancers; these tumours are often surrounded by a dense, poorly vascularized stromal tissue that restricts drug access and confers resistance to standard chemotherapy. In rhabdomyosarcoma, a childhood tumour, Hh signalling is again activated by glypican-5 and its mode of action depends on the constituent HS chains that bind both Hh and its receptor patched (Li et al. 2011). This study also showed that Hh signalling is further augmented by the CS chains on glypican-5.
Of the various members of the Hh family, Shh is the most thoroughly studied in connection with HS/heparin/GAG reactivities. The Shh monomer (19 kDa) is derived from a proprotein by autocatalytic cleavage and interacts with HS/heparin using two binding sites: an 32XBBBXXBX39 CW motif in the unstructured N-terminal region (Rubin et al. 2002; Farshi et al. 2011) and a conformational site made up of five basic residues – K88, R124, R154, R156, K179 – in the central, globular region of the protein (Whalen et al. 2013); the latter site spans about six monosaccharides in heparin. Although the two sites can act independently, molecular modelling and site-specific mutagenesis suggest that Lys 179 (178 in mouse) may be pivotal in establishing functional connectivity between them (Chang et al. 2011). Two appropriately spaced S-domains, with distinct sulphation patterns that complement the different shape and charge characteristics of the CW and central binding sites, may be required for Shh to bind HS with high affinity.
In the secretory pathway, Hh monomers, modified by palmitate at the NT and cholesterol towards the C-terminal region, cluster in the cell membrane as large aggregates in an HS-dependent mechanism (Vyas et al. 2008). GPI-linked glypicans and Shh clusters become localized in lipid rafts, suggesting that glypican HS is involved in the aggregation process. Shh clusters are then released from the cell surface, but the mechanism is unclear. They may dissociate as micelles, in association with lipoprotein particles, as cross-linked complexes or even freely diffusible monomers (Ohlig et al. 2012; Whalen et al. 2013). Shh monomers tend to be selected for short-range signalling and complexes for long-range effects (Ayers et al. 2010; Filmus & Capurro 2014). Shh micelles formed on glypican HSPGs could be released by proteolytic or phospholipase-mediated shedding of the glypican ectodomain (Hufnagel et al. 2006; Ayers et al. 2010). Grobe et al. have shown that Shh clusters can be cross-linked by transglutminase (Dierker et al. 2009a) and then released from the cell surface by ADAM proteases that remove the lipids by acting at the N- and C-termini (Dierker et al. 2009b; Ohlig et al. 2011). N-terminal processing also disrupts the CW motif, which will have the effect of reducing constraints on diffusion in an HS-rich matrix. Various lines of evidence indicate that HS sulphation patterns are key factors determining how Shh multimers are assembled and released (Chang et al. 2011; Ohlig et al. 2012; Whalen et al. 2013). The formation of large Shh complexes is an intriguing and novel characteristic that must serve some basic purposes in signalling. Perhaps Shh clusters maintain protein stability during long-range diffusion and may be essential for the precise orchestration of cell patterning during development.
Heparan sulphate in cell adhesion and ECM organization
The ECM is a dynamic continuum that has an instructive influence on cell growth, motility and differentiation. Growth factor signalling in embryonic and adult tissues is critically dependent on cell attachment to an appropriate matrix. Heparan sulphate plays an important role in the organization of the ECM, and ECM proteins use cell surface HS as a means of conveying information across the cell membrane.
Fibronectin
Fibronectin (FN) is a large, disulphide-linked, dimeric protein, widely distributed in the ECM where, in common with other matrix proteins, it provides support for cell adhesion and traction and guidance for cell migration (for a review, see Schwarzbauer & DeSimone 2011). Heparin and HS induce conformational changes in FN that affect its binding to integrins and its assimilation into the ECM. Fibronectin fibrillogenesis is a cell surface event in which specific interactions with α5/b1 integrin and syndecan-2 are essential steps in the process (Arrington & Yost 2009; Choi et al. 2011). Heparan sulphate appears to unmask cryptic binding sites in FN for PDGFA and VEGF possibly by bringing about a more elongated, open structure of the FN molecule (Mitsi et al. 2006; Smith et al. 2009). The HS-dependent binding of PDGFA to FN is vital for directing the migration of mesendoderm cells at the onset of gastrulation. Interestingly, mouse embryos defective in HS biosynthesis arrest at the gastrula stage (Lin et al. 2000).
There are three separate HS-binding regions in the FN monomer: a low affinity and low specificity Hep 1 site at the NT, a dominant high-affinity site, Hep II, located downstream from the RGD cell-binding domain for α5/b1 integrin and a third site in the centre of the alternatively spliced IIICS region adjacent to the LDV-binding motif for a4b1 integrin (Figure 13, Sharma et al. 1999; Schwarzbauer & DeSimone 2011). The isolated Hep II site binds HS and heparin with comparable affinity to FN (Walker & Gallagher 1996). The co-ordinated binding of the FN Hep II region with syndecan-4 and the RGD site with α5/b1 integrin lead to the assembly of focal adhesions, a complex process that requires substantial reorganization of the cytoskeleton driven by the Rho family of small GTPases (Couchman 2010). The Hep II/HS interaction directs signals into these pathways via the activation of protein kinase C mediated by the unique variable region in the cytoplasmic tail of syndecan-4 (Couchman 2010). The HS-binding site in the IIICS region of FN also works in tandem with its neighbouring a4b1 integrin site to promote cell adhesion and migration of melanoma cells (Mostavi-Pour et al. 2001).
Figure 13.
Cell- and HS-binding regions of fibrillin and fibronectin. The diagram illustrates the similarity in arrangement of the major integrin and HS-binding regions of fibrillin and fibronectin. The co-operative interactions of these sites with cell surface integrins and HSPGs are essential for cell attachment to the ECM and for matrix-driven focal adhesions and signalling.
Most investigations of the GAG-binding characteristics of FN have centred on the Hep II site. In affinity chromatography, Hep 11 showed a strong preference for HS (and heparin) over DS and CS; Hep II interacted exclusively with the S-domains of HS, and dp6 and dp8 saccharides were in the minimal range for a stable interaction (Walker & Gallagher 1996). Affinity increased with saccharide size up to a maximum size of dp14 S-domains with high dependence on N- and, to a lesser extent, 2-O-sulphate groups for efficient binding; further augmentation was derived from the presence of 6-sulphation of GlcNS residues. COO− groups made little contribution to the interaction (Lyon et al. 2000). Binding data were broadly substantiated in cell adhesion assays in which heparin saccharides were used to inhibit cell attachment to surface-adsorbed Hep II. Maximum inhibition was attained with dp14 heparin saccharides, and the prominent role of N-sulphation was confirmed using N-desulphated HS and heparin polymers (Mahalingam et al. 2007).
Hep II is made up of three FN type 3 repeats (III 12–14) and contains two HS-binding sites, with the primary site in the III 13 module and the secondary site in the adjacent III 14 repeat (Barkalow & Schwarzbauer 1991). In the primary site, a cluster of six basic amino acids (R98, R99, R101, R115, K117, R146) arranged in the shape of a ‘cationic cradle’ act synergistically in binding to heparin/HS with mutation of any one of these residues leading to a 10- to 20-fold reduction in affinity (Busby et al. 1995). The first three arginines in the primary site conform to a CW motif and line on one side of the cradle. The primary site and the lower affinity HS binding site in the III 14 repeat appear on the same surface of Hep II where together they form a combined region that extends over 60 Å (Sharma et al. 1999), sufficient to engage an HS dp14 S-domain with a helical translation of approximately 8.5 Å per disaccharide (Mulloy & Forster 2000). In HS itself, this extended site could be occupied by two short, suitably spaced S-domains. Differences in sulphation patterns and/or arrangement of S-domains may explain why HS species of similar overall sulphate content (mucosal and kidney HS) differ significantly in Hep II affinity (Lyon et al. 2000).
It is of interest to reflect on the likelihood of protein conformational effects on the HS reactivity of Hep II with FGF1 and FGF2. The two clusters of positively charged residues that constitute the Hep II site contain a total of 11 basic residues compared with only four in FGF2 (three lys, one arg.) and five in FGF1 (four lys, one arg). However, Hep II binds HS and heparin with only moderate affinity (Kd in the range of 0.1–1.0 μM), about 10- to 100-fold weaker than the interaction with FGF1 and FGF2. Arginine residues, known to prefer co-ordination with sulphate ions (Fromm et al. 1995), predominate in the extended Hep II site. In the FGF sites, mainly composed of lysines, protein folding creates compact interaction domains that may impose steric constraints on lysine side chains. This effect may confer more stringent requirements for HS sequence complementarity coupled with enhanced HS affinity if the local peptide environment increases the electrostatic interaction potential of the side-chain amino groups.
Fibrillin
Fibrillin is a large (350 kDa), modular, connective tissue glycoprotein organized into insoluble microfibrils that form the framework for the assembly of elastin. Fibrillin microfibrils (10–12 nm diameter) are present in blood vessels, ligaments, lungs and other compliant tissues that require elasticity and flexibility to resist stretch and pressure forces. It is now clear that interactions with HS in the pericellular region of secreting cells are an important step in the microfibril formation and for the deposition of tropoelastin (the soluble precursor of elastin) into the ECM (Kielty et al. 2002). Heparan sulphate is important for elastogenesis and repair of damaged connective tissues perhaps by acting as a template for docking the tropoelastin onto the fibrillin architecture (Buczek-Thomas et al. 2002; Tu & Weiss 2008).
Of the four members of the fibrillin family, fibrillin 1 is the major GAG-binding isoform. Its structure is dominated by 43 calcium-binding consensus modules interspersed by cystine-rich TGF beta-binding (TB) protein domains (Figure 13). Multiple heparin/HS interaction sites are distributed along the fibrillin monomer, but although the significance of all these sites is not fully resolved, considerable progress has been made in elucidating the functions of three of them (Tiedemann et al. 2001; Cain et al. 2005). For example, the HS-binding activity in a region located at the NT plays an important role in fibril assembly, cell attachment and cell spreading, whereas a novel, conformation-dependent site towards the C-terminal region is essential for elastin deposition (Cain et al. 2005). Heparin binding in the NT site is impaired by the Marfan mutation T101A. A heparin-/HS-binding site involved in cellular interactions has been detected in the centrally located TB5 module. This module, which contains two subsites each with pairs of critical arginines, plays a key role in cell adhesion to fibrillin (Cain et al. 2008). TB5 is relatively close to TB4, which contains an RGD sequence for integrin recognition. The interaction of cell surface HS with TB5 appears to function in an analogous manner to the HS-binding site in the Hep II region of FN by cooperating with integrins in matrix-driven cell binding and formation of focal adhesions (Bax et al. 2007). This is an important observation. Kielty et al. (Cain et al. 2008) propose that the common arrangement of functionally coupled cell adhesion modules in fibrillin 1 and FN reflects an important evolutionary development in the emergence of an instructive ECM.
The in vivo significance of HS binding in the TB5 region was revealed by the analysis of three rare autosomal dominant disorders of skeletal development (Weill–Marchesani syndrome and acromicric and geleophysic dysplasias). Molecular mapping pinpointed the mutations in the HS-binding sites of TB5, and it was shown experimentally that the mutant proteins had significantly impaired HS affinities (Cain et al. 2012). The authors suggest that such disruptive effects will be a major factor in the molecular pathogenesis of these diseases as a consequence of aberrant cellular interactions in the microfibrillar network.
