

Abstract
Congenital heart disease (CHD) remains a significant health problem, with a growing population of survivors with chronic disease. Despite intense efforts to understand the genetic basis of CHD in humans, the etiology of most CHD is unknown. Furthermore, new models of CHD are required to better understand the development of CHD and to explore novel therapies for this patient population. In this review, we highlight the role that human induced pluripotent stem cell (hiPSC)-derived cardiomyocytes can serve to enhance our understanding of the development, pathophysiology and potential therapeutic targets in CHD. We highlight the use of hiPSC-derived cardiomyocytes to model gene regulatory interactions, cell-cell interactions and tissue interactions contributing to CHD. We further emphasize the importance of using hiPSC-derived cardiomyocytes as personalized research models. The use of hiPSCs presents an unprecedented opportunity to generate disease-specific cellular models, investigate the underlying molecular mechanisms of disease and uncover new therapeutic targets for CHD.
Keywords: congenital heart disease, heart development, human induced pluripotent stem cells, cardiomyocytes, tissue engineering
Introduction
Advances in medical and surgical treatment for infants and children with congenital heart disease (CHD) have achieved survival rates of over 90%, even for patients with the most complex cardiac defects. This survival rate is remarkable, as we continue to have an incomplete understanding of the genetic, molecular and cellular etiologies of these disorders. These patients often develop cardiac, neurodevelopmental or other end-organ sequelae associated with their disease or treatment. New tools and research models are needed to further decipher the pathophysiological mechanisms that contribute to CHD and contribute to the chronic morbidity and demise associated with CHD.
The use of both invertebrate and vertebrate animal model systems have helped define the embryologic processes of heart development and the coordinated transcriptional and signaling networks that govern these processes. Classical genetic model systems, including Drosophila (the fruit fly), zebrafish, mouse, and non-genetic model systems, including the frog (Xenopus laevis) and chick, have been instrumental in understanding the mechanisms of early cardiac patterning and the formation of conserved cardiac structures. More complex mammalian models, including the mouse, rat, pig and sheep, have been used extensively to model cardiac morphogenesis, gene function, and physiology. During the past decade, computer modeling and bioinformatics have become extremely powerful tools for the advancement of our understanding of gene networks, further increasing our understanding of early heart development and regeneration. Despite these tools, we continue to have a limited understanding of the molecular mechanisms leading to congenital heart defects and limited treatment options for adult CHD patients.
Human induced pluripotent stem cells (hiPSCs) offer a unique platform to study the genetic mutations and developmental pathways associated with CHD. This cell-based human model system will allow us to validate previous findings in animal model systems, investigate genetic and epigenetic regulation of human genes implicated in CHD and provide a tool for studying the molecular regulation and cell-cell interactions in normal and abnormal cardiomyocyte (CM) development and maturation. In addition, the hiPSC-CM model system will serve as a platform for preclinical trials of gene therapy or small molecule therapeutics. Lastly, hiPSC-CMs may provide novel forms of cell-based therapies with specific applications for congenital heart disease. Here, we review the molecular mechanisms that govern cardiomyocyte differentiation from human pluripotent stem cells (PSCs) and the potential application of this technology to model CHD.
Genetic Defects and CHD
In current clinical practice, less than 15% of patients have an identified genetic etiology for their CHD. Most of these patients have easily identifiable genetic syndromes that are confirmed by standard genetic testing including karyotypic analysis, fluorescent in situ hybridization (FISH) or array comparative genomic hybridization (CGH) ((1-3); Table 1). An additional subset of patients with syndromic single gene defects are often clinically diagnosed by a recognizable pattern of defects associated with CHD and confirmed by candidate gene sequencing (Table 1). An unclear number of patients have unidentified single gene defects known to be associated with isolated CHD ((2; 3); Figure 1). These patients can be identified by whole exome or candidate gene sequencing which is performed infrequently, although this is gradually becoming more common in clinical practice. The majority of patients with CHD are predicted to have disease that is multifactorial in etiology- those with genetic susceptibility in combination with environmental opportunity to generate a complex interaction of genetics and development (1; 4). This complexity is one of the factors that contribute to the difficulty in the identification of the underlying primary mechanism in the majority of patients with CHD. The use of hiPSCs provides an unprecedented opportunity to dissect the genetic interactions and signaling pathways that contribute to the high incidence of abnormal heart formation during development and ultimately will increase our understanding of the developmental origins of CHD.
Table 1. Selected human genetic syndromes associated with CHD.
| Gene Defect | Incidence/live birthsa | Phenotype (non-cardiac) | % with CHD | Common CHD | CHD Described | |
|---|---|---|---|---|---|---|
|
| ||||||
| Aneuploidy Syndromes | ||||||
|
| ||||||
| Down Syndrome (5-8) | Trisomy 21 | 1:700 | Dev delay, short stature, low tone | 40-50% | AVSD, VSD | ASD, TOF, PDA |
| Turner syndrome (9-13) | Monosomy X (XO) | 1:2500 | Short stature, lymphedema webbed neck | 30-50% | Coarctation, BAV, VSD | HLHS, HTN Aortic dilation |
| Edwards Syndrome (14; 15) | Trisomy 18 | 1:6000 | 80% female, dev delay, arthrogryposis | 90% | ASD, VSD, PDA | Coarctation, HLHS |
|
| ||||||
| Microdeletion Syndromes | ||||||
|
| ||||||
| DiGeorge/VCFS (16-19) | del22p11.2 | 1:4000 | Hypocalcemia, immune deficiency | 80%+ | TOF, TA, IAA (B) | VSD, ASD, Arch anomalies |
| Williams-Beuren (20-22) | del7q11.23 | 1:8000 | Hypercalcemia, Renal disease, dev delay, FTT | 80%+ | SVAS, PPS | Coarct,valve disease, ASD, VSD |
| Jacobsen (23) | del11q23 | 1:100,000 | Abn platelets, dev delay, short stature | 50% | VSD, Ao valve anom | HLHS (5%) Coarct |
|
| ||||||
| Syndromic Single Gene Disorders | ||||||
|
| ||||||
| Noonan (24-29) | PTPN11, BRAF, SOS, KRAS, CBL, RIT1 | 1:1000-1:2500 | Short stature, webbed neck | 80% | Dysplastic PV, ASD, VSD, HCM | AVSD, PDA |
| Alagille (30; 31) | JAG1 NOTCH2 | 1:70,000 | Extrahepatic biliary atresia; vertebral anom | 90%+ | PPS, TOF | ASD, VSD, AS, Coarctation, HLHS |
| Ellis-van Creveld (32; 33) | EVC1; EVC2 | 1:60,000-1:200,000 | Short stature, polydactyly, dental abn. | 50%+ | ASD | Common atrium, AVSD PAPVR |
| Holt-Oram (34; 35) | TBX5 | 1:100,000 | Upper limb malformations | 75% | ASD, VSD | AVSD, conduction defects |
Incidence in the United States
ASD, Atrial Septal Defect; AS, Aortic Stenosis; AVSD, Atrioventricular Septal Defect; BAV, Bicuspid Aortic Valve; HCM, Hypertrophic Cardiomyopathy; HLHS, Hypoplatic Left Heart Syndrome; HTN, Hypertension; IAA, Interrupted Aortic Arch; PAPVR, Partial Anomalous Pulmonary Venous Return; PDA, Patent Ductus Arteriosus; PPS, Peripheral Pulmonary Stenosis; PV, Pulmonary Valve; SVAS, Supravalvular Aortic Stenosis; TA, Tricuspid Atresia; TAPVR, Total Anomalous Pulmonary Venous Return; TOF, Tetralogy of Fallot; VSD, Ventricular Septal Defect.
Figure 1. Modeling single gene defects associated with isolated CHD.
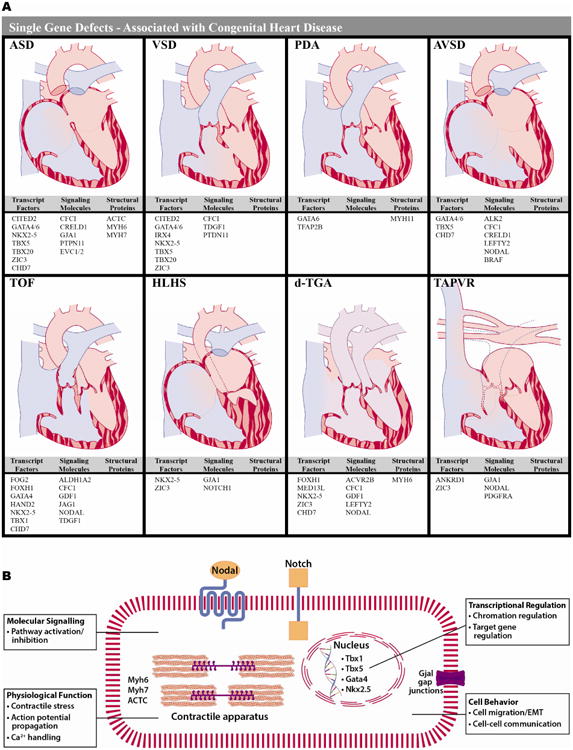
A) Eight forms of CHD are diagrammed and genes associated with that form of CHD are listed below by category (transcription factors; receptors, ligands or signaling molecules; structural proteins). The variability in the types of genes, which can cause a single form of CHD and the numerous forms of CHD that can be caused by disruption of a single gene are highlighted. (2; 3). B) Several examples of the cellular processes for genes associated with CHD are highlighted. These processes can be manipulated using techniques such as gene editing, next-generation sequencing analysis, small molecule manipulation and tissue engineering in hiPSC-CMs.
Cardiac Development
There are significant similarities in the key processes of heart development between species (Figure 2A) (36). Briefly, in mammalian species, precardiac mesoderm is specified during gastrulation and forms a symmetric appearing “crescent” in the anterior and lateral portions of the embryo (37). Bilateral endocardial tubes form from first heart field precursors and fuse in the midline to form a single midline straight heart tube. This tube elongates through the addition of cells from the lateral plate mesoderm, or second heart field (SHF), and loops rightward and caudally to establish the anatomic relationships necessary for chamber formation and septation. Lastly, tissues from outside the cardiac fields contribute to cardiac development. Neural crest cells migrate through branchial arches and pharyngeal mesoderm with cues from SHF signals, to participate in remodeling and septation of the outflow tract. Specialized paraxial mesoderm (proepicardium) in the more ventral region of the embryo migrates to the heart forming the epicardium and contributes to coronary artery, smooth muscle and myocardial development ((38; 39); Figure 2A). The use of model organisms has enhanced our understanding of the conserved mechanisms of cardiac development and the coordinated transcriptional regulation and molecular signaling required for these complex processes. A core set of gene regulatory networks and signaling pathways, including TGFβ/BMP, Wnt, Notch, and Sonic hedgehog (Shh), drive these developmental processes ((39; 40); Figure 2B). The use of hiPSC-CMs will allow us to assemble models of normal and abnormal developmental cellular interactions to assess whether these molecular interactions are conserved in human cardiovascular development.
Figure 2. Molecular regulation of heart development.
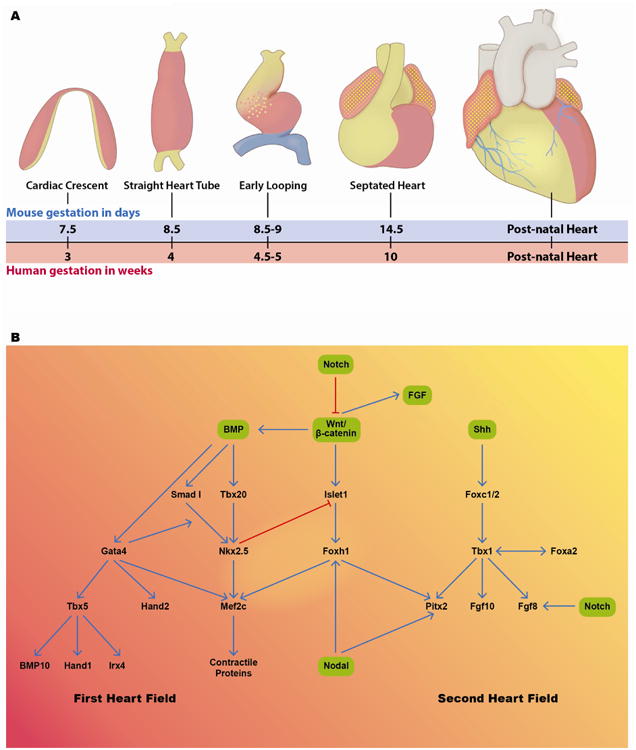
A) Stages of heart development are schematized. After gastrulation, precardiac mesoderm is present in the anterior and lateral regions of the mouse embryo in a crescent pattern. As embryonic development proceeds the embryo folds ventrally and to the midline and the cardiac fields meet and form a straight heart tube. The heart tube begins to loop rightward and caudally in the embryo and then septation begins, setting up the normal anatomic relationships in the heart, seen most rightward in this panel. Red shaded areas represent first heart field derivatives, yellow shaded areas represent areas that are most likely derived from second heart field and blue shaded areas represent areas that are derived from proepicardium (36; 37). B) Schematic representation of gene regulatory interactions between known signaling pathways (green boxes) and transcription factor interactions in the first heart field (left side) and second heart field (right side). A number of signaling molecules and transcription factors play overlapping roles in the two cardiac progenitor cell populations (36; 39; 40).
