

Abstract
Calcium influx elevates mitochondrial oxidant stress (mOS) in dorsal motor nucleus of the vagus (DMV) neurons that are prone to Lewy body pathologies in presymptomatic Parkinson's disease (PD) patients. In experimental PD models, treatment with isradipine, the dihydropyridine with the highest affinity to Cav1.3 channels, prevents subthreshold calcium influx via Cav1.3 channels into midbrain dopamine neurons and protects them from mOS. In DMV neurons, isradipine is also effective in reducing mOS despite overwhelming evidence that subthreshold calcium influx is negligible compared with spike-triggered influx. To solve this conundrum we combined slice electrophysiology, two-photon laser scanning microscopy, mRNA profiling, and computational modeling. We find that the unusually depolarized subthreshold voltage trajectory of DMV neurons is positioned between the relatively hyperpolarized activation curve of Cav1.3 channels and that of other high-voltage activated (HVA) calcium channels, thus creating a functional segregation between Cav1.3 and HVA calcium channels. The HVA channels flux the bulk of calcium during spikes but can only influence pacemaking through their coupling to calcium-activated potassium currents. In contrast, Cav1.3 currents, which we show to be more than an order-of-magnitude smaller than the HVA calcium currents, are able to introduce sufficient inward current to speed up firing. However, Kv4 channels that are constitutively open in the subthreshold range guarantee slow pacemaking, despite the depolarizing action of Cav1.3 and other pacemaking currents. We propose that the efficacy of isradipine in preventing mOS in DMV neurons arises from its mixed effect on Cav1.3 channels and on HVA Cav1.2 channels.
Keywords: calcium dynamics, vagal motoneurons, modeling, Traub model, Cav1.2, Cav1.3, Cav2, Kv4, NALCN, window current, Parkinson's disease, Lewy body neurite pathology, two-photon laser scanning microscopy, calcium imaging, isradipine, lingering, Hodgkin class I excitability, mouse, mitochondrial oxidative stress
the cholinergic motoneurons of the dorsal motor nucleus of the vagus (DMV) are reported to be among the first neurons to exhibit Lewy body and Lewy neurite pathology in Parkinson's disease (PD) (Lewy 1912), before its appearance in dopamine (DA) neurons of the substantia nigra pars compacta (SNc) (Braak et al. 2004). The functional decline of DMV neurons may underlie PD dysautonomia (Miller et al. 2009), and the loss of DMV neurons correlates with the severity of PD (Gai et al. 1992). We have shown that DMV neurons in brain slices taken from the DJ-1 mouse model of PD exhibit higher basal mitochondrial oxidant stress (mOS) than DMV neurons from littermate wild-type mice. Preincubation of these brain slices in isradipine, an antagonist of plasma membrane Cav1 (L-type) calcium (Ca2+) channels, reduced the basal mOS in the DMV neurons of the DJ-1 mice. Isradipine also lowered the cytosolic Ca2+ concentration in DMV neurons (Goldberg et al. 2012). The drop in cytosolic Ca2+ levels, which presumably leads to a drop in mitochondrial Ca2+ levels and a slowing of respiration, reduces basal mOS (Goldberg et al. 2012).
Isradipine was chosen for that study because of its high affinity to Cav1.3 Ca2+ channels (Sinnegger-Brauns et al. 2009) that are activated in the subthreshold range (Lipscombe et al. 2004). This choice was motivated by previous studies in SNc DA neurons that showed that Ca2+ flux through Cav1.3 Ca2+ channels, during subthreshold dendritic Ca2+ oscillations (Guzman et al. 2009, 2010; Puopolo et al. 2007), was a determinant of basal cytosolic Ca2+ levels and basal mOS, both of which were reduced by isradipine.
However, three findings seem to marginalize the contribution of subthreshold Ca2+ influx to basal cytosolic Ca2+ levels in DMV neurons. First, imaging of dendritic Ca2+ concentrations in DMV neurons during autonomous pacemaking failed to reveal substantial Ca2+ influx between spikes. Second, once DMV neurons were silenced by hyperpolarization, further hyperpolarization did not further reduce their cytosolic Ca2+ concentration. Third, measurement of subthreshold Ca2+ currents in DMV neurons demonstrated that they were quite small, in comparison, for example, to leak and voltage-activated sodium (Na+) currents. These findings suggest a more prominent role for the high-voltage activated (HVA) Cav1.2 Ca2+ channels, to which isradipine has an equal affinity (Sinnegger-Brauns et al. 2009), in determining Ca2+ levels and basal mOS in DMV neurons.
In the present study we set out to clarify the specific physiological role of Cav1.2 and Cav1.3 channels in Ca2+ dynamics, as well as the autonomous firing patterns, observed in DMV neurons (Goldberg et al. 2012). Using a combination of 1) slice electrophysiology; 2) two-photon laser scanning microscopy (2PLSM) Ca2+ imaging; 3) mRNA profiling; and 4) numerical modeling, we found that the depolarized autonomous voltage oscillations, due to NALCN, Nav1 Na+, and Kv4 potassium (K+) channel currents, create a functional segregation between Cav1.3 channels and Cav1.2 (as well as other HVA, such as Cav2) Ca2+ channels in DMV neurons. A very small Cav1.3 current suffices to speed up the firing rate, without significantly contributing to the basal Ca2+ concentration, which is attributable almost entirely to spike-triggered Ca2+ influx via HVA Ca2+ channels. This latter influx activates Ca2+ activated K+ currents that underlie afterhyperpolarizations (AHPs) (Sah and McLachlan 1992) that slow the pacemaking (Goldberg et al. 2012). We propose that equal affinity of isradipine to Cav1.2 and Cav1.3 channels reduces spike-triggered Ca2+ influx and prevents the Cav1.3 channels from driving pacemaking, thereby explaining the drug's efficacy in protecting DMV neurons from mOS.
MATERIALS AND METHODS
2PLSM imaging.
The 2PLSM system was described previously (Guzman et al. 2010). Briefly, the two-photon excitation source was a Chameleon Ultra 2 tunable laser system (680–1,080 nm; Coherent Laser Group, Santa Clara, CA). Optical signals were acquired using an 820-nm excitation beam to excite Alexa and Fluo-4 dyes simultaneously. Laser power attenuation was achieved with two Pockels cells electro-optic modulators (model 350-80; Conoptics, Danbury, CT). The fluorescence emission was collected with nondescanned photomultiplier tubes (Prairie Technologies, Madison, WI). A Dodt contrast detector system was used to provide a bright-field transmission image in registration with the fluorescent images.
