

Abstract
Interleukin (IL)-17A is a pro-inflammatory cytokine that markedly enhances inflammatory responses in the lungs by recruiting neutrophils and interacting with other pro-inflammatory mediators. Reducing the expression of IL-17A could attenuate inflammation in the lungs. However, whether VIP exerts its anti-inflammatory effects by regulating the expression of IL-17A has remained unclear. Here, we show that there is a remarkable increase of IL-17A in bronchoalveolar lavage fluid (BALF) and lung tissue of mice with acute lung injury (ALI). Moreover, lipopolysaccharides (LPS) stimulated elevated expression of IL-17A, which was evident by the enhanced levels of mRNA and protein observed. Furthermore, we also found that VIP inhibited LPS-mediated IL-17A expression in a time- and dose-dependent manner in an in vitro model of ALI and that this process might be mediated via the phosphokinase A (PKA) and phosphokinase C (PKC) pathways. Taken together, our results demonstrated that VIP might be an effective protector during ALI by suppressing IL-17A expression.
Keywords: interleukin-17A, lipopolysaccharides, macrophages, vasoactive intestinal peptide
The link between injury and inflammation is well established. Of all the host defence mechanisms, inflammation is obviously the most important because it can become a part of the microbial pathogenesis during certain infections. For example, endothelial and alveolar epithelial injuries observed in acute lung injury (ALI) may create open interface(s) between air and the blood, thereby facilitating the systemic spreading of microorganisms from the lung and initiating a systemic inflammatory response. Substantial amount of evidence indicates that cytokines mediate and control immune reactions as they are mediators of the immune system. However, the mechanism of cytokine-mediated regulation is still unclear.
Previous studies have shown that the interleukin (IL)-17 family is the newest and least understood of the cytokine subclasses. This large family consists of six members: IL-17A, IL-17B, IL-17C, IL-17D, IL-17E (also called IL-25) and IL-17F. IL-17A, also called as IL-17 or CTLA8, which is the first cytokine of this family to be discovered and the most widely investigated, is a pro-inflammatory cytokine (Gu et al. 2013). In general, IL-17A can trigger cascades of events that lead to neutrophil recruitment (Roussel et al. 2010) and inflammation during lung diseases (Tan & Rosenthal 2013) such as asthma (Kim et al. 2014), cystic fibrosis (Tan et al. 2011), pneumonitis (Hasan et al. 2013) and ALI (You et al. 2014). Furthermore, recent evidence supports the idea that endogenous mediators such as IL-17A can finely regulate the inflammatory process. Besides Th17 cells, macrophages can also produce IL-17A (Shimura et al. 2014). Macrophages have a key role in determining the severity of the acute lung injury as they are key effector cells of the inflammatory response during the course of injury (Akbarshahi et al. 2012).
During the last decade, vasoactive intestinal peptide (VIP), a multifunctional peptide, has been clearly identified as a potent anti-inflammatory factor in both innate and adaptive immunity. VIP (Delgado & Ganea 2013), is a 28-residue long peptide hormone which is widely distributed in the central and peripheral nervous systems, the immune system, the respiratory system and the cardiovascular system. It has been shown to provide protection from inflammatory disorders, such as septic shock, rheumatoid arthritis and autoimmune diabetes (Gonzalez-Rey et al. 2007). Furthermore, this multifunctional protein has been found to affect proliferation of epithelial cells (Guan et al. 2006), immunological functions of T cells (Yadav & Goetzl 2008) and inflammatory profile of macrophages (Larocca et al. 2011). VIP also inhibits inflammatory pathways by reducing LPS-induced TNF-α and IL-6 production in macrophages (Jiang et al. 2011). Noticeably, subsequent study has confirmed the inhibitory effect of VIP on pulmonary inflammation in ALI (Sun et al. 2011). However, the mechanism by which VIP exerts its effect has not yet been fully understood. To develop a new and promising therapeutic approach against ALI, we designed a study to test the hypothesis that VIP inhibits pulmonary inflammatory responses in ALI by downregulating macrophage-derived IL-17A.
Materials and methods
Animals and reagents
Specific pathogen-free (SPF) male Swiss mice weighing 20–25 g were obtained from the animal centre in Central South University and maintained in a temperature-controlled room with 70% humidity and 12 h light–dark cycles. Mice underwent an acclimatization period of at least 7 days. All procedures involving animals were approved by the Committee on the Ethics of Animals Experiments of Central South University.
Efficient clearance of apoptotic cells by wound macrophages is a prerequisite for relieving inflammation at an injury site timely. Therefore, macrophages were used to test the relationship between inflammation and IL-17A, in their role as the resident cells, target cells and effector cells during lung inflammation. Mouse RAW 264.7 macrophage cell line was obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China).