Differentiation of hiPSCs to Cardiovascular Lineages
Since the paradigm-shifting discovery highlighting that somatic cells could be reprogrammed to pluripotent cells, hiPSCs have been used to study novel mechanisms of human disease (41-44). Efforts have focused on modeling cardiovascular disease, particularly disease processes intrinsic to CMs (45). Significant progress has been made towards the efficient generation of CMs from human PSCs including both hiPSCs and human embryonic stem cells (hESCs). These stem cell-derived CMs beat spontaneously, they express the expected sarcomeric components and ion channels and exhibit cardiac calcium transients and action potentials. Both mouse and human PSCs have been extensively used to study cardiovascular lineage differentiation in vitro demonstrating conservation of signaling mechanisms. For simplicity, we focus on experiments undertaken using human PSCs.
Initially, the formation of embryoid bodies (EBs) from human PSCs was used to differentiate CMs while current methods use monolayer culture systems where the controlled application of growth factors and small molecules more precisely directs CM differentiation (46-48). In vitro cardiomyocyte differentiation occurs through a stage-specific manner similar to the cardiac developmental program in the embryo (Figure 2). There are three major stages of cardiomyocyte differentiation in vitro: induction of cardiac mesoderm from human PSCs, specification and proliferation of cardiac progenitor cells (CPCs) and differentiation of CPCs to mature CMs (Figure 3). There are at least four major signaling pathways that are involved in cardiac differentiation of human PSCs: TGFβ/Activin/Nodal, BMP, Wnt and FGF. Several additional pathways including p38 MAPK and Notch signaling pathways also modulate cardiac differentiation (49-51). To efficiently direct the differentiation of pluripotent stem cells via mesoderm to cardiovascular cell types requires specific temporal and dose dependent modulation of these pathways (48; 52; 53).
Figure 3. Schematic of cardiac lineage differentiation from human PSCs.
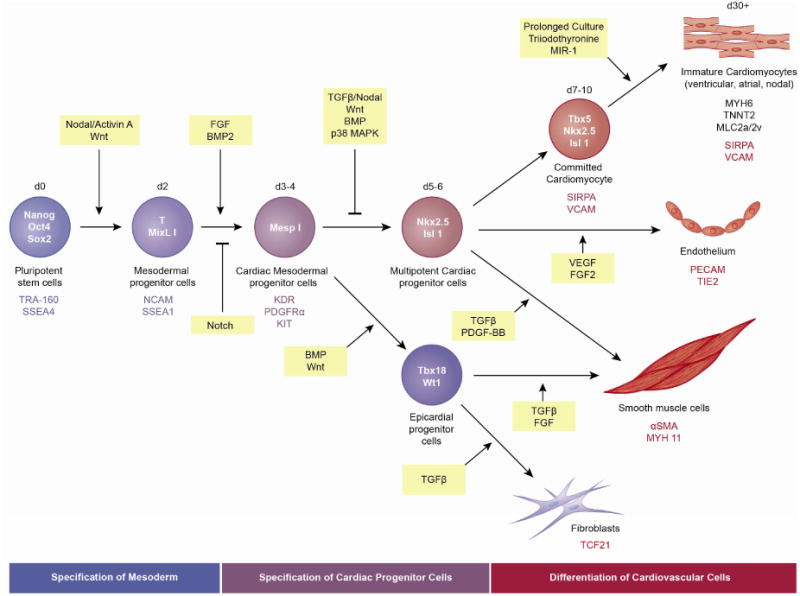
The three primary stages of in vitro CM differentiation from hiPSCs are indicated: induction of cardiac mesoderm, specification of CPCs and differentiation of CMs. Factors involved in directing differentiation of pluripotent stem cells to mesodermal progenitor cells and subsequent cardiovascular lineage cells are indicated. Signaling molecules are in yellow boxes. Transcription factors (within cells) and cell surface markers (below cells) expressed by each cell type are indicated. Genes (structural proteins and cell surface markers) expressed by cardiomyocytes, endothelial cells, smooth muscle cells and fibroblasts are also indicated (below images).
Induction of Cardiac Mesoderm
The earliest identification of a CPC from hESCs emerged from experiments demonstrating that a population of KDRlow/c-kitNeg cells could be generated from hESCs. When cultured as a monolayer, these cells generated more than 50% CMs and when cultured under colony-forming conditions they generated CMs, endothelial cells and vascular smooth muscle cells (50). These findings were consistent with observations in mouse embryos demonstrating that the earliest cardiovascular progenitors could be identified based on expression of Flk1 (KDR), which was upregulated as cells emerged from the primitive streak during gastrulation (54). Further studies demonstrated that cardiac mesoderm is more specifically identified by coexpression of KDR (Flk1) and PDGFRα (platelet derived growth factor α) (52).
Developmental signaling pathways that have a functional role in specification of mesoderm during embryonic development have been manipulated in vitro to promote differentiation of human PSCs to cardiac mesoderm. The modulation of the TGFβ, BMP and the canonical Wnt signaling pathways is critical for promoting cardiac mesoderm differentiation. Murine developmental studies demonstrate that TGFβ signaling, mediated by Smad2 and Smad3, plays an important role in mesoderm specification (55). The sequential exposure to Activin A or Nodal followed by BMP4 induces mesodermal specification and subsequent cardiac differentiation in human PSC cultures (50; 52; 56; 57). Similarly, in mouse ESCs, Nodal induces TGFβ signaling and together these pathways stimulate the formation of KDR+ cardiovascular progenitor cells (58).
Wnt signaling also promotes mesodermal formation from human PSCs in vitro. Experiments using embryoid bodies for differentiation, revealed that the addition of Wnt3a enhanced mesendoderm formation and led to an increase in beating CMs (59). The canonical Wnt ligands, Wnt1, Wnt3a, and Wnt8, are upregulated between days 1-3 (60); the knockdown of β-catenin in the initial stage of differentiation blocks CM specification; and the use of the small molecule CHIR99021, an activator of the Wnt signaling pathway (GSK3β inhibitor), at Day 0 significantly enhances CM differentiation (53). Manipulation of these pathways results in the increased expression of early mesoderm markers such as brachyury (T) and MIXL1 at day 2 and the early cardiac mesoderm markers Mesp1, KDR, PDGFRa, and KIT at day 3-4 (Figure 3 and 4A).
Figure 4. Molecular features of in vitro differentiated cardiomyocytes.
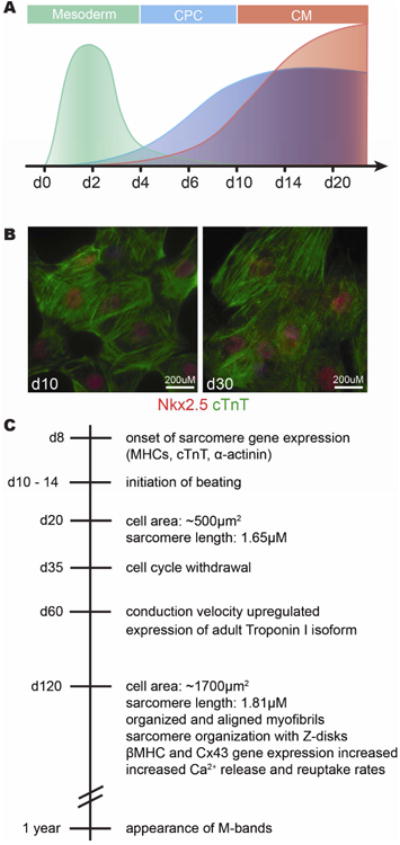
A) Schematic representation of gene expression patterns during the first 20 days of directed CM differentiation demonstrate temporal conservation with patterning events in mouse embryonic development. Mesodermal patterning genes (such as Mesp1 and T) are induced early and peak at day 2 (green). Markers of cardiac progenitor cells (such as Nkx2-5 and Islet1) are expressed beginning between day 4 and 6 of differentiation and are maintained in differentiated CMs (blue). Sarcomeric genes (such as aMHC and cTnT) expressed in differentiated CMs beginning between days 6 and 10 and continue to increase in expression with longer time in culture (red). B) Images of differentiatied CMs at day 10 and day 30 in culture show coexpression of Nkx2-5 (red) and cardiac Troponin T (green). C) Timeline of in vitro differentiation indicating when certain characteristics of mature CMs are acquired. Beating CMs are observed between day 10 -15 and continue to proliferate until about day 35 (88). These day 35 cardiomyocytes are still immature regarding their size, contractility, sarcomeric and mitochondrial structure (90; 92).
Specification of CPCs
In the second stage of human PSC differentiation, cardiac mesodermal cells are specified toward cardiac progenitor cells (CPCs). A number of signaling pathways that were active early during cardiac mesoderm differentiation are inactivated at this stage. TGFβ signaling plays a biphasic role during cardiomyogenesis and is downregulated to promote the differentiation of CPCs. Continued signaling through TGFβ induces cells towards the vascular smooth muscle and endothelial lineages at the expense of CMs (61). The small molecule ITD1 blocks TGFb signaling by inducing the destruction of the type II TGFβ receptor (TGFBR2) and specifically directs hESCs towards CMs (62).
Similar to the biphasic role of TGFβ signaling, Wnt signaling also has a biphasic effect on cardiomyogenesis. A number of experiments have demonstrated that the inhibition of Wnt signaling at this stage is required to promote robust CM differentiation. Further, inhibition of Wnt signaling via DKK1 or the small molecules IWR-1, which stabilizes Axin, part of the β-catenin destruction complex, and IWP-4, which blocks accumulation of β-catenin, increases CM differentiation efficiency (50; 53; 62-65).
Inhibition of BMP signaling via dorsomorphin also promotes further specification of CPCs from mesoderm (52). The dose and duration of BMP signaling during cardiac progenitor cell specification is critical to efficiently generate CMs (66). Witty et al. demonstrated that the optimal dose of the BMP inhibitor Noggin is 12.5 to 50 ng/ml. Above this range or in the presence of BMP4, the generation of cardiac troponin T+ (cTnT) CMs was inhibited. Further, the duration of the BMP dependent stage is approximately 24 hours at day 4 of CM differentiation (66). CPCs are present in monolayer differentiation protocols beginning between day 4 to 6 of differentiation (Figure 4A). Human PSC-derived CPCs can be characterized by expression of key cardiac developmental regulators including Nkx2.5, Gata4, Tbx5, and Islet1.
Human PSC-CPCs are primarily present in a transient state during differentiation protocols designed to direct cells toward beating CMs. Currently it is unclear whether human PSC-CPCs are equivalent to first (FHF) or second heart field (SHF) progenitors cells. Current studies suggest the majority of CPCs derived from hiPSCs express FHF genes while the minority express SHF genes (67). In vitro hiPSC-CPCs express Nkx2-5 by day 7 of CM differentiation (68). During mouse embryonic heart development Nkx2-5 marks cells of both cardiac fields (40). hiPSC-CPCs also robustly express Tbx5 and HCN4; both have been shown to mark cells of the FHF and contribute to CMs in the left ventricle and atria (67; 69-71). Nkx2.5+, Tbx5+ and HCN4+ hiPSC-CPCs differentiate to mostly cTnT+ CMs (67).