Slice preparation.
Experimental procedures adhered to and received prior written approval from the Northwestern University and Hebrew University Animal Care and Use Committees. Three- to eight-week-old C57BL/6 mice of both sexes were deeply anesthetized with ketamine-xylazine and perfused transcardially with ice-cold modified artificial cerebrospinal fluid (ACSF), bubbled with 95% O2-5% CO2, and contained the following (in mM): 2.5 KCl, 26 NaHCO3, 1.25 Na2HPO4, 0.5 CaCl2, 10 MgSO4, 0.4 ascorbic acid, 10 glucose, and 210 sucrose. The cerebellum, pons, and medulla were rapidly removed, blocked in the coronal plane, and sectioned at a thickness of 240 μm in ice-cold modified ACSF. Slices were then submerged in ACSF, bubbled with 95% O2-5% CO2, and containing the following (in mM): 2.5 KCl, 126 NaCl, 26 NaHCO3, 1.25 Na2HPO4, 2 CaCl2, 2 MgSO4, and 10 glucose and stored at room temperature for at least 1 h before recording and/or imaging.
Slice visualization and electrophysiology.
The slices were transferred to the recording chamber mounted on an Olympus BX51 upright, fixed-stage microscope, and perfused with oxygenated ACSF at 32°C. The only exception was that voltage-sensitive Na+ currents were recorded in a HEPES-based solution that contained the following (in mM): 2.5 KCl, 137 NaCl, 1.8 CoCl2, 1 MgCl2, 5.4 tetraethylammonium (TEA)-Cl, 10 4-aminopyridine, 10 glucose, and 10 HEPES (pH 7.4 with HCl). A ×60, 0.9 NA water-immersion objective was used to examine the slice using standard infrared differential interference contrast video microscopy. Patch pipette resistance was typically 3–4.5 MΩ. For whole cell current-clamp recordings and for voltage-clamp measurements of K+ conductances the pipette contained the following (in mM): 135.5 K-MeSO4, 5 KCl, 2.5 NaCl, 5 Na-phosphocreatine, 10 HEPES, 0.2 EGTA, 0.21 Na2GTP, and 2 Mg1.5ATP (pH 7.3 with KOH, 280–290 mosmol/kgH2O). For Ca2+ imaging experiments in conjunction with current-clamp recordings the pipette contained the following (in mM): 135 K-MeSO4, 5 KCl, 5 Na-phosphocreatine, 5 Tris-phosphocreatine, 10 HEPES, 0.1 Fluo-4, 0.05 Alexa Fluor 568, 0.21 Na2GTP, and 2 Mg1.5ATP (pH 7.3 with KOH, 280–290 mosmol/kgH2O). For voltage-clamp Ca2+ imaging the pipette contained the following (in mM): 120 Cs-MeSO3, 15 CsCl, 8 NaCl, 10 TEA, 5 QX-314, 10 HEPES, 0.3 Na2GTP, 3 Mg1.5ATP, 0.1 Fluo-4, and 0.05 Alexa Fluor 568 (pH 7.3 with CsOH, 280–290 mosmol/kgH2O).
Electrophysiological recordings were obtained with a Multiclamp 700B amplifier (Molecular Devices, Sunnyvale, CA). Junction potential, which was 7–8 mV, was not corrected. Signals were digitized at 20–100 kHz and logged onto a personal computer with the Clampex 9.2 software (Molecular Devices) or, in the imaging experiments, using the custom-written shareware package WinFluor (John Dempster, Strathclyde University, Glasgow, Scotland, UK), which automates and synchronizes the 2PLSM imaging and electrophysiological protocols.
Drugs and reagents.
For recording of autonomous discharge the following cocktail of synaptic receptor blockers was used that included the following (in μM): 50 d-APV to block NMDA receptors, 5 NBQX to block AMPA receptors, 10 SR 95531 (gabazine) to block GABAA receptors, 1 CGP 55845 to block GABAB receptors, 10 mecamylamine to block nicotinic receptors, 10 atropine to block muscarinic receptors, and, in some of the experiments, 1 ketanserin to block 5-HT2A receptors. Voltage-sensitive Na+ channels were blocked with 1 μM tetrodotoxin (TTX). Cav1 channels were antagonized with 5 μM of either nifedipine or isradipine or 200 nM calciseptine. The acute effects of solution exchanges or drug applications were measured at least 5 min after wash on, except for dihydropyridine where 10 min were given. The only exception was the application of phrixotoxin-2, a selective Kv4 (A-type) channel blocker. In these experiments we pipetted 20 μl of a 100-μM stock solution into the recording chamber whose volume was ∼1–2 ml, thereby reaching a nominal concentration of 1–2 μM phrixotoxin-2 in the bath until the solution was replaced (Subramaniam et al. 2014) Fluo-4 pentapotassium and Alexa Fluor 568 hydrazide Na+ salts were obtained from Invitrogen (Carlsbad, CA). TTX, phrixotoxin-2, and calciseptine were obtained from Alomone Labs (Jerusalem, Israel). The rest of the drugs and reagents were obtained from Tocris (Ellisville, MO) or Sigma (St. Louis, MO).
RNA extraction from mouse brain slices.
The DMV was dissected out from brain slices taken from five C57BL/6 mice. RNA was extracted from brain slices using the Qiagen (Venlo, The Netherlands) RNAeasy kit, which ensures full representation of all RNA length groups. Briefly, brain slices were homogenized with 500 μl QIAzol lysis buffer, lysed for 5 min, mixed with 100 μl chloroform to allow full neutralization, suspended for 3 min, and centrifuged for 15 min (at 12,000 g and 4°C). The aqueous phase was mixed with 1.5 vol of ethanol, loaded on RNA-binding spin column, and centrifuged for 30 s in 8,400 g. The column was washed once by centrifugation (30 s at 8,400 g) and discarding the flow-through with 700 μl RWT buffer (85% ethanol) and twice with 500 μl RPE buffer (70%; Qiagen), for the removal of DNA and protein remnants, respectively. Finally, spin column was centrifuged for 1 min in 21,067 g to further dry the column. Thirty-five microliters of nuclease-free water were used to elute the RNA, which was immediately put on ice to prevent degradation. RNA concentration was determined by Nanodrop-2000 (Thermo-Scientific, Waltham, MA), and its integrity was assessed via 1% agarose gel electrophoresis followed by identification of two distinct rRNA bands (28S and 18S).
cDNA synthesis from mouse brain slices.
cDNA preparation was performed using the Quanta qScript cDNA Synthesis Kit (Quanta Biosciences, Gaithersburg, MD). One-hundred nanograms of RNA were diluted in 15 μl of nuclease-free water, and mixed with 4 μl 5× reaction buffer and 1 μl of reverse transcriptase (except for the no-RT controls, where reverse transcriptase was not added) for a final 20 μl reaction volume. The mixture was put in a 200-µl PCR tube and placed in a MJ Research PTC 200 Thermal Cycler (GMI, Ramsey, MN) programmed for 5 min in 22°C, 30 min in 42°C, and 5 min in 85°C. cDNA was then diluted 1:10 by addition of 180 ml DDW.