Collagens
Collagens are the most abundant and widespread group of proteins in the ECM. There are 28 members of the collagen family, and their varied molecular architectures reflect an extensive range of functions in the matrix environment. Collagens are modular proteins composed of three polypeptide chains, the α-chains, with at least one region arranged in the form of a triple helix that imparts a rodlike shape to the helical section of the collagen ‘monomer’. In the fibrillar collagens (e.g. collagens I, III and V), the monomers combine in fibrils of different length and diameter that confer shape and stability to tissues such as skin, bone, cartilage, tendon and cornea. Other collagen monomers form networks (e.g. type IV collagen in basement membranes) or bind specifically to the surfaces of collagen fibrils (FACIT collagens) (Ricard-Blum 2011 for review). Heparan sulphate-binding sites are found mainly in fibrillar collagens but also in the helical region of the synaptic acetylcholinesterase (AChE) complex.
Collagen type I
Bernfield et al. demonstrated that a mammary cell surface HSPG (syndecan-1) bound strongly by means of its HS chains to a single saturable site on type I collagen fibrils and that syndecan-1 was an important cell membrane receptor for this collagen type (Koda et al. 1985). The main HS-binding region was later shown to be in the α1-chains towards the N-terminal region of the triple helix; the site was highly selective for heparin and HS (San Antonio et al. 1994). The binding region was defined by a short and novel sequence of 87KGHRGF92 in the α1-chain. In the native type I monomer, which contains two α1-chains and one α2-chain, two such sequences combine to form a single high-affinity heparin-binding site with Kd of approximately 150 nM (San Antonio et al. 1994). The presence of this small cluster of basic residues may affect the local conformation of collagen that favours a strong association with HS and influences the size and shape of the collagen fibril (Sweeney et al. 1998). The heparin-binding region coincides with a site in collagen 1 that is important for the induction of endothelial tube formation in vitro. It seems probable that this site interacts with HS on endothelial cells and is involved in regulating the pro-angiogenic properties of type I collagen.
Collagen types V and XI
Collagens V and XI are genetically related regulatory collagens that perform vital roles in matrix organization and cell–matrix interactions. They are often described as minor fibrillar collagens because of their low abundance in collagen fibres where they co-associate with the major collagens types I and II during nucleation and assembly of heterotypic fibrils. Collagen V co-associates with collagen I in the small, uniform, optically transparent fibrils in the cornea, and collagen XI combines with collagen II to form a dense network of fine fibrils in the pericellular matrix of chondrocytes (Ricard-Blum 2011). Chondrocyte adherence to this dense matrix is dependent on cell surface HS (Vaughan-Thomas et al. 2001). Interactions with ECM and cell surface HSPGs are essential for the common functional properties of collagens V and XI.
Heparan sulphate-/heparin-binding sites in the helical regions
There are several genetic variants of collagens V and XI, but the principle molecular entities are the combination of α1(V)2, α2(V) for collagen V and the heterotrimer a1(XI), a2(XI), a3(XI) for collagen XI. Sequence alignment of the helical sections of the α1-chains indicated the presence of two common regions for GAG recognition in collagens V and XI (Delacoux et al. 1998; Warner et al. 2006; Figure 14a). The main site extends from residues 905Lys to Arg921 and is composed of 905KPGPRGQRGPTGPRGER921. A second site lies closer to the NT of the triple helix and incorporates an XBBXBX CW motif in the sequence 574GKPGRKGRPGADGGR588. This latter site was also identified by rotary shadowing EM of a heparin–BSA conjugate bound to native type XI collagen (Vaughan-Thomas et al. 2001). Homologous regions to both the above sequences are also present in the α2-chains of collagen XI but not in the α2-chains of collagen V. The stoichiometry of the α-chains in collagens V and XI will influence heparin-/HS- binding affinities. For example, in collagen V, the α1(V)3 homotrimer will have a greater affinity than the more frequently encountered α1(V)2, α2(V) heterotrimer (Delacoux et al. 1998).
Figure 14.
(a) Heparan sulphate (HS)/heparin binding sites in the N-terminal domain (NTD) and helical region of the α1 chain of collagen XI. Heparan sulphate-/heparin-binding regions in collagen α1(XI)-chain are found in the 223-residue globular Npp domain in the form of an XBBBXXBX CW motif, in the highly basic variable region, and in two sites in the major triple helix including a similar sequence to the 905–921 residue sequence present in the collagen type α1(V)-chain. The CW motif in the Npp domain is also present in collagen α1(V). The NTD of the α1-chain is retained in the collagen XI heterotrimeric triple helix and projects from the surface of the collagen fibril. The NTDs of the α2- and α3-chains are rapidly removed by proteolysis before assembly of the collagen XI monomer into fibrils. (b) Schematic diagram of the binding sites in synaptic collagen Q. Collagen Q is found only in the neuro-muscular synapse. It contains two (XBBXBX) CW motifs in the triple helix that interact with perlecan HS in the synaptic cleft. The non-collagenous C-terminal region binds to the muscle-specific Musk receptor. The three non-helical N-terminal regions bind four AchE subunits in an asymmetric A12/Q complex that degrades Ach and controls the strength and duration of synaptic transmission.
Mutagenesis of basic residues in the 905–921 sequence of collagen V indicated that three arginine residues (R 912, R918 and R921) are essential for heparin binding with Lys 905 and Arg 909, providing additional points of contact in what is a unique type of motif for the recognition of HS and heparin (Delacoux et al. 2000; Ricard-Blum et al. 2006). The heparin/HS affinity of this sequence is critically dependent on the conformation imposed by the overall structure of the α1-chains; the sequence per se lacks binding activity. Molecular modelling predicted an amphipathic character of the GAG-binding region with the basic residues of the α-chains exposed on the surface of the triple helix. A 12-kDa recombinant fragment (Hep V) of the α1-chain of collagen V, which included the 905–921 binding site, had cell adhesion properties and bound GAGs with high affinity (18 nm and 35 nM for heparin and HS respectively). The binding site accommodated dp8/dp10 heparin/HS saccharides, and the interaction was particularly dependent on 2-O-sulphate groups (Ricard-Blum et al. 2006).
The summation of the potential multivalent interactions between HS and its binding sites in the triple helical regions of both collagens V and XI is likely to be significant for maintaining the organization and resilience of the ECM. Heparan sulphate components of the ECM HSPG perlecan could bind across several collagen fibrils with variable affinities dependent on the lengths and patterns of sulphation of the HS chains. These interactions could contribute significantly to interfibrillar spacing and matrix flexibility and may be advantageous during periods of controlled remodelling during growth, turnover and repair.
Heparan sulphate/heparin binding in the N-terminal domains (NTDs)
All collagens are secreted as triple helical structures with globular N- and C-terminal propeptides that maintain the solubility of collagen monomers during intracellular transport and release. These terminal, non-helical regions are normally removed prior to the formation of insoluble fibrils, but in some instances, the NTDs are retained and although they do not block fibril formation, they have a significant impact on fibril shape and fibril diameter. In collagen types V and XI, these regions also contain important HS-binding sites.
The NTDs of the α1-chains of collagens V and XI are particularly important for controlling the diameter and spacing of small fibrils and for dictating cellular interactions with the ECM. There is one common heparin-/HS-binding site in the NTDs of these collagens located in the so-called amino propeptide (Npp) region towards the NT, and an additional site is in an alternatively spliced region of collagen XI near to the collagenous domain (Figure 14a). The common Npp site is positioned in a beta-strand in residues 147–152 and conforms to the CW XBBBXXBX motif in which the basic amino acids are exclusively lysines (Fallahi et al. 2005; Warner et al. 2006). In contrast, the site in the variable region of collagen XI NTD is a long positively charged sequence containing multiple clusters of arg/lys residues (Warner et al. 2006) somewhat analogous to the unique HS-binding region in SDF1-gamma. This sequence is likely to bind with high affinity to HS. A similar sequence is present in the rare collagen α4V-chain. Unfortunately, there are no published data on the HS/heparin sequences that interact with NTDs at the Npp site or the site in the alternatively spliced region. As noted above, the significance of these regions is reflected in their slow proteolytic processing and persistence on the fibril surface where they remain accessible to the cell surface and ECM HSPGs. Specific collagen-binding heparin or HS sequences could be of considerable value in the design of the fibrillar architecture of biomatrices for tissue reconstruction and repair.
Collagen α4V-chain and the development of the peripheral nervous system
The collagen α4(V)-chain was first identified by Carey et al. as a heparin-binding, collagenous protein synthesized by Schwann cells during the emergence of the peripheral nervous system (PNS) (Chernousov et al. 1996). Molecular cloning revealed it to be a novel member of the collagen V, α-chain subfamily with high-sequence identity to the type V α1- and α3-chains, although notably it lacked the HS-binding site in the Npp region (Rothblum et al. 2000). The α4 (V)-polypeptide is present as a component of triple helical collagen monomers that also contain α1(V)- and α2(V)-chains. The high-affinity HS-/heparin-binding site in a4(V) is located centrally in the NTD region and contains four heparin-binding, consensus motifs of the CW XBBXBX type in a short stretch of only 23 amino acids (Erdman et al. 2002).
In contrast to the more widely expressed α(V)-chain, synthesis of the α4(V)-polypeptide is largely confined to the late embryonic and neonatal nervous system. It is secreted during periods of active Schwann cell migration and induces cell adhesion and spreading through interactions with cell surface HSPGs, principally glypican-1 and syndecan-3. The α4-chain is also adhesive for sensory neurones and a supportive substrate for axon migration (Chernousov et al. 2001). These effects, which are dependent on the high-affinity HS-binding site in the α4(V)-NTD, are essential for the terminal differentiation of Schwann cells, the onset of nerve myelination and the eventual establishment of the PNS (Erdman et al. 2002). It is notable that whilst the α4(V)-NTD can persist in the ECM still attached to the collagen fibril, a significant fraction is released by proteolytic scission at a single site close to the helical region. In vivo, the released component tends to concentrate on cell surface HS and may be an important effector of α4(V) in PNS development (Rothblum et al. 2004). These interesting findings provide new ideas for the development of novel molecular and genetic approaches in the treatment of peripheral nerve disorders.
Collagen Q; the synaptic collagen
Collagen Q (colq) is a triple helical, non-fibrillar collagen (Figure 14b) located exclusively in the cholinergic synapse. It is vital for synaptic organization and the control of synaptic transmission. Mutations in the colq gene cause congenital end-plate deficiency and myasthenic syndrome (Ohno et al. 2013). Colq is the collagenous ‘tail’ of the asymmetric form of AChE that positions the enzyme in the basal lamina by interactions with synaptic HS and with a receptor kinase, MuSK, located specifically in the synaptic muscle membrane (Brandan et al. 1985; Cartaud et al. 2004). Each of the three NTDs of the colq monomer recruits four catalytic subunits of AChE (A12/Q) that associate as enzymatically active tetramers. Acetylcholinesterase regulates the strength and duration of synaptic transmission by controlled degradation of acetylcholine.
