

Abstract
The first outbreaks of bacterial canker of kiwifruit caused by Pseudomonas syringae pv. actinidiae biovar 3 were detected in France in 2010. P. syringae pv. actinidiae causes leaf spots, dieback, and canker that sometimes lead to the death of the vine. P. syringae pv. actinidifoliorum, which is pathogenic on kiwi as well, causes only leaf spots. In order to conduct an epidemiological study to track the spread of the epidemics of these two pathogens in France, we developed a multilocus variable-number tandem-repeat (VNTR) analysis (MLVA). MLVA was conducted on 340 strains of P. syringae pv. actinidiae biovar 3 isolated in Chile, China, France, Italy, and New Zealand and on 39 strains of P. syringae pv. actinidifoliorum isolated in Australia, France, and New Zealand. Eleven polymorphic VNTR loci were identified in the genomes of P. syringae pv. actinidiae biovar 3 ICMP 18744 and of P. syringae pv. actinidifoliorum ICMP 18807. MLVA enabled the structuring of P. syringae pv. actinidiae biovar 3 and P. syringae pv. actinidifoliorum strains in 55 and 16 haplotypes, respectively. MLVA and discriminant analysis of principal components revealed that strains isolated in Chile, China, and New Zealand are genetically distinct from P. syringae pv. actinidiae strains isolated in France and in Italy, which appear to be closely related at the genetic level. In contrast, no structuring was observed for P. syringae pv. actinidifoliorum. We developed an MLVA scheme to explore the diversity within P. syringae pv. actinidiae biovar 3 and to trace the dispersal routes of epidemic P. syringae pv. actinidiae biovar 3 in Europe. We suggest using this MLVA scheme to trace the dispersal routes of P. syringae pv. actinidiae at a global level.
INTRODUCTION
Agricultural systems are continuously afflicted by emerging infectious diseases (1), which can have significant agronomic and economic consequences. A thorough knowledge of the causal agent (propagation and contamination pathways, suitable environmental conditions, host range, and pathogenicity) is essential for determining and implementing efficient disease-management measures. Pathogen genotyping yields precious information for understanding the diversity and population structure of the bacterial organisms responsible for outbreaks. It enables hypotheses about the dispersion routes of bacterial populations or clonal lineages involved in epidemics. Multilocus variable-number tandem-repeat (VNTR) analysis (MLVA) (2) is a powerful and portable genotyping method. It has been demonstrated that MLVA has a higher sensitivity and resolution than any other genotyping methods, such as pulsed-field gel electrophoresis (PFGE) and multilocus sequence type (MLST), applied for an in-depth study of bacteria populations or epidemic outbreaks (3, 4). The aim of MLVA is to use PCR to target the tandem repeats with a motif of more than five nucleotides and to analyze the variability of their pattern in order to discriminate isolates. Generally, VNTR loci evolve according to the stepwise mutation model (SMM) by gain or loss of a single repeat. The evolution of a VNTR is mainly the consequence of DNA polymerase slippage but can also be due to recombination events between repetitions. Large gain or loss of repeats may occasionally occur within VNTR according to the single-step mutation model (SSM) indicating recombination events (3–5). Nowadays, the sequencing of bacterial genomes facilitates the identification of VNTR loci by means of dedicated algorithms and adequate tools such as Tandem Repeats Finder (6) or mreps (7). MLVA was used in an epidemiological survey to trace the routes of Haemophilus influenzae outbreaks (8) or Bacillus anthracis (2, 9) outbreaks. MLVA was applied to monomorphic plant-pathogenic bacteria belonging to different genera and species such as Xylella fastidiosa (10), Xanthomonas citri (11), Ralstonia solanacearum (12), “Candidatus Liberibacter asiaticus” (13), Erwinia amylovora (14), and Xanthomonas arboricola pathovars (15). MLVA was first applied on Pseudomonas syringae by Gironde and Manceau (16) and provided new insights into host specificity of P. syringae pathogenic on brassicaceous and solanaceous plants.
Pseudomonas syringae pv. actinidiae, the causal agent of bacterial canker of kiwifruit (Actinidia spp.), is considered to be a pandemic pathogen and has been isolated around the world over the last 30 years (17, 18). Vanneste et al. (18) suggested classifying these strains into three biovars, biovar 1, biovar 2, and biovar 3, according to phenotypic, pathogenic, and genomic features. Strains of P. syringae pv. actinidiae biovar 1 were isolated in Japan in 1984 and Italy in 1992 (19, 20) and strains of P. syringae pv. actinidiae biovar 2 in South Korea in 1994 (21). P. syringae pv. actinidiae biovar 3 was reported first in China (22) and more recently in Italy in 2008 (23, 24). It was then observed elsewhere in Europe (France and Portugal, 2010; Turkey, Switzerland, and Spain, 2011; Germany and Slovenia, 2013, and Greece, 2014 [18, 25–27]) and outside Europe, in New Zealand and Chile (18). Pseudomonas syringae pv. actinidifoliorum caused only necrotic symptoms on leaves (28); strains of this pathovar were previously described as P. syringae pv. actinidiae biovar 4 (18). P. syringae pv. actinidifoliorum was isolated in New Zealand (18), in Australia (29), and in France in 2011 (28). Two lineages of P. syringae pv. actinidifoliorum were first described in New Zealand strains (30). Recent studies have revealed the presence of this pathovar in France with higher polymorphism, and based on the analysis of four housekeeping genes two additional lineages were reported (28).
Although analyses based on the core genome showed that P. syringae pv. actinidiae biovar 3 strains responsible for the current worldwide outbreaks are monophyletic, genomic analyses based on the accessory genome revealed diversity within these strains (30–32). P. syringae pv. actinidiae biovar 3 strains are found to be monophyletic independently of their geographical origin, when analyzed by MLSA conducted on housekeeping genes (28, 33). Examining the composition of genomic islands in P. syringae pv. actinidiae, such as integrative and conjugative element (ICE)-carrying genes involved in pathogenicity, revealed that strains isolated in Europe are very similar to each other and that epidemics in Europe may have a different source population than epidemics in New Zealand or Chile (30–32).
The aims of this study were the following: (i) to set up a tool to characterize the genetic structure of pathovars causing diseases in kiwifruit; (ii) to gain further insight into the global diversity and population structure of P. syringae pv. actinidiae biovar 3, which is responsible for a worldwide epidemic; (iii) to identify the origin of the P. syringae pv. actinidiae outbreak in France. An MLVA scheme with 11 VNTRs was applied to a collection of 264 strains of P. syringae pv. actinidiae biovar 3 and 29 strains of P. syringae pv. actinidifoliorum isolated in France and to sets of strains of P. syringae pv. actinidiae biovar 3 and P. syringae pv. actinidifoliorum isolated in Australia, China, Italy, and New Zealand. Based on this scheme, P. syringae pv. actinidiae strains isolated in France and in Italy were found to be genetically closely related.
MATERIALS AND METHODS
Bacterial strain collection and DNA extraction.
Overall, 264 strains of P. syringae pv. actinidiae biovar 3 and 29 strains of P. syringae pv. actinidifoliorum, isolated from leaves, canes, flower buds, and roots of Actinidia deliciosa or Actinidia chinensis from different regions in France during the surveys conducted from 2010 to 2013 (28), were included in this study (Table 1). An additional 76 strains of P. syringae pv. actinidiae biovar 3, 1 strain each of P. syringae pv. actinidiae biovars 1 and 2, and 10 strains of P. syringae pv. actinidifoliorum strains isolated outside France were included in our collection (Table 1). Among these P. syringae pv. actinidiae biovar 3 strains, eight strains were initially isolated in China (AHPP1, GC31, HWD3, JF8, JZGMC1, SCHY9, SH8, and WT2) from leaf necrotic spots on four plant species (A. deliciosa, A. chinensis, Paulownia fortunei, Alternanthera philoxeroides) and from one insect (Philagra sp.), in five Chinese provinces (Anhui, Guizhou, Shanghai, Shaanxi, and Sichuan) (Table 1). Samples from plants other than kiwifruit were collected in the vicinity of symptomatic kiwifruit orchards, and the insect was collected on a diseased kiwifruit vine. Bacteria were maintained on KBc-ba agar plates (28) and stored at −80°C in 20% glycerol.
TABLE 1.
Strains of Pseudomonas syringae pv. actinidiae and pv. actinidifoliorum used in this studya
| P. syringae pathovar and strain | Biovar | Host | Yr of isolation | Country (province or region) of isolation | Reference or source | MLVA type |
|---|---|---|---|---|---|---|
| pv. actinidiae | ||||||
| CFBP 4909PT/ICMP 9617 | 1 | A. deliciosa | 1984 | Japan | Takikawa et al., 1989 (19) | |
| ICMP 19071 | 2 | A. chinensis | 1992 | South Korea | Koh et al., 1994 (21), Chapman et al., 2012 (33) | |
| HWD3 | 3 | A. deliciosa | 2012 | China (Shaanxi) | L. Zhu | 1 |
| JF8 | 3 | A. chinensis | 2012 | China (Anhui) | L. Zhu | 1 |
| AHPP1 | 3 | Philagra sp. | 2012 | China (Anhui) | L. Zhu | 2 |
| GC31 | 3 | A. chinensis | 2012 | China (Guizhou) | L. Zhu | 3 |
| JZGMC1 | 3 | Alternanthera philoxeroides | 2013 | China (Anhui) | L. Zhu | 4 |
| SCHY9 | 3 | A. deliciosa | 2012 | China (Sichuan) | L. Zhu | 5 |
| T5 | 3 | Actinidia sp. | 2010 | Italy | J. Vanneste | 6 |
| UOM1 | 3 | Actinidia sp. | 2010 | Italy | J. Vanneste | 7 |
| UOM2 | 3 | Actinidia sp. | 2010 | Italy | J. Vanneste | 7 |
| 1.1 | 3 | Actinidia sp. | 2010 | Italy | J. Vanneste | 8 |
| 10.6 | 3 | Actinidia sp. | 2010 | Italy | J. Vanneste | 8 |
| 17460,1/LSV 46.19 | 3 | A. deliciosa | 2012 | Italy | A. Calzolari | 8 |
| 17704,1/LSV 46.20 | 3 | A. chinensis | 2012 | Italy | A. Calzolari | 8 |
| 2.2 | 3 | Actinidia sp. | 2010 | Italy | J. Vanneste | 8 |