Animal model of LPS-induced acute lung injury
Twenty male Swiss mice were randomly divided into two experiment groups (n = 10), control group and LPS group. Mice in LPS group were treated with a sublethal dose of LPS (10 mg/kg weight, Escherichia coli lipopolysaccharides, 055:B5) intraperitoneal injection for 6 h to produce acute lung injury. Mice in the control group were subjected to the same protocol but received the same volume of intraperitoneal saline instead of LPS.
Histopathological examination
For histopathological studies, the right lower lobe lung was fixed in 4% neutral buffered formalin, dehydrated in a graded series of ethanol, embedded in paraffin, cut into 5-μm-thick serial sections, and stained with haematoxylin–eosin (HE). Microscopic evaluation was performed to characterize lung injury. To prevent bias of observation, semi-quantitative assessment of injury changes was assessed by two observers in a blinded fashion.
The left lower lobe lung was clippered into pieces; added 1 ml ice-cold PBS followed by homogenation on ice, centrifuged at 2000 g for 5 min at 4°C, and the supernatant was stored at −70°C for ELISA measurements. The other lobes of the lung were ground into powder under the liquid N2, added 1 ml TRIzol for RNA extraction.
Bronchoalveolar lavage fluid (BALF) and cell counting
BALF was performed (0.5 ml ice-cold phosphate-buffered saline three times) in two groups. In each mouse, 90% of the total injected volume was recovered consistently. After the lavaged sample from each mouse was centrifuged at 2000 g for 5 min at 4°C, the cell pellets were washed twice and resuspended in 50 μl PBS. Then, total cells, neutrophils and macrophages were counted double-blind using a hemocytometer (Hausser Scientific, Horsham, PA, USA), and the cell-free supernatant was stored at −70°C for biochemical measurements.
Cell culture and experimental protocol
RAW 264.7 cells were grown in DMEM supplemented with 10% FBS, 100 U/ml penicillin, 100 μg/ml streptomycin, and 2 mM l-glutamine (complete medium) at 37°C in a humidified incubator with 5% CO2. Cells allocated to receive LPS (1 μg/ml) or DMEM were used as positive or negative controls and were designated as LPS and control group respectively. To elucidate the effects of different concentrations of VIP on LPS-induced IL-17A expression, cells were pretreated with VIP (10−10, 10−9, 10−8, 10−7, 10−6 M) for 30 min and then stimulated with LPS (1 μg/ml) for 6 h. To elucidate the effects of different time-lasted of VIP on LPS-induced IL-17A expression, cells were pretreated with VIP (10−8 M) for 30 min and then stimulated with LPS (1 μg/ml) for 0, 2, 4, 6, 12 and 24 h. And to make a clear understanding of intracellular signal pathways of VIP, specific inhibitor of PKA (H-89, 10−5 M) and PKC (H-7, 10−5 M) were used in this study.
Reverse transcription–PCR (RT-PCR)
The mRNA expressions of IL-17A in the lung and macrophages were detected by RT-PCR. In brief, the lung tissues were in −80°C, and then, total RNA was isolated from lung homogenates with TRIzol reagent. Total RNA was prepared using TRIzol (Invitrogen Life Technologies, Carlsbad, California, USA), and 2 μg of total RNA was used for the detection of mRNA using a reverse transcription–PCR kit (Fermentas, Glen Burnie, Maryland, USA). Each PCR was performed with a PCR Mix (Bio-Rad Laboratories, Inc., Berkeley, California, USA) in a final volume of 20 μl, forward and backward primers 1 μl each, 10 μl PCR Mix. The primers sequences were as follows: IL-17A (344 bp) forward: 5′-TGTCAATGCGGAGGGAAAG-3′, IL-17A reverse: 5′-GCAGTTTGGGACCCCTTTAC-3′; and GAPDH (580 bp) forward: 5′-AAGCCCATCACCATCTTCCA-3′, GAPDH reverse: 5′-CCTGCTTCACCACCTTCTTG-3′. PCR was performed under the conditions of an initial 3 min at 94°C followed by 35 cycles (30 s at 94°C 30 s at 58°C, 1 min at 72°C), and a final 5 min at 72°C. PCR products after electrophoresis were imaged using Quantity One.
Enzyme-linked immunosorbent assay (ELISA)
The supernatant of BALF was collected and immediately stored at −70°C until the cytokine assay. IL-17A in lung tissue homogenate and BALF were measured with a mouse IL-17A ELISA kit (R&D Systems, Minneapolis, MN, USA) according to the instructions of the manufacturer.