Two XBBXBX CW motifs in the short 166-residue helix mediate interactions of colq with HS (Figure 20). Despite similarities in basic charge in the two sequences (GRKGR and GKRGK), variations in conformation of the helical architecture lead to a significantly higher HS/heparin affinity for the C-terminal site (Deprez et al. 2003). In the colq homotrimer, the two HS-binding sequences each form clusters of positive charge around the triple helix that disrupts the H-bonding pattern and impairs helix stability. This leads to increased local flexibility that favours HS/heparin binding. The GAG-binding specificities of the colq CW motifs have not been investigated, but the presence of two sites with differential binding affinities and potentially distinct sequence preferences is probably important in positioning the catalytic head of the A12/Q complex in the synaptic nerve ending (Kimbell et al. 2004). Colq interacts mainly with the HS chains of perlecan, a component of the dystroglycan complex and one of the major structural elements in the synaptic muscle membrane (Rotundo et al. 2005). Perlecan contains two to three HS chains clustered towards the NT, and the stability of such interactions may be favoured by particular characteristics of perlecan HS (e.g. fine structure and spacing of sulphated regions) that complement the positively charged distribution patterns in the colq triple helix.
Concluding remarks
Heparan sulphate is an information-bearing polymer in which sugar residue sequences, modified by distinct sulphation patterns, confer precise, biologically relevant specificities for protein recognition. These interactions are quite complex, often with the purpose of directing the formation of protein–protein complexes of various types such as ligand dimerization, ligand/receptor engagement and fibril formation. In addition, HS affects protein conformation, stability and diffusion. Heparan sulphate-binding sites in proteins are positively charged regions that appear in conformational or near-linear motifs and are often found at the interface of homodimers or in extended regions of positive charge where ligands and receptors converge in active complexes. Optimizing electrostatic complementarity between proteins and HS is the key to producing interactions of high affinity and specificity. CW motifs are common elements in HS-/heparin-binding sites of proteins but require additional basic residues or clustering of these motifs (e.g. in the collagen triple helix) to construct a functional interaction site. In their original paper, Cardin and Weintraub (1989) predicted that binding of the CW lys/arg residues could induce local conformational changes in proteins that bring additional basic residues into contact with sulphate and COO− groups in heparin, thus stabilizing the interaction and adding a significant dimension to the specificity. In this connection, it is worth re-emphasizing that HS is often a platform for protein–protein interactions and elucidation of the relative positions of two or more protein-binding sites along the polymer chain is an additional challenge in HS analysis. Intuitively, it seems likely that the cell-/tissue-related variations in HS structure reflects the biological need for HS to show considerable selectivity in protein binding. The advances in methods for enzymatic or chemical synthesis of HS/heparin will be a major help in identifying sequences that can be targeted at specific signalling proteins (Xu et al. 2012b; Jayson et al. 2014; Bonnaffé (2011). This is not simply an academic issue as it is self-evident that there are many potential applications of such sequences in drug design and regenerative medicine.
Acknowledgments
I owe a great debt of gratitude to many outstanding postdoctoral scientists, scientific officers and PhD students who have worked with me in the University of Manchester (Paterson Building) during my long (some might say too long!) career in the heparan sulphate field. There are too many names to mention, but their collective contributions have been immense. I must also thank colleagues in the wider proteoglycan community for invaluable advice, stimulating discussion and friendship. It is a great honour for me to be the recipient of the Fell-Muir award from the BSMB. I have enjoyed many happy times at BSMB meetings where good science blends perfectly with equally good beer-in-the-bar. My thanks also to Barbara Mulloy for helpful advice and preparing the models in Figures4 and 5 and Malcolm Lyon for the diagrams in Figure 9. Also thanks to Kai Grobe, Romain Vives, Kay Kielty and Hughes-Lortat-Jacob for clarifying specific points of interest in their publications. Finally, I acknowledge the constant and generous programme grant support from Cancer Research UK (originally the CRC) and additional awards from the MRC and Wellcome Trust.
References
- Ai X, Do AT, Lozynska O, Kusche-Gullberg M, Lindahl U. Emerson CP. Qsulf1 remodels the 6-O sulfation states of cell surface heparan sulfate proteoglycans to promote Wnt signaling. J. Cell Biol. 2003;162:341–351. doi: 10.1083/jcb.200212083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ai X, Kitazawa T, Do AT, Kusche-Gullberg M, Labosky PA. Emerson CP., Jr SULF1 and SULF2 regulate heparan sulfate-mediated GDNF signaling for esophageal innervation. Development. 2007;134:3327–3338. doi: 10.1242/dev.007674. [DOI] [PubMed] [Google Scholar]
- Alfano I, Vora P, Mummery R, Mulloy B. Rider C. The major determinant of the heparin binding of glial cell-line-derived neurotrophic factor is near the N-terminus and is dispensable for receptor binding. Biochem. J. 2007;404:131–140. doi: 10.1042/BJ20061747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen BL. Rapraeger AC. Spatial and temporal expression of heparan sulfate in mouse development regulates FGF and FGF receptor assembly. J. Cell Biol. 2003;163:637–648. doi: 10.1083/jcb.200307053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen BL, Filla MS. Rapraeger AC. Role of heparan sulfate as a tissue-specific regulator of FGF-4 and FGF receptor recognition. J. Cell Biol. 2003;155:845–858. doi: 10.1083/jcb.200106075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrington CB. Yost HJ. Extra-embryonic syndecan 2 regulates organ primordia migration and fibrillogenesis throughout the zebrafish embryo. Development. 2009;136:3143–3152. doi: 10.1242/dev.031492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asai T, Watanabe K, Ichihara-Tanaka K, et al. Identification of heparin-binding sites in midkine and their role in neurite-promotion. Biochem. Biophys. Res. Commun. 1997;236:66–70. doi: 10.1006/bbrc.1997.6905. [DOI] [PubMed] [Google Scholar]
- Ashikari-Hada S, Habuchi H, Kariya Y, Itoh N, Reddi AH. Kimata K. Characterization of growth factor-binding structures in heparin/heparan sulfate using an octasaccharide library. J. Biol. Chem. 2004;279:12346–12354. doi: 10.1074/jbc.M313523200. [DOI] [PubMed] [Google Scholar]
- Ashikari-Hada S, Habuchi H, Kariya Y. Kimata K. Heparin regulates VEGF165-dependent mitogenic activity, tube formation and its receptor phosphorylation of human endothelial cells: comparison of the effects of heparin and modified heparins. J. Biol. Chem. 2005;280:31508–31515. doi: 10.1074/jbc.M414581200. [DOI] [PubMed] [Google Scholar]
- Ayers KL, Gallet A, Staccini-Lavenant L. Thérond LP. The long-range activity of hedgehog is regulated in the apical extracellular space by the glypican dally and the hydrolase notum. Dev. Cell. 2010;18:605–620. doi: 10.1016/j.devcel.2010.02.015. [DOI] [PubMed] [Google Scholar]
- Baloh R, Enomoto H, Johnson E. Milbrandt J. The GDNF family ligands and receptors – implications for neural development. Curr. Opin. Neurobiol. 2000;10:103–110. doi: 10.1016/s0959-4388(99)00048-3. [DOI] [PubMed] [Google Scholar]
- Barkalow FJ. Schwarzbauer JE. Localization of the major heparin-binding site in fibronectin. J. Biol. Chem. 1991;266:7812–7818. [PubMed] [Google Scholar]
- Barnett MW, Fisher CE, Perona-Wright G. Davies JA. Signalling by glial cell line-derived neurotrophic factor (GDNF) requires heparan sulfate glycosaminoglycan. J. Cell Sci. 2002;115:4495–4503. doi: 10.1242/jcs.00114. [DOI] [PubMed] [Google Scholar]
- Bax DV, Mahalingham Y, Cain S, et al. Cell adhesion to fibrillin-1: identification of an Arg-Gly-Asp-dependent synergy region and a heparin-binding site that regulates focal adhesion formation. J. Cell Sci. 2007;120:1383–1392. doi: 10.1242/jcs.003954. [DOI] [PubMed] [Google Scholar]
- Beenken A. Mohammadi M. The FGF family: biology, pathophysiology and therapy. Nat. Rev. Drug Discovery. 2009;8:235–253. doi: 10.1038/nrd2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bespalov MM, Sidorova YA, Tumova S, et al. Heparan sulfate proteoglycan syndecan-3 is a novel receptor for GDNF, neurturin, and artemin. J. Cell Biol. 2011;192:153–169. doi: 10.1083/jcb.201009136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birchmeier C, Birchmeier W, Gherardi E. Vande Woude GF. Met, metastasis, motility and more. Nat. Rev. Mol. Cell Biol. 2003;4:915–925. doi: 10.1038/nrm1261. [DOI] [PubMed] [Google Scholar]
- Bonnaffé D. Bioactive synthetic heparan sulfate and heparin derivatives: from long fragments mimetics to chimeras. C. R. Chim. 2011;14:59–73. [Google Scholar]
- Brandan E, Maldonado M, Garrido J. Inestrosa NC. Anchorage of collagen-tailed acetylcholinesterase to the extracellular matrix is mediated by heparan sulfate proteoglycans. J. Cell Biol. 1985;101:985–992. doi: 10.1083/jcb.101.3.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown A, Robinson CJ, Gallagher JT. Blundell TL. Cooperative heparin-mediated oligomerization of fibroblast growth factor-1 (FGF1) precedes recruitment of FGFR2 to ternary complexes. Biophys. J. 2013;104:1720–1730. doi: 10.1016/j.bpj.2013.02.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buczek-Thomas JA, Chu CL, Rich CB, Stone PJ, Foster JA. Nugent MA. Heparan sulfate depletion within pulmonary fibroblasts: implications for elastogenesis and repair. J. Cell. Physiol. 2002;192:294–303. doi: 10.1002/jcp.10135. [DOI] [PubMed] [Google Scholar]