Quantitative real-time PCR and primers.
For each reaction, iTaq Universal SYBR green Supermix 2 × (7.5 ul; Bio-Rad, Hercules, CA) was used for both the target and reference genes employed for normalization together with 0.75 μl of left and right primers (10 mM of each), and 6 μl of cDNA on a Bio-Rad CFX96 Touch Real-Time PCR Detection System. Amplification was performed in triplicates for each primer pair and tested samples, for 95°C (3 min), 95°C (15 s) and 40 repeats of 60°c for 30 s, followed by increasing the temperature from 67.0 to 94.6°C in 0.3°C increments every 5 s to create a melting curve. The data were obtained using Bio-Rad CFX Manager 3.0 software. Also, for each primer pair a dilution curve was created to calculate primer efficiency that was then used to recalculate the expression results. Fixed values were normalized to either 18S as a housekeeping gene or the neuronal-specific TUBB3 as a neuronal marker. The primers (Sigma) used to identify all known transcriptional variants of the Cav channels and reference genes were as listed in Table 1.
Table 1.
Primer sequences
| Gene Name/Primer Direction | Sequence |
|---|---|
| Cav1.2 | |
| Forward | CGAAGGTACATCCCCAAGAA |
| Backward | CGATTTTGAAGAGGCAGCTC |
| Cav1.3 | |
| Forward | ATCCCTACACCGAAGCTCCT |
| Backward | TCGGCAATCTCATGTTTTGT |
| Cav2.1 | |
| Forward | GACAGGGAGCGCAGACAC |
| Backward | GGTGGCCAGCTTGTTATTCT |
| Cav2.2 | |
| Forward | GGTGGAAGGGGATAAGGAAA |
| Backward | GGTTGTCTGCATCCTCTGGT |
| Cav2.3 | |
| Forward | ACACCTTCCCTGCAGCTATC |
| Backward | GCGAATGCCATTGTACATCA |
| 18S | |
| Forward | ACAACAAGCTGCGTGAGGAC |
| Backward | CAAAGGCCCAGAGACTCATT |
| TUBB3 | |
| Forward | CGCCTTTGGACACCTATTCA |
| Backward | TGCAGGCAGTCACAATTCTC |
Data and statistical analysis.
Electrophysiological data were analyzed and curve fitting was done using custom-made code on custom-made (Winfluor, Dr. John Dempster, University of Strathclyde; NUPver, Nicholas Schwarz, Northwestern University; Oscilloscope, Dr. Charles Wilson, University of Texas at San Antonio) and commercial (Matlab; The Mathworks, Natick, MA) software. The two-tailed Wilcoxon signed-rank test (SRT) was used to test for changes of medians in matched-paired comparisons. The nonparametric Kruskal-Wallis one-way ANOVA test was used to compare the relative expression of Cav channel mRNA. The null hypothesis of equal medians was rejected if the P value was <0.05.
Computational modeling.
To study the Ca2+ dynamics exhibited by the DMV cells we converted the Traub model, a single compartment model of a nonautonomously spiking neuron, exactly as it appears in Ermentrout (1998), into an autonomous pacemaker by replacing the externally applied current term with two voltage-activated Ca2+ currents and by shifting all voltages by +20 mV to replicate the very depolarized potentials of the DMV pacemaker that result from the voltage-insensitive NALCN cation current that they express (Goldberg et al. 2012). The two Ca2+ conductances differed only in their half-activation voltages and in their maximal conductance: IL with a half activation of V1/2L = −40 mV and maximal conductance of ḡL representing the Cav1.3 current and IN with a half activation of V1/2N = −20 mV and maximal conductance of ḡN representing the HVA currents such as the Cav1.2 and Cav2 currents. The units of the maximal conductance are mS/cm2.
The Ca2+ currents were given by
where ECa = 120 mV and VSX = 8 mV for both X = L, N (Goldberg et al. 2009). The equation for the concentration of free Ca2+ in the cell is given by
where [Ca] are in units of nM, r = 600 cm−1, and τCa = 50 ms. The units of the Faraday constant are kC/mol and z = 2 for Ca2+ ions.
RESULTS
Blockade of Cav1 channels reduces Ca2+ influx per spike and speeds up pacemaking.
Motoneurons of the DMV are autonomous pacemakers that discharge at a median rate of ∼1 spike/s in the presence of synaptic blockers. The basal cytosolic Ca2+ concentration in DMV neurons is generated almost exclusively by Ca2+ influx associated with this spiking (Goldberg et al. 2012). Because Cav1 antagonists reduce this basal concentration (Goldberg et al. 2012), these drugs should also reduce the amount of Ca2+ influx per individual spike. To test this we conducted 2PLSM imaging of somatic Ca2+ in DMV neurons while injecting various holding currents to either hyperpolarize or depolarize the cells (Fig. 1A). As described previously (Goldberg et al. 2012), this generated a linear dependence of ΔF/F0 on firing rate. Application of calciseptine, a Cav1-selective neurotoxin, significantly reduced the slope indicating that Cav1 channels contribute to the amount of Ca2+ influx that occurs with each spike. Previous studies have shown that Ca2+ influx via Cav1.2 channels generates the slow AHP (sAHP) in DMV neurons (Goldberg et al. 2012; Sah and McLachlan 1992). We have previously shown that Ca2+ influx through Cav2.2 channels (another HVA Ca2+ channel) activates the small-conductance Ca2+-activated K+ (SK) current that underlies medium AHP (mAHP) that follows each individual spike and slows DMV pacemaking significantly (Goldberg et al. 2012). In contrast, in several neuronal types the sAHP usually affects mostly evoked discharge rates (Gustafsson and Wigstrom 1983; Hotson and Prince 1980; Schwindt et al. 1988). Nevertheless, we tested whether antagonizing Cav1 channels affected pacemaking. Application of 5 μM nifedipine modestly raised the firing rate of all seven DMV neurons tested (median firing rate increase from 1.05 to 1.28 spikes/s, P < 0.05; SRT, Fig. 2A). Therefore, Cav1 Ca2+ channels contribute to spike-triggered Ca2+ influx and may contribute to the slowing of pacemaking in DMV neurons by activating AHPs.