| 4.2 | 3 | Actinidia sp. | 2010 | Italy | J. Vanneste | 8 |
| 4.4 | 3 | Actinidia sp. | 2010 | Italy | J. Vanneste | 8 |
| 4.6 | 3 | Actinidia sp. | 2010 | Italy | J. Vanneste | 8 |
| E7 | 3 | Actinidia sp. | 2010 | Italy (Contarino) | J. Vanneste | 8 |
| H1.2 | 3 | Actinidia sp. | 2010 | Italy | J. Vanneste | 8 |
| H1.3 | 3 | Actinidia sp. | 2010 | Italy | J. Vanneste | 8 |
| H1.4 | 3 | Actinidia sp. | 2010 | Italy | J. Vanneste | 8 |
| 2.1 | 3 | Actinidia sp. | 2010 | Italy | J. Vanneste | 9 |
| CFBP 8100 | 3 | A. deliciosa | 2013 | France (Midi-Pyrénées) | Cunty et al., 2014 (28) | 10 |
| LSV 36.45 | 3 | A. deliciosa | 2010 | France (Rhône-Alpes) | This study | 10 |
| LSV 37.37 | 3 | A. deliciosa | 2011 | France (Midi-Pyrénées) | This study | 10 |
| LSV 37.64 | 3 | A. chinensis | 2011 | France (Aquitaine) | This study | 10 |
| LSV 38.08 | 3 | A. deliciosa | 2011 | France (PACA) | This study | 10 |
| LSV 38.13 | 3 | A. deliciosa | 2011 | France (Aquitaine) | This study | 10 |
| LSV 38.14 | 3 | A. deliciosa | 2011 | France (Aquitaine) | This study | 10 |
| LSV 38.79 | 3 | A. deliciosa | 2011 | France (PACA) | This study | 10 |
| LSV 38.80 | 3 | A. deliciosa | 2011 | France (PACA) | This study | 10 |
| LSV 39.36 | 3 | A. deliciosa | 2011 | France (Aquitaine) | This study | 10 |
| LSV 40.52 | 3 | A. deliciosa | 2012 | France (Aquitaine) | This study | 10 |
| LSV 40.53 | 3 | A. deliciosa | 2012 | France (Aquitaine) | This study | 10 |
| LSV 41.06 | 3 | A. deliciosa | 2012 | France (Aquitaine) | This study | 10 |
| LSV 41.18 | 3 | A. deliciosa | 2012 | France (Aquitaine) | This study | 10 |
| LSV 41.56 | 3 | A. deliciosa | 2012 | France (Aquitaine) | This study | 10 |
| LSV 42.61 | 3 | A. deliciosa | 2013 | France (Aquitaine) | This study | 10 |
| LSV 42.64 | 3 | A. deliciosa | 2013 | France (Midi-Pyrénées) | This study | 10 |
| LSV 42.72 | 3 | A. deliciosa | 2013 | France (Aquitaine) | This study | 10 |
| LSV 43.34 | 3 | A. deliciosa | 2013 | France (Midi-Pyrénées) | This study | 10 |
| LSV 43.56 | 3 | A. deliciosa | 2013 | France (Aquitaine) | This study | 10 |
| LSV 44.53 | 3 | A. deliciosa | 2013 | France (Rhône-Alpes) | This study | 10 |
| LSV 37.29 | 3 | A. chinensis | 2011 | France (Aquitaine) | This study | 11 |
| LSV 37.32 | 3 | A. chinensis | 2011 | France (Aquitaine) | This study | 11 |
| LSV 41.10 | 3 | A. deliciosa | 2012 | France (Aquitaine) | This study | 11 |
| LSV 41.12 | 3 | A. deliciosa | 2012 | France (Midi-Pyrénées) | This study | 11 |
| LSV 39.12 | 3 | A. deliciosa | 2011 | France (Midi-Pyrénées) | This study | 12 |
| LSV 38.04 | 3 | A. chinensis | 2011 | France (Aquitaine) | This study | 13 |
| LSV 43.67 | 3 | A. deliciosa | 2013 | France (Aquitaine) | This study | 13 |
| LSV 44.06 | 3 | A. deliciosa | 2013 | France (Aquitaine) | This study | 13 |
| LSV 36.46 | 3 | A. deliciosa | 2010 | France (Rhône-Alpes) | This study | 14 |
| LSV 36.47 | 3 | A. deliciosa | 2010 | France (Rhône-Alpes) | This study | 14 |
| LSV 40.61 | 3 | A. deliciosa | 2012 | France (Rhône-Alpes) | This study | 14 |
| LSV 42.23 | 3 | A. deliciosa | 2012 | France (Rhône-Alpes) | This study | 14 |
| CFBP 7910 | 3 | A. deliciosa | 2012 | France (Aquitaine) | Cunty et al., 2014 (28) | 15 |
| LSV 37.13 | 3 | A. deliciosa | 2011 | France (Midi-Pyrénées) | This study | 15 |
| LSV 41.32 | 3 | A. deliciosa | 2012 | France (Aquitaine) | This study | 15 |
| LSV 43.54 | 3 | A. deliciosa | 2013 | France (Aquitaine) | This study | 15 |
| LSV 43.66 | 3 | A. deliciosa | 2013 | France (Aquitaine) | This study | 15 |
| LSV 44.07 | 3 | A. deliciosa | 2013 | France (Aquitaine) | This study | 15 |
| LSV 44.56 | 3 | A. deliciosa | 2013 | France (Rhône-Alpes) | This study | 16 |
| LSV 37.21 | 3 | A. chinensis | 2011 | France (Aquitaine) | This study | 17 |
| E4 | 3 | Actinidia sp. | 2010 | Italy (Contarino) | J. Vanneste | 18 |
| E-AB | 3 | Actinidia sp. | 2010 | Italy (Contarino) | J. Vanneste | 18 |
| ICMP 19439 | 3 | A. deliciosa | 2010 | Chile | Butler et al., 2013 (30) | 19 |
| ICMP 19455 | 3 | A. deliciosa | 2010 | Chile | Butler et al., 2013 (30) | 19 |
| ICMP 19457 | 3 | A. deliciosa | 2010 | Chile | Butler et al., 2013 (30) | 19 |
| LSV 42.70 | 3 | A. deliciosa | 2013 | France (Aquitaine) | This study | 20 |
| LSV 43.25 | 3 | A. deliciosa | 2013 | France (Aquitaine) | This study | 20 |
| LSV 43.38 | 3 | A. deliciosa | 2013 | France (Aquitaine) | This study | 20 |
| CFBP 7287/LSV 40.47 | 3 | A. deliciosa | 2008 | Italy (Latina) | Balestra et al., 2009 (29), Vanneste et al., 2013 (18) | 21 |
| CFBP 8031 | 3 | A. deliciosa | 2011 | France (Rhône-Alpes) | Cunty et al., 2014 (28) | 21 |
| CFBP 8036 | 3 | A. deliciosa | 2011 | France (Rhône-Alpes) | Cunty et al., 2014 (28) | 21 |
| CFBP 8055 | 3 | A. deliciosa | 2011 | France (Aquitaine) | Cunty et al., 2014 (28) | 21 |
| CFBP 8102 | 3 | A. deliciosa | 2013 | France (Aquitaine) | Cunty et al., 2014 (28) | 21 |
| LSV 39.24 | 3 | A. deliciosa | 2011 | France (Aquitaine) | This study | 21 |
| LSV 41.28 | 3 | A. deliciosa | 2012 | France (Aquitaine) | This study | 21 |
| LSV 41.51 | 3 | A. deliciosa | 2012 | France (Aquitaine) | This study | 21 |
| LSV 42.22 | 3 | A. deliciosa | 2012 | France (Rhône-Alpes) | This study | 21 |
| CFBP 8059 | 3 | A. deliciosa | 2012 | France (Aquitaine) | Cunty et al., 2014 (28) | 22 |
| LSV 41.57 | 3 | A. chinensis | 2012 | France (Aquitaine) | This study | 22 |
| LSV 43.37 | 3 | A. chinensis | 2013 | France (Aquitaine) | This study | 22 |
| LSV 43.77 | 3 | A. deliciosa | 2013 | France (Aquitaine) | This study | 23 |
| LSV 43.36 | 3 | A. deliciosa | 2013 | France (Aquitaine) | This study | 24 |
| LSV 42.77 | 3 | A. deliciosa | 2013 | France (Aquitaine) | This study | 25 |
| LSV 40.81 | 3 | A. deliciosa | 2012 | France (Aquitaine) | This study | 26 |
| LSV 40.63 | 3 | A. deliciosa | 2012 | France (Rhône-Alpes) | This study | 27 |
| LSV 41.19 | 3 | A. deliciosa | 2012 | France (Aquitaine) | This study | 27 |
| LSV 44.61 | 3 | A. deliciosa | 2013 | France (Rhône-Alpes) | This study | 27 |
| CFBP 8097 | 3 | A. deliciosa | 2013 | France (Aquitaine) | Cunty et al., 2014 (28) | 28 |
| LSV 41.22 | 3 | A. deliciosa | 2012 | France (Aquitaine) | This study | 28 |
| WT2 | 3 | Paulownia fortunei | 2013 | China (Anhui) | L. Zhu | 29 |
| SH8 | 3 | A. chinensis | 2013 | China (Shanghai) | L. Zhu | 30 |
| LSV 43.55 | 3 | A. deliciosa | 2013 | France (Aquitaine) | This study | 31 |
| LSV 37.75 | 3 | A. deliciosa | 2011 | France (Aquitaine) | This study | 32 |
| LSV 39.76 | 3 | A. deliciosa | 2011 | France (Aquitaine) | This study | 33 |
| 3.2 | 3 | Actinidia sp. | 2010 | Italy | J. Vanneste | 34 |
| 1.2 | 3 | Actinidia sp. | 2010 | Italy | J. Vanneste | 35 |
| 3.8 | 3 | Actinidia sp. | 2010 | Italy | J. Vanneste | 36 |
| SP3 | 3 | Actinidia sp. | 2010 | Italy | J. Vanneste | 36 |
| 21726,1/LSV 46.22 | 3 | A. deliciosa | 2013 | Italy | A. Calzolari | 37 |
| 21736,1/LSV 46.23 | 3 | A. deliciosa | 2013 | Italy | A. Calzolari | 37 |
| 10638 | 3 | A. chinensis | 2010 | New Zealand | J. Vanneste | 38 |
| 10787 | 3 | A. deliciosa | 2010 | New Zealand | J. Vanneste | 38 |
| 11266 | 3 | A. deliciosa | 2010 | New Zealand | J. Vanneste | 38 |
| 11268 | 3 | A. deliciosa | 2010 | New Zealand | J. Vanneste | 38 |
| 11282 | 3 | A. deliciosa | 2010 | New Zealand | J. Vanneste | 38 |
| 11283 | 3 | Actinidia sp. | 2010 | New Zealand | J. Vanneste | 38 |
| 11287 | 3 | Actinidia sp. | 2010 | New Zealand | J. Vanneste | 38 |
| 11290 | 3 | Actinidia sp. | 2010 | New Zealand | J. Vanneste | 38 |
| 11293 | 3 | A. deliciosa | 2010 | New Zealand | J. Vanneste | 38 |
| 11298 | 3 | A. deliciosa | 2011 | New Zealand | J. Vanneste | 38 |
| 13093 | 3 | A. arguta | 2011 | New Zealand | J. Vanneste | 38 |
| CFBP 7811/10627 | 3 | A. chinensis | 2010 | New Zealand | Vanneste et al., 2013 (18) | 38 |
| 6.6 | 3 | Actinidia sp. | 2010 | Italy | J. Vanneste | 39 |
| T4 | 3 | Actinidia sp. | 2010 | Italy | J. Vanneste | 39 |
| T6 | 3 | Actinidia sp. | 2010 | Italy | J. Vanneste | 39 |
| 1.A | 3 | Actinidia sp. | 2010 | Italy | J. Vanneste | 40 |
| 1.B | 3 | Actinidia sp. | 2010 | Italy | J. Vanneste | 40 |
| 1.D | 3 | Actinidia sp. | 2010 | Italy | J. Vanneste | 40 |
| 1.E | 3 | Actinidia sp. | 2010 | Italy | J. Vanneste | 40 |
| 16803,1/LSV 46.18 | 3 | A. deliciosa | 2012 | Italy | A. Calzolari | 40 |
| 2.9 | 3 | Actinidia sp. | 2010 | Italy | J. Vanneste | 40 |
| 21375,1/LSV 46.21 | 3 | A. deliciosa | 2013 | Italy | A. Calzolari | 40 |
| 2E | 3 | Actinidia sp. | 2010 | Italy | J. Vanneste | 40 |
| 4.1 | 3 | Actinidia sp. | 2010 | Italy | J. Vanneste | 40 |
| CFBP 7285/LSV 39.66 | 3 | Actinidia sp. | 2008 | Italy (Treviso) | Balestra et al., 2009 (29), Vanneste et al., 2013 (18) | 40 |
| CORE | 3 | Actinidia sp. | 2010 | Italy | J. Vanneste | 40 |
| CRA-FRU 11.46 | 3 | A. chinensis | 2010 | Italy (Latina) | Scortichini | 40 |
| CRA-FRU 8.15 | 3 | Actinidia sp. | 2009 | Italy (Latina) | Scortichini | 40 |
| D3-b | 3 | Actinidia sp. | 2010 | Italy (Agrintesa) | J. Vanneste | 40 |
| D4 | 3 | Actinidia sp. | 2010 | Italy (Agrintesa) | J. Vanneste | 40 |
| H2.1 | 3 | Actinidia sp. | 2010 | Italy | J. Vanneste | 40 |
| H2.2 | 3 | Actinidia sp. | 2010 | Italy | J. Vanneste | 40 |
| H2.3 | 3 | Actinidia sp. | 2010 | Italy | J. Vanneste | 40 |
| L1 | 3 | Actinidia sp. | 2010 | Italy | J. Vanneste | 40 |
| L2 | 3 | Actinidia sp. | 2010 | Italy | J. Vanneste | 40 |
| L3 | 3 | Actinidia sp. | 2010 | Italy | J. Vanneste | 40 |
| Psa Ic | 3 | Actinidia sp. | 2010 | Italy | J. Vanneste | 40 |
| LSV 44.46 | 3 | A. deliciosa | 2013 | France (Rhône-Alpes) | This study | 41 |
| CFBP 7906 | 3 | A. deliciosa | 2011 | France (Rhône-Alpes) | Cunty et al., 2014 (28) | 42 |
| CFBP 8026 | 3 | A. deliciosa | 2010 | France (Rhône-Alpes) | Cunty et al., 2014 (28) | 42 |
| CFBP 8047 | 3 | A. deliciosa | 2010 | France (Rhône-Alpes) | Cunty et al., 2014 (28) | 42 |
| CFBP 8062 | 3 | Actinidia sp. | 2012 | France (PACA) | Cunty et al., 2014 (28) | 42 |
| CFBP 8092 | 3 | A. deliciosa | 2013 | France (Rhône-Alpes) | Cunty et al., 2014 (28) | 42 |