- Bullock S, Fletcher J, Beddington R. Wilson V. Renal agenesis in mice homozygous for a gene trap mutation in the gene encoding heparan sulfate 2-sulfotransferase. Genes Dev. 1998;12:1894–1906. doi: 10.1101/gad.12.12.1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulow HE. Hobert O. The molecular diversity of glycosaminoglycans shapes animal development. Annu. Rev. Cell Dev. Biol. 2006;22:375–407. doi: 10.1146/annurev.cellbio.22.010605.093433. [DOI] [PubMed] [Google Scholar]
- Busby TF, Argraves WS, Brew SA, Pechik I, Gilliland GL, Ingham KC. Heparin binding by fibronectin module III-13 involves six discontinuous basic residues brought together to form a cationic cradle. J. Biol. Chem. 1995;270:18558–18562. doi: 10.1074/jbc.270.31.18558. [DOI] [PubMed] [Google Scholar]
- Cain SA, Baldock C, Gallagher JT, et al. Fibrillin-1 interactions with heparin. Implications for microfibril and elastic fiber assembly. J. Biol. Chem. 2005;280:30526–30537. doi: 10.1074/jbc.M501390200. [DOI] [PubMed] [Google Scholar]
- Cain SA, Baldwin AK, Mahalingham Y, et al. Heparan sulfate regulates fibrillin-1 N- and C-terminal interactions. J. Biol. Chem. 2008;283:27017–27027. doi: 10.1074/jbc.M803373200. [DOI] [PubMed] [Google Scholar]
- Cain SA, McGovern A, Baldwin A, Baldock C, Kielty CM. Fibrillin-1 mutations causing Weill-Marchesani syndrome and acromicric and geleophysic dysplasias disrupt heparan sulfate interactions. PLoS ONE. 2012;7:e48634. doi: 10.1371/journal.pone.0048634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capila I. Linhardt RJ. Heparin–protein interactions. Agnew. Chem. Ed. Engl. 2002;41:391–412. doi: 10.1002/1521-3773(20020201)41:3<390::aid-anie390>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- Cardin AD. Weintraub HJR. Molecular modelling of protein-glycosaminoglycan interactions. Atherosclerosis. 1989;9:21–32. doi: 10.1161/01.atv.9.1.21. [DOI] [PubMed] [Google Scholar]
- Carey DJ. N-syndecan: structure and function of a transmembrane heparan sulfate proteoglycan. Perspect. Dev. Neurobiol. 1996;3:331–346. [PubMed] [Google Scholar]
- Cartaud A, Strochlic L, Guerra M, et al. MuSK is required for anchoring acetylcholinesterase at the neuromuscular junction. J. Cell Biol. 2004;165:505–515. doi: 10.1083/jcb.200307164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casu B. Lindahl U. Structure and biological interactions of heparin and heparan sulfate. Adv. Carbohydr. Chem. Biochem. 2001;57:159–206. doi: 10.1016/s0065-2318(01)57017-1. [DOI] [PubMed] [Google Scholar]
- Caterson B. Fell-Muir Lecture: Chondroitin sulphate glycosaminoglycans: fun for some and confusion for others. Int. J. Exp. Pathol. 2012;93:1–10. doi: 10.1111/j.1365-2613.2011.00807.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catlow KR, Deakin JA, Wei Z, et al. Interactions of hepatocyte growth factor/scatter factor with various glycosaminoglycans reveal an important interplay between the presence of iduronate and sulphate density. J. Biol. Chem. 2008;283:5235–5248. doi: 10.1074/jbc.M706589200. [DOI] [PubMed] [Google Scholar]
- Chang SC, Mulloy B, Magee AI. Couchman JR. Two distinct sites in sonic Hedgehog combine for heparan sulfate interactions and cell signalling functions. J. Biol. Chem. 2011;286:44391–44402. doi: 10.1074/jbc.M111.285361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernousov MA, Stahl RC. Carey DJ. Schwann cells secrete a novel collagen-like adhesive protein that binds N-syndecan. J. Biol. Chem. 1996;271:3844–13853. doi: 10.1074/jbc.271.23.13844. [DOI] [PubMed] [Google Scholar]
- Chernousov MA, Stahl RC. Carey DJ. Schwann cell type V collagen inhibits axonal outgrowth and promotes Schwann cell migration via distinct adhesive activities of the collagen and noncollagen domains. J. Neurosci. 2001;21:6125–6135. doi: 10.1523/JNEUROSCI.21-16-06125.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y, Chung H, Jung H, Couchman JR. Oh ES. Syndecans as cell surface receptors: unique structure equates with functional diversity. Matrix Biol. 2011;30:93–99. doi: 10.1016/j.matbio.2010.10.006. [DOI] [PubMed] [Google Scholar]
- Chuang CY, Lord MS, Melrose J, et al. Heparan sulfate-dependent signalling of fibroblast growth factor 18 by chondrocyte-derived perlecan. Biochemistry. 2010;49:5524–5532. doi: 10.1021/bi1005199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clore GM, Appella E, Yamada M, Matsushima K. Gronenborn AM. Three-dimensional structure of interleukin 8 in solution. Biochemistry. 1990;29:1689–1696. doi: 10.1021/bi00459a004. [DOI] [PubMed] [Google Scholar]
- Couchman JR. Transmembrane signalling proteoglycans. Annu. Rev. Cell Dev. Biol. 2010;26:89–114. doi: 10.1146/annurev-cellbio-100109-104126. [DOI] [PubMed] [Google Scholar]
- Couchman JR. Pataki CA. An introduction to proteoglycans and their localisation. J. Histochem. Cytochem. 2012;60:885–897. doi: 10.1369/0022155412464638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai E, Liu LY, Wang H, et al. Inhibition of chemokine-glycosaminoglycan interactions in donor tissue reduces mouse allograft vasculopathy and transplant rejection. PLoS ONE. 2010;6:5. doi: 10.1371/journal.pone.0010510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies JA, Yates EA. Turnbull JE. Structural determinants of heparan sulfate modulation of GDNF signalling. Growth Factors. 2003;21:109–119. doi: 10.1080/08977190310001621005. [DOI] [PubMed] [Google Scholar]
- Deakin JD, Blaum BS, Gallagher JT, Uhrin D. Lyon M. The binding properties of minimal oligosaccharides reveal a common heparan sulfate/dermatan sulfate-binding site in hepatocyte growth factor/scatter factor that can accommodate a wide variety of sulfation patterns. J. Biol. Chem. 2008;284:6311–6321. doi: 10.1074/jbc.M807671200. [DOI] [PubMed] [Google Scholar]
- Dejima K, Kanai MI, Akiyama T, Levings DC, Nakato H. Novel contact-dependent bone morphogenetic protein (BMP) signalling mediated by heparan sulfate proteoglycans. J. Biol. Chem. 2011;286:17103–17111. doi: 10.1074/jbc.M110.208082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delacoux F, Fichard A, Gourjon C, Garrone R. Ruggiero F. Molecular features of the collagen V heparin binding site. J. Biol. Chem. 1998;273:15069–15076. [PubMed] [Google Scholar]
- Delacoux F, Fichard A, Cogne S, Garrone R. Ruggiero F. Unraveling the amino acid sequence crucial for heparin binding to collagen V. J. Biol. Chem. 2000;275:29377–29382. doi: 10.1074/jbc.M004724200. [DOI] [PubMed] [Google Scholar]
- Delehedde M, Lyon M, Vidyasagar R, McDonnell TJ. Fernig D. Hepatocyte growth factor/scatter factor binds to small heparin-derived oligosaccharides and stimulates proliferation of human HaCaT cells. J. Biol. Chem. 2002;277:12456–12462. doi: 10.1074/jbc.M111345200. [DOI] [PubMed] [Google Scholar]
- Deprez P, Inestrosa NC. Krejci E. Two different heparin-binding domains in the triple-helical domain of colq, the collagen tail subunit of synaptic acetylcholinesterase. J. Biol. Chem. 2003;278:23233–23242. doi: 10.1074/jbc.M301384200. [DOI] [PubMed] [Google Scholar]
- Dhoot GK, Gustafsson MK, Ai X, Sun W, Standiford DM. Emerson CP. Regulation of Wnt signaling and embryo patterning by an extracellular sulfatase. Science. 2001;293:1663–1666. doi: 10.1126/science.293.5535.1663. [DOI] [PubMed] [Google Scholar]
- Dierker T, Dreier R, Migone M, Hamer S. Grobe K. Heparan sulfate and transglutaminase activity are required for the formation of covalently cross-linked hedgehog oligomers. J. Biol. Chem. 2009a;284:32562–32571. doi: 10.1074/jbc.M109.044867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dierker T, Dreier R, Petersen A, Bordych C. Grobe K. Heparan sulfate-modulated, metalloprotease-mediated sonic hedgehog release from producing cells. J. Biol. Chem. 2009b;284:8013–8022. doi: 10.1074/jbc.M806838200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiGabriele AD, Lax I, Chen DI, et al. Structure of a heparin-linked biologically-active dimer of fibroblast growth factor. Nature. 1998;393:812–817. doi: 10.1038/31741. [DOI] [PubMed] [Google Scholar]
- Duchesne L, Octeau V, Bearon RN, et al. Transport of fibroblast growth factor 2 in the pericellular matrix is controlled by the spatial distribution of its binding sites in heparan sulfate. PLoS Biol. 2012;10:e1001361. doi: 10.1371/journal.pbio.1001361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdman R, Stahl RC, Rothblum K, Chernousov MA. Carey DJ. Schwann cell adhesion to a novel heparan sulfate binding site in the N-terminal domain of α4 type V collagen is mediated by syndecan-3. J. Biol. Chem. 2002;277:7619–7625. doi: 10.1074/jbc.M111311200. [DOI] [PubMed] [Google Scholar]
- Esko JD. Lindahl U. Molecular diversity of heparan sulfate. J. Clin. Invest. 2001;108:169–173. doi: 10.1172/JCI13530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faham S, Hileman RE, Fromm JR, Linhardt RJ. Rees DC. Heparin structure and interactions with basic fibroblast growth factor. Science. 1996;271:1116–1120. doi: 10.1126/science.271.5252.1116. [DOI] [PubMed] [Google Scholar]
- Fallahi A, Kroll B, Warner LR. Oxford RJ. Structural model of the amino propeptide of collagen XI α1 chain with similarity to the LNS domains. Protein Sci. 2005;14:1526–1537. doi: 10.1110/ps.051363105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farshi P, Ohlig F, Pickhinke U, et al. Dual roles of the Cardin-Weintraub motif in multimeric Sonic hedgehog. J. Biol. Chem. 2011;286:23608–23619. doi: 10.1074/jbc.M110.206474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrara N. Vascular endothelial growth factor: basic science and clinical progress. Endocr. Rev. 2004;25:581–611. doi: 10.1210/er.2003-0027. [DOI] [PubMed] [Google Scholar]
- Ferreras C, Rushton G, Cole GL, et al. Endothelial heparan sulfate 6-O-sulfation levels regulate angiogenic responses of endothelial cells to fibroblast growth factor 2 and vascular endothelial growth factor. J. Biol. Chem. 2012;287:36132–36146. doi: 10.1074/jbc.M112.384875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filmus J. Capurro M. The role of glypicans in Hedgehog signalling. Matrix Biol. 2014;35:248–252. doi: 10.1016/j.matbio.2013.12.007. [DOI] [PubMed] [Google Scholar]
- Forster M. Mulloy B. Computational approaches to the identification of heparin binding sites on the surfaces of proteins. Biochem. Soc. Trans. 2006;34:431–434. doi: 10.1042/BST0340431. [DOI] [PubMed] [Google Scholar]