Statistical analysis
All data were expressed as means ± SEM. Statistical analysis was performed with SigmaPlot software (SPSS, Inc., Chicago, lllinois, USA). Statistical differences were determined by one-way analysis of variance (one-way anova), followed by a Tukey's test for multiple comparisons, and P < 0.05 was considered to be statistically significant. The protocol was in accordance with ethical policies of the International Journal of Experimental Pathology and approved by the local ethical committee.
Results
Enhanced inflammation in the lungs of mice with ALI
Pulmonary histological changes were observed under a microscope, 6 h after LPS injection. Normal pulmonary tissue structure and alveoli were seen in the control group. Infiltration of inflammatory cells and alveolar damage appeared after LPS administration (Figure 1a). Furthermore, analysis of leucocytes in BALF showed that the total cell count was greater in the ALI group [(35.57 ± 3.27) × 106] than in the control group [(21.48 ± 1.39) × 106]. After LPS administration, the number of neutrophils markedly increased in the ALI group [(16.71 ± 2.14) × 106] as compared to the control [(3.01 ± 0.38) × 106] (Figure 1b, P < 0.01). Significant differences were observed in the pulmonary histological changes and in the number of neutrophils in BALF between the control and ALI groups. The pathological changes observed were consistent with ALI and were indicative of enhanced infiltration by inflammatory cells, which in turn enhanced the inflammatory response.
Figure 1.
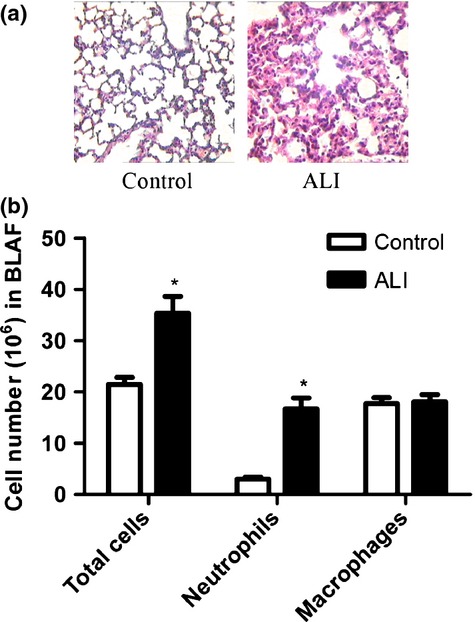
Enhanced inflammation in the lungs of mice with ALI. (a) Representative histological lung sections (HE staining) from each group. Images are shown at 250 × magnification. (b) Cell counts in BALF. LPS increases inflammatory cells in BALF in mice with ALI. Each bar represents the mean ± SEM (n = 5). *P < 0.05 compared with control group.
IL-17A expression in the lungs of mice with ALI
As shown in Figure 2, lungs of mice with ALI exhibited increased IL-17A mRNA levels (Figure 2a, P < 0.05). The LPS-induced increase in IL-17A mRNA expression was associated with similar increases in protein levels in pulmonary tissue (Figure 2b, P < 0.05). IL-17A in BALF (Figure 2c, P < 0.01) was dramatically increased after intraperitoneal administration of LPS. The results were indicative of a close association between IL-17A expression and enhanced inflammation in the lungs of mice with ALI.
Figure 2.
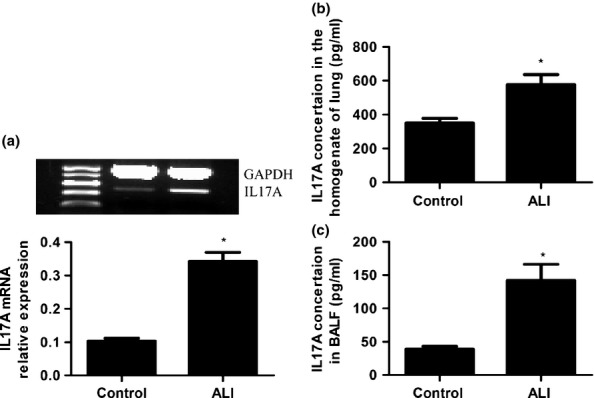
IL-17A expression in the lungs (a, b) and BALF (c) of mice with ALI. A: Relative to saline-treated mice, IL-17A mRNA was increased after LPS administration. b and c: Compared with control mice, IL-17A was increased in the lung (b) and BALF (c) of mice with ALI. Data are expressed as means ± SEM. *P < 0.05 compared with control (n = 5 per group).