| LSV 36.67 | 3 | Actinidia sp. | 2010 | France (Rhône-Alpes) | This study | 42 |
| LSV 36.68 | 3 | Actinidia sp. | 2010 | France (Rhône-Alpes) | This study | 42 |
| LSV 37.24 | 3 | A. deliciosa | 2011 | France (Rhône-Alpes) | This study | 42 |
| LSV 43.30 | 3 | A. deliciosa | 2013 | France (Rhône-Alpes) | This study | 42 |
| D1 | 3 | Actinidia sp. | 2010 | Italy (Agrintesa) | J. Vanneste | 43 |
| CFBP 8060 | 3 | A. deliciosa | 2012 | France (Aquitaine) | Cunty et al., 2014 (28) | 44 |
| LSV 39.28 | 3 | A. deliciosa | 2011 | France (Aquitaine) | This study | 45 |
| LSV 40.67 | 3 | A. chinensis | 2012 | France (Aquitaine) | This study | 45 |
| LSV 41.34 | 3 | A. chinensis | 2012 | France (Aquitaine) | This study | 45 |
| LSV 41.36 | 3 | A. deliciosa | 2012 | France (Aquitaine) | This study | 45 |
| LSV 41.37 | 3 | A. deliciosa | 2012 | France (Aquitaine) | This study | 45 |
| LSV 41.50 | 3 | A. deliciosa | 2012 | France (Aquitaine) | This study | 45 |
| LSV 42.69 | 3 | A. deliciosa | 2013 | France (Aquitaine) | This study | 45 |
| LSV 43.53 | 3 | A. deliciosa | 2013 | France (Aquitaine) | This study | 45 |
| CFBP 8065 | 3 | A. chinensis | 2012 | France (Aquitaine) | Cunty et al., 2014 (28) | 46 |
| LSV 37.76 | 3 | A. deliciosa | 2011 | France (Aquitaine) | This study | 46 |
| LSV 38.19 | 3 | A. chinensis | 2011 | France (Aquitaine) | This study | 46 |
| LSV 39.03 | 3 | A. deliciosa | 2011 | France (Aquitaine) | This study | 46 |
| LSV 42.45 | 3 | A. deliciosa | 2013 | France (Midi-Pyrénées) | This study | 47 |
| LSV 42.58 | 3 | A. deliciosa | 2013 | France (Aquitaine) | This study | 48 |
| LSV 42.67 | 3 | A. deliciosa | 2013 | France (Aquitaine) | This study | 48 |
| LSV 42.81 | 3 | A. deliciosa | 2013 | France (Aquitaine) | This study | 48 |
| LSV 43.05 | 3 | A. deliciosa | 2013 | France (Aquitaine) | This study | 48 |
| LSV 43.15 | 3 | A. deliciosa | 2013 | France (Aquitaine) | This study | 48 |
| LSV 37.31 | 3 | A. chinensis | 2011 | France (Aquitaine) | This study | 49 |
| LSV 41.08 | 3 | A. deliciosa | 2012 | France (Aquitaine) | This study | 49 |
| LSV 41.09 | 3 | A. deliciosa | 2012 | France (Aquitaine) | This study | 49 |
| LSV 39.26 | 3 | A. chinensis | 2011 | France (Centre) | This study | 50 |
| LSV 41.35 | 3 | A. deliciosa | 2012 | France (Aquitaine) | This study | 50 |
| LSV 44.22 | 3 | A. deliciosa | 2013 | France (Aquitaine) | This study | 50 |
| CFBP 8056 | 3 | A. chinensis | 2012 | France (Aquitaine) | Cunty et al., 2014 (28) | 51 |
| CFBP 8063 | 3 | A. chinensis | 2012 | France (Aquitaine) | Cunty et al., 2014 (28) | 52 |
| CFBP 8089 | 3 | A. deliciosa | 2013 | France (Aquitaine) | Cunty et al., 2014 (28) | 52 |
| LSV 41.13 | 3 | A. deliciosa | 2012 | France (Aquitaine) | This study | 52 |
| LSV 36.69 | 3 | A. deliciosa | 2010 | France (Rhône-Alpes) | This study | 53 |
| LSV 42.59 | 3 | A. deliciosa | 2013 | France (Aquitaine) | This study | 53 |
| LSV 43.69 | 3 | A. deliciosa | 2013 | France (Aquitaine) | This study | 54 |
| CFBP 7286/LSV 40.46 | 3 | A. chinensis | 2008 | Italy (Latina) | Balestra et al., 2009 (24), Vanneste et al., 2013 (18) | 55 |
| CFBP 8025 | 3 | A. chinensis | 2010 | France (Aquitaine) | Cunty et al., 2014 (28) | 55 |
| CFBP 8027 | 3 | A. deliciosa | 2010 | France (Rhône-Alpes) | Cunty et al., 2014 (28) | 55 |
| CFBP 8028 | 3 | A. deliciosa | 2010 | France (Rhone-Alpes) | Cunty et al., 2014 (28) | 55 |
| CFBP 8029 | 3 | A. chinensis | 2010 | France (Aquitaine) | Cunty et al., 2014 (28) | 55 |
| CFBP 8030 | 3 | A. chinensis | 2010 | France (Aquitaine) | Cunty et al., 2014 (28) | 55 |
| CFBP 8032 | 3 | Actinidia sp. | 2011 | France (Aquitaine) | Cunty et al., 2014 (28) | 55 |
| CFBP 8033 | 3 | A. chinensis | 2011 | France (Aquitaine) | Cunty et al., 2014 (28) | 55 |
| CFBP 8034 | 3 | A. deliciosa | 2011 | France (Rhône-Alpes) | Cunty et al., 2014 (28) | 55 |
| CFBP 8035 | 3 | A. deliciosa | 2011 | France (Rhône-Alpes) | Cunty et al., 2014 (28) | 55 |
| CFBP 8037 | 3 | A. deliciosa | 2011 | France (Aquitaine) | Cunty et al., 2014 (28) | 55 |
| CFBP 8052 | 3 | A. deliciosa | 2011 | France (Midi-Pyrénées) | Cunty et al., 2014 (28) | 55 |
| CFBP 8053 | 3 | A. chinensis | 2011 | France (Midi Pyrénées) | Cunty et al., 2014 (28) | 55 |
| CFBP 8054 | 3 | A. deliciosa | 2011 | France (Midi-Pyrénées) | Cunty et al., 2014 (28) | 55 |
| CFBP 8057 | 3 | A. deliciosa | 2012 | France (Rhône-Alpes) | Cunty et al., 2014 (28) | 55 |
| CFBP 8058 | 3 | A. chinensis | 2012 | France (Rhône-Alpes) | Cunty et al., 2014 (28) | 55 |
| CFBP 8061 | 3 | A. deliciosa | 2012 | France (Rhône-Alpes) | Cunty et al., 2014 (28) | 55 |
| CFBP 8064 | 3 | A. deliciosa | 2012 | France (Poitou-Charentes) | Cunty et al., 2014 (28) | 55 |
| CFBP 8066 | 3 | A. deliciosa | 2012 | France (Midi Pyrénées) | Cunty et al., 2014 (28) | 55 |
| CFBP 8087 | 3 | A. deliciosa | 2013 | France (Aquitaine) | Cunty et al., 2014 (28) | 55 |
| CFBP 8088 | 3 | A. deliciosa | 2013 | France (Aquitaine) | Cunty et al., 2014 (28) | 55 |
| CFBP 8090 | 3 | A. deliciosa | 2013 | France (Aquitaine) | Cunty et al., 2014 (28) | 55 |
| CFBP 8091 | 3 | A. deliciosa | 2013 | France (Midi-Pyrénées) | Cunty et al., 2014 (28) | 55 |
| CFBP 8094 | 3 | A. deliciosa | 2013 | France (Aquitaine) | Cunty et al., 2014 (28) | 55 |
| CFBP 8095 | 3 | A. deliciosa | 2013 | France (Aquitaine) | Cunty et al., 2014 (28) | 55 |
| CFBP 8096 | 3 | A. deliciosa | 2013 | France (Midi-Pyrénées) | Cunty et al., 2014 (28) | 55 |
| CFBP 8098 | 3 | A. deliciosa | 2013 | France (Aquitaine) | Cunty et al., 2014 (28) | 55 |
| CFBP 8099 | 3 | A. deliciosa | 2013 | France (Aquitaine) | Cunty et al., 2014 (28) | 55 |
| CFBP 8101 | 3 | A. deliciosa | 2013 | France (Aquitaine) | Cunty et al., 2014 (28) | 55 |
| CFBP 8103 | 3 | A. deliciosa | 2013 | France (Aquitaine) | Cunty et al., 2014 (28) | 55 |
| CFBP 8108 | 3 | A. deliciosa | 2013 | France (Poitou-Charentes) | Cunty et al., 2014 (28) | 55 |
| CFBP 8109 | 3 | A. deliciosa | 2013 | France (Poitou-Charentes) | Cunty et al., 2014 (28) | 55 |
| CFBP 8110 | 3 | A. deliciosa | 2013 | France (Poitou-Charentes) | Cunty et al., 2014 (28) | 55 |
| LSV 37.14 | 3 | A. chinensis | 2011 | France (Aquitaine) | This study | 55 |
| LSV 37.17 | 3 | A. chinensis | 2011 | France (Aquitaine) | This study | 55 |
| LSV 37.18 | 3 | A. chinensis | 2011 | France (Aquitaine) | This study | 55 |
| LSV 37.19 | 3 | A. chinensis | 2011 | France (Aquitaine) | This study | 55 |
| LSV 37.25 | 3 | A. deliciosa | 2011 | France (Aquitaine) | This study | 55 |
| LSV 37.26 | 3 | A. deliciosa | 2011 | France (Aquitaine) | This study | 55 |
| LSV 37.33 | 3 | A. chinensis | 2011 | France (Midi-Pyrénées) | This study | 55 |
| LSV 37.34 | 3 | A. chinensis | 2011 | France (Aquitaine) | This study | 55 |
| LSV 37.36 | 3 | A. deliciosa | 2011 | France (Aquitaine) | This study | 55 |
| LSV 37.38 | 3 | A. deliciosa | 2011 | France (Rhône-Alpes) | This study | 55 |
| LSV 37.41 | 3 | A. deliciosa | 2011 | France (Midi-Pyrénées) | This study | 55 |
| LSV 37.42 | 3 | A. chinensis | 2011 | France (Midi-Pyrénées) | This study | 55 |
| LSV 37.43 | 3 | A. deliciosa | 2011 | France (Aquitaine) | This study | 55 |
| LSV 37.51 | 3 | A. chinensis | 2011 | France (Aquitaine) | This study | 55 |
| LSV 37.52 | 3 | A. deliciosa | 2011 | France (Aquitaine) | This study | 55 |
| LSV 37.55 | 3 | A. deliciosa | 2011 | France (Aquitaine) | This study | 55 |
| LSV 37.58 | 3 | A. deliciosa | 2011 | France (Rhône-Alpes) | This study | 55 |
| LSV 37.63 | 3 | A. chinensis | 2011 | France (Aquitaine) | This study | 55 |
| LSV 37.65 | 3 | A. deliciosa | 2011 | France (Aquitaine) | This study | 55 |
| LSV 37.66 | 3 | A. arguta | 2011 | France (Aquitaine) | This study | 55 |
| LSV 37.68 | 3 | A. deliciosa | 2011 | France (Aquitaine) | This study | 55 |
| LSV 37.69 | 3 | A. deliciosa | 2011 | France (Midi-Pyrénées) | This study | 55 |
| LSV 37.73 | 3 | A. deliciosa | 2011 | France (Aquitaine) | This study | 55 |
| LSV 37.77 | 3 | A. deliciosa | 2011 | France (Aquitaine) | This study | 55 |
| LSV 37.78 | 3 | A. deliciosa | 2011 | France (Aquitaine) | This study | 55 |
| LSV 37.79 | 3 | A. chinensis | 2011 | France (Aquitaine) | This study | 55 |
| LSV 37.80 | 3 | A. chinensis | 2011 | France (Aquitaine) | This study | 55 |
| LSV 38.01 | 3 | A. deliciosa | 2011 | France (Rhône-Alpes) | This study | 55 |
| LSV 38.02 | 3 | A. deliciosa | 2011 | France (Rhône-Alpes) | This study | 55 |
| LSV 38.03 | 3 | A. chinensis | 2011 | France (Aquitaine) | This study | 55 |
| LSV 38.05 | 3 | A. chinensis | 2011 | France (Aquitaine) | This study | 55 |
| LSV 38.06 | 3 | A. deliciosa | 2011 | France (Aquitaine) | This study | 55 |
| LSV 38.09 | 3 | A. deliciosa | 2011 | France (Aquitaine) | This study | 55 |
| LSV 38.11 | 3 | A. chinensis | 2011 | France (Aquitaine) | This study | 55 |
| LSV 38.12 | 3 | A. chinensis | 2011 | France (Aquitaine) | This study | 55 |
| LSV 38.15 | 3 | A. chinensis | 2011 | France (Aquitaine) | This study | 55 |
| LSV 38.16 | 3 | A. chinensis | 2011 | France (Rhône-Alpes) | This study | 55 |
| LSV 38.22 | 3 | A. deliciosa | 2011 | France (Aquitaine) | This study | 55 |
| LSV 39.02 | 3 | A. deliciosa | 2011 | France (Midi-Pyrénées) | This study | 55 |
| LSV 39.06 | 3 | A. chinensis | 2011 | France (Aquitaine) | This study | 55 |
| LSV 39.11 | 3 | A. deliciosa | 2011 | France (Aquitaine) | This study | 55 |
| LSV 39.13 | 3 | A. chinensis | 2011 | France (Midi-Pyrénées) | This study | 55 |
| LSV 39.14 | 3 | A. chinensis | 2011 | France (Aquitaine) | This study | 55 |
| LSV 39.23 | 3 | A. deliciosa | 2011 | France (Aquitaine) | This study | 55 |
| LSV 39.25 | 3 | A. deliciosa | 2011 | France (Aquitaine) | This study | 55 |
| LSV 39.29 | 3 | A. deliciosa | 2011 | France (Aquitaine) | This study | 55 |
| LSV 39.30 | 3 | A. chinensis | 2011 | France (Aquitaine) | This study | 55 |
| LSV 39.38 | 3 | A. chinensis | 2011 | France (Aquitaine) | This study | 55 |
| LSV 39.39 | 3 | A. deliciosa | 2011 | France (Aquitaine) | This study | 55 |
| LSV 39.40 | 3 | A. deliciosa | 2011 | France (Aquitaine) | This study | 55 |
| LSV 39.41 | 3 | A. deliciosa | 2011 | France (Aquitaine) | This study | 55 |