Islet1 and Nkx2.5 are similarly expressed during in vitro cardiovascular differentiation. Bu et al. used a lineage tracing strategy to irreversibly mark Islet1 expressing cells and their descendents during in vitro differentiation of hESCs. These authors demonstrated that Islet1 marks a multipotent CPC that gives rise to CMs, smooth muscle and endothelial cells (72). This observation is consistent with the role for Islet1 in the developing mouse heart where it is critical for CPCs derived from the second heart field that can give rise to all three cardiovascular lineages (73). These results support the notion that in vitro derived CPCs could include SHF progenitors. In the mouse PSC differentiation system, Buikema et al. used reporters under the control of a Nkx2-5 enhancer and an Islet1 dependent anterior heart field-specific enhancer of Mef2c to demonstrate the presence of distinct FHF and SHF progenitor cells (74). A similar approach has not been undertaken with hiPSCs. Other SHF markers, including Tbx1 and FGFs, are detected by qPCR in hiPSC-CPCs (unpublished data). Whether these represent a distinct progenitor cell population with unique contributions to mature cardiovascular cell types is yet to be determined.
In addition to CPCs that are primarily directed towards the CM lineage, a number of other cardiovascular progenitors can be generated in vitro from human PSCs. Cao et al. demonstrated that hiPSC-CPCs could be stably maintained and expanded under conditions where the GSK3β, BMP, and Activin/Nodal signaling pathways were simultaneously inhibited. These cells retained the ability to differentiate into CMs (when treated with BMP4 and the Wnt antagonist IWR-1), smooth muscle cells (when treated with PDGF-BB and TGFβ1), and endothelial cells (when treated with VEGF plus FGF2) (75). CD34+, CD31+ endothelial progenitor cells can be efficiently produced by GSK3 inhibition at the mesodermal stage (76). Additionally, the temporal manipulation of BMP and Wnt signaling promotes the generation of a WT1+Tbx18+ epicardial cell population (66). Stimulation of the BMP and Wnt signaling pathways during CPC specification (differentiation days 4 to 6) promotes the formation of epicardial progenitor cells. In contrast, the inhibition of these pathways during this phase is required for efficient differentiation of CMs. Human PSC-derived epicardial cells undergo epithelial-mesenchymal transition (EMT) and give rise to smooth muscle cells and fibroblasts upon activation of the TGFβ1 and bFGF signaling pathways (66).
Differentiation of CMs
In the last stage of differentiation, CPCs proliferate and form immature CMs. At this stage, several cardiac transcription factors, including Nkx2-5, Tbx5 and Gata4 cooperate to activate the transcription of cardiac structural genes including: cardiac troponins, myosin heavy chain, myosin light chain, and desmin (47). The upregulation of cardiac structural genes occurs between days 7 and 10 in monolayer culture systems (Figure 3 and 4A). Nkx2-5 is coexpressed in cTnT+ cells at day 10 of differentiation and continues to be expressed in cTnT+ cells at day 30 and beyond (Figure 4B). The most commonly used differentiation protocols yield a mixed pool of ventricular, atrial and nodal cell types based on sarcomeric gene expression and electrophysiologic properties (77; 78). Enrichment of specific cardiac cell types has been achieved in some experimental settings. The BMP antagonist Grem2 was recently shown to preferentially drive differentiation to atrial CMs (79). Modulation of retinoic acid (RA) signaling in combination with Noggin has similarly been shown to drive ventricular or atrial CM differentiation (80). Although specific protocols vary, regarding the use of small molecules and growth factors, it is clear that the temporal modulation of the Activin/Nodal, BMP and Wnt signaling pathways can efficiently produce CMs in vitro.
Human iPSC-CMs harbor many features of native CMs including spontaneous beating, expression of sarcomeric proteins and ion channels and the generation of cardiac action potentials. Upon transplantation into infarcted hearts of mice, rats and guinea pigs, they engraft, electrically couple with host CMs, and enhance cardiac mechanical function (81-85). hESC-CMs contributed significantly to remuscularization and were electrically coupled with host CMs when engrafted in a primate model of myocardial ischemia, however incomplete maturation of the transplanted cells was observed over a three month period (86). Despite this progress, hiPSC-CMs remain morphologically and functionally immature, more similar to fetal rather that adult CMs. Multiple strategies have been recently undertaken to promote the differentiation and maturation of CMs derived from hiPSCs in vitro, including long-term culture, 3-dimensional tissue engineering, mechanical and electrical stimulation and treatment with neurohormonal factors (87). hiPSC-CMs maintained in long-term culture demonstrate the following characteristics: decreased proliferation, a shift towards an adult-like expression pattern (increased expression of MLC2v, adult isoforms of cardiac Troponin I, Na+ and K+ channel genes, etc.), changes in cell shape, increased sarcomere alignment and organization, and more mature electrophysiological properties ((88-92); Figure 4C). Further molecular, proteomic and functional characterization of hiPSC-derived and isolated adult CMs is underway.
Cardiovascular Disease Modeling using hiPSCs
Generating hiPSCs from patients with gene mutations provides an unprecedented opportunity to study disease causing pathophysiology in the affected individual on a molecular, cellular and even a tissue level. Moreover, for the first time these hiPSC model systems allow the opportunity to dissect the genetic regulation and developmental disruption as well as to develop and test directed treatments without exposing the subject to risk. hiPSC-CMs have been successfully used to model and test drug responsiveness for a variety of inherited cardiovascular disorders (Table 2; (45; 93)). hiPSC-CM models of arrhythmic disorders recapitulate cardiac channel function and electrophysiological features of Long QT syndrome and catecholaminergic polymorphic ventricular tachycardia (CPVT) (94-107). Morphologic, contractile and electrical phenotypes observed in hiPSC-CMs generated from hypertrophic cardiomyopathy (HCM) and dilated cardiomyopathy (DCM) patients are consistent with clinical phenotypes (108-116). Lastly, some aspects of the phenotypes for arrhythmogenic right ventricular dysplasia (ARVD), LEOPARD syndrome and Pompe's disease are recapitulated in hiPSC-CMs (99; 117-120).
Table 2. Inherited Cardiac Diseases Modeled in hiPSCs.
| Disease Name | Gene | hiPSC-CM phenotype | References |
|---|---|---|---|
|
| |||
| Congenital Heart Disease (CHD) | |||
|
| |||
| Hypoplastic Left Heart Syndrome (HLHS) | NKX2.5 CX43 | Decreased differentiation efficiency, reduced transcription factor expression, myofibrillar disorganization, altered Ca2+ transients and responses to caffeine and Beta-adrenergic agonists. | (125; 127) |
|
| |||
| Genetic Conditions Associated with CHD | |||
|
| |||
| Trisomy 21 | ETS2 ERG | Alteration in expression of transcription factors. Increased ETS2 and ERG expression in cardiac mesoderm may mediate abnormal endocardial cushion formation. Conservation of epigenetic modifications has been demonstrated. | (128; 129) |
| Leopard Syndrome | PTPN11 | Increased cell surface area, increased nuber of cells with organized sarcomere, increased nuclear expression of NFATC4. | (119) |
| Williams Syndrome/Elastin Deficiency/Supravalvar Aortic Stenosis (SVAS) | Microdel 7q11.23/ELN | Increased proliferation, decreased expression of differentiated SMC markers, reduced response to vasoactive agonists, reduced Ca2+ flux. | (123; 124) |
|
| |||
| Cardiomyopathies | |||
|
| |||
| Dilated Cardiomyopathy | TNNT2 (cardiac troponin T) | Irregular organization of the sarcomere (increased number of disorganized cells), reduced contractile force, altered Ca2+ regulation, and reduced β1-adrenergic stress tolerance | (108) |
| LMNA (Lamin A/C) | Accelerated nuclear senescence and apoptotic activity with pacing. Rescued by ERK1/2 signaling blockade. | (109) | |
| DES (Desmin) | Isolated aggregation of desmin particles. Altered Ca2+ uptake. Reduced response to β-agonists. | (110) | |
| Hypertrophic Cardiomyopathy | MHY7 | Enlarged cardiomyocytes, increased myofibril content with disordered sarcomeres, elevated diastolic Ca2+, and impaired Ca2+ handling. Rescued by β-blockers or verapamil. | (111; 112) |
|
| |||
| Channelopathies | |||
|
| |||
| Long QT Syndrome Type I | KCNQ1 | Prolongation of action potential and field potential. Abnormal protein localization in some variants. Develop arrhythmias with β-adrenergic stimulation, rescued by β-blockers. | (94; 106) |
| Long QT Syndrome Type II | KCNH2 | Prolonged action and field potentials. Develop arrhythmias with β-adrenergic stimulation, rescued by β-blockers. | (95; 107) |
| Long QT Syndrome Type III | SCN5A | Gain of function in sodium ion channels, recapitulated with voltage-dependent inactivation of sodium channels. Faster pacing and mexilitine rescued. | (99; 104) |
| Long QT Syndrome Type 8 (Timothy Syndrome) | CACNA1C | Prolonged action potential, arrhythmias, excess Ca2+ influx, abnormal Ca2+ transients, rescued by Roscovitine, a CA(v)1.2 activator. | (105) |
| Catecholaminergic polymorphic ventricular tachycardia (CPVT) | RYR2 CASQ2 | Increased susceptibility to arrhythmia due to delayed after-depolarizations. Improved with flecainide or thapsigargin (SERCA2a pump inhibitor). | (107) |
|
| |||
| Other | |||
|
| |||
| Arrhythmogenic right ventricular dysplasia (ARVDC) | PKP2, PKP | Abnormal nuclear translocation of PKG. Low β-catenin expression and activity. Increased induction of lipogenesis and apoptosis, particularly in ISl1+derived cells. Increased lipid content in lipogenic medium. | (99; 120) |
| Barth Syndrome | TAZ tafazzin | Deficient sarcomere assembly and diminished contractile function. Improved with reduction of reactive oxygen species. | (122) |
| Pompe's Disease | GAA (α–glucosidase) | Exhibited decreased GAA activity and reduced metabolism. Large glycogen-containing lysozomes. Normal contractility. Abnormal glycosylation of lysosomal associated membrane proteins. | (117; 118) |
| Friedreich's Ataxia | FXN (frataxin) | Impaired mitochrondrial function, mitochrondrial abnormalities including decrease membrane potential | (114-116) |
A small number of congenital syndromes with cardiovascular phenotypes caused by mutations in single genes have been studied using the hiPSC platform (Table 2). LEOPARD Syndrome, which causes HCM in 80% of patients due to a mutation in PTPN11 was the first cardiovascular disease modeled with hiPSCs (119; 121). This study demonstrated hypertrophy of single CMs as well as increased nuclear NFATc4 accumulation and altered RAS/MAPK signaling, a potential target pathway for therapeutic interventions in this disorder. hiPSC-CMs from the mitochrondrial cardiomyopathy associated with Barth syndrome, which is caused by a mutation in the gene encoding tafazzin (a mitochrondrial phospholipid acyltransferase), show deficient sarcomere assembly and weak contractility (122). Wang et al. used the hiPSC-CM system to demonstrate a link between mitochrondrial function, sarcomere assembly and contractile function. hiPSCs have also been generated from patients with isolated elastin mutations and Williams-Beuren syndrome, which is caused by a microdeletion on chromosome 7q11.23, a region that includes the elastin gene. Elastin deficient hiPSC-SMCs show increased proliferation, decreased expression of differentiated SMC markers, reduced response to vasoactive agonists and reduced calcium flux (123; 124). These two studies implicated ERK1/2 and mTOR signaling in increased SMC proliferation suggesting pathways appropriate for therapeutic intervention. These studies support the notion that disease phenotypes can be recapitulated in hiPSC-derived cardiovascular cells and that hiPSCs will provide a valuable platform to identify and screen novel therapeutic targets for disease and patient specific conditions.
hiPSCs as a Model for CHD
hiPSC technology provides an opportunity to enhance our understanding of the genetic, molecular and cellular mechanisms contributing to CHD. The use of hiPSCs has several advantages over animal model systems. These include direct applicability to human disease and unique human phenotypes; the ability to study single cell genomics and epigenetics; scalability to increase detection of low level or transient signaling molecules; the ability to perturb and study developmental interactions that occur at the molecular and cellular levels; and the ability to study tissue organization and interactions using hiPSCs on bioengineered matrices. Tools currently available include: rapid gene editing, RNAseq, single cell qPCR and CHIPseq. Given the ease of gene editing and scalability of this model, as well as the direct applicability of therapeutic testing, it is likely that additional tools for facilitating work with hiPSC-CMs will be developed in the near future. The use of hiPSCs, together with these technologies will compliment existing approaches using animal models to address developmental mechanisms in CHD that have been yet to be defined.