VIP decreased LPS-induced IL-17A expression in macrophages
As macrophages are both resident cells in lungs and target cells during inflammation, they were used to elucidate the effect of VIP on IL-17A expression. VIP (10−8 M-10−6 M) inhibited LPS-induced IL-17A mRNA expression in a dose-dependent manner (Figure 3a, P < 0.01). As 10−8 M of VIP efficiently inhibited IL-17A mRNA expression, this concentration was used for subsequent experiments. Compared to LPS group under the same conditions, LPS-stimulated IL-17A mRNA expression was strikingly attenuated by VIP at 4 h, 6 h, 12 h and 24 h (Figure 3c, P < 0.05). The VIP-induced attenuation was most evident at 6 h. VIP (10−8 M-10−6 M) inhibited LPS-induced IL-17A secretion in a dose-dependent manner (Figure 3b, P < 0.01). Compared to LPS group without VIP treatment, LPS-induced IL-17A secretion was attenuated by VIP at 4 h, 6 h, 12 h and 24 h (Figure 3d, P < 0.01).
Figure 3.
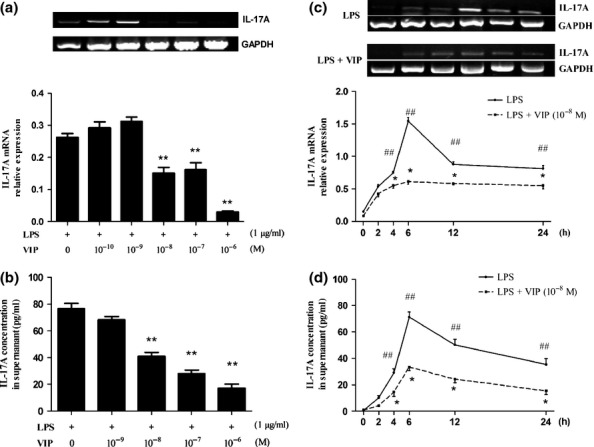
VIP decreased LPS-induced mRNA expression of IL-17A (a, c) and protein expression of IL-17A (b, d) in macrophages. a and b: Among different concentrations of VIP-primed RAW264.7 cells, 10−6 M VIP was the most significant inhibitory concentration on IL-17A mRNA (a) and IL-17 protein (b) expression. c and d: mRNA expression of IL-17A (c) and protein expression of IL-17A (d) in LPS-treated RAW 264.7 cells peaked at 6 h, and the inhibition of VIP on LPS-induced IL-17A mRNA (c) and IL-17A protein (d) were the most significant. Data are expressed as means ± SEM, n = 5. ##P < 0.01 compared with 0 h group; *P < 0.05, **P < 0.01 compared with LPS group.
PKC and PKA pathways were involved in VIP-mediated inhibition of LPS-induced IL-17A expression in macrophages
To determine whether PKC or PKA affects the ability of VIP to suppress LPS-induced IL-17A expression in macrophages, H-7, a selective PKC inhibitor, and H-89, a selective PKA inhibitor, were used. As the results in Figure 4a,b show, the effect of VIP was remarkably reversed after H-7 and H-89 application. These findings indicated that both PKA and PKC pathways might be involved in mediating the inhibitory effect of VIP on LPS-induced IL-17A expression.
Figure 4.
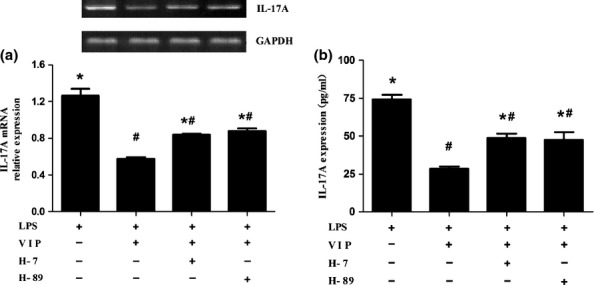
PKC and PKA pathways were involved in VIP-mediated inhibition of LPS-induced IL-17A expression in macrophages. (a and b) Suppression of VIP on LPS-induced IL-17A mRNA (a) and IL-17A (b) was remarkably reversed after H-7 and H-89 application respectively. Data are means ± SEM, n = 5. *P < 0.05 compared with LPS+VIP group. #P < 0.05 compared with LPS group.
Discussion
The results presented here strongly indicate a protective role of VIP against LPS-stimulated ALI. VIP acted by downregulating macrophage-derived IL-17A expression in an in vitro model. Furthermore, VIP might downregulate IL-17A expression via the PKA- and PKC-dependent pathways. Therefore, the significantly elevated level of IL-17A that has been associated with the administration of LPS was suppressed by VIP. This effect was confirmed at the protein and mRNA levels by the considerable decrease of IL-17A expression observed by adding VIP before LPS stimulation. Hence, the results demonstrated the anti-inflammatory effect of pretreatment with VIP on the elevation of IL-17A levels after LPS-stimulated lung injuries.