| LSV 39.44 | 3 | A. deliciosa | 2011 | France (Midi-Pyrénées) | This study | 55 |
| LSV 39.45 | 3 | A. deliciosa | 2011 | France (Midi-Pyrénées) | This study | 55 |
| LSV 40.10 | 3 | Actinidia sp. | 2011 | France (Aquitaine) | This study | 55 |
| LSV 40.11 | 3 | Actinidia sp. | 2011 | France (Aquitaine) | This study | 55 |
| LSV 40.13 | 3 | Actinidia sp. | 2011 | France (Midi-Pyrénées) | This study | 55 |
| LSV 40.20 | 3 | A. deliciosa | 2011 | France (Aquitaine) | This study | 55 |
| LSV 40.22 | 3 | A. deliciosa | 2011 | France (Rhône-Alpes) | This study | 55 |
| LSV 40.23 | 3 | A. deliciosa | 2011 | France (Rhône-Alpes) | This study | 55 |
| LSV 40.30 | 3 | A. chinensis | 2011 | France (Aquitaine) | This study | 55 |
| LSV 40.33 | 3 | A. deliciosa | 2011 | France (Rhône-Alpes) | This study | 55 |
| LSV 40.34 | 3 | A. deliciosa | 2011 | France (Rhône-Alpes) | This study | 55 |
| LSV 40.58 | 3 | A. deliciosa | 2012 | France (Rhône-Alpes) | This study | 55 |
| LSV 40.62 | 3 | A. deliciosa | 2012 | France (Rhône-Alpes) | This study | 55 |
| LSV 40.64 | 3 | A. deliciosa | 2012 | France (Rhône-Alpes) | This study | 55 |
| LSV 40.65 | 3 | A. deliciosa | 2012 | France (Rhône-Alpes) | This study | 55 |
| LSV 40.66 | 3 | A. deliciosa | 2012 | France (Rhône-Alpes) | This study | 55 |
| LSV 40.69 | 3 | A. deliciosa | 2012 | France (Rhône-Alpes) | This study | 55 |
| LSV 40.70 | 3 | A. deliciosa | 2012 | France (Rhône-Alpes) | This study | 55 |
| LSV 40.71 | 3 | A. deliciosa | 2012 | France (Rhône-Alpes) | This study | 55 |
| LSV 40.73 | 3 | A. deliciosa | 2012 | France (Aquitaine) | This study | 55 |
| LSV 41.15 | 3 | A. deliciosa | 2012 | France (Aquitaine) | This study | 55 |
| LSV 41.16 | 3 | A. deliciosa | 2012 | France (Aquitaine) | This study | 55 |
| LSV 41.17 | 3 | A. deliciosa | 2012 | France (Midi-Pyrénées) | This study | 55 |
| LSV 41.20 | 3 | A. deliciosa | 2012 | France (Aquitaine) | This study | 55 |
| LSV 41.23 | 3 | A. deliciosa | 2012 | France (Aquitaine) | This study | 55 |
| LSV 41.24 | 3 | A. deliciosa | 2012 | France (Aquitaine) | This study | 55 |
| LSV 41.25 | 3 | A. deliciosa | 2012 | France (Aquitaine) | This study | 55 |
| LSV 41.26 | 3 | A. chinensis | 2012 | France (Aquitaine) | This study | 55 |
| LSV 41.29 | 3 | A. deliciosa | 2012 | France (Aquitaine) | This study | 55 |
| LSV 41.38 | 3 | A. deliciosa | 2012 | France (Aquitaine) | This study | 55 |
| LSV 41.39 | 3 | A. deliciosa | 2012 | France (Aquitaine) | This study | 55 |
| LSV 41.41 | 3 | A. deliciosa | 2012 | France (Aquitaine) | This study | 55 |
| LSV 41.42 | 3 | A. deliciosa | 2012 | France (Aquitaine) | This study | 55 |
| LSV 41.49 | 3 | A. deliciosa | 2012 | France (Midi-Pyrénées) | This study | 55 |
| LSV 41.52 | 3 | A. deliciosa | 2012 | France (Aquitaine) | This study | 55 |
| LSV 41.55 | 3 | A. deliciosa | 2012 | France (Aquitaine) | This study | 55 |
| LSV 41.62 | 3 | A. deliciosa | 2012 | France (Aquitaine) | This study | 55 |
| LSV 41.67 | 3 | A. deliciosa | 2012 | France (Midi-Pyrénées) | This study | 55 |
| LSV 42.16 | 3 | A. deliciosa | 2012 | France (Aquitaine) | This study | 55 |
| LSV 42.20 | 3 | A. deliciosa | 2012 | France (Rhône-Alpes) | This study | 55 |
| LSV 42.44 | 3 | A. deliciosa | 2013 | France (Midi-Pyrénées) | This study | 55 |
| LSV 42.63 | 3 | A. deliciosa | 2013 | France (Midi-Pyrénées) | This study | 55 |
| LSV 42.68 | 3 | A. deliciosa | 2013 | France (Aquitaine) | This study | 55 |
| LSV 42.71 | 3 | A. deliciosa | 2013 | France (Aquitaine) | This study | 55 |
| LSV 42.76 | 3 | A. deliciosa | 2013 | France (Aquitaine) | This study | 55 |
| LSV 42.78 | 3 | A. deliciosa | 2013 | France (Aquitaine) | This study | 55 |
| LSV 42.79 | 3 | A. deliciosa | 2013 | France (Aquitaine) | This study | 55 |
| LSV 42.80 | 3 | A. deliciosa | 2013 | France (Aquitaine) | This study | 55 |
| LSV 43.07 | 3 | A. deliciosa | 2013 | France (Pays de la Loire) | This study | 55 |
| LSV 43.16 | 3 | A. deliciosa | 2013 | France (Aquitaine) | This study | 55 |
| LSV 43.17 | 3 | A. deliciosa | 2013 | France (Aquitaine) | This study | 55 |
| LSV 43.26 | 3 | A. deliciosa | 2013 | France (Aquitaine) | This study | 55 |
| LSV 43.27 | 3 | A. deliciosa | 2013 | France (Aquitaine) | This study | 55 |
| LSV 43.29 | 3 | A. deliciosa | 2013 | France (Rhône-Alpes) | This study | 55 |
| LSV 43.35 | 3 | A. deliciosa | 2013 | France (Midi-Pyrénées) | This study | 55 |
| LSV 43.57 | 3 | A. deliciosa | 2013 | France (Aquitaine) | This study | 55 |
| LSV 43.58 | 3 | A. deliciosa | 2013 | France (Aquitaine) | This study | 55 |
| LSV 43.59 | 3 | A. deliciosa | 2013 | France (Aquitaine) | This study | 55 |
| LSV 43.68 | 3 | A. deliciosa | 2013 | France (Aquitaine) | This study | 55 |
| LSV 43.75 | 3 | A. deliciosa | 2013 | France (Aquitaine) | This study | 55 |
| LSV 43.78 | 3 | A. deliciosa | 2013 | France (Aquitaine) | This study | 55 |
| LSV 44.08 | 3 | A. deliciosa | 2013 | France (Aquitaine) | This study | 55 |
| LSV 44.18 | 3 | A. deliciosa | 2013 | France (Aquitaine) | This study | 55 |
| LSV 44.23 | 3 | A. deliciosa | 2013 | France (Aquitaine) | This study | 55 |
| LSV 44.31 | 3 | A. deliciosa | 2013 | France (Rhône-Alpes) | This study | 55 |
| LSV 44.47 | 3 | A. deliciosa | 2013 | France (Rhône-Alpes) | This study | 55 |
| LSV 44.48 | 3 | A. deliciosa | 2013 | France (Rhône-Alpes) | This study | 55 |
| LSV 44.49 | 3 | A. deliciosa | 2013 | France (Rhône-Alpes) | This study | 55 |
| LSV 44.52 | 3 | A. deliciosa | 2013 | France (Rhône-Alpes) | This study | 55 |
| LSV 44.54 | 3 | A. deliciosa | 2013 | France (Rhône-Alpes) | This study | 55 |
| LSV 44.55 | 3 | A. deliciosa | 2013 | France (Rhône-Alpes) | This study | 55 |
| LSV 44.62 | 3 | A. deliciosa | 2013 | France (Rhône-Alpes) | This study | 55 |
| ICMP 18744/CRA-FRU 11.41 | 3 | A. deliciosa | 2010 | Italy (Rome) | Butler et al., 2013 (30) | 55 |
| pv. actinidifoliorum | ||||||
| CFBP 7907 | A. deliciosa | 2011 | France (Pays de la Loire) | Cunty et al., 2014 (28) | A | |
| CFBP 8048 | A. deliciosa | 2011 | France (Pays de la Loire) | Cunty et al., 2014 (28) | A | |
| CFBP 8051 | A. deliciosa | 2011 | France (Pays de la Loire) | Cunty et al., 2014 (28) | A | |
| CFBP 8067 | A. deliciosa | 2012 | France (Pays de la Loire) | Cunty et al., 2014 (28) | A | |
| CFBP 8085 | A. deliciosa | 2012 | France (Pays de la Loire) | Cunty et al., 2014 (28) | A | |
| CFBP 7951/ICMP 18807 | A. deliciosa | 2011 | New Zealand | Butler et al., 2013 (30), Cunty et al., 2014 (28) | B | |
| CFBP 8044/ICMP 19440 | A. chinensis | 2010 | Australia | Chapman et al., 2012 (33), Cunty et al., 2014 (28) | C | |
| CFBP 8045/ICMP 19486 | A. chinensis | 2010 | Australia | Chapman et al., 2012 (33), Cunty et al., 2014 (28) | C | |
| CFBP 8046/ICMP 19441 | A. chinensis | 2010 | Australia | Chapman et al., 2012 (33), Cunty et al., 2014 (28) | C | |
| CFBP 8043 | A. deliciosa | 2011 | France (Pays de la Loire) | Cunty et al., 2014 (28) | D | |
| LSV 43.74 | A. deliciosa | 2013 | France (Mayenne) | This study | D | |
| CFBP 8161 | A. deliciosa | 2013 | France (Centre) | Cunty et al., 2014 (28) | E | |
| CFBP 7909 | A. deliciosa | 2012 | France (Poitou-Charentes) | Cunty et al., 2014 (28) | F | |
| CFBP 8039 | A. deliciosa | 2011 | France (Aquitaine) | Cunty et al., 2014 (28) | F | |
| CFBP 8086 | A. deliciosa | 2012 | France (Pays de la Loire) | Cunty et al., 2014 (28) | F | |
| CFBP 8106 | A. deliciosa | 2013 | France (Pays de la Loire) | Cunty et al., 2014 (28) | F | |
| LSV 43.40 | A. deliciosa | 2013 | France (Aquitaine) | This study | F | |
| CFBP 8107 | A. deliciosa | 2013 | France (Aquitaine) | Cunty et al., 2014 (28) | G | |
| LSV 44.20 | A. deliciosa | 2013 | France (Pays de la Loire) | This study | H | |
| CFBP 7812/ICMP19098 | A. chinensis | 2010 | New Zealand | Vanneste et al., 2013 (18) | I | |
| CFBP 7903/ICMP18882 | A. chinensis | 2010 | New Zealand | Chapman et al., 2012 (33), Cunty et al., 2014 (28) | I | |
| CFBP 7904/ICMP 18883 | A. chinensis | 2010 | New Zealand | Chapman et al., 2012 (33), Cunty et al., 2014 (28) | I | |
| LSV 43.28 | A. chinensis | 2013 | France (Limousin) | This study | J | |
| LSV 43.43 | A. deliciosa | 2013 | France (Limousin) | This study | J | |
| LSV 43.44 | A. deliciosa | 2013 | France (Limousin) | This study | J | |
| LSV 43.65 | A. deliciosa | 2013 | France (Mayenne) | This study | K | |
| CFBP 7901/ICMP 18803 | A. chinensis | 2010 | New Zealand | Chapman et al., 2012 (33), Cunty et al., 2014 (28) | L | |
| CFBP 7902/ICMP 18804 | A. chinensis | 2010 | New Zealand | Chapman et al., 2012 (33), Cunty et al., 2014 (28) | L | |
| CFBP 7950/ICMP 18806 | A. deliciosa | 2011 | New Zealand | Butler et al., 2013 (30), Cunty et al., 2014 (28) | M | |
| CFBP 8160 | A. deliciosa | 2013 | France (Centre) | Cunty et al., 2014 (28) | N | |
| CFBP 8038 | A. deliciosa | 2011 | France (Poitou-Charentes) | Cunty et al., 2014 (28) | O | |
| CFBP 8041 | A. deliciosa | 2011 | France (Pays de la Loire) | Cunty et al., 2014 (28) | O | |
| CFBP 8042 | A. deliciosa | 2011 | France (Pays de la Loire) | Cunty et al., 2014 (28) | O | |
| CFBP 8105 | A. deliciosa | 2013 | France (Pays de la Loire) | Cunty et al., 2014 (28) | O | |
| CFBP 7908 | A. deliciosa | 2011 | France (Aquitaine) | Cunty et al., 2014 (28) | P | |
| CFBP 8040 | A. deliciosa | 2011 | France (Pays de la Loire) | Cunty et al., 2014 (28) | P | |
| CFBP 8049 | A. deliciosa | 2011 | France (Pays de la Loire) | Cunty et al., 2014 (28) | P | |
| CFBP 8050 | A. deliciosa | 2011 | France (Pays de la Loire) | Cunty et al., 2014 (28) | P | |
| CFBP 8104 | A. deliciosa | 2013 | France (Poitou-Charentes) | Cunty et al., 2014 (28) | P |
Haplotypes are indicated with a number for pv. actinidiae biovar 3 strains and with a letter for pv. actinidifoliorum strains. Abbreviations: superscript PT, pathotype strain; CFBP, Collection Française de Bactéries associées aux Plantes; CRA-FRU, Centro di Ricerca Agronomica per la Fruti; ICMP, International Collection of Microorganisms from Plants; LSV, Laboratoire de la Santé des Végétaux; PACA, Provence-Alpes-Côte-d'Azur.