There are certainly limitations to using the hiPSC system to model CHD. These challenges include the complex inheritance, gene dosage effects, and phenotypic variation of CHD, as well as the four dimensional interaction of form and function that occurs with the initiation of the heartbeat and the establishment of circulation in early embryonic development. At the present time, it is not possible to model the complexity of these interactions at the molecular and cellular level as a whole accurately in vitro. Nonetheless, studies in hiPSCs will help dissect these processes and contribute to our understanding of human CHD in ways animal models have had a limited impact. Several examples are noted below.
One of the most challenging and highly fatal forms of congenital heart disease is Hypoplastic Left Heart Syndrome (HLHS). It has multiple etiologies, including multiple associated genetic and anatomic conditions. Two studies were recently published characterizing hiPSC-derived CM from HLHS patients. In one study, cardiomyocyte differentiation and the expression of Nkx2-5, Hand1, Hand2 and Tbx2 transcription factors were reduced in CMs derived from hiPSCs generated from HLHS patients (125). This observation is consistent with in vivo findings of decreased Nkx2-5+ in CPCs and decreased CD34+ in endothelial cell progenitors as well as decreased differentiated cells in midgestational fetuses with HLHS (126). In the second study, the HLHS hiPSC-CMs additionally showed decreased myofibrillar organization and different calcium transient patterns and electrophysiological responses to caffeine and β-adrenergic antagonists compared to control hiPSC-CMs (127). Since there have been no genetic animal models available to study this form of CHD, the availability of hiPSC-derived CMs provides unique information about the molecular variation in CMs associated with this rare but devastating form of CHD.
Congenital heart defects occur in more than 40% of patients with Down Syndrome. The most frequently observed defects are atrio-ventricular septal defect (AVSD) and ventricular septal defects (VSD) (7; 8). A recent study of monozygotic twins discordant for trisomy 21 identified domains of differential gene expression along all the chromosomes in fibroblasts isolated from the twins (128). The pattern of gene expression dysregulation domains (GEDDs) is conserved in hiPSCs generated from the twin's fibroblasts. This study also demonstrated that GEDDs are correlated with epigenetic modifications, specifically the differential enrichment of H3K4me3. The preservation of gene expression and epigenetic modification differences in hiPSCs validates the concept that hiPSC-CPCs or CMs could be used to identified molecular mechanisms contributing to CHD in Down Syndrome patients. In a second study, the CM differentiation potential and function was examined in hESC lines exhibiting complete Trisomy 21 (T21) (129). Expression of genes involved in mesodermal induction and early cardiac specification including SHF genes was perturbed in T21 hESCs during CM differentiation. Expression of Notch1, Tbx20, Islet1 and Tbx1 were downregulated during CM differentiation while Gata4 and Wnt11 were upregulated. Two transcription factors, ETS2 and ERG, located on human chromosome 21 are overexpressed in T21 hESCs during CM differentiation and when either are knocked down, the expression of Islet1 and Gata4 is restored. ETS2 and ERG have been associated with the development of the cardiac cushion in mouse (130; 131). Together these results suggest ETS2 and ERG are candidate genes for congenital heart defects observed in Down Syndrome patients. Additionally, an abnormal electrophysiological phenotype was reported. These studies highlight the use of hiPSCs to identify novel genes critical to development of CHD as well as functional differences in CMs.
Modeling Molecular Interactions in CHD
Genetic mouse models have enhanced our understanding of transcriptional regulation and cardiac morphogenesis and yet a limited number of direct transcriptional target genes have been identified. Chromatin immunoprecipitation coupled to next generation sequencing (ChIP-Seq) is a powerful technique commonly used to identify transcriptional target genes, but is limited by the requirement of a large number of cells to efficiently pulldown the transcription factor of interest and associated DNA fragments. Therefore, identification of direct target genes of early cardiac regulators has been limited by the small number of cells in the developing heart and the difficulty in isolating cells from the embryonic heart. The hiPSC-cardiovascular differentiation system is scalable making the identification of direct targets feasible. ChIP-Seq has been successfully utilized in both mouse and human ESC differentiation systems to map the temporal alterations in chromatin structure that distinguish the key cardiac transcriptional regulators from other genes (132; 133). Recently, a large-scale genomic study identified de novo mutations in multiple genes encoding histone modifying proteins in CHD patients (134). This information coupled with the chromatin mapping performed during CM differentiation could reveal new pathways to target for treatment. Additionally, this technique could be applied to CHD linked to single gene mutations, including the cardiac defects associated with Holt-Oram syndrome, a cardiac-limb syndrome caused by mutations in TBX5 ((34), Table1). Tbx5 directly interacts with Nkx2-5 and Mef2c in a context dependent fashion during heart development (69; 135; 136) but few of the target genes are known. Tbx5, along with Nkx2-5 and Mef2c are robustly expressed in CPCs derived from hiPSCs. The genetic targets of Tbx5 depend on the cofactor (Nkx2-5 versus Mef2c) and could be determined in a temporal manner during hiPSC-CM differentiation using ChIP-Seq. This strategy could significantly enhance our knowledge of transcriptional regulators in normal and abnormal cardiac development.
Analysis of Molecular Networks in CHD
Signaling and transcriptional regulation of cardiac specification, cardiac looping, chamber formation and septation are complex. The temporal and spatial balance of these networks is frequently disrupted in congenital heart disease resulting in a range of phenotypic outcomes in patients (137; 138). Three interacting cardiac transcription factors, Gata4, Tbx5, and Nkx2-5, are particularly important dosage sensitive regulators of heart formation (69; 135; 139; 140). For example, Gata4 sequence varients have been identified in families with diverse CHD lesions, including septal defects (ASD, VSD, AVSD) and cyanotic heart disease (TOF) (Figure 1; (2; 141). A Gata4 missense mutation disrupts the Gata4-Tbx5 interaction but does not affect the Gata4-Nkx2-5 interaction (139). Large scale gene expression analysis could be performed on hiPSC-CPCs carrying patient-specific Gata4 mutations or gene edited Gata4 mutations to identify differences in the gene regulatory networks that lead to phenotypic variation. In addition to understanding perturbations in molecular networks associated with single gene mutations, genome wide expression analysis can be performed on patient specific hiPSC-CPCs with unknown genetics to identify specific molecular signaling perturbations. Additionally, since CPCs are heterogenous in nature and distinct molecular markers are limited, the use of single cell RNA-seq analysis could more fully define the relationship between first heart field (FHF) and second heart field (SHF) progenitor cells. This approach was successfully used to classify lung epithelial cells into distinct groups and define the lineage progression of lung progenitor cells (142). Alternatively, heterogenous cardiovascular cell populations could be isolated using fluorescent-labeled reporters (to designate atrial, ventricular, pacemaker, endothelial lineages, etc.) prior to gene expression analysis to define molecular networks specific to these cell types. Large-scale mapping of transcription factor networks is now commonly used to distinguish molecularly distinct cell types, to identify cell-type specific regulatory units and to identify lineage hierarchies (143; 144). An enhanced understanding of the molecular identity of and signaling networks in CPCs would contribute to our understanding of normal heart development and CHD pathogenesis.
Cell-Cell Interactions in CHD
Signaling between the myocardium and endocardium by TGFβ, Notch, and Erbb3 pathways controls cardiac valve formation and is integral to normal heart formation (145). Mutations in all of these pathways contribute to a variety of CHD (2), including aortic valve defects (30; 146), ventricular septal, atrioventricular and pulmonary valve defects (147). Signaling through these pathways induces endocardial cells to undergo an epithelial to mesenchymal transition (EMT) required for valve formation. A number of cellular events are required for cells to undergo the complete process of EMT: extracellular stimulation, transcription-linked signaling, loss of apicobasal polarity, decreased cell adhesion and cytoskeletal remodeling. EMT can be assayed in vitro by assessing changes in cell morphology (cell shape changes, loss of apicobasal polarity and cell-cell adhesion) and gene expression changes (upregulation of Snail1, Twist, Vimentin and N-cadherin instead of E-cadherin) in hiPSCs. Zhang et al. demonstrated the ability of hiPSCs to undergo EMT in vitro during the induction of mesoderm. This study further demonstrated that the extracellular matrix (ECM) positively regulates EMT based on the upregulation of the mesenchymal genes N-cadherin, Snail1/2, and Vimentin (57). The signaling events, cellular processes (morphology changes and migration ability) and ECM interactions that modulate EMT during valve formation could be explored using hiPSC-derived endothelial progenitor cells. For example, regarding Notch signaling, Kobayashi showed decreased Notch/Hey signaling in HLHS hiPSC-CPCs (125). Consistently, prior studies in mouse models have demonstrated impaired EMT in atrioventricular explants from Hey1/HeyL, Hey2, and Notch1 deficient mice (147). Whether the defect in EMT is conserved in human disease cell lines could be tested using endothelial progenitors generated from HLHS iPSCs. Further, the ability of small molecule pathway modulators to rescue this phenotype could be assayed and may provide insights into potential therapeutic targets. Additionally, these experiments could be performed with hiPSCs generated from patients with copy number variants (large deletions or amplifications of DNA segments) that impact these signaling pathways where the role in EMT is unknown (148).
Tissue Interactions in CHD
Most hiPSC differentiation systems are limited regarding the study of tissue interactions since they are a two dimensional model. Advances in heart tissue engineering have made it possible to engineer 3D heart tissues, but the technology has been limited by the high numbers of cardiovascular cells the scaffold requires. Using hiPSCs, it is now possible to generate large numbers of normal or disease specific cells from patients in vitro and to study them on three-dimensional matrices. Matrices include a whole heart extracellular matrix (ECM) scaffold generated by the use of detergents to decellularize intact hearts (Figure 5). Ott et al. generated the first bioartificial heart by reseeding rat heart ECM with isolated rat neonatal cardiomyocytes (149). Decellularization of whole heart is advantageous for two reasons: it preserves the heart structure and native heart ECM proteins. The use of decellularized heart ECM is beneficial for studying the differentiation of hiPSC-derived cardiovascular cells, cell maturation, cell migration and cell-matrix or cell-cell interactions, such as myocardial-vascular interactions. Recently, Lu et al. repopulated decellularized mouse hearts with human iPSC-CPCs demonstrating that the seeded cardiovascular progenitors differentiated into CMs, smooth muscle cells and endothelial cells with high efficiency. They further demonstrated that the heart ECM could promote proliferation, specific cell differentiation and myofilament formation (150). The use of the decellularized heart ECM may be further expanded to study tissue specific mechanisms that contribute to cardiac morphogenesis.
Figure 5. The use of hiPSCs to model CHD.
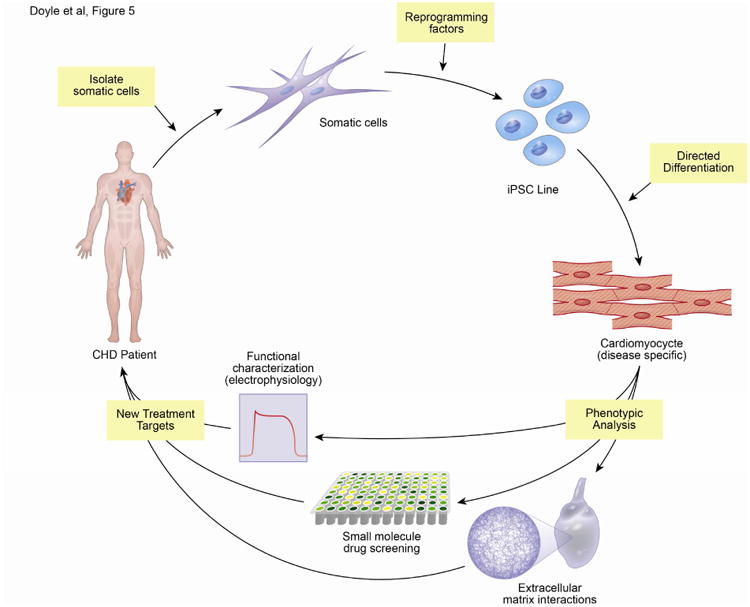
The diagram represents the process of isolating somatic cells (blood or fibroblasts) from patients, reprogramming the cells using four factors (Oct3/4, Sox2, Klf4, c-Myc or LIN28) to generate iPSC lines, directed differentiation to CMs, and phenotypic assays performed on hiPSC-CMs to characterize the pathophysiology of individual CHD with the goal of understanding disease mechanisms and informing new therapeutic options. A wide variety of phenotypic analysis could be carried out. The schematic highlights functional characterization of hiPSC-CMs, small molecular perturbation of pathways, identification of drug targets and the interaction of hiPSC-CMs or CPCs with the extracellular matrix.