Fig. 1.
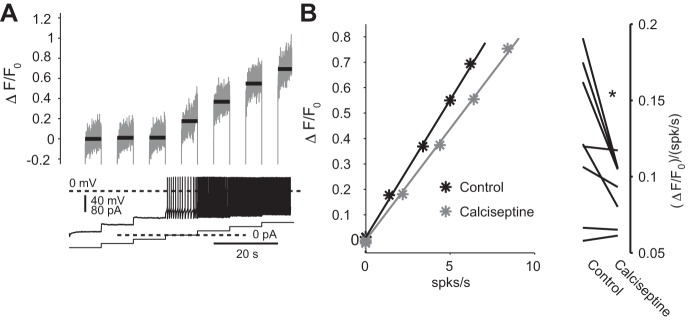
Antagonizing Cav1 Ca2+ channels reduces the amount of Ca2+ influx per spike. A, bottom: a sequence of 10-s current injections from −60 to +60 pA either hyperpolarize and silence discharge or depolarize and increase discharge. Two-photon laser scanning microscopy (2PLSM) Fluo-4 imaging of the soma reveals that the level of fluorescence in the silenced cell is relatively insensitive to the degree of hyperpolarization. In contrast, the fluorescence increases linearly as the cell speeds up. Imaging was conducted in the final 5 s of each current pulse, and black bar indicates the average level of fluorescence. B: plotting fluorescence levels as a function of the firing rate also reveals a linear relationship. Application of 200 nM calciseptine, a Cav1 channel-specific neurotoxin, reduces the slope of the dependence of fluorescence on firing rate indicating that the amount of Ca2+ flux per spike is reduced in the presence of this toxin. *P < 0.05, signed-rank test (SRT).
Fig. 2.
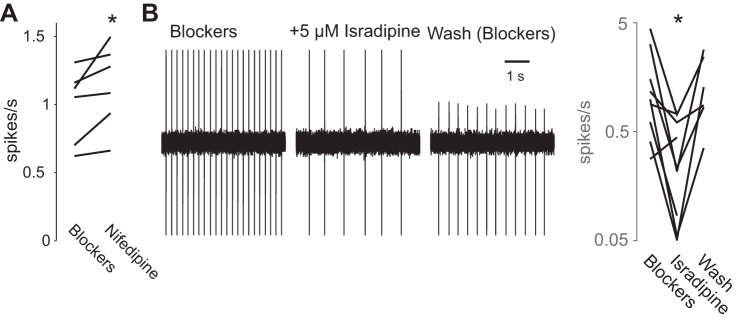
Acute application of various dihydropyridines affects the rate of pacemaking differentially. A: application of 5 μM nifedipine slightly, but significantly, speeds up pacemaking in dorsal motor nucleus of the vagus (DMV) neurons. B: acute application of 5 μM isradipine significantly and reversibly slows pacemaking in DMV neuron (shown on a logarithmic scale). *P < 0.05, SRT. To better gauge the autonomous firing rate of the DMV neurons, the slices were bathed in artificial cerebrospinal fluid (at an elevated temperature) that included a cocktail of glutamatergic, GABAergic, cholinergic, and serotonergic receptor antagonists.
Acute isradipine application slows pacemaking.
In contrast to the robust suprathreshold Ca2+ influx through HVA Ca2+ channels, the subthreshold Ca2+ influx in between spikes (that must flow through Cav1.3 channels because they are the only noninactivating low-voltage activated Ca2+ channels) is small or nonexistent (Goldberg et al. 2012). If Cav1.3 channels do not flux much Ca2+, then what is their physiological role? To answer this question we tested the effect of isradipine, which is the dihydropyridine with the highest affinity to Cav1.3 channels, on autonomous firing in DMV neurons recorded in the loose patch configuration. We have previously shown that preincubation of DMV slices with 200 nM isradipine does not affect the basal firing rates (Goldberg et al. 2012). However, it is possible that application of a higher concentration would lead to a different effect by antagonizing Cav1.3 channels acutely. Indeed, acute application of 5 μM isradipine reduced the median firing rate from 0.99 to 0.23 spikes/s (P < 0.01, SRT, n = 9). Furthermore, in all six cells in which we attempted to wash off the effect of isradipine the firing rate recovered from a median of 0.22 to 1.08 spikes/s (P < 0.05, SRT; Fig. 2B). The caveat in these experiments is that voltage-dependent persistent sodium currents are necessary and sufficient to drive pacemaking of DMV motoneurons (Goldberg et al. 2012), and there is a possibility that 5 µM isradipine also antagonizes them. However, direct measurements of the persistent TTX-sensitive current (in the presence of an equimolar replacement of calcium chloride with cobalt chloride to block Cav currents) in DMV neurons demonstrated (Goldberg et al. 2012) that acute application of 5 μM isradipine did not antagonize the persistent sodium current in DMV neurons (data not shown).
A functional segregation between the Cav1.3-like and HVA-like Ca2+ currents in a model of a slow autonomous pacemaker.