Bacterial strains were grown on KBc-ba agar plates at 25°C for 24 h. Single colonies were suspended in sterile distilled water, and bacterial suspensions were adjusted to 1 × 106 CFU ml−1. Aliquots (1.5 ml bacterial suspension) were heated at 100°C for 15 min. The bacterial lysates were then centrifuged at 10,000 × g for 10 min to obtain a clear nucleic acid-containing supernatant. The supernatant samples were stored at −20°C until further analysis.
MLSA.
In order to study the phylogeny of the eight strains isolated in China, a multilocus sequence analysis (MLSA) was conducted on these eight strains and a set of eight P. syringae pv. actinidiae and five P. syringae pv. actinidifoliorum strains representative of the phylogenetic lineages previously described (28). Partial sequences of four housekeeping genes, gapA, gltA (also known as cts), gyrB, and rpoD, which code for glyceraldehyde-3-phosphate dehydrogenase, citrate synthase, DNA gyrase B, and sigma factor 70, respectively, of the eight strains of P. syringae pv. actinidiae isolated in China were amplified using primers designed by Sarkar and Guttman (34) and Hwang et al. (35). PCRs were carried out as previously indicated (28). The two strands of the PCR products were sequenced by Genoscreen (Lille, France). Sequence analyses were performed using Geneious 8.0.4 software (Biomatters, Auckland, New Zealand) and the BioEdit program (36).
The sequences were concatenated according to the alphabetic order of the gene. The concatenated data set was 3,159 bp long (gapA from bp 1 to 675, gltA from bp 676 to 1671, gyrB from bp 1672 to 2346, rpoD from bp 2347 to 3159). A neighbor-joining tree was built with the MEGA 5.1 program using the Jukes-Cantor distance methods with the DNA sequences for the four housekeeping genes. The P. syringae pv. tomato strain CFBP 2212 was included as an outgroup to root the tree, and bootstrap analyses were done with 1,000 replicates. The tree was visualized with the MEGA 5.1 program.
VNTR locus extraction, primer design, and PCR amplification.
In silico detection of VNTR loci was done by analyzing the genomic sequences of the P. syringae pv. actinidifoliorum strain ICMP 18807 and P. syringae pv. actinidiae strain ICMP 18744, available on NCBI (Bioprojects PRJNA199894 and PRJNA199875) with the Tandem Repeats Finder program (http://tandem.bu.edu), using the following parameters: region length of 30 to 1,000 bp, unit length of 5 to 12 bp, at least six tandem repeats (TR), and a similarity of at least 80% among the repeats. Primers were designed in the TR flanking region of each VNTR locus retained with Primer3 software (37) to generate amplicons of less than 450 bp. VNTRs were named according to the contig (numerical number) and the number of the strain (I or II corresponding to P. syringae pv. actinidifoliorum strain ICMP 18807 or P. syringae pv. actinidiae strain ICMP 18744, respectively) (Table 2) on which they were found.
TABLE 2.
Description of the 11 VNTR markers and PCR conditionsa
| Name | Tandem repeat sequence | Flanking region size (bp) | Forward primer | Reverse primer | Tm (°C) | PCR pool |
|---|---|---|---|---|---|---|
| TR10I | CCTGCA | 118 | F-AGTCTCTGCGCCTCAGGAT | GTCTGGAAAAATCCAGTGCC | 53 | 1 |
| TR14I | TTGATG | 105 | P-CTGGAAAACGTCCTGAGCAT | ACTCGGTTTGCCTGACTCAC | 55 | 1 |
| TR15I | GGCTGGTGCGTCT | 138 | F-TCGAGAGGAACACCAATGTG | TTTTGCAGACGATGTTTCCA | 53 | 2 |
| TR30I | AGCTACA | 98 | P-GCGTTACTTTGAGCGGAGTC | CACATATTCGGGTAGGTCGG | 53 | 2 |
| TR1II | AGGCCGAA | 230 | F-TGCCTGAGTACCTTTACCGG | CACCCAGCTCGACAATCAAG | 59 | 3 |
| TR2II | TAGTTGAGG | 231b | H-GTCATAACGGGTGAGAGTGC | ACGGCCCTTGAAAGTGACTA | 59 | 3 |
| TR3II | TGGAGGGCT | 127 | N-CGTGAGGCTCTGACTTTCTG | AAATCCGGGCTGTTTATCGC | 59 | 3 |
| TR39II | TCGAAAA | 145 | P-CGGTGGACTTGAAGAACACG | CACCCTGAACTGATTGCACC | 59 | 3 |
| TR11II | AATTGTATCTG | 136 | F-GATTGGTGACGTTGCGATGA | TTGTTGCCCTACACGCTCTA | 60 | 4 |
| TR19II | GCTTGTA | 164 | H-CCCAGAAAGAATGCGGACTG | AGCAGGAGATGGAAGAGCTG | 60 | 4 |
| TR64II | TTGAGCT | 103 | P-GTTGGCGGGTATGTGTCTG | CACCACGCTTCTTCTTGCAG | 60 | 4 |
Labeling dye abbreviations: F, 6-FAM; P, PET; H, HEX; N, NED. TRI, VNTR designed on P. syringae pv. actinidifoliorum strain ICMP 18807; TRII, VNTR designed on P. syringae pv. actinidiae biovar 3 strain ICMP 18844.
Flanking region size only for P. syringae pv. actinidiae biovar 3.
The potential VNTR loci were first amplified in simplex PCR carried out in a final volume of 20 μl containing 0.25 U of GoTaq Flexi DNA polymerase (Promega, Fitchburg, WI, USA), 1× colorless GoTaq Flexi buffer, 1.5 mM MgCl2, 62.5 μM each deoxynucleotide phosphate, 0.125 μM each primer (Table 2), and 1 μl of boiled extract. PCRs were performed on a Veriti 96-well thermal cycler (Applied Biosystems, Courtaboeuf, France) using a thermal cycling program of 5 min at 95°C followed by 32 cycles of 30 s at 95°C and 30 s at melting temperature (Tm) (Table 2) and ending at 72°C for 10 min. PCR products were separated by horizontal 1.5% agarose gel electrophoresis in Tris-borate-EDTA (TBE) buffer and staining with ethidium bromide (5 μg/ml). The DNA bands were visualized with Gel Doc XR+ Imager (Bio-Rad), and the amplicon size was estimated using the 100-bp DNA Molecular-Weight Marker XIV (100-bp ladder) (Roche Applied Science).
The VNTRs retained after agarose gel electrophoresis were labeled using labeled primers with a fluorescent dye at the 5′ end: F for 6-carboxyfluorescein (FAM), P for PET, H for HEX, and N for NED (Table 2). The PCRs were performed under the same conditions as those described above. Amplified products were diluted to one-eighth with sterile distilled water. Then, 2.5-μl aliquots were mixed with 9.45 μl of Hi-Di formamide (Sigma-Aldrich, Saint Quentin, France) and 0.15 μl of Genscan 500 Liz internal line size standard (Applied Biosystems). Capillary electrophoreses were performed using an ABI PRISM 3130 platform (ANAN platform of the SFR Quasav, Angers, France).
Multilocus VNTR analysis genotyping.
The output data from capillary electrophoresis were processed in order to assess repeats number of each VNTR using Geneious 8.0.4 software (Biomatters). The size of the flanking region for each VNTR was identified, and the size of the tandem repeat was converted into a repeat number (Table 3). The amplicons of each VNTR generated by strains ICMP 18807 and ICMP 18744 were sequenced in order to verify that the calculated number of tandem repeats indeed corresponded to actual sequence length.
TABLE 3.
Characteristics of the 11 VNTRs for P. syringae pv. actinidiae biovar 3 and and pv. actinidifoliorum strainsa
| VNTR locus |
P. syringae pv. actinidiae biovar 3 (n = 340) |
P. syringae pv. actinidifoliorum (n = 39) |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No. of haplotypes | Range of repeats | Simpson's index | Allelic richness |
No. of haplotypes | Range of repeats | Simpson's index | Allelic richness | ||||||
| Total (n = 340) | France (n = 264) | Italy (n = 53) | New Zealand (n = 12) | China (n = 8) | Chile (n = 3) | ||||||||
| TR10I | 11 | 7–21 | 0.47 | 7.43 | 2.28 | 2.41 | 1.00 | 3.37 | 1.00 | 10 | 6–16 | 0.90 | 10.0 |
| TR14I | 3 | 5–8 | 0.06 | 2.45 | 1.24 | 1.00 | 1.00 | 1.00 | 1.00 | 5 | 5–9 | 0.44 | 5.00 |
| TR15I | 3 | 2–4 | 0.02 | 1.82 | 1.04 | 1.21 | 1.00 | 1.00 | 1.00 | 1 | 2 | 0.00 | 1.00 |
| TR30I | 3 | 1–4 | 0.06 | 2.25 | 1.00 | 1.21 | 1.00 | 1.00 | 1.00 | 2 | 2–11 | 0.05 | 2.00 |
| TR1II | 3 | 2–4 | 0.13 | 2.76 | 1.44 | 1.00 | 1.00 | 1.88 | 1.00 | 1 | 1 | 0.00 | 1.00 |
| TR2II | 2 | 2–3 | 0.32 | 2.00 | 1.00 | 1.30 | 1.00 | 1.88 | 1.00 | NA | NA | NA | NA |
| TR3II | 3 | 3–5 | 0.01 | 1.43 | 1.00 | 1.11 | 1.00 | 1.62 | 1.00 | 1 | 1 | 0.00 | 1.00 |
| TR39II | 9 | 5–17 | 0.17 | 4.65 | 1.11 | 1.21 | 1.00 | 3.73 | 1.00 | 1 | 2 | 0.00 | 1.00 |
| TR11II | 3 | 2–4 | 0.15 | 2.77 | 1.38 | 1.46 | 1.00 | 1.97 | 1.00 | 1 | 1 | 0.00 | 1.00 |
| TR19II | 7 | 4–10 | 0.14 | 4.48 | 1.31 | 1.22 | 1.00 | 2.87 | 1.00 | 2 | 1–2 | 0.05 | 2.00 |
| TR64II | 5 | 1–7 | 0.04 | 2.17 | 1.02 | 1.00 | 1.00 | 3.20 | 1.00 | 1 | 1 | 0.00 | 1.00 |
n, number of strains; TRI, VNTR designed on P. syringae pv. actinidifoliorum strain ICMP 18807; TRII, VNTR designed on P. syringae pv. actinidiae biovar 3 ICMP 18744; NA, not analyzed.
Bioinformatic analysis.
The phylogenetic relation between the strains was inferred using a minimum spanning tree (MST) with BioNumerics (version 6.5; Applied Maths, St-Martens-Latem, Belgium). The MST was generated using the categorical coefficient and the maximum number of single-locus variants as a priority rule. Equal weight was assigned to each VNTR. Clonal complexes grouped single-locus variants (SLVs), e.g., haplotypes that differed from one another by only one locus. Simpson's index of diversity (38) ranging from 0 to 1 and allelic richness were calculated using BioNumerics (version 6.5; Applied Maths) and FSTAT 2.9.3 (http://www2.unil.ch/popgen/softwares/fstat.htm) (39), respectively, in order to assess the discriminatory power for each VNTR.