In addition to the decellularized heart matrix, a number of other tissue engineering constructs have been developed to assay CM function. For example, the “heart-on-a-chip” technology can be used to measure contractile stress, action potential propagation, and cellular architecture. To generate the “heart-on-a-chip”, CMs are seeded onto a deformable elastic thin filament with micropatterned fibronection where they self-organize into an electromechanically coupled monolayer (muscular thin filaments or MTF). The contractility of the tissue is observed by the deformations of the MTF (151; 152). hiPSC-CMs seeded onto micropatterned fibronectin align and adopt regular shapes allowing more precise comparisons of sarcomeric architecture between cells. This technology has been used to model and rescue mitochondrial associated cardiomyopathy in Barth Syndrome (122). Many adult patients with surgically corrected CHD have cardiac complications, including arrhythmias and ventricular dysfunction. The use of the “heart-on-a-chip” or other similar technologies could help identify the distinct function that developmental regulators (such as Gata4 or Tbx20) have in adult CMs. Using these techniques described above coupled with the advantages of a human cellular model system, large scale cell expansion, differentiation, and advanced physiology techniques hiPSCs offer a significant improvement in our understanding of the role of gene regulation, transcriptional downstream targets, signaling molecules, cell-cell interactions, and tissue interactions on the genesis of CHD.
Summary
CHD remains a significant health problem, with a growing population of survivors with chronic disease. Despite significant efforts to understand the genetic basis of CHD in humans and the molecular control of heart development in animal model systems, an understanding of the etiology of most CHD is unclear. This is due in part to the genetic complexity of CHD, the limitations of animal model systems, the phenotypic pleotrophy of CHD, as well as the interaction of genetics and the environment during development. Furthermore, many of the developmental patterning genes have distinct roles in adult heart function that have not been fully elucidated. Currently, ongoing research efforts such as genomic sequencing and new experimental models including hiPSCs are enhancing our understanding of the causes and mechanisms of CHD. The ability to study genetic and molecular regulation during in vitro differentiation of human cells makes hiPSC differentiation a unique system for uncovering novel molecular interactions that contribute to CHD. We have highlighted some of the potential strategies for using hiPSC technology to investigate cellular processes that contribute to heart development and cardiomyocyte function. The ability to generate patient specific cell lines that allow for the investigation of molecular mechanisms linking genotype with phenotype in CHD is a powerful strategy that will contribute to our understanding of the mechanisms that contribute to CHD.
Acknowledgments
We thank Cynthia DeKay and Erik Munsen for assistance with graphics. This work was supported by funding from the NHLBI (Etv2 R01: 1R01HL122576) and NHLBI Progenitor Cell Biology Consortium (U01: 5U01HL100407).
Footnotes
Disclosures: The authors indicate no potential conflicts of interest.
References
- 1.Syrmou A, Tzetis M, Fryssira H, Kosma K, Oikonomakis V, Giannikou K, et al. Array comparative genomic hybridization as a clinical diagnostic tool in syndromic and nonsyndromic congenital heart disease. Pediatr Res. 2013 Jun;73(6):772–6. doi: 10.1038/pr.2013.41. [DOI] [PubMed] [Google Scholar]
- 2.Fahed AC, Gelb BD, Seidman JG, Seidman CE. Genetics of congenital heart disease: the glass half empty. Circ Res. 2013 Feb 15;112(4):707–20. doi: 10.1161/CIRCRESAHA.112.300853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pierpont ME, Basson CT, Benson DW, Gelb BD, Giglia TM, Goldmuntz E, et al. Genetic basis for congenital heart defects: current knowledge: a scientific statement from the American Heart Association Congenital Cardiac Defects Committee, Council on Cardiovascular Disease in the Young: endorsed by the American Academy of Pediatrics. Circulation. 2007 Jun 12;115(23):3015–38. doi: 10.1161/CIRCULATIONAHA.106.183056. [DOI] [PubMed] [Google Scholar]
- 4.Ferencz C, Boughman JA, Neill CA, Brenner JI, Perry LW. Congenital cardiovascular malformations: questions on inheritance Baltimore- Washington Infant Study Group. J Am Coll Cardiol. 1989 Sep;14(3):756–63. doi: 10.1016/0735-1097(89)90122-8. [DOI] [PubMed] [Google Scholar]
- 5.Pueschel SM. Clinical aspects of Down syndrome from infancy to adulthood. Am J Med Genet Suppl. 1990:752–6. doi: 10.1002/ajmg.1320370708. [DOI] [PubMed] [Google Scholar]
- 6.Goldhaber SZ, Brown WD, Sutton MG. High frequency of mitral valve prolapse and aortic regurgitation among asymptomatic adults with Down's syndrome. JAMA. 1987 Oct 2;258(13):1793–5. [PubMed] [Google Scholar]
- 7.Freeman SB, Taft LF, Dooley KJ, Allran K, Sherman SL, Hassold TJ, et al. Population-based study of congenital heart defects in Down syndrome. Am J Med Genet. 1998 Nov 16;80(3):213–7. [PubMed] [Google Scholar]
- 8.Källén B, Mastroiacovo P, Robert E. Major congenital malformations in Down syndrome. Am J Med Genet. 1996 Oct 16;65(2):160–6. doi: 10.1002/(SICI)1096-8628(19961016)65:2<160::AID-AJMG16>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 9.Prandstraller D, Mazzanti L, Picchio FM, Magnani C, Bergamaschi R, Perri A, et al. Turner's syndrome: cardiologic profile according to the different chromosomal patterns and long-term clinical follow-Up of 136 nonpreselected patients. Pediatr Cardiol. 1999;20(2):108–12. doi: 10.1007/s002469900416. [DOI] [PubMed] [Google Scholar]
- 10.Mazzanti L, Cacciari E. Congenital heart disease in patients with Turner's syndrome. Italian Study Group for Turner Syndrome (ISGTS) J Pediatr. 1998 Nov;133(5):688–92. doi: 10.1016/s0022-3476(98)70119-2. [DOI] [PubMed] [Google Scholar]
- 11.Mazzanti L, Prandstraller D, Tassinari D, Rubino I, Santucci S, Picchio FM, et al. Heart disease in Turner's syndrome. Helv Paediatr Acta. 1988 Aug;43(1-2):25–31. [PubMed] [Google Scholar]
- 12.Lin AE, Lippe BM, Geffner ME, Gomes A, Lois JF, Barton CW, et al. Aortic dilation, dissection, and rupture in patients with Turner syndrome. J Pediatr. 1986 Nov;109(5):820–6. doi: 10.1016/s0022-3476(86)80700-4. [DOI] [PubMed] [Google Scholar]
- 13.Lin AE, Lippe B, Rosenfeld RG. Further delineation of aortic dilation, dissection, and rupture in patients with Turner syndrome. Pediatrics. 1998 Jul;102(1):e12. doi: 10.1542/peds.102.1.e12. [DOI] [PubMed] [Google Scholar]
- 14.Van Praagh S, Truman T, Firpo A, Bano-Rodrigo A, Fried R, McManus B, et al. Cardiac malformations in trisomy-18: a study of 41 postmortem cases. J Am Coll Cardiol. 1989 Jun;13(7):1586–97. doi: 10.1016/0735-1097(89)90353-7. [DOI] [PubMed] [Google Scholar]
- 15.Matsuoka R, Misugi K, Goto A, Gilbert EF, Ando M. Congenital heart anomalies in the trisomy 18 syndrome, with reference to congenital polyvalvular disease. Am J Med Genet. 1983 Apr;14(4):657–68. doi: 10.1002/ajmg.1320140409. [DOI] [PubMed] [Google Scholar]
- 16.Ryan AK, Goodship JA, Wilson DI, Philip N, Levy A, Seidel H, et al. Spectrum of clinical features associated with interstitial chromosome 22q11 deletions: a European collaborative study. J Med Genet. 1997 Oct;34(10):798–804. doi: 10.1136/jmg.34.10.798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McDonald-McGinn DM, Kirschner R, Goldmuntz E, Sullivan K, Eicher P, Gerdes M, et al. The Philadelphia story: the 22q11.2 deletion: report on 250 patients. Genet Couns. 1999;10(1):11–24. [PubMed] [Google Scholar]
- 18.Johnson TR, Goldmuntz E, McDonald-McGinn DM, Zackai EH, Fogel MA. Cardiac magnetic resonance imaging for accurate diagnosis of aortic arch anomalies in patients with 22q11.2 deletion. Am J Cardiol. 2005 Dec 15;96(12):1726–30. doi: 10.1016/j.amjcard.2005.07.084. [DOI] [PubMed] [Google Scholar]
- 19.McDonald-McGinn DM, LaRossa D, Goldmuntz E, Sullivan K, Eicher P, Gerdes M, et al. The 22q11.2 deletion: screening, diagnostic workup, and outcome of results; report on 181 patients. Genet Test. 1997;1(2):99–108. doi: 10.1089/gte.1997.1.99. [DOI] [PubMed] [Google Scholar]
- 20.Eronen M, Peippo M, Hiippala A, Raatikka M, Arvio M, Johansson R, Kähkönen M. Cardiovascular manifestations in 75 patients with Williams syndrome. J Med Genet. 2002 Aug;39(8):554–8. doi: 10.1136/jmg.39.8.554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bruno E, Rossi N, Thüer O, Córdoba R, Alday LE. Cardiovascular findings, and clinical course, in patients with Williams syndrome. Cardiol Young. 2003 Dec;13(6):532–6. [PubMed] [Google Scholar]
- 22.Wu YQ, Sutton VR, Nickerson E, Lupski JR, Potocki L, Korenberg JR, et al. Delineation of the common critical region in Williams syndrome and clinical correlation of growth, heart defects, ethnicity, and parental origin. Am J Med Genet. 1998 Jun 16;78(1):82–9. doi: 10.1002/(sici)1096-8628(19980616)78:1<82::aid-ajmg17>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 23.Grossfeld PD, Mattina T, Lai Z, Favier R, Jones KL, Cotter F, Jones C. The 11q terminal deletion disorder: a prospective study of 110 cases. Am J Med Genet A. 2004 Aug 15;129A(1):51–61. doi: 10.1002/ajmg.a.30090. [DOI] [PubMed] [Google Scholar]
- 24.Tartaglia M, Kalidas K, Shaw A, Song X, Musat DL, van der Burgt I, et al. PTPN11 mutations in Noonan syndrome: molecular spectrum, genotype-phenotype correlation, and phenotypic heterogeneity. Am J Hum Genet. 2002 Jun;70(6):1555–63. doi: 10.1086/340847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tartaglia M, Mehler EL, Goldberg R, Zampino G, Brunner HG, Kremer H, et al. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat Genet. 2001 Dec;29(4):465–8. doi: 10.1038/ng772. [DOI] [PubMed] [Google Scholar]
- 26.Tartaglia M, Pennacchio LA, Zhao C, Yadav KK, Fodale V, Sarkozy A, et al. Gain-of-function SOS1 mutations cause a distinctive form of Noonan syndrome. Nat Genet. 2007 Jan;39(1):75–9. doi: 10.1038/ng1939. [DOI] [PubMed] [Google Scholar]
- 27.Pandit B, Sarkozy A, Pennacchio LA, Carta C, Oishi K, Martinelli S, et al. Gain-of-function RAF1 mutations cause Noonan and LEOPARD syndromes with hypertrophic cardiomyopathy. Nat Genet. 2007 Aug;39(8):1007–12. doi: 10.1038/ng2073. [DOI] [PubMed] [Google Scholar]
- 28.Sarkozy A, Carta C, Moretti S, Zampino G, Digilio MC, Pantaleoni F, et al. Germline BRAF mutations in Noonan, LEOPARD, and cardiofaciocutaneous syndromes: molecular diversity and associated phenotypic spectrum. Hum Mutat. 2009 Apr;30(4):695–702. doi: 10.1002/humu.20955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bertola DR, Yamamoto GL, Almeida TF, Buscarilli M, Jorge AA, Malaquias AC, et al. Further evidence of the importance of RIT1 in Noonan syndrome. Am J Med Genet A. 2014 Nov;164A(11):2952–7. doi: 10.1002/ajmg.a.36722. [DOI] [PubMed] [Google Scholar]