The fact that 5 μM isradipine slows pacemaking while 5 μM nifedpine speeds it up is probably due to the facts that 1) the isradipine is the dihydropyridine with the highest affinity to Cav1.3 channels; and that conversely 2) nifedipine more-or-less completely blocks Cav1.2 channels whereas isradipine does so only partially. Thus, Cav1.3 channels that contribute only a small inward current to DMV motoneurons (Goldberg et al. 2012) are capable of speeding up pacemaking while fluxing little Ca2+ into the cell (Goldberg et al. 2012). In contrast, Cav1.2 and Cav2 channels generate large Ca2+ currents and flux much more Ca2+ into the cell that slows pacemaking by activating AHPs (Goldberg et al. 2012). How can such a small Cav1.3 current robustly speed up pacemaking? We hypothesized that the answer to this question lies in the fact that the membrane voltage of DMV motoneurons during autonomous pacemaking is very depolarized, which we have previously shown to arise from the action of the “leak” NALCN channels (Goldberg et al. 2012). These channels should place the Cav1.3 current in a position to dictate how closely the cell's steady-state I–V curve approaches its zero current line. The proximity to the zero current line presumably causes the membrane potential to linger for longer just beneath spike threshold and consequently lengthens the interspike intervals (ISIs). Thus, because the DMV motoneurons' empirical negative conductance region is within a few picoamperes of the zero current point (Goldberg et al. 2012), even a very small Cav1.3 current, also on the order of a few picoamperes, should suffice to dramatically influence the lingering. In contrast, according to this hypothesis the HVA currents, as large as they are, are not activated in this voltage region and therefore cannot influence the pacemaking directly. Instead, they do so via their coupling to K+ channels that underlie AHPs, as discussed above.
To test this hypothesis, we constructed a model of a slow pacemaking neuron (Ermentrout 1998) that like the DMV neurons possesses an N-shaped steady-state I–V curve with a negative conductance region. We also shifted the voltage of the trajectories to correspond to the relatively depolarized voltage oscillations of DMV neurons (Goldberg et al. 2012). The autonomous pacemaking of the model was achieved by including in the model two voltage-activated Ca2+ currents that shared the same functional shape but differed in that one half-activated at −20 mV (representing the HVA currents, denoted IN) and the other at −40 mV (representing the Cav1.3 current, denoted IL). We also assumed that the maximal conductance of IN was 25 times larger than the maximal conductance of IL (Fig. 3). Under these conditions the model cell fired autonomously and displayed Ca2+ transients with amplitudes of ∼75 nM. Abolishing IN reduced the amplitude to < 20 nM but did not abolish pacemaking. In contrast, abolishing IL alone arrested pacemaking and reduced the Ca2+ concentration to less than 1 nM (Fig. 3A).
Fig. 3.

A 2-Ca2+ channel single-compartment model of pacemaking DMV neurons. A: model neuron discharges rhythmically and each action potential is accompanied by a rapid influx of Ca2+ that decays with time (red). The green arrow marks a small yet visible rise in Ca2+ just before the spike. Elimination of the HVA-like Ca2+ current (IN) eliminated the rapid influx and reveals much smaller subthreshold oscillations (blue, note the change is scale). Elimination of the Cav1.3-like Ca2+ current (IL) arrests pacemaking and creates a stable resting potential. Ca2+ concentration all but vanishes (black). B, left: steady-state I–V curves of the 3 various parameter regimes in A, with corresponding color-coding. B, top right: zoom-in on the region of the I–V curves nearest to the zero-current line (dashed black line). The green arrow marks the value of the stable resting potential. B, bottom right: the corresponding steady-state I–V curves for the 2 calcium currents. C: voltage trajectory for 2 values of IL. The larger current passes further away from the zero-current line and does so faster (as indicated by the density of the corresponding marks) resulting in a shorter interstimulus interval.
How does pacemaking depend on IL despite its maximal conductance being 25 times smaller than the maximal conductance of IN? The answer to this question is given by considering the steady-state I–V curve of the model and the value of the two Ca2+ conductances in the voltage region where the I–V curve approaches the zero current line. Despite being larger, the voltage dependence of IN prevents it from making a significant contribution in this subthreshold range. Therefore, abolishing IN hardly moves the I–V curve any closer to the zero current line (Fig. 3B). In contrast, IL is strongly activated at this region, and therefore despite being 25 times smaller than IN, abolishing IL raises the I–V such that it crosses the zero current line. The zero crossing establishes a steady-state voltage, which arrests pacemaking. IL also controls the degree of lingering in the model. For example, doubling the amplitude of its conductance, pushes the I–V curve further from the zero current line, thereby increasing the inward current near threshold that speeds up the trajectory of the membrane voltage in that region (Fig. 3C).
Voltage dependency of somatic Ca2+ currents and dendritic Ca2+ concentrations in DMV neurons fit the functional segregation model.
In the model, in order for the bulk of Ca2+ influx to result from IN, the maximal conductance of IN needed to be more than an order of magnitude larger than the maximal conductance of IL (as mentioned above, we used a factor of 25). To quantify Ca2+ currents and the rises in Ca2+ concentrations that they generated in the model, we measured them in response to a simulated voltage ramp. These simulations were conducted either for IL alone or for the total current (IL + IN). Indeed, the currents and associated concentrations were roughly 20 times larger when both currents were activated compared with when only IL was activated. If such a discrepancy in the sizes of the Cav1.3 and HVA currents was present in real DMV neurons, it should be detectable by conducting the same voltage-ramp experiment on actual DMV neurons. We applied somatic voltage ramps to cesium (Cs+)-loaded DMV neurons and measured the total current and the 5 μM isradipine-sensitive subthreshold current as well as the dendritic Ca2+ signals associated with them. Accurately determining the isradipine-sensitive subthreshold response required averaging measurements from several cells. These measurements revealed that an isradipine-sensitive somatic Ca2+ current is indeed activated in the subthreshold range, which explains why applications of 5 μM isradipine slowed pacemaking (Fig. 2B). Moreover this current and the associated dendritic Ca2+ signals were indeed ∼20 times smaller then the full-blown responses (Fig. 4A), in agreement with the model prediction. Because the isradipine-sensitive subthreshold current is generated by Cav1.3 channels, these results suggest that DMV neurons express much less Cav1.3 channels compared with other HVA Cav channels. We tested this with quantitative RT-PCR analysis that revealed a differential tissue-level expression in the DMV of the various channels, when normalized either to housekeeping genes or to the tubulin marker TUBB3, which is specific to neurons (P < 0.001, Kruskal-Wallis one-way ANOVA). The transcript levels of Cav1.3 channel were ∼20-fold less than the sum of the transcript levels of all the other HVA channels (Fig. 4B).
Fig. 4.