The genetic population structure of P. syringae pv. actinidiae biovar 3 and P. syringae pv. actinidifoliorum was analyzed using a discriminant analysis of principal components (DAPC), a clustering method without a priori, which did not make any assumption as to the population genetic models (40). The optimal number of clusters was determined by running k-means with increasing values of k and comparing the different clustering solutions using the Bayesian information criterion (BIC) (40). The value of k related to the lowest value of BIC is ideally the optimal number of clusters. This analysis was performed using the “adegenet” package in R (40).
Nucleotide sequence accession numbers.
The partial sequences of the PCR products were deposited in GenBank under accession numbers KP677392 to KP677423.
RESULTS
The strains of P. syringae pv. actinidiae biovar 3 isolated from various plants and an insect are monomorphic in multilocus sequence analysis.
The eight P. syringae pv. actinidiae strains isolated in China clustered within the lineage that groups exclusively P. syringae pv. actinidiae biovar 3 strains isolated during the latest outbreaks (in France, CFBP 8047, CFBP 8063, and CFBP 8064; in New Zealand, CFBP 7811; and in Italy, CFBP 7287) (Fig. 1). This clustering is supported by a strong bootstrap value (99%). These strains of P. syringae pv. actinidiae biovar 3 isolated in China presented the same biochemical features as the other P. syringae pv. actinidiae biovar 3 strains (data not shown), although these strains were isolated from five different organisms (four plant species, Actinidia deliciosa, A. chinensis, Paulownia fortunei, and Alternanthera philoxeroides, and one insect, Philagra sp.) and in five different regions (Anhui, Guizhou, Shaanxi, Shanghai, and Sichuan) (Table 1). They all grouped in a single lineage based on MLSA.
FIG 1.

Neighbor-joining tree constructed with the concatenated partial sequences of four housekeeping genes (gapA, gltA, gyrB, and rpoD, respectively) for 15 P. syringae pv. actinidiae and 6 P. syringae pv. actinidifoliorum strains. The percentage of bootstrap scores obtained for 1,000 replicates is indicated at each node. b, biovar; L, lineage. Symbols represent the geographical origin of the strain: white circle, Japan; black circle, China; gray circle, France; black square, New Zealand; gray square, Italy; white square, South Korea; white triangle, Australia.
Multilocus VNTR analysis on a worldwide collection of P. syringae pv. actinidiae and P. syringae pv. actinidifoliorum.
In silico analysis of the genomic sequences of the P. syringae pv. actinidifoliorum strain ICMP 18807 and P. syringae pv. actinidiae strain ICMP 18744 led to the finding of 64 potential VNTR loci. These 64 VNTRs were first tested on a set of eight strains representative of P. syringae pv. actinidiae biovar 1 (CFBP 4909), biovar 2 (ICMP 19071) and biovar 3 (CFBP 8050, CFBP 8051) and P. syringae pv. actinidifoliorum (CFBP 7951, CFBP 8043, CFBP 8050, CFBP 8051). Thirteen VNTRs did not generate an amplicon for any pathovar, 40 VNTRs were monomorphic for all pathovars, and only 11 VNTRs were polymorphic. The final set of polymorphic VNTRs retained for MLVA consisted of four VNTRs designed on the genome sequence of P. syringae pv. actinidifoliorum strain ICMP 18807 and seven VNTRs designed on the genome sequence of P. syringae pv. actinidiae ICMP 18744 (Table 2).
The flanking sequences of the 11 VNTRs were analyzed in silico from the genomic resources of P. syringae pv. actinidiae and P. syringae pv. actinidifoliorum strains available on public databases. The analysis revealed that the flanking regions were well conserved for all VNTRs, except for VNTR TR2II. Insertions were detected in the VNTR TR2II flanking regions for strains of P. syringae pv. actinidiae biovar 1, P. syringae pv. actinidiae biovar 2, and P. syringae pv. actinidifoliorum, but not for P. syringae pv. actinidiae biovar 3. Thus, VNTR TR2II was used to explore the diversity of P. syringae pv. actinidiae biovar 3 strains only.
The global minimum spanning tree (MST) (Fig. 2) revealed the ability of the set of the 10 VNTRs (all except VNTR TR2II) to discriminate pathovars actinidiae and actinidifoliorum and, within P. syringae pv. actinidiae, biovars 1, 2, and 3. Among the 381 P. syringae pv. actinidiae and P. syringae pv. actinidifoliorum strains analyzed, 64 haplotypes were revealed. The pathovar actinidifoliorum differed from P. syringae pv. actinidiae at six VNTR loci. Biovar 1 differed from biovar 2 at four VNTR loci and was distinguished from biovar 3 by seven VNTR loci.
FIG 2.
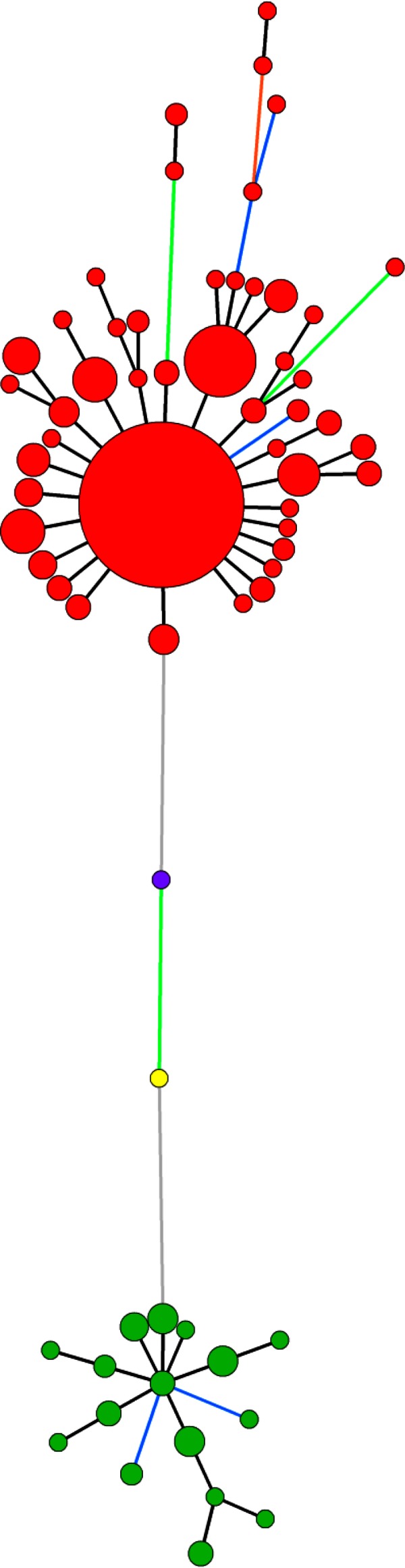
Minimum spanning tree (MST) based on the genotyping of 10 VNTRs for the entire strain collection (P. syringae pv. actinidiae biovar 1, yellow; P. syringae pv. actinidiae biovar 2, purple; P. syringae pv. actinidiae biovar 3, red; and P. syringae pv. actinidifoliorum, green). Each circle represents a haplotype, and the circle size is proportional to the number of strains sharing the same haplotype. The line color represents the number of the loci that are different between two haplotypes (black, 1; blue, 2; orange, 4; green, 5; green, 6; gray, 7).
Multilocus VNTR analysis on P. syringae pv. actinidiae biovar 3.
The genetic polymorphism revealed by the MLVA scheme was linked to the geographical origin of the strains. All the 11 VNTRs were polymorphic for the 340 P. syringae pv. actinidiae biovar 3 strains. The polymorphism of each VNTR varied from 2 to 11 haplotypes, and the higher polymorphic VNTRs were TR10I, TR39II, and TR19II, which generated 11, 9, and 7 haplotypes, respectively (Table 3). The values of Simpson's index varied from 0.01 to 0.47, and allelic richness values ranged from 1.43 to 7.43 (Table 3). The allelic richness differed according to the geographical origin of the strains. The strains isolated in China showed the highest allelic richness values for 8 of the 11 VNTRs. The VNTRs' allele frequencies were evaluated for each geographical origin of the P. syringae pv. actinidiae biovar 3 strains (Fig. 3A). Strains isolated in Italy and in France showed clearly different allele frequencies only at VNTR TR2II. Strains isolated in Europe differed from strains isolated in New Zealand and Chile at VNTRs TR10I, TR2II, and TR39II. The 12 strains isolated in New Zealand are genetically homogeneous. The three strains isolated in Chile were also genetically homogeneous and differed by only one locus (TR39II) from strains isolated in New Zealand. Strains isolated in Europe and in New Zealand differed from strains isolated in China at VNTRs TR10I, TR30I, TR19II, TR39II, and TR64II. All strains isolated in China had a single original haplotype at VNTR locus TR30I, the allele 1 corresponding to one repeat (Fig. 3A; see also Table S1 in the supplemental material).
FIG 3.

Allele frequencies in populations of different geographical origins of P. syringae pv. actinidiae biovar 3 (A) and P. syringae pv. actinidifoliorum (B). For each VNTR, one color corresponds to one haplotype.
A total of 55 haplotypes were distinguished by pooling the genotyping data of the 11 VNTRs (Table 1). The relationships identified between the haplotypes and the geographical origin of the strains are shown in the MST (Fig. 4A). The eight strains isolated in China produced seven unique different haplotypes. Specific haplotypes grouped strains from New Zealand and Chile. Among the 53 strains isolated in Italy, 3 strains (CFBP 7286, ICMP 18744, and CFBP 7287) shared two haplotypes with 164 strains isolated in France (Fig. 4A), and 12 different haplotypes were distinguished within the 50 other strains isolated in Italy. Thirty-four haplotypes were found within the 264 strains isolated in France.
FIG 4.

Minimum spanning tree (MST) based on the genotyping of 11 VNTRs of 340 P. syringae pv. actinidiae biovar 3 strains (A) and of 10 VNTRs of 39 P. syringae pv. actinidifoliorum strains (B). Colors refer to the geographical origin of the strains (green, France; red, Italy; purple, New Zealand; yellow, China; light pink, Chile; pink, Australia). The numbers refer to the haplotypes reported in Table 1. Each circle represents a haplotype, and the circle size is proportional to the number of strains sharing the same haplotype. The color of the line between circles represents the number of loci that differ between two haplotypes (black, 1 locus; blue, 2 loci; orange, 4 loci; green, 5 loci). The gray areas represent clonal complexes.
One haplotype was identified as the potential founder of the recent epidemics in Europe. P. syringae pv. actinidiae biovar 3 strains formed one clonal complex, two doubletons, and four singletons. The main clonal complex included all strains isolated in France, 51 strains isolated in Italy, and all strains isolated in New Zealand and in Chile. In the center of this clonal complex, the haplotype no. 55 (Table 1) grouped the majority of strains (156 strains) isolated in France, and the strains CFBP 7286 and ICMP 18744, isolated in Italy, exhibited the same haplotype (Fig. 4 A). It was the most frequent haplotype, which included 46.47% of P. syringae pv. actinidiae biovar 3 strains and was surrounded by the largest number of SLVs, suggesting that it is the founder of the epidemic in France. One singleton (haplotype no. 37) included two strains isolated in Italy (LSV 46.22 and LSV 46.23), which differed from haplotype no. 40 at two loci (TR15I and TR30I) by two and one repetitions (see Table S1 in the supplemental material), respectively. The two other doubletons and the three other singletons are separated from the main one by at least four loci. They included all strains isolated in China. This genetic distance precludes drawing conclusions about genealogic links between strains isolated in China and Europe from our collection.
The MST did not split isolates from different plant species (A. deliciosa, A. chinensis, Actinidia arguta, Actinidia sp., Paulownia fortunei, Alternanthera philoxeroides) into different haplotypes. Strains from China were genetically highly diverse whatever the organism and place of isolation. One clonal complex included strains SH8 and WT2, isolated from A. chinensis in Shanghai and from P. fortunei in Anhui, respectively. The other clonal complex included strains AHPP1, HWD3, and JF8, isolated from various organisms in Anhui, Shaanxi, and Anhui, respectively. Three singletons each included one strain isolated in Chinese provinces: Guizhou (GC31), Anhui (JZGMC1), and Sichuan (SCHY9). The strain isolated from Paulownia fortunei was genetically closely related (only one differential locus) to the strain SH8 isolated from A. chinensis. The strain JZGMC1 isolated from Alternanthera philoxeroides was genetically closely related (only two differential loci) to strains isolated in Italy from Actinidia sp. or in China (SCHY9) from A. deliciosa. The strain isolated from the insect (AHPP1) is genetically closely related (only one differential locus) to two strains (HWD3 and JF8) isolated from A. deliciosa and A. chinensis.
Discriminant analysis in principal components on P. syringae pv. actinidiae biovar 3 data.