- 30.Li L, Krantz ID, Deng Y, Genin A, Banta AB, Collins CC, et al. Alagille syndrome is caused by mutations in human Jagged1, which encodes a ligand for Notch1. Nat Genet. 1997 Jul;16(3):243–51. doi: 10.1038/ng0797-243. [DOI] [PubMed] [Google Scholar]
- 31.McElhinney DB, Krantz ID, Bason L, Piccoli DA, Emerick KM, Spinner NB, Goldmuntz E. Analysis of cardiovascular phenotype and genotype-phenotype correlation in individuals with a JAG1 mutation and/or Alagille syndrome. Circulation. 2002 Nov 12;106(20):2567–74. doi: 10.1161/01.cir.0000037221.45902.69. [DOI] [PubMed] [Google Scholar]
- 32.Ruiz-Perez VL, Ide SE, Strom TM, Lorenz B, Wilson D, Woods K, et al. Mutations in a new gene in Ellis-van Creveld syndrome and Weyers acrodental dysostosis. Nat Genet. 2000 Mar;24(3):283–6. doi: 10.1038/73508. [DOI] [PubMed] [Google Scholar]
- 33.Ruiz-Perez VL, Tompson SW, Blair HJ, Espinoza-Valdez C, Lapunzina P, Silva EO, et al. Mutations in two nonhomologous genes in a head-to-head configuration cause Ellis-van Creveld syndrome. Am J Hum Genet. 2003 Mar;72(3):728–32. doi: 10.1086/368063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Basson CT, Bachinsky DR, Lin RC, Levi T, Elkins JA, Soults J, et al. Mutations in human TBX5 [corrected] cause limb and cardiac malformation in Holt-Oram syndrome. Nat Genet. 1997 Jan;15(1):30–5. doi: 10.1038/ng0197-30. [DOI] [PubMed] [Google Scholar]
- 35.Basson CT, Huang T, Lin RC, Bachinsky DR, Weremowicz S, Vaglio A, et al. Different TBX5 interactions in heart and limb defined by Holt-Oram syndrome mutations. Proc Natl Acad Sci U S A. 1999 Mar 16;96(6):2919–24. doi: 10.1073/pnas.96.6.2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Amsterdam: Academic Press; 2010. Heart development and regeneration set [Internet] Available from: http://www.sciencedirect.com/science/book/9780123813329. [Google Scholar]
- 37.Martinsen B, Lohr JL. Cardiac Development. Handbook of Cardiac Anatomy, Physiology, and Devices. 2009 [Google Scholar]
- 38.Brade T, Pane LS, Moretti A, Chien KR, Laugwitz KL. Embryonic heart progenitors and cardiogenesis. Cold Spring Harb Perspect Med. 2013 Oct;3(10):a013847. doi: 10.1101/cshperspect.a013847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bruneau BG. Signaling and transcriptional networks in heart development and regeneration. Cold Spring Harb Perspect Biol. 2013 Mar;5(3):a008292. doi: 10.1101/cshperspect.a008292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Buckingham M, Meilhac S, Zaffran S. Building the mammalian heart from two sources of myocardial cells. Nat Rev Genet. 2005 Nov;6(11):826–35. doi: 10.1038/nrg1710. [DOI] [PubMed] [Google Scholar]
- 41.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006 Aug 25;126(4):663–76. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 42.Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007 Nov 30;131(5):861–72. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 43.Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007 Dec 21;318(5858):1917–20. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- 44.Takahashi K, Yamanaka S. Induced pluripotent stem cells in medicine and biology. Development. 2013 Jun;140(12):2457–61. doi: 10.1242/dev.092551. [DOI] [PubMed] [Google Scholar]
- 45.Matsa E, Burridge PW, Wu JC. Human stem cells for modeling heart disease and for drug discovery. Sci Transl Med. 2014 Jun 4;6(239):239ps6. doi: 10.1126/scitranslmed.3008921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Burridge PW, Keller G, Gold JD, Wu JC. Production of de novo cardiomyocytes: human pluripotent stem cell differentiation and direct reprogramming. Cell Stem Cell. 2012 Jan 6;10(1):16–28. doi: 10.1016/j.stem.2011.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mummery CL, Zhang J, Ng ES, Elliott DA, Elefanty AG, Kamp TJ. Differentiation of human embryonic stem cells and induced pluripotent stem cells to cardiomyocytes: a methods overview. Circ Res. 2012 Jul 20;111(3):344–58. doi: 10.1161/CIRCRESAHA.110.227512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Burridge PW, Matsa E, Shukla P, Lin ZC, Churko JM, Ebert AD, et al. Chemically defined generation of human cardiomyocytes. Nat Methods. 2014 Aug;11(8):855–60. doi: 10.1038/nmeth.2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Graichen R, Xu X, Braam SR, Balakrishnan T, Norfiza S, Sieh S, et al. Enhanced cardiomyogenesis of human embryonic stem cells by a small molecular inhibitor of p38 MAPK. Differentiation. 2008 Apr;76(4):357–70. doi: 10.1111/j.1432-0436.2007.00236.x. [DOI] [PubMed] [Google Scholar]
- 50.Yang L, Soonpaa MH, Adler ED, Roepke TK, Kattman SJ, Kennedy M, et al. Human cardiovascular progenitor cells develop from a KDR+ embryonic-stem-cell-derived population. Nature. 2008 May 22;453(7194):524–8. doi: 10.1038/nature06894. [DOI] [PubMed] [Google Scholar]
- 51.Yu J, Thomson JA. Pluripotent stem cell lines. Genes Dev. 2008 Aug 1;22(15):1987–97. doi: 10.1101/gad.1689808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kattman SJ, Witty AD, Gagliardi M, Dubois NC, Niapour M, Hotta A, et al. Stage-Specific Optimization of Activin/Nodal and BMP Signaling Promotes Cardiac Differentiation of Mouse and Human Pluripotent Stem Cell Lines. Cell Stem Cell. 2011 Feb 4;8(2):228–40. doi: 10.1016/j.stem.2010.12.008. [DOI] [PubMed] [Google Scholar]
- 53.Lian X, Hsiao C, Wilson G, Zhu K, Hazeltine LB, Azarin SM, et al. Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling. Proc Natl Acad Sci U S A. 2012 Jul 3;109(27):E1848–57. doi: 10.1073/pnas.1200250109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ema M, Takahashi S, Rossant J. Deletion of the selection cassette, but not cis-acting elements, in targeted Flk1-lacZ allele reveals Flk1 expression in multipotent mesodermal progenitors. Blood. 2006 Jan 1;107(1):111–7. doi: 10.1182/blood-2005-05-1970. [DOI] [PubMed] [Google Scholar]
- 55.Dunn NR, Vincent SD, Oxburgh L, Robertson EJ, Bikoff EK. Combinatorial activities of Smad2 and Smad3 regulate mesoderm formation and patterning in the mouse embryo. Development. 2004 Apr;131(8):1717–28. doi: 10.1242/dev.01072. [DOI] [PubMed] [Google Scholar]
- 56.Laflamme MA, Chen KY, Naumova AV, Muskheli V, Fugate JA, Dupras SK, et al. Cardiomyocytes derived from human embryonic stem cells in pro-survival factors enhance function of infarcted rat hearts. Nat Biotechnol. 2007 Sep;25(9):1015–24. doi: 10.1038/nbt1327. [DOI] [PubMed] [Google Scholar]
- 57.Zhang Y, Wang X, Xu X, Wang J, Liu X, Chen Y. Distinct microRNA expression signatures in human right atrial and ventricular myocardium. Mol Cell Biochem. 2012 Aug 14; doi: 10.1007/s11010-012-1417-5. [DOI] [PubMed] [Google Scholar]
- 58.Cai B, Li J, Wang J, Luo X, Ai J, Liu Y, et al. microRNA-124 Regulates Cardiomyocyte Differentiation of Bone Marrow-Derived Mesenchymal Stem Cells Via Targeting STAT3 Signaling. Stem Cells. 2012 Aug;30(8):1746–55. doi: 10.1002/stem.1154. [DOI] [PubMed] [Google Scholar]
- 59.Tran TH, Wang X, Browne C, Zhang Y, Schinke M, Izumo S, Burcin M. Wnt3a-induced mesoderm formation and cardiomyogenesis in human embryonic stem cells. Stem Cells. 2009 Aug;27(8):1869–78. doi: 10.1002/stem.95. [DOI] [PubMed] [Google Scholar]
- 60.Paige SL, Osugi T, Afanasiev OK, Pabon L, Reinecke H, Murry CE. Endogenous Wnt/beta-catenin signaling is required for cardiac differentiation in human embryonic stem cells. PLoS One. 2010;5(6):e11134. doi: 10.1371/journal.pone.0011134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cai W, Guzzo RM, Wei K, Willems E, Davidovics H, Mercola MA. Nodal-to-TGFβ cascade exerts biphasic control over cardiopoiesis. Circ Res. 2012 Sep 14;111(7):876–81. doi: 10.1161/CIRCRESAHA.112.270272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Willems E, Spiering S, Davidovics H, Lanier M, Xia Z, Dawson M, et al. Small-molecule inhibitors of the Wnt pathway potently promote cardiomyocytes from human embryonic stem cell-derived mesoderm. Circ Res. 2011 Aug 5;109(4):360–4. doi: 10.1161/CIRCRESAHA.111.249540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hudson JE, Zimmermann WH. Tuning Wnt-signaling to enhance cardiomyogenesis in human embryonic and induced pluripotent stem cells. J Mol Cell Cardiol. 2011 Sep;51(3):277–9. doi: 10.1016/j.yjmcc.2011.06.011. [DOI] [PubMed] [Google Scholar]
- 64.Ren Y, Lee MY, Schliffke S, Paavola J, Amos PJ, Ge X, et al. Small molecule Wnt inhibitors enhance the efficiency of BMP-4-directed cardiac differentiation of human pluripotent stem cells. J Mol Cell Cardiol. 2011 Sep;51(3):280–7. doi: 10.1016/j.yjmcc.2011.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Karakikes I, Senyei GD, Hansen J, Kong CW, Azeloglu EU, Stillitano F, et al. Small molecule-mediated directed differentiation of human embryonic stem cells toward ventricular cardiomyocytes. Stem Cells Transl Med. 2014 Jan;3(1):18–31. doi: 10.5966/sctm.2013-0110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Witty AD, Mihic A, Tam RY, Fisher SA, Mikryukov A, Shoichet MS, et al. Generation of the epicardial lineage from human pluripotent stem cells. Nat Biotechnol. 2014 Oct;32(10):1026–35. doi: 10.1038/nbt.3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Später D, Abramczuk MK, Buac K, Zangi L, Stachel MW, Clarke J, et al. A HCN4+ cardiomyogenic progenitor derived from the first heart field and human pluripotent stem cells. Nat Cell Biol. 2013 Sep;15(9):1098–106. doi: 10.1038/ncb2824. [DOI] [PubMed] [Google Scholar]
- 68.Elliott DA, Braam SR, Koutsis K, Ng ES, Jenny R, Lagerqvist EL, et al. NKX2-5(eGFP/w) hESCs for isolation of human cardiac progenitors and cardiomyocytes. Nat Methods. 2011;8(12):1037–40. doi: 10.1038/nmeth.1740. [DOI] [PubMed] [Google Scholar]
- 69.Bruneau BG, Nemer G, Schmitt JP, Charron F, Robitaille L, Caron S, et al. A murine model of Holt-Oram syndrome defines roles of the T-box transcription factor Tbx5 in cardiogenesis and disease. Cell. 2001 Sep 21;106(6):709–21. doi: 10.1016/s0092-8674(01)00493-7. [DOI] [PubMed] [Google Scholar]
- 70.Takeuchi JK, Ohgi M, Koshiba-Takeuchi K, Shiratori H, Sakaki I, Ogura K, et al. Tbx5 specifies the left/right ventricles and ventricular septum position during cardiogenesis. Development. 2003 Dec;130(24):5953–64. doi: 10.1242/dev.00797. [DOI] [PubMed] [Google Scholar]
- 71.Liang X, Wang G, Lin L, Lowe J, Zhang Q, Bu L, et al. HCN4 Dynamically Marks the First Heart Field and Conduction System Precursors. Circulation Research. 2013 Aug;113(4):399–407. doi: 10.1161/CIRCRESAHA.113.301588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bu L, Jiang X, Martin-Puig S, Caron L, Zhu S, Shao Y, et al. Human ISL1 heart progenitors generate diverse multipotent cardiovascular cell lineages. Nature. 2009 Jul 2;460(7251):113–7. doi: 10.1038/nature08191. [DOI] [PubMed] [Google Scholar]