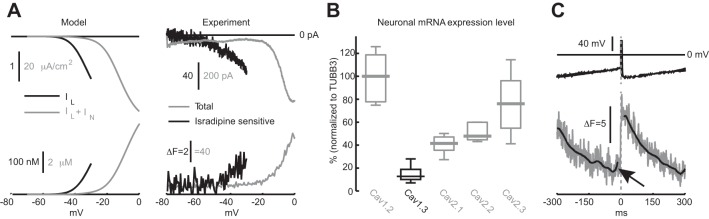
Ca2+ responses to voltage ramps and the spike-triggered waveform of Ca2+ in DMV neurons are consistent with the 2 Ca2+ channel model. A, left: voltage-dependence of IL (black) and IL + IN (gray) and the corresponding Ca2+ rises they induce in response to a slow voltage ramp in the model. A, right: measurement of the total (gray) and isradipine-sensitive (black) somatic Ca2+ currents and the corresponding raw dendritic fluorescence from DMV neurons in response to a slow voltage ramp. Ramp speed was 100 mV/s for the full range (gray trace) and 50 mV/s for the subthreshold range (black one). Black trace is the average of 5 measurements from 3 cells. Note the difference in scale between black and gray traces both in the model and in the experiment. The internal pipette solution included 5 mM QX-314 to block Nav channels. B: neuronal Cav channel mRNA expression normalized to TUBB3 in the DMV. The expression level is expressed in percentages of the median value of the expression of Cav1.2 mRNA. C: spike-triggered average of Ca2+ oscillations (as in Fig. 6C, different cell) reveals a slight rise in dendritic Ca2+ concentration (70 μm from the soma) just before the spike (marked by black arrow; compare to red trace in A).
We have previously reported that the dendritic Ca2+ transient measured in current clamp in DMV neurons rose rapidly with each spike and then decayed exponentially (Goldberg et al. 2012). This shape contrasted with that observed in SNc DA neurons that exhibited substantial Ca2+ influx before the spike (Guzman et al. 2009). The current model, nevertheless, predicted a very small albeit visible increase in Ca2+ before the occurrence of a spike (Fig. 3A, green arrow), despite the size of IL relative to IN. This discrepancy may have stemmed from the distal location of the dendritic spike-triggered changes in fluorescence, which were typically >50 μm from the soma (Fig. 4C), vs. the measurements of Ca2+ currents, which despite being done in Cs+-loaded cells, probably represented current flowing through proximally located channels. Similarly the fluorescence measurements conducted during the voltage ramps (Fig. 4A) were taken from proximal dendrites (20–50 μm from the soma). Thus it is possible that relative complement of Cav1.3 is even smaller more distally, thereby reducing the putative subthreshold calcium entry via these channels. Nevertheless, upon closer scrutiny of the experimental data, we found some examples of DMV neurons in which a small subthreshold component was visible in the spike-triggered average of the dendritic Ca2+ oscillations (Fig. 4C).
A window A current slows pacemaking in DMV neurons.
The voltage-clamp recordings, Ca2+ imaging, and mRNA profiling support our conclusion that Cav1.3 is an order-of-magnitude less abundant than other HVA Ca2+ currents in DMV motoneurons. Using the model we explained how a small Cav1.3-like current can nevertheless significantly modulate the slow pacemaking of the model neuron. However, this modulation relied on the ability of the model neuron to exhibit class I Hodgkin excitability (Hodgkin 1948) meaning that it can exhibit very long ISIs (or lingering) near the transition point (bifurcation) of its voltage to oscillations (Ermentrout 1996). We therefore need to identify the biophysical mechanism that confers such behavior in DMV motoneurons. Lingering behavior depends on two biophysical mechanisms. First, the cell must have a steady-state negative conductance region. Our previous work has shown that DMV cells exhibit a negative conductance region due to the action of the persistent Na+ current (Goldberg et al. 2012). However, while the negative conductance region is necessary, it may not be sufficient, because the oscillations might become very fast even with a small deviation from the bifurcation point. Thus the second mechanism that serves to slow pacemaking is the outward fast inactivating (A-type) K+ current. This current is activated when the cell depolarizes towards spike threshold and resists the depolarization, thereby causing the cell to slow its ramp up to spike threshold (Connor and Stevens 1971; Ermentrout and Terman 2010).
The influence of the A-type current on the autonomous spike rate and membrane voltage trajectory near spike threshold is evident from biophysical measurements of its voltage activation and inactivation curves. These curves reveal a “window current” in the subthreshold region (centered around −50 mV) just below spike threshold (Fig. 5A), at a much more depolarized voltage range than was previously reported in the rat (Sah and McLachlan 1992). Whole cell current-clamp measurements in DMV neurons strongly suggest that A-type currents indeed temper the autonomous firing rate of DMV motoneurons, because dialysis of the intracellular milieu after rupturing of the membrane always significantly reduces the A current (P < 0.01, SRT; Fig. 5B), which is accompanied by a speed-up of autonomous pacemaking (P < 0.01, SRT; Fig. 5C). To verify that antagonizing A currents affect the firing rate in an intact cell, we tested the effect of phrixotoxin-2, a selective antagonist of Kv4 channels that give rise to A currents, on the discharge rate of DMV neurons recorded in the loose patch mode. Application of nominally 1–2 μM phrixotoxin-2 to the bath (see materials and methods) (Subramaniam et al. 2014) transiently increased the firing rate of these neuron by 21% (P < 0.05, SRT; Fig. 5D).
Fig. 5.
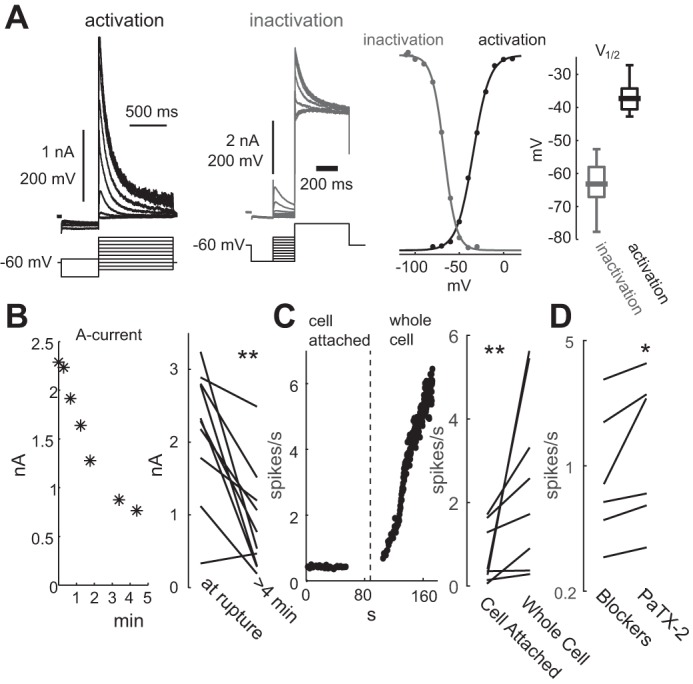
A-type Kv4 K+ currents slow pacemaking of DMV neurons. A, left: example of measurements of the activation (black) and inactivation (gray) of the transient K+ current in the presence of TTX. A, right: normalized Boltzman sigmoidal functions describing the voltage dependence of activation and inactivation of the Kv4 current, and the distribution of the voltages of half activation (n = 13) and inactivation (n = 15), reveal a window Kv4 current that spans the typical subthreshold range of the voltage trajectory of DMV neurons during pacemaking. B, left: Time course of the reduction in the peak A current as a function of time after membrane rupture in whole cell configuration. B, right: comparison of the peak A current at membrane rupture vs. past 4 min from rupture. C, left: time course of the instantaneous firing rate of a DMV neuron in cell-attached mode before membrane rupture and after whole cell configuration was achieved. B, right: comparison of autonomous discharge rate before and after rupturing the membrane. D: brief application of nominally 1–2 μM phrixotoxin-2 (PaTX-2), a selective Kv4 antagonist, transiently increases the firing rate of DMV neurons recorded in the loose-patch configuration (shown on a logarithmic scale). *P < 0.05, **P < 0.01, SRT.