ALI is a life-threatening disease. Despite improvements in the care of critically ill patients, the hospital mortality rate of ALI continues to remain high (Brown et al. 2011). Interfering by IL-17A that can alleviate lung injury has been reported. For example, IL-17A Ab neutralization reduced neutrophil influx during the early lung injury response (Lo et al. 2010). You et al. reported that specific antioxidants or hormonal drugs significantly reduced the expression of IL-17A and inhibited the increase in some parameters of ALI in rats (You et al. 2014). These findings indicated that this cytokine is involved in the ALI model. It is also well-known that VIP protects against lung injury. However, it was unclear whether this VIP protection against lung injury is mediated by regulation of IL-17A expression. The present study addressed this hitherto uninvestigated question.
Several structurally and functionally specialized members of the IL-17 family participate in both acute and chronic inflammatory responses. IL-17A is involved in various inflammatory conditions. For example, considerably elevated IL-17A levels can be detected in the lumen and BALF in conditions such as asthma and cystic fibrosis. Moreover, IL-17A seemed to contribute to systemic inflammation, and therefore, control of IL-17A production could attenuate inflammation and tissue injury associated with some infectious diseases (Guabiraba et al. 2013). To date, more attention has focused on the pro-inflammatory properties of IL-17A (Kurimoto et al. 2013). It was reported that IL-17A expression is elevated during bacterial, viral and fungal infections, subsequently resulting in tissue inflammation (Yan et al. 2014). Additionally, significantly elevated IL-17A levels were also observed during LPS-induced ALI (You et al. 2014), and this was consistent with the findings of us. We found that 6 h after LPS administration, inflammatory response was observable in the lungs of mice with ALI, while the levels of IL-17A mRNA and IL-17A protein were significantly increased in pulmonary tissue and BALF respectively.
Interestingly, VIP exerts a biphasic effect on modulating inflammatory cytokines. Commonly, VIP inhibited IL-6, IL-8, TNF-α and IL-12 in macrophages or monocytes (Martinez et al. 1998a; Xin & Sriram 1998; Delgado et al. 1999; Delgado & Ganea 2003). However, VIP was also reported to enhance IL-6 secretion in unstimulated peritoneal macrophages as early as in the 1990s (Martinez et al. 1998b). Ling et al. found that VIP increases TNF-α-induced production of IL-6 and IL-8 via activating the NF-κB pathway in human proximal renal tubular epithelial cells (Huang et al. 2012). Moreover, it was also reported that VIP increases pro-inflammatory osteotropic cytokines such as IL-6 in an osteoblastic cell line (Persson & Lerner 2005). The complex functions of VIP may be due to the presence of its specific receptors, VPAC1 and VPAC2 (Temerozo et al. 2013). VPAC1 expressed in the lung, CNS and other tissues (Ishihara et al. 1992; Usdin et al. 1994; Screedharan et al. 1995). VPAC2 was discovered in the respiratory tract where it was identified in bronchial epithelial cells and immune cells (Groneberg et al. 2001). The pro-inflammatory effect of VIP was mediated mainly by VPAC2, whereas an anti-inflammatory mediated by VPAC1 was reported in acute pancreatitis (Kojima et al. 2005).
In fact, mouse macrophages only express VPAC1 and stimulation has little effect on this pattern. Our present study showed that pretreatment with VIP prior to LPS stimulation led to significant reduction of IL-17A mRNA and protein levels in macrophages. This result is consistent with the report by Juarranz Y et al., who found that collagen-induced arthritis mice treated with VIP showed a decrease in mRNA expression of IL-17 in the joint (Juarranz et al. 2005). This phenomenon may be explained by the possibility that VIP exerts its anti-inflammatory effect through VPAC1. In contrast, enhancement of the VIP–VPAC1 axis signalling can skew the CD4 T-cell response towards a Th17-rich pro-inflammatory type (Yadav et al. 2008). Furthermore, it has been proposed that VPAC1 is constitutively expressed in T cells and downregulated during Th-cell stimulation, while VPAC2 is upregulated during stimulation (Groneberg et al. 2006). All of the above-mentioned studies indicate that the effects of VIP are influenced not only by the particular cell type but also by the microenvironment. Therefore, it remains to be seen whether the effect of VIP on regulating IL-17A is dependent on VPAC1 in macrophages.