- 73.Moretti A, Caron L, Nakano A, Lam JT, Bernshausen A, Chen Y, et al. Multipotent embryonic isl1+ progenitor cells lead to cardiac, smooth muscle, and endothelial cell diversification. Cell. 2006 Dec 15;127(6):1151–65. doi: 10.1016/j.cell.2006.10.029. [DOI] [PubMed] [Google Scholar]
- 74.Buikema JW, Zwetsloot PP, Doevendans PA, Sluijter JP, Domian IJ. Expanding mouse ventricular cardiomyocytes through GSK-3 inhibition. Curr Protoc Cell Biol. 2013;61:23.9.1–23.9.10. doi: 10.1002/0471143030.cb2309s61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cao X, Wang J, Wang Z, Du J, Yuan X, Huang W, et al. MicroRNA profiling during rat ventricular maturation: A role for miR-29a in regulating cardiomyocyte cell cycle reentry. FEBS Lett. 2013 May 21;587(10):1548–55. doi: 10.1016/j.febslet.2013.01.075. [DOI] [PubMed] [Google Scholar]
- 76.Lian X, Bao X, Al-Ahmad A, Liu J, Wu Y, Dong W, et al. Efficient differentiation of human pluripotent stem cells to endothelial progenitors via small-molecule activation of WNT signaling. Stem Cell Reports. 2014 Nov 11;3(5):804–16. doi: 10.1016/j.stemcr.2014.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.He JQ, Ma Y, Lee Y, Thomson JA, Kamp TJ. Human embryonic stem cells develop into multiple types of cardiac myocytes: action potential characterization. Circ Res. 2003 Jul 11;93(1):32–9. doi: 10.1161/01.RES.0000080317.92718.99. [DOI] [PubMed] [Google Scholar]
- 78.Xu C, Police S, Rao N, Carpenter MK. Characterization and enrichment of cardiomyocytes derived from human embryonic stem cells. Circ Res. 2002 Sep 20;91(6):501–8. doi: 10.1161/01.res.0000035254.80718.91. [DOI] [PubMed] [Google Scholar]
- 79.Tanwar V, Bylund JB, Hu J, Yan J, Walthall JM, Mukherjee A, et al. Gremlin 2 promotes differentiation of embryonic stem cells to atrial fate by activation of the JNK signaling pathway. Stem Cells. 2014 Jul;32(7):1774–88. doi: 10.1002/stem.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhang Q, Jiang J, Han P, Yuan Q, Zhang J, Zhang X, et al. Direct differentiation of atrial and ventricular myocytes from human embryonic stem cells by alternating retinoid signals. Cell Res. 2011 Apr;21(4):579–87. doi: 10.1038/cr.2010.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Caspi O, Huber I, Kehat I, Habib M, Arbel G, Gepstein A, et al. Transplantation of human embryonic stem cell-derived cardiomyocytes improves myocardial performance in infarcted rat hearts. J Am Coll Cardiol. 2007 Nov 6;50(19):1884–93. doi: 10.1016/j.jacc.2007.07.054. [DOI] [PubMed] [Google Scholar]
- 82.van Laake LW, Passier R, Monshouwer-Kloots J, Nederhoff MG, Ward-van Oostwaard D, Field LJ, et al. Monitoring of cell therapy and assessment of cardiac function using magnetic resonance imaging in a mouse model of myocardial infarction. Nat Protoc. 2007;2(10):2551–67. doi: 10.1038/nprot.2007.371. [DOI] [PubMed] [Google Scholar]
- 83.Laflamme MA, Gold J, Xu C, Hassanipour M, Rosler E, Police S, et al. Formation of human myocardium in the rat heart from human embryonic stem cells. Am J Pathol. 2005 Sep;167(3):663–71. doi: 10.1016/S0002-9440(10)62041-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Fernandes S, Naumova AV, Zhu WZ, Laflamme MA, Gold J, Murry CE. Human embryonic stem cell-derived cardiomyocytes engraft but do not alter cardiac remodeling after chronic infarction in rats. J Mol Cell Cardiol. 2010 Dec;49(6):941–9. doi: 10.1016/j.yjmcc.2010.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Shiba Y, Fernandes S, Zhu WZ, Filice D, Muskheli V, Kim J, et al. Human ES-cell-derived cardiomyocytes electrically couple and suppress arrhythmias in injured hearts. Nature. 2012 Sep 13;489(7415):322–5. doi: 10.1038/nature11317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chong JJ, Murry CE. Cardiac regeneration using pluripotent stem cells-Progression to large animal models. Stem Cell Res. 2014 Nov;13(3PB):654–665. doi: 10.1016/j.scr.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yang X, Pabon L, Murry CE. Engineering adolescence: maturation of human pluripotent stem cell-derived cardiomyocytes. Circ Res. 2014 Jan 31;114(3):511–23. doi: 10.1161/CIRCRESAHA.114.300558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Snir M, Kehat I, Gepstein A, Coleman R, Itskovitz-Eldor J, Livne E, Gepstein L. Assessment of the ultrastructural and proliferative properties of human embryonic stem cell-derived cardiomyocytes. Am J Physiol Heart Circ Physiol. 2003 Dec;285(6):H2355–63. doi: 10.1152/ajpheart.00020.2003. [DOI] [PubMed] [Google Scholar]
- 89.Kamakura T, Makiyama T, Sasaki K, Yoshida Y, Wuriyanghai Y, Chen J, et al. Ultrastructural maturation of human-induced pluripotent stem cell-derived cardiomyocytes in a long-term culture. Circ J. 2013;77(5):1307–14. doi: 10.1253/circj.cj-12-0987. [DOI] [PubMed] [Google Scholar]
- 90.Lundy SD, Zhu WZ, Regnier M, Laflamme MA. Structural and functional maturation of cardiomyocytes derived from human pluripotent stem cells. Stem Cells Dev. 2013 Jul 15;22(14):1991–2002. doi: 10.1089/scd.2012.0490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ivashchenko CY, Pipes GC, Lozinskaya IM, Lin Z, Xu X, Needle S, et al. Human induced pluripotent stem cell derived cardiomyocytes exhibit temporal changes in phenotype. Am J Physiol Heart Circ Physiol. 2013 Jul 5; doi: 10.1152/ajpheart.00819.2012. [DOI] [PubMed] [Google Scholar]
- 92.Bedada FB, Chan SS, Metzger SK, Zhang L, Zhang J, Garry DJ, et al. Acquisition of a quantitative, stoichiometrically conserved ratiometric marker of maturation status in stem cell-derived cardiac myocytes. Stem Cell Reports. 2014 Oct 14;3(4):594–605. doi: 10.1016/j.stemcr.2014.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sallam K, Kodo K, Wu JC. Modeling inherited cardiac disorders. Circ J. 2014;78(4):784–94. doi: 10.1253/circj.cj-14-0182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Egashira T, Yuasa S, Suzuki T, Aizawa Y, Yamakawa H, Matsuhashi T, et al. Disease characterization using LQTS-specific induced pluripotent stem cells. Cardiovasc Res. 2012 Sep 1;95(4):419–29. doi: 10.1093/cvr/cvs206. [DOI] [PubMed] [Google Scholar]
- 95.Matsa E, Rajamohan D, Dick E, Young L, Mellor I, Staniforth A, Denning C. Drug evaluation in cardiomyocytes derived from human induced pluripotent stem cells carrying a long QT syndrome type 2 mutation. Eur Heart J. 2011 Apr;32(8):952–62. doi: 10.1093/eurheartj/ehr073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yazawa M, Dolmetsch RE. Modeling Timothy syndrome with iPS cells. J Cardiovasc Transl Res. 2013 Feb;6(1):1–9. doi: 10.1007/s12265-012-9444-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Matsa E, Dixon JE, Medway C, Georgiou O, Patel MJ, Morgan K, et al. Allele-specific RNA interference rescues the long-QT syndrome phenotype in human-induced pluripotency stem cell cardiomyocytes. Eur Heart J. 2014 Apr;35(16):1078–87. doi: 10.1093/eurheartj/eht067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Okata S, Yuasa S, Yamane T, Furukawa T, Fukuda K. The generation of induced pluripotent stem cells from a patient with KCNH2 G603D, without LQT2 disease associated symptom. J Med Dent Sci. 2013;60(1):17–22. [PubMed] [Google Scholar]
- 99.Ma D, Wei H, Zhao Y, Lu J, Li G, Sahib NB, et al. Modeling type 3 long QT syndrome with cardiomyocytes derived from patient-specific induced pluripotent stem cells. Int J Cardiol. 2013 Oct 15;168(6):5277–86. doi: 10.1016/j.ijcard.2013.08.015. [DOI] [PubMed] [Google Scholar]
- 100.Fatima A, Kaifeng S, Dittmann S, Xu G, Gupta MK, Linke M, et al. The disease-specific phenotype in cardiomyocytes derived from induced pluripotent stem cells of two long QT syndrome type 3 patients. PLoS One. 2013;8(12):e83005. doi: 10.1371/journal.pone.0083005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Mehta A, Sequiera GL, Ramachandra CJ, Sudibyo Y, Chung Y, Sheng J, et al. Retrafficking of hERG reverses long QT syndrome 2 phenotype in human iPS-derived cardiomyocytes. Cardiovasc Res. 2014 Jun 1;102(3):497–506. doi: 10.1093/cvr/cvu060. [DOI] [PubMed] [Google Scholar]
- 102.Bellin M, Casini S, Davis RP, D'Aniello C, Haas J, Ward-van Oostwaard D, et al. Isogenic human pluripotent stem cell pairs reveal the role of a KCNH2 mutation in long-QT syndrome. EMBO J. 2013 Dec 11;32(24):3161–75. doi: 10.1038/emboj.2013.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Malan D, Friedrichs S, Fleischmann BK, Sasse P. Cardiomyocytes obtained from induced pluripotent stem cells with long-QT syndrome 3 recapitulate typical disease-specific features in vitro. Circ Res. 2011 Sep 30;109(8):841–7. doi: 10.1161/CIRCRESAHA.111.243139. [DOI] [PubMed] [Google Scholar]
- 104.Terrenoire C, Wang K, Tung KW, Chung WK, Pass RH, Lu JT, et al. Induced pluripotent stem cells used to reveal drug actions in a long QT syndrome family with complex genetics. J Gen Physiol. 2013 Jan;141(1):61–72. doi: 10.1085/jgp.201210899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Yazawa M, Hsueh B, Jia X, Pasca AM, Bernstein JA, Hallmayer J, Dolmetsch RE. Using induced pluripotent stem cells to investigate cardiac phenotypes in Timothy syndrome. Nature. 2011 Feb 9;471(7337):230–4. doi: 10.1038/nature09855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Moretti A, Bellin M, Welling A, Jung CB, Lam JT, Bott-Flügel L, et al. Patient-specific induced pluripotent stem-cell models for long-QT syndrome. N Engl J Med. 2010 Oct 7;363(15):1397–409. doi: 10.1056/NEJMoa0908679. [DOI] [PubMed] [Google Scholar]
- 107.Itzhaki I, Maizels L, Huber I, Zwi-Dantsis L, Caspi O, Winterstern A, et al. Modelling the long QT syndrome with induced pluripotent stem cells. Nature. 2011 Jan 16; doi: 10.1038/nature09747. [DOI] [PubMed] [Google Scholar]
- 108.Sun N, Yazawa M, Liu J, Han L, Sanchez-Freire V, Abilez OJ, et al. Patient-specific induced pluripotent stem cells as a model for familial dilated cardiomyopathy. Sci Transl Med. 2012 Apr 18;4(130):130ra47. doi: 10.1126/scitranslmed.3003552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Siu CW, Lee YK, Ho JC, Lai WH, Chan YC, Ng KM, et al. Modeling of lamin A/C mutation premature cardiac aging using patient-specific induced pluripotent stem cells. Aging (Albany NY) 2012 Nov;4(11):803–822. doi: 10.18632/aging.100503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Tse HF, Ho JC, Choi SW, Lee YK, Butler AW, Ng KM, et al. Patient-specific induced-pluripotent stem cells-derived cardiomyocytes recapitulate the pathogenic phenotypes of dilated cardiomyopathy due to a novel DES mutation identified by whole exome sequencing. Hum Mol Genet. 2013 Apr 1;22(7):1395–403. doi: 10.1093/hmg/dds556. [DOI] [PubMed] [Google Scholar]