DISCUSSION
Functional segregation between the Cav1.3 and the HVA Ca2+ currents.
DMV neurons express two kinds of L-type Ca2+ channel: Cav1.2 and Cav1.3 (Goldberg et al. 2012), that differ in their voltage dependency with the half-activation voltage of Cav1.3 channels being ∼20 mV more hyperpolarized (Lipscombe et al. 2004). In the present study we have shown that DMV neurons manipulate this voltage difference to bestow Cav1.3 channels with complementary (if not opposing) role to that of Cav1.2 channels and at least one other HVA channel, the Cav2.2 (N-type) channel. The cells do so by expressing a substantial “leak” NALCN channel that positions their subthreshold membrane trajectories in between the half-activation voltages of the Cav1.3 channel and the HVA Ca2+ channels. This segregation causes the majority of the Ca2+ influx into the cytosol to enter via the HVA channels, with a negligible contribution from subthreshold flux via Cav1.3 channels. Conversely, the HVA currents can only influence the cells' autonomous firing rate via their coupling to the K+ currents that underlie the AHPs: Cav1.2 channels activate the sAHP and Cav2.2 channels activate SK channels that underlie the mAHP (Goldberg et al. 2012; Sah and McLachlan 1992). The end result is a slowing of the firing rate. In contrast, Cav1.3 channels are able to speed up the firing rate of the cells by controlling the proximity of the cells' N-shaped steady-state I–V curve to their zero (ionic) current point. In doing so, Cav1.3 channels determine the degree of subthreshold voltage lingering and the duration of the ISI. In our analysis, we used a wide range of ages (3–8 wk), spanning immature to young adults. This could give rise to a potential confound as we may have overlooked any age dependence of Ca2+ regulation in these cells. However, the three biophysical properties that are essential for our argument, namely: 1) an N-shaped steady state I–V curve; 2) a depolarized subthreshold region due to NALCN channels; and 3) having the majority of Ca2+ ions entering during spiking, were similar across all these ages. Hence, although this is a legitimate concern, we do not see any evidence of age-dependent Ca2+ regulation.
Our physiological estimate of the relative sizes of the Cav1.3 current vs. the HVA currents shows that the former is over an order of magnitude smaller than the latter. We reached this conclusion by comparing the predictions concerning the Ca2+ currents and the Ca2+ cytosolic concentration that arose from our simplified one-compartment model to the empirically measured Ca2+ currents and Ca2+ concentration. Molecular profiling revealed that the transcript level of neuronal Cav1.3 mRNA was an order of magnitude less than the total transcript level of mRNA of the other HVA channels, in close agreement with the physiological and imaging estimates.
Multiple conductances activated in the subthreshold range guarantee robust pacemaking in DMV neurons.
Cav1.3 channels are not alone in influencing the subthreshold trajectory of pacemaking. Persistent Na+ and HCN currents are other depolarizing currents that drive pacemaking in these neurons (Goldberg et al. 2012). Despite the driving to spike threshold by at least three pacemaking currents, DMV neurons fire at a relatively slow rate of ∼1 spike/s. We have shown that this slowing of the discharge rate is attributable at least in part to the constitutive activity of a Kv4 channels (although we cannot rule out a contribution from Kv1 channels) that give rise to an A-type window current that is conveniently targeted precisely to the subthreshold voltage range. Previous measurements using sharp electrodes in the rat found the A-type window current to reside at a much more hyperpolarized voltage range: even after correcting for junction potential the difference is at least 20 mV (Sah and McLachlan 1992). The difference is attributable to our use of a different recording technique. Whole cell patch electrodes afford better voltage control than do high resistance sharp electrodes.
Clinical implications.
We have shown the Cav1 channels contribute directly to the Ca2+ influx that occurs with each spike. Because Ca2+ influx into the weakly buffered DMV neurons generates basal mOS, Cav1 channels must contribute to it. Unlike the case of SNc DA neurons, Cav1.3 channels themselves do not flux much Ca2+ into DMV cells. However, by virtue of the ability of Cav1.3 channels to speed up spiking they too could contribute indirectly to Ca2+ influx during DMV neuron discharge and consequently increase mOS. It is also possible that Ca2+ influx via Cav1.3 channels has direct access to the endoplasmic reticulum and from there to the mitochondria. In such a scenario, it is possible that the Cav1.3 current is particularly efficacious in driving basal mOS despite it small amplitude. Because the clinically approved drug isradipine has equal affinity to both Cav1.2 and Cav1.3 Ca2+ channels (Sinnegger-Brauns et al. 2009), it should antagonize either of these putative mechanisms. As we have shown previously, isradipine is effective at lowering basal mOS in DMV neurons in wild-type as well as in the transgenic DJ-1 knockout mouse model of PD. Our present results suggest that the beneficial effect of isradipine in treating early stage PD, and particularly the symptoms of dysautonomia (Miller et al. 2009), arises from its equal affinity to Cav1.2 and Cav1.3 Ca2+ channels. Antagonizing these channels reduces Ca2+ influx during spiking via both channels, and prevents Cav1.3 Ca2+ channels from driving pacemaking in DMV motoneurons. Because nifedipine failed to slow down pacemaking in DMV motoneurons, it would seem that dihydropyridines that have a lower affinity to Cav1.3 channels, while still able to reduce spike-related Ca2+ influx, will not have the added benefit of preventing subthreshold Ca2+ influx from exacerbating mOS in neurons known to be vulnerable in PD (Goldberg et al. 2012; Guzman et al. 2010; Sanchez-Padilla et al. 2014).