Considerable research effort has been invested into elucidating the rather complicated and confusing function of VIP. The PKC and PKA pathways are possibly involved in VIP-related multiple intracellular events. VIP induced surfactant protein A expression in ATII cells and attenuated liver ischaemia/reperfusion injury in mice through the activation of the PKC/c-Fos pathway (Li et al. 2010) and the PKA pathway (Ji et al. 2013) respectively. In addition, VIP could increase VEGF expression to promote proliferation of brain vascular endothelial cells after ischaemic insult via the cAMP/PKA pathway in vitro (Yang et al. 2013). The absence of PKC led to a remarkable decrease in the proliferation in CD4(+) T cells, which resulted in a marked decrease in the colonic CD4(+) T cells that are capable of producing IL-17A (Nagahama et al. 2008). Moreover, the levels of pro-inflammatory cytokines were reduced when the PKC pathway was blocked (Leppanen et al. 2014). Previous report has shown that inflammation was alleviated by reducing the LPS-induced IL-17A properties of lipoic acid, which was mediated by the cAMP/PKA signalling cascade (Salinthone et al. 2010). This study revealed that the repressing effect of VIP on LPS-activated IL-17A expression could be reversed by blocking the PKA and PKC pathways.
In summary, the results elucidated the modulatory effect of VIP on expression of IL-17A and the mechanism by which this process resulted in protection during ALI. Our findings offer new insights into the processes underlying pulmonary inflammation in ALI and will also be useful for identifying potential new therapeutic targets to treat this devastating disorder.
Acknowledgments
This study was supported by National Natural Science Foundation of China (No. 81170059), Hunan Provincial Natural Science Foundation of China (14JJ2040) and Hunan Provincial Innovation Foundation for Postgraduate (CX2013B094).
Conflict of interest
No conflict of interests declared.
References
- Akbarshahi H, Menzel M, Posaric Bauden M, et al. Enrichment of murine CD68 + CCR2 + and CD68 + CD206 + lung macrophages in acute pancreatitis-associated acute lung injury. PLoS ONE. 2012;7:e42654. doi: 10.1371/journal.pone.0042654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown LM, Calfee CS, Matthay MA, et al. A simple classification model for hospital mortality in patients with acute lung injury managed with lung protective ventilation. Crit. Care Med. 2011;39:2645–2651. doi: 10.1097/CCM.0b013e3182266779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado M. Ganea D. Vasoactive intestinal peptide inhibits IL-8 production in human monocytes. Biochem. Biophys. Res. Common. 2003;301:825–832. doi: 10.1016/s0006-291x(03)00059-7. [DOI] [PubMed] [Google Scholar]
- Delgado M. Ganea D. Vasoactive intestinal peptide: a neuropeptide with pleiotropic immune functions. Amino Acids. 2013;45:25–39. doi: 10.1007/s00726-011-1184-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado M, Pozo D, Martinez C, et al. Vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide inhibit endotoxin-induced TNF-alpha production by macrophages: in vitro and in vivo studies. J. Immunol. 1999;162:2358–2367. [PubMed] [Google Scholar]
- Gonzalez-Rey E, Varela N, Chorny A, et al. Therapeutical approaches of vasoactive intestinal peptide as a pleiotropic immunomodulator. Curr. Pharm. Des. 2007;13:1113–1139. doi: 10.2174/138161207780618966. [DOI] [PubMed] [Google Scholar]
- Groneberg DA, Hartmann P, Dinh QT, et al. Expression and distribution of vasoactive intestinal polypeptide receptor VPAC(2) mRNA in human airways. Lab. Invest. 2001;81:749–755. doi: 10.1038/labinvest.3780283. [DOI] [PubMed] [Google Scholar]
- Groneberg DA, Rabe KF. Fischer A. Novel concepts of neuropeptide-based drug therapy: vasoactive intestinal polypeptide and its receptors. Eur. J. Pharmacol. 2006;533:182–194. doi: 10.1016/j.ejphar.2005.12.055. [DOI] [PubMed] [Google Scholar]
- Gu C, Wu L. Li X. IL-17 family: cytokines, receptors and signaling. Cytokine. 2013;64:477–485. doi: 10.1016/j.cyto.2013.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guabiraba R, Besnard AG, Marques RE, et al. IL-22 modulates IL-17A production and controls inflammation and tissue damage in experimental dengue infection. Eur. J. Immunol. 2013;43:1529–1544. doi: 10.1002/eji.201243229. [DOI] [PubMed] [Google Scholar]