The DAPC led to structure the 340 P. syringae pv. actinidiae biovar 3 strains into four clusters (Fig. 5), which were plotted in the ordination space with the horizontal and vertical axis explaining 60% and 21% of the total variability among clusters, respectively. The eigenvalues showed that the genetic structure was captured by the first two principal components (Fig. 5). The horizontal axis explained most of the total variability and distributed the four clusters into two groups. The first group consisted of clusters 2 and 3 including five strains isolated in China (AHPP1, HDW3, JF8, SH8, and WT2) (cluster 2) and three strains isolated in China (GC31, JZGMC1, and SCHY9) and all the strains isolated in Chile and New Zealand (cluster 3). Clearly distant on the horizontal axis, the second group gathered clusters 1 and 4, both of which mixed strains isolated in France and in Italy.
FIG 5.

Discriminant analysis of principal components of 340 P. syringae pv. actinidiae b3 strains. The scatterplot shows a projection of the four genetic clusters retained from BIC values onto axis 1 (horizontal axis) and axis 2 (vertical axis). The eigenvalues showed that the genetic structure was captured by the first two principal components. The dots represent the individuals, and the clusters are shown as inertia ellipses. Clusters 1 and 4 grouped strains isolated in France and in Italy; cluster 2 grouped some strains isolated in China (AHPP1, HWD3, JF8, SH8, WT2), and cluster 3 grouped some strains isolated in China (GC31, JZGMC1, SCHY9) and all strains isolated in New Zealand and Chile.
Multilocus VNTR analysis on P. syringae pv. actinidifoliorum.
Among the set of 10 VNTRs, only 4 (TR10I, TR14I, TR30I, and TR19II) generated polymorphism within P. syringae pv. actinidifoliorum strains (Table 3), while the other 6 VNTRs were monomorphic. The polymorphism of these five VNTRs varied from 2 to 10 haplotypes; the higher-polymorphism VNTRs were TR10I and TR14I, which generated 10 and 5 haplotypes, respectively. The Simpson's index and allelic richness values ranged from 0.05 to 0.90 and from 2.00 to 10.00, respectively. The distribution of the allele frequencies for the four VNTRs differed according to the geographical origin of the strains (Fig. 3B), especially for VNTR TR10I, which was specific to each of the three countries of origin of the strains.
No strains isolated in different geographic areas shared the same haplotype. A total of 16 haplotypes were revealed after pooling the genotyping data of the four VNTRs (Table 1). The relationships identified between the haplotypes and the geographical origin of strains were shown by the MST (Fig. 4B). All strains isolated in Australia had the same haplotype. The seven strains isolated in New Zealand were split among four haplotypes, and the 29 strains isolated in France were split among 11 haplotypes. All haplotypes differed by one or two loci of the five polymorphic loci. One clonal complex grouped the strains isolated in all geographical locations, and two singletons characterized the strains isolated in France. When running the DAPC (data not shown), no clear clustering was found, suggesting that within our data set of strains of P. syringae pv. actinidifoliorum there is no evident population structure.
DISCUSSION
The multilocus VNTR analysis scheme revealed high diversity within P. syringae pv. actinidiae strains.
MLVA is an inexpensive resolving tool, which is widely used to study genetic diversity and to deduce patterns of the spread of genetically monomorphic bacterial pathogens (41). Here, we report on an MLVA-based genotyping scheme targeting 11 carefully selected VNTRs for surveillance of the genetically monomorphic P. syringae pv. actinidiae biovar 3 responsible for the recent outbreaks of kiwifruit bacterial canker. The VNTR 2II has insertions, with variable length, in the flanking region for strains of P. syringae pv. actinidiae biovar 1 and biovar 2. According to this information, we propose to remove the VNTR 2II and to use a set of 10 VNTRs adapted to reveal the diversity within biovars 1 and 2 (data not shown) for the surveillance of these biovars.
Among the 11 VNTRs, 9 were considered to be microsatellites (the tandem-repeat motif is about six to nine nucleotides) and 2 to be minisatellites (the tandem-repeat motif is greater than nine nucleotides) (Tables 2 and 3) (3). Microsatellites are known to evolve faster than minisatellites and to be more polymorphic (42). Here the most polymorphic VNTRs are microsatellites, as expected. MLVA is a method of high resolution, which distinguishes strains of P. syringae pv. actinidiae biovar 3 from different origins. Various genomic analyses have concluded that P. syringae pv. actinidiae biovar 3 remains a highly monomorphic pathogen at the genomic level (30–32).
The multilocus VNTR analysis scheme composed of 11 VNTRs made it possible to gain further insight into the global diversity of the recent worldwide epidemic pathogen P. syringae pv. actinidiae biovar 3.
The developed MLVA scheme made it possible to distinguish 55 haplotypes within 340 strains of P. syringae pv. actinidiae biovar 3. The 340 strains grouped into one clonal complex, two doubletons, and four singletons (Fig. 4A) when the classical criterion of one allelic mismatch was used. All the strains isolated in Chile, France, and New Zealand and 51 of the strains in Italy grouped into one major clonal complex and one singleton consisting of two strains isolated in Italy. The Italian singleton (haplotype no. 37) is a double-locus variant of this major clonal complex as well as the closest singleton (haplotype no. 4) that groups strains isolated in China. Moreover, the major clonal complex grouped almost all the haplotypes of P. syringae pv. actinidiae biovar 3, with many overrepresented haplotypes, which is a strong epidemiological signature of a recent emergence of P. syringae pv. actinidiae biovar 3. The low genetic diversity revealed for P. syringae pv. actinidiae biovar 3 strains isolated from Chile, France, Italy, and New Zealand could be correlated with the status of the emerging epidemic pathogen for P. syringae pv. actinidiae biovar 3 as described in the case of Xanthomonas citri pv. citri in Viet Nam (43).
The strains of P. syringae pv. actinidiae biovar 3 presented in this study were sampled over 4 years since the beginning of the outbreak in France (Table 1). This time lapse was long enough to generate diversity within P. syringae pv. actinidiae biovar 3 but was not long enough to separate different genetic clusters or clonal complexes according to their geographical origins.
More diversity was revealed among the eight strains isolated in China than among the 3, 264, 53, and 12 strains isolated in Chile, France, Italy, and New Zealand, respectively. This high level of diversity observed within strains isolated in China indicates that they were sampled from a pool of P. syringae pv. actinidiae biovar 3 strains with a longer period of diversification than strains isolated in Europe, New Zealand, or Chile. A thorough analysis of the diversity of P. syringae pv. actinidiae biovar 3 strains in China would be useful to link the strains from China to those recently isolated in epidemics outside China. This could support the hypothesis that the common ancestor of P. syringae pv. actinidiae biovar 3 strains could originate from China, which is the diversification area for kiwifruit as well (30–32, 44).
The structure of the P. syringae pv. actinidiae biovar 3 obtained in the MST (Fig. 4A) was compared with the year and the host of isolation of the strains isolated in Europe, but no correlation was found (data not shown). No P. syringae pv. actinidiae biovar 3 structuring was identified in our collection of strains isolated from different organisms and different locations in China. Strains isolated from diverse organisms in diverse provinces in China share the same haplotype (i.e., haplotype no. 1), but strains isolated from one host (A. chinensis) or from one province (Anhui) are separated into several haplotypes (i.e., haplotypes no. 1, no. 3, and no. 30 and haplotypes no. 1, no. 2, no. 4, and no. 29, respectively).
We suggest that P. syringae pv. actinidiae biovar 3 is an intrapathovar subgroup highly virulent on Actinidia spp. This subgroup would be widely distributed in China with epiphytic capacity, which permits the dispersion of bacterial cells in the plant canopy with the assistance of insects present on the leaf surface. The strains isolated from Paulownia fortunei and Alternanthera philoxeroides were isolated from leaf necrotic spots. We do not have additional information about the pathogenicity of these strains on these plant species, and we do not consider these plants to be reservoirs or susceptible plants at that stage, but we would suggest that P. syringae pv. actinidiae biovar 3 could develop epiphytically on aerial parts of other plants than Actinidia spp.
The epidemics of P. syringae pv. actinidiae biovar 3 in France and in Italy share the same origin.
The recent outbreaks in Chile, Europe, and New Zealand could have three different origins (32). In the present study, we did not find any common haplotype for strains isolated in Chile, China, France, and New Zealand. In contrast, several strains isolated in Italy (CFBP 7286, ICMP 18744, and CFBP 7287) displayed haplotypes that are identical to those of strains isolated in France. One of these haplotypes corresponds to the most frequent haplotype and is shared by most of the strains from our collection isolated in Europe. As most SLVs are radially linked to this central haplotype, it may be proposed as the founder of this clonal complex as suggested by Feil et al. (45). Furthermore, haplotype no. 37 grouped two strains isolated in Italy, which are distant from the haplotype at two different loci (TR15I and TR30I) by two and one repetitions, respectively. This represents just an additional evolutionary step (i.e., a mutation at a locus irrespective of the mutation model retained) and the haplotype linking haplotypes no. 37 and no. 40 is probably missing in the sample. Haplotype no. 37 is related to the major clonal complex and belongs to the same “epidemic” population, which would diverge more in Italy than in France, where it had been introduced earlier.
DAPC confirms that the strains isolated in France and Italy are genetically linked. The DAPC clustering method grouped these strains in mixture into two clusters that are genetically closely related and distinguishable from the two other clusters, which include five strains isolated in China (AHPP1, HDW3, JF8, SH8, and WT2), three strains isolated in China (GC31, JZGMC1, and SCHY9), and all the strains isolated in New Zealand and Chile, respectively. This is in contrast to the results obtained with the MST, which showed that strains isolated in Chile and New Zealand were genetically closer to strains isolated in Europe than to strains isolated in China. The algorithm used to build MST assigned the same weight to each locus, whereas the algorithm implemented in DAPC gives more weight to loci with more alleles. The strains isolated in Chile and New Zealand shared two distinct alleles, which had been identified only in strains isolated in China. This sharing of a rare allele could explain why the strains isolated in Chile and New Zealand grouped preferentially with strains isolated in China in the analysis with DAPC. The DAPC analysis provides complementary information to the MST analysis in the determination of the origin of epidemic populations in New Zealand and Chile. The use of MST and DAPC on MLVA data validate the hypothesis that the epidemics observed in Europe, New Zealand, and Chile originated from China, independently. This conclusion is in accordance with those drawn from single nucleotide polymorphism (SNP) analysis (30–32).
The French epidemic may have originated in Italy through the importation of infected plants for planting material. P. syringae pv. actinidiae biovar 3 was detected in Europe first in Italy in 2008 (23, 24) and 2 years later in France, in 2010 (18). P. syringae pv. actinidiae has been registered by the European and Mediterranean Plant Protection Organization (EPPO) in the A2 list of pests recommended for regulation as quarantine pests (http://www.eppo.int/QUARANTINE/listA2.htm). The exchange of contaminated plant material between countries generally favored the long-distance spread of plant-pathogenic bacteria (1). In November 2012, the Commission of the European Union (EU) ordered surveys to assess the presence of P. syringae pv. actinidiae and set up measures to limit its propagation. Consequently, pollen and plants originating from third countries (no EU countries) must be accompanied by a P. syringae pv. actinidiae-free phytosanitary certificate to be imported in the EU. P. syringae pv. actinidiae, like all Pseudomonas syringae pathovars, has an epiphytic life on leaves (46, 47) and is also systemic through the xylem vessels (48). The main pathway of dissemination of P. syringae pv. actinidiae within and between orchards is the dispersal of bacterial exudates, oozing from cankers, favored by wind and rain (49). Agronomical techniques that induce wounds can favor the propagation of the disease. It was not confirmed that the pollen could be a pathway of P. syringae pv. actinidiae dispersion, even if P. syringae pv. actinidiae was already found on pollen (47). Furthermore, pollen used for artificial pollination in France is mainly locally produced. Spadaro et al. (50) suggested that P. syringae pv. actinidiae was probably introduced in Piemonte by infected propagation material. In France, imported kiwifruit plant material originated from Italy and New Zealand. The sharing of haplotypes between strains isolated in France and Italy and the precedence of the Italian epidemics support the hypothesis of the Italian origin of the epidemics in France.
The present multilocus VNTR analysis scheme is not adapted to P. syringae pv. actinidifoliorum.
We previously described four lineages within a P. syringae pv. actinidifoliorum strain collection isolated in Australia, France, and New Zealand, based on an MLSA (28). The MLVA scheme that we developed and have described here enables us to explore more thoroughly the diversity of P. syringae pv. actinidifoliorum, thus revealing 16 haplotypes with five polymorphic VNTRs. The MST built with the results of the genotyping revealed that the strains split according to their geographical origin.
Although strains isolated in Australia, France, and New Zealand are distinct, P. syringae pv. actinidifoliorum did not show any genetic structuring. The phylogeny of P. syringae pv. actinidifoliorum revealed by MLSA described four lineages within P. syringae pv. actinidifoliorum (28), which did not fit with the absence of structuring defined by MLVA. No evident population structure was revealed with the DAPC method. These observations could be due to size homoplasy and to low sampling (3 strains isolated in Australia, 29 in France, and 7 in New Zealand). Even if the MLVA scheme revealed more polymorphism than observed with data from MLSA, strains from different MLSA lineages do not share a MLVA haplotype. We concluded that development of an MLVA-based method for assessing the structuring of P. syringae pv. actinidifoliorum would need to increase the number of polymorphic VNTRs and to be conducted on MLSA lineages separately because of the long phylogenetic distances observed between lineages (28).