GRANTS
This work was supported by grants from the Hartman Foundation, IDP Foundation, Picower Foundation, National Institute of Neurological Disorders and Stroke (P50-NS-047085 and T32-NS-041234), and the Department of Defense (W81XWH-11-1-051; to D. J. Surmeier); by a grant from the Israel Science Foundation (ISF; to H. Soreq); and by grants from the ISF (No. 154/14) and the Michael J. Fox Foundation (to J. A. Goldberg).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: G.C., E.L.-K., and J.A.G. performed experiments; G.C., E.L.-K., A.S., R.S., H.S., D.J.S., and J.A.G. approved final version of manuscript; A.S. and J.A.G. analyzed data; A.S., H.S., D.J.S., and J.A.G. interpreted results of experiments; R.S., H.S., D.J.S., and J.A.G. conception and design of research; D.J.S. and J.A.G. edited and revised manuscript; J.A.G. prepared figures; J.A.G. drafted manuscript.
REFERENCES
- Braak H, Ghebremedhin E, Rub U, Bratzke H, Del Tredici K. Stages in the development of Parkinson's disease-related pathology. Cell Tissue Res 318: 121–134, 2004. [DOI] [PubMed] [Google Scholar]
- Connor JA, Stevens CF. Prediction of repetitive firing behaviour from voltage clamp data on an isolated neurone soma. J Physiol 213: 31–53, 1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ermentrout B. Linearization of F-I curves by adaptation. Neural Comput 10: 1721–1729, 1998. [DOI] [PubMed] [Google Scholar]
- Ermentrout B. Type I membranes, phase resetting curves, and synchrony. Neural Comput 8: 979–1001, 1996. [DOI] [PubMed] [Google Scholar]
- Ermentrout GB, Terman DH. Mathematical Foundations of Neuroscience. New York: Springer-Verlag, 2010. [Google Scholar]
- Gai WP, Blumbergs PC, Geffen LB, Blessing WW. Age-related loss of dorsal vagal neurons in Parkinson's disease. Neurology 42: 2106–2111, 1992. [DOI] [PubMed] [Google Scholar]
- Goldberg JA, Guzman JN, Estep CM, Ilijic E, Kondapalli J, Sanchez-Padilla J, Surmeier DJ. Calcium entry induces mitochondrial oxidant stress in vagal neurons at risk in Parkinson's disease. Nat Neurosci 15: 1414–1421, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg JA, Teagarden MA, Foehring RC, Wilson CJ. Nonequilibrium calcium dynamics regulate the autonomous firing pattern of rat striatal cholinergic interneurons. J Neurosci 29: 8396–8407, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafsson B, Wigstrom H. Hyperpolarization following long-lasting tetanic activation of hippocampal pyramidal cells. Brain Res 275: 159–163, 1983. [DOI] [PubMed] [Google Scholar]
- Guzman JN, Sanchez-Padilla J, Chan CS, Surmeier DJ. Robust pacemaking in substantia nigra dopaminergic neurons. J Neurosci 29: 11011–11019, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman JN, Sanchez-Padilla J, Wokosin D, Kondapalli J, Ilijic E, Schumacker PT, Surmeier DJ. Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature 468: 696–700, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkin AL. The local electric changes associated with repetitive action in a nonmedullated axon. J Physiol 107: 165–181, 1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotson JR, Prince DA. A calcium-activated hyperpolarization follows repetitive firing in hippocampal neurons. J Neurophysiol 43: 409–419, 1980. [DOI] [PubMed] [Google Scholar]
- Lewy FH. Paralysis agitans. Pathologische anatomie I. In: Handbuch der Neurologie, edited by Lewandowski B. Berlin, Germany: Springer, 1912, p. 920–933. [Google Scholar]
- Lipscombe D, Helton TD, Xu W. L-type calcium channels: the low down. J Neurophysiol 92: 2633–2641, 2004. [DOI] [PubMed] [Google Scholar]
- Miller VM, Kenny RA, Oakley AE, Hall R, Kalaria RN, Allan LM. Dorsal motor nucleus of vagus protein aggregates in Lewy body disease with autonomic dysfunction. Brain Res 1286: 165–173, 2009. [DOI] [PubMed] [Google Scholar]
- Puopolo M, Raviola E, Bean BP. Roles of subthreshold calcium current and sodium current in spontaneous firing of mouse midbrain dopamine neurons. J Neurosci 27: 645–656, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sah P, McLachlan EM. Potassium currents contributing to action potential repolarization and the afterhyperpolarization in rat vagal motoneurons. J Neurophysiol 68: 1834–1841, 1992. [DOI] [PubMed] [Google Scholar]
- Sanchez-Padilla J, Guzman JN, Ilijic E, Kondapalli J, Galtieri DJ, Yang B, Schieber S, Oertel W, Wokosin D, Schumacker PT, Surmeier DJ. Mitochondrial oxidant stress in locus coeruleus is regulated by activity and nitric oxide synthase. Nat Neurosci 17: 832–840, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwindt PC, Spain WJ, Foehring RC, Stafstrom CE, Chubb MC, Crill WE. Multiple potassium conductances and their functions in neurons from cat sensorimotor cortex in vitro. J Neurophysiol 59: 424–449, 1988. [DOI] [PubMed] [Google Scholar]
- Sinnegger-Brauns MJ, Huber IG, Koschak A, Wild C, Obermair GJ, Einzinger U, Hoda JC, Sartori SB, Striessnig J. Expression and 1,4-dihydropyridine-binding properties of brain L-type calcium channel isoforms. Mol Pharmacol 75: 407–414, 2009. [DOI] [PubMed] [Google Scholar]
- Subramaniam M, Althof D, Gispert S, Schwenk J, Auburger G, Kulik A, Fakler B, Roeper J. Mutant alpha-synuclein enhances firing frequencies in dopamine substantia nigra neurons by oxidative impairment of A-type potassium channels. J Neurosci 34: 13586–13599, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]