- Frese MA, Milz F, Dick M, Lamanna WC. Dierks T. Characterization of the human sulfatase Sulf1 and its high affinity heparin/heparan sulfate interaction domain. J. Biol. Chem. 2009;284:28033–28044. doi: 10.1074/jbc.M109.035808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fromm JR, Hileman RE, Caldwell EEO, Weiler JM. Linhardt RJ. Differences in the interaction of heparin with arginine and lysine and the importance of these basic amino acids in the binding of heparin to acidic fibroblast growth factor. ArchBiochem Biophys. 1995;323:279–287. doi: 10.1006/abbi.1995.9963. [DOI] [PubMed] [Google Scholar]
- Fuster MM, Wang L, Castagnola J, et al. Genetic alteration of endothelial heparan sulfate selectively inhibits tumour angiogenesis. J. Cell Biol. 2007;177:539–549. doi: 10.1083/jcb.200610086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher JT. Heparan sulfate: growth control with a restricted sequence menu. J. Clin. Invest. 2001;108:357–361. doi: 10.1172/JCI13713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher JT, Lyon M. Heparan sulphate. Molecular structure and interactions with growth factors and morphogens. In: Iozzo RV, editor. Proteoglycans; Structure, Biology and Molecular Interactions. New York, NY – Basel: Marcel Dekker Inc; 2000. pp. 27–60. [Google Scholar]
- Gallagher JT. Walker A. Molecular distinctions between heparan sulphate and heparin. Analysis of sulphation patterns indicates that heparan sulphate and heparin are separate families of N-sulphated polysaccharides. Biochem. J. 1985;23:665–674. doi: 10.1042/bj2300665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giraldez AJ, Copley RR. Cohen SM. HSPG modification by the secreted enzyme Notum shapes the Wingless morphogen gradient. Dev. Cell. 2002;2:667–676. doi: 10.1016/s1534-5807(02)00180-6. [DOI] [PubMed] [Google Scholar]
- Goetz R. Mohammadi M. Exploring mechanisms of FGF signalling through the lens of structural biology. Nat. Rev. Mol. Cell Biol. 2013;14:166–180. doi: 10.1038/nrm3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goger B, Halden Y, Rek A, et al. Different affinities of glycosaminoglycan oligosaccharides for monomeric and dimeric interleukin-8: a model for chemokine regulation at inflammatory sites. Biochemistry. 2001;41:1640–1646. doi: 10.1021/bi011944j. [DOI] [PubMed] [Google Scholar]
- Goodger SJ, Robinson CJ, Murphy KJ, et al. Evidence that heparin saccharides promote FGF2 mitogenesis through two distinct mechanisms. J. Biol. Chem. 2008;283:13001–13008. doi: 10.1074/jbc.M704531200. [DOI] [PubMed] [Google Scholar]
- Grünewald FS, Prota AE, Giese A. Ballmer-Hofer K. Structure–function analysis of VEGF receptor activation and the role of co-receptors in angiogenic signalling. Biochim. Biophys. Acta. 2010;1804:567–580. doi: 10.1016/j.bbapap.2009.09.002. [DOI] [PubMed] [Google Scholar]
- Guerrini M, Agulles T, Bisio A, et al. Minimal heparin/heparan sulfate sequences for binding to fibroblast growth factor-1. Biochem. Biophys. Res. Commun. 2002;292:222–230. doi: 10.1006/bbrc.2002.6634. [DOI] [PubMed] [Google Scholar]
- Guimond S, Maccarana M, Olwin BB, Lindahl U. Rapraeger AC. Activating and inhibitory heparin sequences for FGF-2 (basic FGF). Distinct requirements for FGF-1, FGF-2, and FGF-4. J. Biol. Chem. 1993;268:23906–23914. [PubMed] [Google Scholar]
- Habuchi H, Suzuki S, Saito T, et al. Structure of a heparan sulphate oligosaccharide that binds to basic fibroblast growth factor. Biochem. J. 1992;285:805–813. doi: 10.1042/bj2850805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handel TM, Johnson Z, Crown SE, Lau EK. Proudfoot AE. Regulation of protein function by glycosaminoglycans – as exemplified by chemokines. Annu. Rev. Biochem. 2005;74:385–410. doi: 10.1146/annurev.biochem.72.121801.161747. [DOI] [PubMed] [Google Scholar]
- Hartmann G, Prospero T, Brinkmann V, et al. Engineered mutants of HGF/SF with reduced binding to heparan sulphate proteoglycans, decreased clearance and enhanced activity in vivo. Curr. Biol. 1998;8:125–134. doi: 10.1016/s0960-9822(98)70059-4. [DOI] [PubMed] [Google Scholar]
- Hileman RE, Fromm JR, Weiler JM. Linhardt RJ. Glycosaminoglycan-protein interactions: definition of consensus sites in glycosaminoglycan binding regions. BioEssays. 1998;20:156–167. doi: 10.1002/(SICI)1521-1878(199802)20:2<156::AID-BIES8>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Holley RJ, Pickford CE, Rushton G, et al. Influencing hematopoietic differentiation of mouse embryonic stem cells using soluble heparin and heparan sulfate saccharides. J. Biol. Chem. 2011;286:6241–6252. doi: 10.1074/jbc.M110.178483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes O, Pillozzi S, Deakin JA, et al. Insights into the structure/function of hepatocyte growth factor/scatter factor from studies with individual domains. J. Mol. Biol. 2007;367:395–408. doi: 10.1016/j.jmb.2006.12.061. [DOI] [PubMed] [Google Scholar]
- Hufnagel L, Kreuger J, Cohen MC. Shraiman BI. On the role of glypicans in the process of morphogen gradient formation. Dev. Biol. 2006;300:512–522. doi: 10.1016/j.ydbio.2006.08.076. [DOI] [PubMed] [Google Scholar]
- Iwasaki W, Nagata K, Hatanaka H, et al. Solution structure of midkine, a new heparin-binding growth factor. EMBO J. 1997;16:6936–6946. doi: 10.1093/emboj/16.23.6936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayson GC, Miller GJ, Hansen SU, Barath M, Gardiner JM. Avizienyte E. The development of anti-angiogenic heparan sulphate oligosaccharides. Biochem. Soc. Trans. 2014;42:1596–1600. doi: 10.1042/BST20140229. [DOI] [PubMed] [Google Scholar]
- Kadomatsu K, Kishida S. Tsubota S. The heparin-binding growth factor midkine: the biological activities and candidate receptors. J. Biochem. 2013;153:511–521. doi: 10.1093/jb/mvt035. [DOI] [PubMed] [Google Scholar]
- Kakugawa S, Langton PF, Zebisch M, et al. Notum deacylates Wnt proteins to suppress signalling activity. Nature. 2015;519:187–192. doi: 10.1038/nature14259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneda N, Talukder AH, Nishiyama H, Koizumi S. Muramatsu T. Midkine, a heparin-binding growth/differentiation factor, exhibits nerve cell adhesion and guidance activity for neurite outgrowth in vitro. J. Biochem. 1996;119:1150–1156. doi: 10.1093/oxfordjournals.jbchem.a021361. [DOI] [PubMed] [Google Scholar]
- Kemp LE, Mulloy B. Gherardi E. Signalling by HGF/SF and Met: the role of heparin sulphate co-receptors. Biochem. Soc. Trans. 2006;34:414–417. doi: 10.1042/BST0340414. [DOI] [PubMed] [Google Scholar]
- Kielty CM, Baldock C, Lee D, Rock MJ, Ashworth JL. Shuttleworth A. Fibrillin: from microfibril assembly to biomechanical function. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2002;357:207–217. doi: 10.1098/rstb.2001.1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilpeläinen I, Kaksonen M, Kinnunen T, et al. Heparin-binding growth-associated molecule contains two heparin-binding β-sheet domains that are homologous to the thrombospondin type I repeat. J. Biol. Chem. 2000;275:13564–13570. doi: 10.1074/jbc.275.18.13564. [DOI] [PubMed] [Google Scholar]
- Kimbell LM, Ohno K, Engel AG. Rotundo RL. C-terminal and heparin-binding domains of collagenic tail subunit are both essential for anchoring acetylcholinesterase at the synapse. J. Biol. Chem. 2004;279:10997–11005. doi: 10.1074/jbc.M305462200. [DOI] [PubMed] [Google Scholar]
- Kinnunen T, Nolo R, Maccarana M, Lindahl U. Rauvala H. Neurite outgrowth in brain neurons induced by heparin-binding growth-associated molecule (HB-GAM) depends on the specific interaction of HB-GAM with heparan sulphate. J. Biol. Chem. 1996;271:2243–2248. doi: 10.1074/jbc.271.4.2243. [DOI] [PubMed] [Google Scholar]
- Kinnunen T, Kaksonen M, Saarinen J, Kalkkinen N, Peng HB. Rauvala H. Cortactin-src kinase signalling pathway is involved in N-syndecan-dependent neurite outgrowth. J. Biol. Chem. 1998;273:10702–10708. doi: 10.1074/jbc.273.17.10702. [DOI] [PubMed] [Google Scholar]
- Koda JE, Rapraeger A. Bernfield M. Heparan sulfate proteoglycans from mouse mammary epithelial cells. Basal extracellular proteoglycan binds specifically to native type I collagen fibrils. J. Biol. Chem. 1985;260:8157–8162. [PubMed] [Google Scholar]
- Koopmann W. Krangel MS. Identification of a glycosaminoglycan-binding site in chemokine macrophage inflammatory protein-1α. J. Biol. Chem. 1997;272:10103–10109. doi: 10.1074/jbc.272.15.10103. [DOI] [PubMed] [Google Scholar]
- Kramer KL. Yost HJ. Ectodermal syndecan-2 mediates left–right axis formation in migrating mesoderm as a cell-nonautonomous Vg1 cofactor. Dev. Cell. 2002;2:115–124. doi: 10.1016/s1534-5807(01)00107-1. [DOI] [PubMed] [Google Scholar]
- Kreuger J. Kjellen L. Heparan sulfate biosynthesis–regulation and variability. J. Histochem. Cytochem. 2012;60:898–907. doi: 10.1369/0022155412464972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreuger J, Salmivirta M, Sturiale L, Gimenez-Gallego G. Lindahl U. Sequence analysis of heparan sulfate epitopes with graded affinities for fibroblast growth factors 1 and 2. J. Biol. Chem. 2001;276:30744–30752. doi: 10.1074/jbc.M102628200. [DOI] [PubMed] [Google Scholar]
- Kreuger J, Spillmann D, Li JP. Lindahl U. Interactions between heparan sulphate and proteins: the concept of specificity. J. Cell Biol. 2006;174:323–327. doi: 10.1083/jcb.200604035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krilleke D, DeErkenez A, Schubert W, et al. Molecular mapping and functional characterization of the VEGF164 heparin-binding domain. J. Biol. Chem. 2007;282:28045–28056. doi: 10.1074/jbc.M700319200. [DOI] [PubMed] [Google Scholar]
- Krilleke D, Ng Y-S. Shima DT. The heparin-binding domainconfersdiverse functions of VEGF-A in development and disease:a structure-function study. Biochem. Soc. Trans. 2009;37:1201–1206. doi: 10.1042/BST0371201. [DOI] [PubMed] [Google Scholar]
- Kusche-Gullberg M, Nybakken K, Perrimon N. Lindahl U. Drosophila heparan sulfate, a novel design. J. Biol. Chem. 2012;287:21950–21956. doi: 10.1074/jbc.M112.350389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laguri C, Sadir R, Rueda P, et al. The novel CXCL12gamma isoform encodes an unstructured cationic domain which regulates bioactivity and interaction with both glycosaminoglycans and CXCR4. PLoS ONE. 2007;2:e1110. doi: 10.1371/journal.pone.0001110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander A. Proteoglycans: master regulators of molecular encounter? Matrix Biol. 1998;17:465–472. doi: 10.1016/s0945-053x(98)90093-2. [DOI] [PubMed] [Google Scholar]