- Guan CX, Zhang M, Qin XQ, et al. Vasoactive intestinal peptide enhances wound healing and proliferation of human bronchial epithelial cells. Peptides. 2006;27:3107–3114. doi: 10.1016/j.peptides.2006.08.004. [DOI] [PubMed] [Google Scholar]
- Hasan SA, Eksteen B, Reid D, et al. Role of IL-17A and neutrophils in fibrosis in experimental hypersensitivity pneumonitis. J. Allergy Clin. Immunol. 2013;131:1663–1673. doi: 10.1016/j.jaci.2013.01.015. [DOI] [PubMed] [Google Scholar]
- Huang L, Tang Y, Qin J, et al. Vasoactive intestinal peptide enhances TNF-α-induced IL-6 and IL-8 synthesis in human proximal renal tubular epithelial cells by NF-κB-dependent mechanism. Inflammation. 2012;35:1154–1160. doi: 10.1007/s10753-011-9423-4. [DOI] [PubMed] [Google Scholar]
- Ishihara T, Shigemoto R, Mori K, et al. Functional expression and tissue distribution of a novel receptor for vasoactive intestinal polypeptide. Neuron. 1992;8:811–819. doi: 10.1016/0896-6273(92)90101-i. [DOI] [PubMed] [Google Scholar]
- Ji H, Zhang Y, Liu Y, et al. Vasoactive intestinal peptide attenuates liver ischemia/reperfusion injury in mice via the cyclic adenosine monophosphate-protein kinase a pathway. Liver Transpl. 2013;19:945–956. doi: 10.1002/lt.23681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W, Tang W, Geng Q, et al. Inhibition of Toll-like receptor 4 with vasoactive intestinal peptide attenuates liver ischemia-reperfusion injury. Transplant. Proc. 2011;43:1462–1467. doi: 10.1016/j.transproceed.2011.01.191. [DOI] [PubMed] [Google Scholar]
- Juarranz Y, Abad C, Martinez C, et al. Protective effect of vasoactive intestinal peptide on bone destruction in the collagen-induced arthritis model of rheumatoid arthritis. Arthritis. Res. Ther. 2005;7:R1034–R1045. doi: 10.1186/ar1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HY, Lee HJ, Chang YJ, et al. Interleukin-17-producing innate lymphoid cells and the NLRP3 inflammasome facilitate obesity-associated airway hyperreactivity. Nat. Med. 2014;20:54–61. doi: 10.1038/nm.3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima M, Ito T, Oono T, et al. VIP attenuation of the severity of experimental pancreatitis is due to VPAC1 receptor-mediated inhibition of cytokine production. Pancreas. 2005;30:62–70. [PubMed] [Google Scholar]
- Kurimoto E, Miyahara N, Kanehiro A, et al. IL-17A is essential to the development of elastase-induced pulmonary inflammation and emphysema in mice. Respir. Res. 2013;14:5. doi: 10.1186/1465-9921-14-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larocca L, Hauk V, Calafat M, et al. Modulation of macrophage inflammatory profile in pregnant nonobese diabetic (NOD) mice. Mol. Cell. Endocrinol. 2011;333:112–118. doi: 10.1016/j.mce.2010.11.035. [DOI] [PubMed] [Google Scholar]
- Leppanen T, Tuominen RK. Moilanen E. Protein kinase C and its inhibitors in the regulation of inflammation: inducible nitric oxide synthase as an example. Basic Clin. Pharmacol. Toxicol. 2014;114:37–43. doi: 10.1111/bcpt.12139. [DOI] [PubMed] [Google Scholar]
- Li L, She H. Yue S. Vasoactive intestinal peptide induces surfactant protein A expression in ATII cells through activation of PKC/c-Fos pathway. Peptides. 2010;31:2046–2051. doi: 10.1016/j.peptides.2010.07.017. [DOI] [PubMed] [Google Scholar]
- Lo RS, Dumoutier L, Couillin I, et al. IL-17A-producing gammadelta T and Th17 lymphocytes mediate lung inflammation but not fibrosis in experimental silicosis. J Immunol. 2010;184:6367–6377. doi: 10.4049/jimmunol.0900459. [DOI] [PubMed] [Google Scholar]
- Martinez C, Delgado M, Pozo D, et al. Vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide modulate endotoxin-induced IL-6 production by murine peritoneal macrophages. J Leukoc Biol. 1998a;63:591–601. doi: 10.1002/jlb.63.5.591. [DOI] [PubMed] [Google Scholar]
- Martinez C, Delgado M, Pozo D, et al. VIP and PACAP enhance IL-6 release and mRNA levels in resting peritoneal macrophages: in vitro and in vivo studies. J. Neuroimmunol. 1998b;85:155–167. doi: 10.1016/s0165-5728(98)00018-6. [DOI] [PubMed] [Google Scholar]