- 111.Lan F, Lee AS, Liang P, Sanchez-Freire V, Nguyen PK, Wang L, et al. Abnormal calcium handling properties underlie familial hypertrophic cardiomyopathy pathology in patient-specific induced pluripotent stem cells. Cell Stem Cell. 2013 Jan 3;12(1):101–13. doi: 10.1016/j.stem.2012.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Han L, Li Y, Tchao J, Kaplan AD, Lin B, Li Y, et al. Study familial hypertrophic cardiomyopathy using patient-specific induced pluripotent stem cells. Cardiovasc Res. 2014 Nov 1;104(2):258–69. doi: 10.1093/cvr/cvu205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Eschenhagen T, Mummery C, Knollmann BC. Modelling sarcomeric cardiomyopathies in the dish: from human heart samples to iPSC cardiomyocytes. Cardiovasc Res. 2015 Apr 1;105(4):424–38. doi: 10.1093/cvr/cvv017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Liu J, Verma PJ, Evans-Galea MV, Delatycki MB, Michalska A, Leung J, et al. Generation of induced pluripotent stem cell lines from Friedreich ataxia patients. Stem Cell Rev. 2011 Sep;7(3):703–13. doi: 10.1007/s12015-010-9210-x. [DOI] [PubMed] [Google Scholar]
- 115.Lee YK, Ho PW, Schick R, Lau YM, Lai WH, Zhou T, et al. Modeling of Friedreich ataxia-related iron overloading cardiomyopathy using patient-specific-induced pluripotent stem cells. Pflugers Arch. 2014 Sep;466(9):1831–44. doi: 10.1007/s00424-013-1414-x. [DOI] [PubMed] [Google Scholar]
- 116.Hick A, Wattenhofer-Donzé M, Chintawar S, Tropel P, Simard JP, Vaucamps N, et al. Neurons and cardiomyocytes derived from induced pluripotent stem cells as a model for mitochondrial defects in Friedreich's ataxia. Dis Model Mech. 2013 May;6(3):608–21. doi: 10.1242/dmm.010900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Huang HP, Chen PH, Hwu WL, Chuang CY, Chien YH, Stone L, et al. Human Pompe disease-induced pluripotent stem cells for pathogenesis modeling, drug testing and disease marker identification. Hum Mol Genet. 2011 Dec 15;20(24):4851–64. doi: 10.1093/hmg/ddr424. [DOI] [PubMed] [Google Scholar]
- 118.Raval KK, Tao R, White BE, De Lange WJ, Koonce CH, Yu J, et al. Pompe disease results in a Golgi-based glycosylation deficit in human induced pluripotent stem cell-derived cardiomyocytes. J Biol Chem. 2014 Dec 8; doi: 10.1074/jbc.M114.628628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Carvajal-Vergara X, Sevilla A, D'Souza SL, Ang YS, Schaniel C, Lee DF, et al. Patient-specific induced pluripotent stem-cell-derived models of LEOPARD syndrome. Nature. 2010 Jun 10;465(7299):808–12. doi: 10.1038/nature09005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kim C, Wong J, Wen J, Wang S, Wang C, Spiering S, et al. Studying arrhythmogenic right ventricular dysplasia with patient-specific iPSCs. Nature. 2013 Feb 7;494(7435):105–10. doi: 10.1038/nature11799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Josowitz R, Carvajal-Vergara X, Lemischka IR, Gelb BD. Induced pluripotent stem cell-derived cardiomyocytes as models for genetic cardiovascular disorders. Curr Opin Cardiol. 2011 May;26(3):223–9. doi: 10.1097/HCO.0b013e32834598ad. [DOI] [PubMed] [Google Scholar]
- 122.Wang G, McCain ML, Yang L, He A, Pasqualini FS, Agarwal A, et al. Modeling the mitochondrial cardiomyopathy of Barth syndrome with induced pluripotent stem cell and heart-on-chip technologies. Nat Med. 2014 Jun;20(6):616–23. doi: 10.1038/nm.3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kinnear C, Chang WY, Khattak S, Hinek A, Thompson T, de Carvalho Rodrigues D, et al. Modeling and rescue of the vascular phenotype of Williams-Beuren syndrome in patient induced pluripotent stem cells. Stem Cells Transl Med. 2013 Jan;2(1):2–15. doi: 10.5966/sctm.2012-0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ge X, Ren Y, Bartulos O, Lee MY, Yue Z, Kim KY, et al. Modeling supravalvular aortic stenosis syndrome with human induced pluripotent stem cells. Circulation. 2012 Oct 2;126(14):1695–704. doi: 10.1161/CIRCULATIONAHA.112.116996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kobayashi J, Yoshida M, Tarui S, Hirata M, Nagai Y, Kasahara S, et al. Directed differentiation of patient-specific induced pluripotent stem cells identifies the transcriptional repression and epigenetic modification of NKX2-5, HAND1, and NOTCH1 in hypoplastic left heart syndrome. PLoS One. 2014;9(7):e102796. doi: 10.1371/journal.pone.0102796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Gaber N, Gagliardi M, Patel P, Kinnear C, Zhang C, Chitayat D, et al. Fetal reprogramming and senescence in hypoplastic left heart syndrome and in human pluripotent stem cells during cardiac differentiation. Am J Pathol. 2013 Sep;183(3):720–34. doi: 10.1016/j.ajpath.2013.05.022. [DOI] [PubMed] [Google Scholar]
- 127.Jiang Y, Habibollah S, Tilgner K, Collin J, Barta T, Al-Aama JY, et al. An induced pluripotent stem cell model of hypoplastic left heart syndrome (HLHS) reveals multiple expression and functional differences in HLHS-derived cardiac myocytes. Stem Cells Transl Med. 2014 Apr;3(4):416–23. doi: 10.5966/sctm.2013-0105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Letourneau A, Santoni FA, Bonilla X, Sailani MR, Gonzalez D, Kind J, et al. Domains of genome-wide gene expression dysregulation in Down's syndrome. Nature. 2014 Apr 17;508(7496):345–50. doi: 10.1038/nature13200. [DOI] [PubMed] [Google Scholar]
- 129.Bosman A, Letourneau A, Sartiani L, Del Lungo M, Ronzoni F, Kuziakiv R, et al. Perturbations of Heart Development and Function in Cardiomyocytes from hESC with Trisomy 21. Stem Cells. 2015 Jan 31; doi: 10.1002/stem.1961. [DOI] [PubMed] [Google Scholar]
- 130.Schachterle W, Rojas A, Xu SM, Black BL. ETS-dependent regulation of a distal Gata4 cardiac enhancer. Dev Biol. 2012 Jan 15;361(2):439–49. doi: 10.1016/j.ydbio.2011.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Majka SM, McGuire PG. Regulation of urokinase expression in the developing avian heart: a role for the Ets-2 transcription factor. Mech Dev. 1997 Nov;68(1-2):127–37. doi: 10.1016/s0925-4773(97)00138-x. [DOI] [PubMed] [Google Scholar]
- 132.Wamstad JA, Alexander JM, Truty RM, Shrikumar A, Li F, Eilertson KE, et al. Dynamic and coordinated epigenetic regulation of developmental transitions in the cardiac lineage. Cell. 2012 Sep 28;151(1):206–20. doi: 10.1016/j.cell.2012.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Paige SL, Thomas S, Stoick-Cooper CL, Wang H, Maves L, Sandstrom R, et al. A temporal chromatin signature in human embryonic stem cells identifies regulators of cardiac development. Cell. 2012 Sep 28;151(1):221–32. doi: 10.1016/j.cell.2012.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Zaidi S, Choi M, Wakimoto H, Ma L, Jiang J, Overton JD, et al. De novo mutations in histone-modifying genes in congenital heart disease. Nature. 2013 Jun 13;498(7453):220–3. doi: 10.1038/nature12141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Hiroi Y, Kudoh S, Monzen K, Ikeda Y, Yazaki Y, Nagai R, Komuro I. Tbx5 associates with Nkx2-5 and synergistically promotes cardiomyocyte differentiation. Nat Genet. 2001 Jul;28(3):276–80. doi: 10.1038/90123. [DOI] [PubMed] [Google Scholar]
- 136.Ghosh TK, Song FF, Packham EA, Buxton S, Robinson TE, Ronksley J, et al. Physical interaction between TBX5 and MEF2C is required for early heart development. Mol Cell Biol. 2009 Apr;29(8):2205–18. doi: 10.1128/MCB.01923-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Kathiresan S, Srivastava D. Genetics of human cardiovascular disease. Cell. 2012 Mar 16;148(6):1242–57. doi: 10.1016/j.cell.2012.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Bruneau BG. The developmental genetics of congenital heart disease. Nature. 2008 Feb 21;451(7181):943–8. doi: 10.1038/nature06801. [DOI] [PubMed] [Google Scholar]
- 139.Garg V, Kathiriya IS, Barnes R, Schluterman MK, King IN, Butler CA, et al. GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5. Nature. 2003 Jul 24;424(6947):443–7. doi: 10.1038/nature01827. [DOI] [PubMed] [Google Scholar]
- 140.Pu WT, Ishiwata T, Juraszek AL, Ma Q, Izumo S. GATA4 is a dosage-sensitive regulator of cardiac morphogenesis. Dev Biol. 2004 Nov 1;275(1):235–44. doi: 10.1016/j.ydbio.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 141.Rajagopal SK, Ma Q, Obler D, Shen J, Manichaikul A, Tomita-Mitchell A, et al. Spectrum of heart disease associated with murine and human GATA4 mutation. J Mol Cell Cardiol. 2007 Dec;43(6):677–85. doi: 10.1016/j.yjmcc.2007.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Treutlein B, Brownfield DG, Wu AR, Neff NF, Mantalas GL, Espinoza FH, et al. Reconstructing lineage hierarchies of the distal lung epithelium using single-cell RNA-seq. Nature. 2014 May 15;509(7500):371–5. doi: 10.1038/nature13173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Neph S, Stergachis AB, Reynolds A, Sandstrom R, Borenstein E, Stamatoyannopoulos JA. Circuitry and dynamics of human transcription factor regulatory networks. Cell. 2012 Sep 14;150(6):1274–86. doi: 10.1016/j.cell.2012.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Gong W, Koyano-Nakagawa N, Li T, Garry DJ. Inferring dynamic gene regulatory networks in cardiac differentiation through the integration of multidimensional data. BMC Bioinformatics. 2015 Dec;16(1) doi: 10.1186/s12859-015-0460-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Lim J, Thiery JP. Epithelial-mesenchymal transitions: insights from development. Development. 2012 Oct;139(19):3471–86. doi: 10.1242/dev.071209. [DOI] [PubMed] [Google Scholar]
- 146.Garg V, Muth AN, Ransom JF, Schluterman MK, Barnes R, King IN, et al. Mutations in NOTCH1 cause aortic valve disease. Nature. 2005 Sep 8;437(7056):270–4. doi: 10.1038/nature03940. [DOI] [PubMed] [Google Scholar]
- 147.Fischer A, Steidl C, Wagner TU, Lang E, Jakob PM, Friedl P, et al. Combined loss of Hey1 and HeyL causes congenital heart defects because of impaired epithelial to mesenchymal transition. Circ Res. 2007 Mar 30;100(6):856–63. doi: 10.1161/01.RES.0000260913.95642.3b. [DOI] [PubMed] [Google Scholar]
- 148.Greenway SC, Pereira AC, Lin JC, DePalma SR, Israel SJ, Mesquita SM, et al. De novo copy number variants identify new genes and loci in isolated sporadic tetralogy of Fallot. Nat Genet. 2009 Aug;41(8):931–5. doi: 10.1038/ng.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Ott HC, Matthiesen TS, Goh SK, Black LD, Kren SM, Netoff TI, Taylor DA. Perfusion-decellularized matrix: using nature's platform to engineer a bioartificial heart. Nat Med. 2008 Feb;14(2):213–21. doi: 10.1038/nm1684. [DOI] [PubMed] [Google Scholar]
- 150.Lu TY, Lin B, Kim J, Sullivan M, Tobita K, Salama G, Yang L. Repopulation of decellularized mouse heart with human induced pluripotent stem cell-derived cardiovascular progenitor cells. Nat Commun. 2013;42307 doi: 10.1038/ncomms3307. [DOI] [PubMed] [Google Scholar]
- 151.Grosberg A, Alford PW, McCain ML, Parker KK. Ensembles of engineered cardiac tissues for physiological and pharmacological study: heart on a chip. Lab Chip. 2011 Dec 21;11(24):4165–73. doi: 10.1039/c1lc20557a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Feinberg AW, Feigel A, Shevkoplyas SS, Sheehy S, Whitesides GM, Parker KK. Muscular thin films for building actuators and powering devices. Science. 2007 Sep 7;317(5843):1366–70. doi: 10.1126/science.1146885. [DOI] [PubMed] [Google Scholar]