- Lander AD, Nie Q. Wan FY. Do morphogen gradients arise by diffusion? Dev. Cell. 2002;2:785–796. doi: 10.1016/s1534-5807(02)00179-x. [DOI] [PubMed] [Google Scholar]
- Lau EK, Paavola CD, Johnson Z, et al. Identification of the glycosaminoglycan binding site of the CC chemokine, MCP-1: implications for structure and function in vivo. J. Biol. Chem. 2004;279:22294–22305. doi: 10.1074/jbc.M311224200. [DOI] [PubMed] [Google Scholar]
- Ledin J, Staatz W, Li JP, et al. Heparan sulfate structure in mice with genetically modified heparan sulfate production. J. Biol. Chem. 2004;279:42732–42741. doi: 10.1074/jbc.M405382200. [DOI] [PubMed] [Google Scholar]
- Li F, Nandini CD, Hattori T, et al. Structure of pleiotrophin- and hepatocyte growth factor-binding sulfated hexasaccharide determined by biochemical and computational approaches. J. Biol. Chem. 2010;285:27673–27685. doi: 10.1074/jbc.M110.118703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Shi W, Capurro M. Filmus J. Glypican-5 stimulates rhabdomyosarcoma cell proliferation by activating Hedgehog signalling. J. Cell Biol. 2011;192:691–704. doi: 10.1083/jcb.201008087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lietha D, Chirgadze DY, Mulloy B, Blundell TL. Gherardi E. Crystal structures of NK1–heparin complexes reveal the basis for NK1 activity and enable engineering of potent agonists of the MET receptor. EMBO J. 2001;20:5543–5555. doi: 10.1093/emboj/20.20.5543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X. Functions of heparan sulfate proteoglycans in cell signaling during development. Development. 2004;131:6009–6021. doi: 10.1242/dev.01522. [DOI] [PubMed] [Google Scholar]
- Lin X. Perrimon N. Role of heparan sulfate proteoglycans in cell-cell signaling in Drosophila. Matrix Biol. 2000;19:303–307. doi: 10.1016/s0945-053x(00)00073-1. [DOI] [PubMed] [Google Scholar]
- Lin X, Wei G, Shi Z, et al. Disruption of gastrulation and heparan sulfate biosynthesis in EXT1-deficient mice. Dev. Biol. 2000;224:299–311. doi: 10.1006/dbio.2000.9798. [DOI] [PubMed] [Google Scholar]
- Lindahl U. Li JP. Interactions between heparin sulphate and proteins-design and functional implications. Int. Rev. Cell Mol. Biol. 2009;276:105–159. doi: 10.1016/S1937-6448(09)76003-4. [DOI] [PubMed] [Google Scholar]
- Lindahl U, Kusche M, Lidholt K. Oscarsson LG. Biosynthesis of heparin and heparan sulphate. Ann. N.Y. Acad. Sci. 1989;556:36–50. doi: 10.1111/j.1749-6632.1989.tb22488.x. [DOI] [PubMed] [Google Scholar]
- Lortat-Jacob H. The molecular basis and functional implications of chemokine interactions with heparan sulphate. Curr. Opin. Struct. Biol. 2009;19:543–548. doi: 10.1016/j.sbi.2009.09.003. [DOI] [PubMed] [Google Scholar]
- Lortat-Jacob H, Turnbull JE. Grimaud JA. Molecular organization of the interferon gamma-binding domain in heparan sulphate. Biochem. J. 1995;310:497–505. doi: 10.1042/bj3100497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lortat-Jacob H, Grosdidier A. Imberty A. Structural diversity of heparan sulfate binding domains in chemokines. Proc Natl Acad Sci USA. 2002;99:1229–1234. doi: 10.1073/pnas.032497699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y, Ye S, Kan M. McKeehan WL. Control of fibroblast growth factor (FGF7)- and FGF1-induced mitogenesis and downstream signalling by distinct heparin octasaccharide motifs. J. Biol. Chem. 2006;281:21052–21061. doi: 10.1074/jbc.M601559200. [DOI] [PubMed] [Google Scholar]
- Lyon M, Steward WP, Hampson I. Gallagher JT. Identification of an extended N-acetylated sequence adjacent to the protein-linkage region of heparan sulphate. Biochem. J. 1987;242:493–498. doi: 10.1042/bj2420493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyon M, Deakin J. Gallagher JT. Liver heparan sulphate structure: a novel molecular design. J. Biol. Chem. 1994;269:11208–11215. [PubMed] [Google Scholar]
- Lyon M, Rushton GR, Askari JA, Humphries M. Gallagher JT. Elucidation of the structural features of heparan sulfate important for interaction with the Hep-2 domain of fibronectin. J. Biol. Chem. 2000;275:4599–4606. doi: 10.1074/jbc.275.7.4599. [DOI] [PubMed] [Google Scholar]
- Lyon M, Deakin J. Gallagher JT. The mode of action of heparan and dermatan sulfates in the regulation of hepatocyte growth factor/scatter factor. J. Biol. Chem. 2002;277:1040–1046. doi: 10.1074/jbc.M107506200. [DOI] [PubMed] [Google Scholar]
- Lyon M, Deakin JD, Lietha D, Gherardi E. Gallagher JT. The interactions of hepatocyte growth factor/scatter factor and Its NK1 and NK2 variants with glycosaminoglycans using a modified gel mobility shift assay. J. Biol. Chem. 2004;279:43560–43567. doi: 10.1074/jbc.M408510200. [DOI] [PubMed] [Google Scholar]
- Maccarana M, Casu B. Lindahl U. Minimal sequence in heparin/heparan sulphate required for binding basic fibroblast growth factor. J. Biol. Chem. 1993;268:23898–23905. [PubMed] [Google Scholar]
- Maeda N. Noda M. Involvement of receptor-like protein tyrosine phosphatase ζ/RPTPβ and its ligand pleiotrophin/heparin-binding growth-associated molecule (HB-GAM) in neuronal migration. J. Cell Biol. 1998;142:203–216. doi: 10.1083/jcb.142.1.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahalingam Y, Gallagher JT. Couchman JR. Cellular adhesion responses to the heparin-binding (hepii) domain of fibronectin require heparan sulfate with specific properties. J. Biol. Chem. 2007;282:3221–3230. doi: 10.1074/jbc.M604938200. [DOI] [PubMed] [Google Scholar]
- Makarenkova HP, Hoffman MP, Beenken A, et al. Differential interactions of FGFs with heparan sulfate control gradient formation and branching morphogenesis. Sci. Signal. 2009;2:ra55. doi: 10.1126/scisignal.2000304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamluk R, Gechtman Z, Kutcher ME, Gasiunas N, Gallagher JT. Klagsbrun M. Neuropilin-1 binds vascular endothelial growth factor 165, placenta growth factor-2, and heparin via its b1b2 domain. J. Biol. Chem. 2002;277:24818–24825. doi: 10.1074/jbc.M200730200. [DOI] [PubMed] [Google Scholar]
- Matsuo I. Kimura-Yoshida C. Extracellular distribution of diffusible growth factors controlled by heparan sulfate proteoglycans during mammalian embryogenesis. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2014;369:20130545. doi: 10.1098/rstb.2013.0545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayo KH, Ilyina E, Roongta V, et al. Heparin binding to platelet factor-4. An NMR and site-directed mutagenesis study: arginine residues are crucial for binding. Biochem. J. 1995;312:357–365. doi: 10.1042/bj3120357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon AP, Ingham PW. Tabin CJ. Developmental roles and clinical significance of hedgehog signaling. Curr. Top. Dev. Biol. 2003;53:1–114. doi: 10.1016/s0070-2153(03)53002-2. [DOI] [PubMed] [Google Scholar]
- Merry CL, Lyon M, Deakin JA, Hopwood JJ. Gallagher JT. Highly sensitive sequencing of the sulphated domains of heparan sulphate. J. Biol. Chem. 1999;274:18455–18462. doi: 10.1074/jbc.274.26.18455. [DOI] [PubMed] [Google Scholar]
- Merry CL, Bullock SL, Swan DC, et al. The molecular phenotype of heparan sulfate in the Hs2st−/−mutant mouse. J. Biol. Chem. 2001;276:35429–35434. doi: 10.1074/jbc.M100379200. [DOI] [PubMed] [Google Scholar]
- Mitsi M, Hong Z, Costello CE. Nugent MA. Heparin-mediated conformational changes in fibronectin expose vascular endothelial growth factor binding sites. Biochemistry. 2006;45:10319–10328. doi: 10.1021/bi060974p. [DOI] [PubMed] [Google Scholar]
- Mobli M, Nilsson M. Almond A. The structural plasticity of heparan sulfate NA-domains and hence their role in mediating multivalent interactions is confirmed by high-accuracy 15N-NMR relaxation studies. Glycoconj. J. 2008;25:401–414. doi: 10.1007/s10719-007-9081-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammadi M, Olsen SK. Ibrahimi OA. Structural basis for fibroblast growth factor receptor activation. Cytokine Growth Factor Rev. 2005;16:107–137. doi: 10.1016/j.cytogfr.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Mostavi-Pour Z, Askari JA, Whittard JD. Humphries MJ. Identification of a novel heparin-binding site in the alternatively-spliced IICS region of fibronectin: roles of integrins and proteoglycans in cell adhesion to fibronectin splice variants. Matrix Biol. 2001;20:63–73. doi: 10.1016/s0945-053x(00)00131-1. [DOI] [PubMed] [Google Scholar]
- Mulloy B. The specificity of interactions between proteins and sulfated polysaccharides. An. Acad. Bras. Ciênc. 2005;77:651–664. doi: 10.1590/s0001-37652005000400007. [DOI] [PubMed] [Google Scholar]
- Mulloy B. Structure and physiochemical characterisation of heparin. In: Lever R, Mulloy B, Page CP, editors. Heparin – A Century of Progress. Berlin, Heidelberg: Springer Verlag; 2012. pp. 77–98. [Google Scholar]
- Mulloy B. Forster MJ. Conformation and dynamics of heparin and heparan sulfate. Glycobiology. 2000;10:1147–1156. doi: 10.1093/glycob/10.11.1147. [DOI] [PubMed] [Google Scholar]
- Mulloy B. Linhardt RJ. Order out of complexity – protein structures that interact with heparin. Curr. Opin. Struct. Biol. 2001;11:623–628. doi: 10.1016/s0959-440x(00)00257-8. [DOI] [PubMed] [Google Scholar]
- Muramatsu H, Inui T, Kimura T, et al. Localization of heparin-binding, neurite outgrowth and antigenic regions in midkine molecule. Biochem. Biophys. Res. Commun. 1994;203:1131–1139. doi: 10.1006/bbrc.1994.2300. [DOI] [PubMed] [Google Scholar]
- Murphy KJ, Merry CLR, Lyon M, Thompson JE, Roberts IS. Gallagher JT. A new model for the domain structure of heparan sulfate based on the novel specificity of K5 lyase. J. Biol. Chem. 2004;279:27239–27245. doi: 10.1074/jbc.M401774200. [DOI] [PubMed] [Google Scholar]
- Naimy H, Buczek-Thomas JA, Nugent MA, Leymarie N. Zaia J. Highly sulfated nonreducing end-derived heparan sulfate domains bind fibroblast growth factor-2 with high affinity and are enriched in biologically active fractions. J. Biol. Chem. 2011;286:19311–19319. doi: 10.1074/jbc.M110.204693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netelenbos T, van den Born J, Kessler FL, Zweegman S, Huijgens PC. Dräger AM. In vitro model for hematopoietic progenitor cell homing reveals endothelial heparan sulfate proteoglycans as direct adhesive ligands. J. Leukoc. Biol. 2003;72:353–362. doi: 10.1189/jlb.1202593. [DOI] [PubMed] [Google Scholar]