- Nagahama K, Ogawa A, Shirane K, et al. Protein kinase C theta plays a fundamental role in different types of chronic colitis. Gastroenterology. 2008;134:459–469. doi: 10.1053/j.gastro.2007.11.005. [DOI] [PubMed] [Google Scholar]
- Persson E. Lerner UH. The neuropeptide VIP potentiates IL-6 production induced by proinflammatory osteotropic cytokines in calvarial osteoblasts and the osteoblastic cell line MC3T3-E1. Biochem. Biophys. Res. Common. 2005;335:705–711. doi: 10.1016/j.bbrc.2005.07.135. [DOI] [PubMed] [Google Scholar]
- Roussel L, Houle F, Chan C, et al. IL-17 promotes p38 MAPK-dependent endothelial activation enhancing neutrophil recruitment to sites of inflammation. J. Immunol. 2010;184:4531–4537. doi: 10.4049/jimmunol.0903162. [DOI] [PubMed] [Google Scholar]
- Salinthone S, Yadav V, Schillace RV, et al. Lipoic acid attenuates inflammation via cAMP and protein kinase A signaling. PLoS ONE. 2010;5:1–10. doi: 10.1371/journal.pone.0013058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Screedharan SP, Huang JX, Cheung MC, et al. Structure, expression and chromosomal localization of the type I human vasoactive intestinal peptide receptor gene. Proc. Natl. Acad. Sci. U. S. A. 1995;92:2939–2943. doi: 10.1073/pnas.92.7.2939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimura E, Shibui A, Narushima S, et al. Potential role of myeloid cell/eosinophil-derived IL-17 in LPS-induced endotoxin shock. Biochem. Biophys. Res. Commun. 2014;453:1–6. doi: 10.1016/j.bbrc.2014.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun GY, Guan CX, Zhou Y, et al. Vasoactive intestinal peptide re-balances TREM-1/TREM-2 ratio in acute lung injury. Regul. Pept. 2011;167:56–64. doi: 10.1016/j.regpep.2010.11.008. [DOI] [PubMed] [Google Scholar]
- Tan HL. Rosenthal M. IL-17 in lung disease: friend or foe? Thorax. 2013;68:788–790. doi: 10.1136/thoraxjnl-2013-203307. [DOI] [PubMed] [Google Scholar]
- Tan HL, Regamey N, Brown S, et al. The Th17 pathway in cystic fibrosis lung disease. Am. J. Respir. Crit. Care Med. 2011;184:252–258. doi: 10.1164/rccm.201102-0236OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temerozo JR, Joaguim R, Regis EG, et al. Macrophage resistance to HIV-1 infection is enhanced by the neuropeptides VIP and PACAP. PLoS ONE. 2013;8:e67701. doi: 10.1371/journal.pone.0067701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usdin TB, Bonner TI. Mezey E. Two receptor for vasoactive intestinal polypeptide with similar specificity and complementary distributions. Endocrinology. 1994;135:2662–2680. doi: 10.1210/endo.135.6.7988457. [DOI] [PubMed] [Google Scholar]
- Xin Z. Sriram S. Vasoactive intestinal peptide inhibits IL-12 and nitric oxide production in murine macrophages. J. Neuroimmunol. 1998;89:206–212. doi: 10.1016/s0165-5728(98)00140-4. [DOI] [PubMed] [Google Scholar]
- Yadav M. Goetzl EJ. Vasoactive intestinal peptide-mediated Th17 differentiation: an expanding spectrum of vasoactive intestinal peptide effects in immunity and autoimmunity. Ann. N. Y. Acad. Sci. 2008;1144:83–89. doi: 10.1196/annals.1418.020. [DOI] [PubMed] [Google Scholar]
- Yadav M, Rosenbaum J. Goetzl EJ. Cutting edge: vasoactive intestinal peptide (VIP) induces differentiation of Th17 cells with a distinctive cytokine profile. J. Immunol. 2008;180:2772–2776. doi: 10.4049/jimmunol.180.5.2772. [DOI] [PubMed] [Google Scholar]
- Yan JW, Wang YJ, Peng WJ, et al. Therapeutic potential of interleukin-17 in inflammation and autoimmune diseases. Expert Opin. Ther. Targets. 2014;18:29–41. doi: 10.1517/14728222.2013.843669. [DOI] [PubMed] [Google Scholar]
- Yang J, Shi QD, Song TB, et al. Vasoactive intestinal peptide increases VEGF expression to promote proliferation of brain vascular endothelial cells via the cAMP/PKA pathway after ischemic insult in vitro. Peptides. 2013;42:105–111. doi: 10.1016/j.peptides.2013.01.007. [DOI] [PubMed] [Google Scholar]
- You QH, Zhang D, Niu CC, et al. Expression of IL-17A and IL-17F in lipopolysaccharide-induced acute lung injury and the counteraction of anisodamine or methylprednisolone. Cytokine. 2014;66:78–86. doi: 10.1016/j.cyto.2013.12.019. [DOI] [PubMed] [Google Scholar]