- Ng YS, Krilleke D. Shima DT. VEGF function in vascular pathogenesis. Exp. Cell Res. 2006;312:527–537. doi: 10.1016/j.yexcr.2005.11.008. [DOI] [PubMed] [Google Scholar]
- Ohlig S, Farshi P, Pickhinke U, et al. Sonic hedgehog shedding results in functional activation of the solubilized protein. Dev. Cell. 2011;20:764–774. doi: 10.1016/j.devcel.2011.05.010. [DOI] [PubMed] [Google Scholar]
- Ohlig S, Pickhinke U, Sirko S, et al. An emerging role of Sonic hedgehog shedding as a modulator of heparan sulfate interactions. J. Biol. Chem. 2012;287:43708–43719. doi: 10.1074/jbc.M112.356667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno K, Ito M, Kawakami Y, Krejci E. Engel AG. Specific binding of collagen Q to the neuromuscular junction is exploited to cure congenital myasthenia and to explore bases of myasthenia gravis. Chem. Biol. Interact. 2013;203:335–340. doi: 10.1016/j.cbi.2012.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ori A, Wilkinson MC. Fernig DG. The heparanome and regulation of cell function: structures, functions and challenges. Front Biosci. 2008;13:4309–4338. doi: 10.2741/3007. [DOI] [PubMed] [Google Scholar]
- Parkash V, Leppänen V-M, Virtanen H, et al. The structure of the glial cell line-derived neurotrophic factor-coreceptor complex: insights into RET signalling and heparin binding. J. Biol. Chem. 2008;283:35164–35172. doi: 10.1074/jbc.M802543200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel VN, Likar KM, Zisman-Rozen S, et al. Specific heparan sulfate structures modulate FGF10-mediated submandibular gland epithelial morphogenesis and differentiation. J. Biol. Chem. 2008;283:9308–9317. doi: 10.1074/jbc.M709995200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrini L. Role of heparan sulfate in fibroblast growth factor signalling: a structural view. Curr. Opin. Struct. Biol. 2001;11:629–634. doi: 10.1016/s0959-440x(00)00258-x. [DOI] [PubMed] [Google Scholar]
- Pellegrini L, Burke DF, von Delft F, Mulloy B. Blundell TL. Crystal structure of fibroblast growth factor receptor ectodomain bound to ligand and heparin. Nature. 2000;407:1029–1034. doi: 10.1038/35039551. [DOI] [PubMed] [Google Scholar]
- Proudfoot AE, Handel TM, Johnson Z, et al. Glycosaminoglycan binding and oligomerization are essential for the in vivo activity of certain chemokines. Proc. Natl Acad. Sci. USA. 2003;100:1885–1890. doi: 10.1073/pnas.0334864100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pye D, Vives R, Turnbul JE, Hyde P. Gallagher JT. Heparan sulphate oligosaccharides require 6-O sulphation for promotion of basic fibroblast factor mitogenic activity. J. Biol. Chem. 1998;273:1183–1192. doi: 10.1074/jbc.273.36.22936. [DOI] [PubMed] [Google Scholar]
- Qu X, Carbe C, Tao C, et al. Lacrimal gland development and Fgf10-Fgfr2b signalling are controlled by 2-O- and 6-O-sulfated heparan sulphate. J. Biol. Chem. 2011;286:14435–14444. doi: 10.1074/jbc.M111.225003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman R, Sasisekharan V. Sasisekharan R. Structural insights into biological roles of protein-glycosaminoglycan interactions. Chem. Biol. 2005;12:267–277. doi: 10.1016/j.chembiol.2004.11.020. [DOI] [PubMed] [Google Scholar]
- Ramani VC, Yang Y, Ren Y, Nan L. Sanderson R. Heparanase plays a dual role in driving hepatocyte growth factor (HGF) signaling by enhancing HGF expression and activity. J. Biol. Chem. 2011;286:6490–6499. doi: 10.1074/jbc.M110.183277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapraeger AC, Krufka A. Olwin BB. Requirement of heparan sulfate for bFGF-mediated fibroblast growth and myoblast differentiation. Science. 1991;252:1705–1708. doi: 10.1126/science.1646484. [DOI] [PubMed] [Google Scholar]
- Raulo E, Chernousov MA, Carey DJ, Nolo R. Rauvala H. Isolation of a neuronal cell surface receptor of heparin binding growth-associated molecule (HB-GAM). Identification as N-syndecan (syndecan-3) J. Biol. Chem. 1994;269:12999–13004. [PubMed] [Google Scholar]
- Rauvala H. An 18 kDa heparin-binding protein of developing brain that is distinct from fibroblast growth factors. EMBO J. 1989;8:2933–2941. doi: 10.1002/j.1460-2075.1989.tb08443.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricard-Blum S. The collagen family. Cold Spring Harb. Perspect. Biol. 2011;3:a004978. doi: 10.1101/cshperspect.a004978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricard-Blum S, Beraud M, Raynal N, Farndale RW. Ruggiero F. Structural requirements for heparin/heparan sulfate binding to type V collagen. J. Biol. Chem. 2006;281:25195–25204. doi: 10.1074/jbc.M603096200. [DOI] [PubMed] [Google Scholar]
- Rickard SM, Mummery RS, Mulloy B. Rider CC. The binding of human glial cell line–derived neurotrophic factor to heparin and heparan sulfate: importance of 2-O-sulfate groups and effect on its interaction with its receptor, GFRα1. Glycobiology. 2003;13:419–426. doi: 10.1093/glycob/cwg046. [DOI] [PubMed] [Google Scholar]
- Rider CC. Heparin/heparan sulphate binding in the TGF-beta cytokine superfamily. Biochem. Soc. Trans. 2006;34:458–461. doi: 10.1042/BST0340458. [DOI] [PubMed] [Google Scholar]
- Robinson CR. Stringer SE. The splice variants of vascular endothelial growth factor (VEGF) and their receptors. J. Cell Sci. 2006;114:853–865. doi: 10.1242/jcs.114.5.853. [DOI] [PubMed] [Google Scholar]
- Robinson CJ, Harmer NJ, Goodger SJ, Blundell TL. Gallagher JT. Cooperative dimerization of fibroblast growth factor 1 (FGF1) upon a single heparin saccharide may drive the formation of 2:2:1 FGF1-FGFR2c-heparin ternary complexes. J. Biol. Chem. 2005;280:42274–42282. doi: 10.1074/jbc.M505720200. [DOI] [PubMed] [Google Scholar]
- Robinson CR, Mulloy B, Gallagher JT. Stringer SE. VEGF165 – binding sites within heparan sulphate encompass two highly-sulphated domains and can be liberated by K5lyase. J. Biol. Chem. 2006;281:1731–1740. doi: 10.1074/jbc.M510760200. [DOI] [PubMed] [Google Scholar]
- Rothblum K, Tyler WA, Stahl RC. Carey DJ. Schwann cells synthesize Type V collagen that contains a novel α4 chain: molecular cloning, biochemical characterization, and high affinity heparin binding of α4 (v) collagen. J. Biol. Chem. 2000;275:28208–28215. doi: 10.1074/jbc.M003922200. [DOI] [PubMed] [Google Scholar]
- Rothblum K, Stahl RC. Carey DJ. Constitutive release of α4 type V collagen N-terminal domain by Schwann cells and binding to cell surface and extracellular matrix heparan sulfate proteoglycans. J. Biol. Chem. 2004;279:51282–51288. doi: 10.1074/jbc.M408837200. [DOI] [PubMed] [Google Scholar]
- Rotundo RL, Rossi SG, Kimbell LM, Ruiz C. Marrero E. Targeting acetylcholinesterase to the neuromuscular synapse. Chem. Biol. Interact. 2005;157–158:15–21. doi: 10.1016/j.cbi.2005.10.007. [DOI] [PubMed] [Google Scholar]
- Rubin JB, Choi Y. Segal RA. Cerebellar proteoglycans regulate sonic hedgehog responses during development. Development. 2002;129:2223–2232. doi: 10.1242/dev.129.9.2223. [DOI] [PubMed] [Google Scholar]
- Rudd TR. Yates EA. A highly efficient tree structure for the biosynthesis of heparan sulfate accounts for the commonly observed disaccharides and suggests a mechanism for domain synthesis. Mol. BioSyst. 2012;8:1499–1506. doi: 10.1039/c2mb25019e. [DOI] [PubMed] [Google Scholar]
- Rueda P, Balabanian K, Lagane B, et al. The CXCL12gamma chemokine displays unprecedented structural and functional properties that make it a paradigm of chemoattractant proteins. PLoS ONE. 2008;3:e2543. doi: 10.1371/journal.pone.0002543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rueda P, Richart A, Récalde A, et al. Homeostatic and tissue reparation defaults in mice carrying selective genetic invalidation of CXCL12/proteoglycan interactions. Circulation. 2012;126:1882–1895. doi: 10.1161/CIRCULATIONAHA.112.113290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruhrberg C, Gerhardt H, Golding M, et al. Spatially restricted patterning cues provided by heparin-binding VEGF-A control blood vessel branching morphogenesis. Genes Dev. 2002;16:2684–2698. doi: 10.1101/gad.242002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadir R, Baleux F, Grosdidier A, Imberty A. Lortat-Jacob H. Characterization of the stromal cell-derived factor-1alpha-heparin complex. J. Biol. Chem. 2001;276:8288–8296. doi: 10.1074/jbc.M008110200. [DOI] [PubMed] [Google Scholar]
- Sadir R, Imberty A, Baleux F. Lortat-Jacob H. Heparan sulfate/heparin oligosaccharides protect stromal cell-derived factor-1 (SDF-1)/CXCL12 against proteolysis induced by CD26/dipeptidyl peptidase IV. J. Biol. Chem. 2004;279:43854–43860. doi: 10.1074/jbc.M405392200. [DOI] [PubMed] [Google Scholar]
- San Antonio JD, Lander AD, Karnovski MJ, Slayter HS. Mapping the heparin-binding sites on type I collagen monomers and fibrils. J. Cell Biol. 1994;125:1179–1188. doi: 10.1083/jcb.125.5.1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlessinger J, Plotnikov AN, Ibrahimi OA, et al. Crystal structure of a ternary FGF-FGFR–heparin complex reveals a dual role for heparin in FGFR binding and dimerization. Mol. Cell. 2000;6:743–750. doi: 10.1016/s1097-2765(00)00073-3. [DOI] [PubMed] [Google Scholar]
- Schwarzbauer JE. DeSimone DW. Fibronectins, their fibrillogenesis, and in vivo functions. Cold Spring Harb. Perspect. Biol. 2011;3:a005041. doi: 10.1101/cshperspect.a005041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seffouh A, Milz F, Przybylski C, et al. HSulf sulfatases catalyze processive and oriented 6-O-desulfation of heparan sulfate that differentially regulates fibroblast growth factor activity. FASEB J. 2013;27:2431–2439. doi: 10.1096/fj.12-226373. [DOI] [PubMed] [Google Scholar]
- Selleck SB. Genetic dissection of proteoglycan function in Drosophila and C. elegans. Semin. Cell Dev. Biol. 2001;12:127–134. doi: 10.1006/scdb.2000.0242. [DOI] [PubMed] [Google Scholar]
- Severin IC, Gaudry JP, Johnson Z, et al. Characterization of the chemokine CXCL11-heparin interaction suggests two different affinities for glycosaminoglycans. J. Biol.Chem. 2010;285:17713–17724. doi: 10.1074/jbc.M109.082552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah MM, Sakurai H, Sweeney DE, et al. Hs2st mediated kidney mesenchyme induction regulates early ureteric bud branching. Dev. Biol. 2010;339:354–365. doi: 10.1016/j.ydbio.2009.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A, Askari AJ, Humphries MJ, Jones EY. Stuart DI. Crystal structure of a heparin-and integrin-binding segment of human fibronectin. EMBO J. 1999;18:1468–1479. doi: 10.1093/emboj/18.6.1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X. Zaia J. Organ-specific heparan sulphate structural phenotypes. J. Biol. Chem. 2009;284:11806–11814. doi: 10.1074/jbc.M809637200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shintani Y, Takashima S, Asano Y, et al. Glycosaminoglycan modification of neuropilin-1 modulates VEGFR2 signaling. EMBO J. 2006;25:3045–3055. doi: 10.1038/sj.emboj.7601188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shute J. Glycosaminoglycan and chemokine/growth factor interactions. In: Lever R, Mulloy B, Page CP, editors. Heparin – A Century of Progress. Berlin, Heidelberg: Springer Verlag; 2012. pp. 307–324. [Google Scholar]
- Skidmore MA, Guimond SE, Rudd TR, Fernig DG, Turnbull JE. Yates EA. The activities of heparan sulfate and its analogue heparin are dictated by biosynthesis, sequence, and conformation. Connect. Tissue Res. 2008;49:140–144. doi: 10.1080/03008200802148595. [DOI] [PubMed] [Google Scholar]
- Smith EM, Mitsi M. Nugent MA. PDGF-A interactions with fibronectin reveal a critical role for heparan sulfate in directed cell migration during Xenopus gastrulation. PNAS. 2009;106:21683–21688. doi: 10.1073/pnas.0902510106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillmann D, Witt D. Lindahl U. Defining the interleukin binding domain of heparin sulphate. J. Biol. Chem. 1998;273:15847–15893. doi: 10.1074/jbc.273.25.15487. [DOI] [PubMed] [Google Scholar]
- Staples GO, Shi X. Zaia J. Extended N-sulfated domains reside at the nonreducing end of heparan sulfate chains. J. Biol. Chem. 2010;285:18336–18343. doi: 10.1074/jbc.M110.101592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterner E, Masuko S, Li G, et al. Fibroblast growth factor-based signaling through synthetic heparan sulfate blocks copolymers studied using high cell density three-dimensional cell printing. J. Biol. Chem. 2014;289:9754–9765. doi: 10.1074/jbc.M113.546937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stringer SE. Gallagher JT. Specific binding of the chemokine platelet factor 4 to heparan sulfate. J. Biol. Chem. 1997;272:20508–20514. doi: 10.1074/jbc.272.33.20508. [DOI] [PubMed] [Google Scholar]
- Stringer SE, Forster MJ, Mulloy B, Bishop CR, Graham GJ. Gallagher JT. Characterization of the binding site on heparan sulfate for macrophage inflammatory protein 1a. Blood. 2002;100:1543–1550. [PubMed] [Google Scholar]
- Stringer SE, Nelson MS. Gupta P. Identification of an MIP-1α–binding heparan sulfate oligosaccharide that supports long-term in vitro maintenance of human LTC-ICs. Blood. 2003;101:2243–2245. doi: 10.1182/blood-2002-08-2588. [DOI] [PubMed] [Google Scholar]
- Sugahara K. Kitagawa H. Recent advances in the study of the biosynthesis and functions of sulfated glycosaminoglycans. Curr. Opin. Struct. Biol. 2000;10:518–527. doi: 10.1016/s0959-440x(00)00125-1. [DOI] [PubMed] [Google Scholar]
- Sugaya N, Habuchi H, Nagai N, Ashikari-Hada S. Kimata K. 6-O-sulfation of heparan sulfate differentially regulates various FGFs-dependent signalling in culture. J. Biol. Chem. 2008;283:10366–10376. doi: 10.1074/jbc.M705948200. [DOI] [PubMed] [Google Scholar]
- Sweeney SM, Guy CA, Fields GB. San Antonio JD. Defining the domains of type I collagen involved in heparin-binding and endothelial tube formation. Proc. Natl Acad. Sci. USA. 1998;90:7275–7280. doi: 10.1073/pnas.95.13.7275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiedemann K, Bätge B, Muller PK. Reinhardt DP. Interactions of fibrillin-1 with heparin/heparan sulphate: implications for microfibrillar assembly. J. Biol. Chem. 2001;276:36035–36042. doi: 10.1074/jbc.M104985200. [DOI] [PubMed] [Google Scholar]
- Tu Y. Weiss AS. Glycosaminoglycan-mediated coacervation of tropoelastin abolishes the critical concentration, accelerates coacervate formation, and facilitates spherule fusion: implications for tropoelastin microassembly. Biomacromolecules. 2008;9:1739–1744. doi: 10.1021/bm7013153. [DOI] [PubMed] [Google Scholar]
- Turnbull JE. Gallagher JT. Molecular organization of heparan sulphate from human skin fibroblasts. Biochem. J. 1990;265:715–724. doi: 10.1042/bj2650715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbull JE. Gallagher JT. Distribution of iduronate 2-sulphate residues in heparan sulphate. Evidence for an ordered polymeric structure. Biochem. J. 1991;273:553–559. doi: 10.1042/bj2730553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbull JE, Fernig DG, Ke Y, Wilkinson MC. Gallagher JT. Identification of the basic fibroblast growth factor binding sequence in fibroblast heparan sulfate. J. Biol. Chem. 1992;267:10337–10341. [PubMed] [Google Scholar]
- Uniewicz KA, Ori A, Xu R, et al. Differential scanning fluorimetry measurement of protein stability changes upon binding to glycosaminoglycans: a screening test for binding specificity. Anal. Chem. 2010;82:3796–3802. doi: 10.1021/ac100188x. [DOI] [PubMed] [Google Scholar]
- Vander Kooi CW, Jusino MA, Perman B, Neau DB, Bellamy HD. Leahy DJ. Structural basis for ligand and heparin binding to neuropilin domains. PNAS. 2007;104:6152–6157. doi: 10.1073/pnas.0700043104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan-Thomas A, Young RD, Phillips AC. Duance VC. Characterization of type XI collagen-glycosaminoglycan interactions. J. Biol. Chem. 2001;276:5303–5309. doi: 10.1074/jbc.M008764200. [DOI] [PubMed] [Google Scholar]
- Vives RR, Sadir R, Imberty A, Rencurosi A. Lortat-Jacob H. A kinetics and modelling study of RANTES (9–68) binding to heparin reveals a mechanism of cooperative oligomerization. Biochemistry. 2002;41:14779–14789. doi: 10.1021/bi026459i. [DOI] [PubMed] [Google Scholar]
- Viviano BL, Paine-Saunders S, Gasiunas N, Gallagher JT. Saunders S. Domain-specific modification of heparan sulfate by Qsulf1 modulates the binding of the bone morphogenetic protein antagonist noggin. J. Biol. Chem. 2004;279:5604–5611. doi: 10.1074/jbc.M310691200. [DOI] [PubMed] [Google Scholar]
- Vyas N, Goswami D, Manonmani A, et al. Nanoscale organization of hedgehog is essential for long-range signalling. Cell. 2008;133:1214–1227. doi: 10.1016/j.cell.2008.05.026. [DOI] [PubMed] [Google Scholar]
- Walker A. Gallagher JT. Structural domains of heparan sulphate for specific recognition of the C-terminal heparin-binding domain of human plasma fibronectin (HEP II) Biochem. J. 1996;317:871–877. doi: 10.1042/bj3170871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker A, Turnbull JE. Gallagher JT. Specific heparan sulfate saccharides mediate the activity of basic fibroblast growth factor. J. Biol. Chem. 1994;269:931–935. [PubMed] [Google Scholar]
- Warner LR, Brown RJ, Yingst SMC. Oxford JT. Isoform-specific heparan sulfate binding within the amino-terminal noncollagenous domain of collagen α1 (XI) J. Biol. Chem. 2006;281:39507–39516. doi: 10.1074/jbc.M608551200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whalen DM, Malinauskas T, Gilbert RJC. Siebold C. Structural insights into proteoglycan-shaped Hedgehog signalling. PNAS. 2013;110:16420–16425. doi: 10.1073/pnas.1310097110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitelock J. Melrose J. Heparan sulphate proteoglycans in healthy and diseased systems. Wiley Interdiscip. Rev. Syst. Biol. Med. 2011;3:739–751. doi: 10.1002/wsbm.149. [DOI] [PubMed] [Google Scholar]
- Xu D. Esko JD. Demystifying heparan sulfate-protein interactions. Annu. Rev. Biochem. 2014;83:129–157. doi: 10.1146/annurev-biochem-060713-035314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu D, Fuster MM, Lawrence R. Esko JD. Heparan sulfate regulates VEGF165- and VEGF121-mediated vascular hyperpermeability. J. Biol. Chem. 2011;286:737–745. doi: 10.1074/jbc.M110.177006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu R, Ori A, Rudd TR, et al. Diversification of the structural determinants of fibroblast growth factor–heparin interactions: implications for binding specificity. J. Biol. Chem. 2012a;287:40061–40073. doi: 10.1074/jbc.M112.398826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Wang Z, Liu R, Bridges AS, Huang X. Liu J. Directing the biological activities of heparan sulfate oligosaccharides using a chemoenzymatic approach. Glycobiology. 2012b;22:96–106. doi: 10.1093/glycob/cwr109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan D. Lin X. Shaping morphogen gradients by proteoglycans. Cold. Spring. Harb. Perspect. Biol. 2009;1:a002493. doi: 10.1101/cshperspect.a002493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yayon A, Klagsbrun M, Esko JD, Leder P. Ornitz DM. Cell surface, heparin-like molecules are required for binding of basic fibroblast growth factor to its high affinity receptor. Cell. 1991;64:841–848. doi: 10.1016/0092-8674(91)90512-w. [DOI] [PubMed] [Google Scholar]
- Yu Y, Sweeney MD, Saad OM, et al. Chemokine-glycosaminoglycan binding: specificity for CCR2 ligand binding to highly sulfated oligosaccharides using FTICR mass spectrometry. J. Biol. Chem. 2005;280:32200–32208. doi: 10.1074/jbc.M505738200. [DOI] [PubMed] [Google Scholar]
- Zhang X, Chen L, Bancroft DP, Lai CK. Maione TE. Crystal structure of recombinant human platelet factor 4. Biochemistry. 1994;33:8361–8366. doi: 10.1021/bi00193a025. [DOI] [PubMed] [Google Scholar]
- Zhou H, Mazzulla MJ, Kaufman JD, et al. The solution structure of the N-terminal domain of hepatocyte growth factor reveals a potential heparin-binding site. Structure. 1998;6:109–116. doi: 10.1016/s0969-2126(98)00012-4. [DOI] [PubMed] [Google Scholar]
- Zioncheck TF, Richardson L, Liu J, et al. Sulfated oligosaccharides promote hepatocyte growth factor association and govern its mitogenic activity. J. Biol. Chem. 1995;270:16871–16878. doi: 10.1074/jbc.270.28.16871. [DOI] [PubMed] [Google Scholar]
- Zou P, Zou K, Muramatsu H, et al. Glycosaminoglycan structures required for strong binding to midkine, a heparin-binding growth factor. Glycobiology. 2003;13:35–42. doi: 10.1093/glycob/cwg001. [DOI] [PubMed] [Google Scholar]