Development of a new tool for epidemiological monitoring of kiwifruit canker.
In conclusion, we have developed a reliable set of tools that combines MLSA and MLVA schemes that are useful for exploring diversity among P. syringae pv. actinidiae strains isolated in France. This MLVA scheme is a good candidate for tracing the dispersal routes of P. syringae pv. actinidiae in other places in the world where kiwifruit canker spreads. It would be interesting to test this MLVA scheme by assessing the genetic structuring of P. syringae pv. actinidiae biovar 1 and P. syringae pv. actinidiae biovar 2 as well.
Supplementary Material
ACKNOWLEDGMENTS
We thank Perrine Portier and Géraldine Taghouti at the International Centre for Microbial Resources and Plant-associated Bacteria (CIRM-CFBP), International Collection of Microorganisms from Plants (ICMP), J. L. Vanneste of Plant & Food Research Limited, Ruakura Research Center, New Zealand, M. Scortichini and S. Loreti of CRA and CRA-PAV, Rome, Italy, Gorgio Balestra of DAFNE, Viterbo, Italy, A. Calzolari, Servizio Fitosanitario, Bologna, Italy, and Liwu Zhu of the Key Laboratory of Pomology, Anhui Agricultural University, Hefei, China, for providing bacterial strains. We also thank Celine Rousseau, Martial Briand, and Gilles Hunault for their help with bioinformatical and statistical analysis. We are also grateful to the SFR Quasav and the ANAN platform, in Angers, France, for sequencing facilities.
A. Cunty is supported by a fellowship provided by Anses and the Region Pays de la Loire, France.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.01688-15.
REFERENCES
- 1.Anderson PK, Cunningham AA, Patel NG, Morales FJ, Epstein PR, Daszak P. 2004. Emerging infectious diseases of plants: pathogen pollution, climate change and agrotechnology drivers. Trends Ecol Evol 19:535–544. doi: 10.1016/j.tree.2004.07.021. [DOI] [PubMed] [Google Scholar]
- 2.Keim P, Klevytska A, Price L, Schupp J, Zinser G, Smith K, Hugh-Jones M, Okinaka R, Hill K, Jackson P. 1999. Molecular diversity in Bacillus anthracis. J Appl Microbiol 87:215–217. doi: 10.1046/j.1365-2672.1999.00873.x. [DOI] [PubMed] [Google Scholar]
- 3.Lindstedt BA. 2005. Multiple-locus variable number tandem repeats analysis for genetic fingerprinting of pathogenic bacteria. Electrophoresis 26:2567–2582. doi: 10.1002/elps.200500096. [DOI] [PubMed] [Google Scholar]
- 4.Elberse KE, Nunes S, Sá-Leão R, van der Heide HG, Schouls LM. 2011. Multiple-locus variable number tandem repeat analysis for Streptococcus pneumoniae: comparison with PFGE and MLST. PLoS One 6:e19668. doi: 10.1371/journal.pone.0019668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vogler AJ, Keys C, Nemoto Y, Colman RE, Jay Z, Keim P. 2006. Effect of repeat copy number on variable-number tandem repeat mutations in Escherichia coli O157: H7. J Bacteriol 188:4253–4263. doi: 10.1128/JB.00001-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Benson G. 1999. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 27:573. doi: 10.1093/nar/27.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kolpakov R, Bana G, Kucherov G. 2003. mreps: efficient and flexible detection of tandem repeats in DNA. Nucleic Acids Res 31:3672–3678. doi: 10.1093/nar/gkg617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Belkum A, Scherer S, Van Leeuwen W, Willemse D, Van Alphen L, Verbrugh H. 1997. Variable number of tandem repeats in clinical strains of Haemophilus influenzae. Infect Immun 65:5017–5027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Keim P, Price L, Klevytska A, Smith K, Schupp J, Okinaka R, Jackson P, Hugh-Jones M. 2000. Multiple-locus variable-number tandem repeat analysis reveals genetic relationships within Bacillus anthracis. J Bacteriol 182:2928–2936. doi: 10.1128/JB.182.10.2928-2936.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Coletta-Filho H, Takita MA, de Souza AA, Aguilar-Vildoso CI, Machado MA. 2001. Differentiation of strains of Xylella fastidiosa by a variable number of tandem repeat analysis. Appl Environ Microbiol 67:4091–4095. doi: 10.1128/AEM.67.9.4091-4095.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bui TN, Verniere C, Jarne P, Brisse S, Guerin F, Boutry S, Gagnevin L, Pruvost O. 2009. From local surveys to global surveillance: three high-throughput genotyping methods for epidemiological monitoring of Xanthomonas citri pv. citri pathotypes. Appl Environ Microbiol 75:1173–1184. doi: 10.1128/AEM.02245-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.N′Guessan CA, Brisse S, Le Roux-Nio A-C, Poussier S, Koné D, Wicker E. 2013. Development of variable number of tandem repeats typing schemes for Ralstonia solanacearum, the agent of bacterial wilt, banana Moko disease and potato brown rot. J Microbiol Methods 92:366–374. doi: 10.1016/j.mimet.2013.01.012. [DOI] [PubMed] [Google Scholar]
- 13.Matos LA, Hilf ME, Chen J, Folimonova SY. 2013. Validation of ‘variable number of tandem repeat'-based approach for examination of ‘Candidatus Liberibacter asiaticus' diversity and its applications for the analysis of the pathogen populations in the areas of recent introduction. PLoS One 8:e78994. doi: 10.1371/journal.pone.0078994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bühlmann A, Dreo T, Rezzonico F, Pothier JF, Smits TH, Ravnikar M, Frey JE, Duffy B. 2014. Phylogeography and population structure of the biologically invasive phytopathogen Erwinia amylovora inferred using minisatellites. Environ Microbiol 16:2112–2125. doi: 10.1111/1462-2920.12289. [DOI] [PubMed] [Google Scholar]
- 15.Cesbron S, Pothier J, Gironde S, Jacques M-A, Manceau C. 2014. Development of multilocus variable-number tandem repeat analysis (MLVA) for Xanthomonas arboricola pathovars. J Microbiol Methods 100:84–90. doi: 10.1016/j.mimet.2014.02.017. [DOI] [PubMed] [Google Scholar]
- 16.Gironde S, Manceau C. 2012. Housekeeping gene sequencing and multilocus variable-number tandem-repeat analysis to identify subpopulations within Pseudomonas syringae pv. maculicola and Pseudomonas syringae pv. tomato that correlate with host specificity. Appl Environ Microbiol 78:3266–3279. doi: 10.1128/AEM.06655-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scortichini M, Marcelletti S, Ferrante P, Petriccione M, Firrao G. 2012. Pseudomonas syringae pv. actinidiae: a re-emerging, multi-faceted, pandemic pathogen. Mol Plant Pathol 13:631–640. doi: 10.1111/j.1364-3703.2012.00788.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vanneste JL, Yu J, Cornish DA, Tanner DJ, Windner R, Chapman JR, Taylor RK, Mackay JF, Dowlut S. 2013. Identification, virulence, and distribution of two biovars of Pseudomonas syringae pv. actinidiae in New Zealand. Plant Dis 97:708–719. doi: 10.1094/PDIS-07-12-0700-RE. [DOI] [PubMed] [Google Scholar]
- 19.Takikawa Y, Serizawa S, Ichikawa T, Tsuyumu S, Goto M. 1989. Pseudomonas syringae pv. actinidiae pv nov: the causal bacterium of canker of kiwifruit in Japan. Ann Phytopathol Soc Jpn 55:437–444. doi: 10.3186/jjphytopath.55.437. [DOI] [Google Scholar]
- 20.Scortichini M. 1994. Occurrence of Pseudomonas syringae pv. actinidiae on kiwifruit in Italy. Plant Pathol 43:1035–1038. doi: 10.1111/j.1365-3059.1994.tb01654.x. [DOI] [Google Scholar]
- 21.Koh Y, Cha BJ, Chung HJ, Lee DH. 1994. Outbreak and spread of bacterial canker of kiwifruit. Korean J Plant Pathol 10:68–72. [Google Scholar]
- 22.Wang Z, Tang X, Liu S. 1992. Identification of the pathogenic bacterium for bacterial canker on Actinidia in Sichuan. J Southwest Agric University http://en.cnki.com.cn/Article_en/CJFDTOTAL-XNND199206007.htm. [Google Scholar]
- 23.Ferrante P, Scortichini M. 2009. Identification of Pseudomonas syringae pv. actinidiae as causal agent of bacterial canker of yellow kiwifruit (Actinidia chinensis Planchon) in central Italy. J Phytopathol 157:768–770. doi: 10.1111/j.1439-0434.2009.01550.x. [DOI] [Google Scholar]
- 24.Balestra GM, Mazzaglia A, Quattrucci A, Renzi M, Rossetti A. 2009. Current status of bacterial canker spread on kiwifruit in Italy. Australasian Plant Dis Notes 4:34–36. [Google Scholar]
- 25.EPPO. 2013. First report of Pseudomonas syringae pv. actinidiae in Germany. EPPO Rep Serv 9:2013/185. [Google Scholar]
- 26.Dreo T, Pirc M, Ravnikar M, Zezlina I, Poliakoff F, Rivoal C, Nice F, Cunty A. 2014. First report of Pseudomonas syringae pv. actinidiae, the causal agent of bacterial canker of kiwifruit in Slovenia. Plant Dis 98:1578–1578. [DOI] [PubMed] [Google Scholar]
- 27.Holeva MC, Glynos PE, Karafla CD. 2015. First report of bacterial canker of kiwifruit caused by Pseudomonas syringae pv. actinidiae in Greece. Plant Dis 99(5):150113075012005. doi: 10.1094/PDIS-07-14-0738-PDN. [DOI] [Google Scholar]
- 28.Cunty A, Poliakoff F, Rivoal C, Cesbron S, Saux FL, Lemaire C, Jacques M, Manceau C, Vanneste J. 2014. Characterization of Pseudomonas syringae pv. actinidiae (P. syringae pv. actinidiae) isolated from France and assignment of P. syringae pv. actinidiae biovar 4 to a de novo pathovar: Pseudomonas syringae pv. actinidifoliorum pv. nov. Plant Pathol 64:582–596. doi: 10.1111/ppa.12297. [DOI] [Google Scholar]
- 29.EPPO. 2011. First report of Pseudomonas syringae pv. actinidiae in Australia, p 2130. EPPO Rep Serv 6:2011/130. [Google Scholar]
- 30.Butler MI, Stockwell PA, Black MA, Day RC, Lamont IL, Poulter RT. 2013. Pseudomonas syringae pv. from recent outbreaks of kiwifruit bacterial canker belong to different clones that originated in China. PLoS One 8(2):e57464. doi: 10.1371/journal.pone.0057464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mazzaglia A, Studholme DJ, Taratufolo MC, Cai R, Almeida NF, Goodman T, Guttman DS, Vinatzer BA, Balestra GM. 2012. Pseudomonas syringae pv. actinidiae (PSA) isolates from recent bacterial canker of kiwifruit outbreaks belong to the same genetic lineage. PLoS One 7:e36518. doi: 10.1371/journal.pone.0036518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McCann HC, Rikkerink EH, Bertels F, Fiers M, Lu A, Rees-George J, Andersen MT, Gleave AP, Haubold B, Wohlers MW. 2013. Genomic analysis of the kiwifruit pathogen Pseudomonas syringae pv. actinidiae provides insight into the origins of an emergent plant disease. PLoS Pathog 9:e1003503. doi: 10.1371/journal.ppat.1003503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chapman JR, Taylor RK, Weir BS, Romberg MK, Vanneste JL, Luck J, Alexander BJ. 2012. Phylogenetic relationships among global populations of Pseudomonas syringae pv. actinidiae. Phytopathology 102:1034–1044. doi: 10.1094/PHYTO-03-12-0064-R. [DOI] [PubMed] [Google Scholar]
- 34.Sarkar SF, Guttman DS. 2004. Evolution of the core genome of Pseudomonas syringae, a highly clonal, endemic plant pathogen. Appl Environ Microbiol 70:1999–2012. doi: 10.1128/AEM.70.4.1999-2012.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hwang MS, Morgan RL, Sarkar SF, Wang PW, Guttman DS. 2005. Phylogenetic characterization of virulence and resistance phenotypes of Pseudomonas syringae. Appl Environ Microbiol 71:5182–5191. doi: 10.1128/AEM.71.9.5182-5191.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hall TA. 1999. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser 41:95–98. [Google Scholar]
- 37.Rozen S, Skaletsky H. 2000. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol 132:365–386. [DOI] [PubMed] [Google Scholar]
- 38.Hunter PR, Gaston MA. 1988. Numerical index of the discriminatory ability of typing systems: an application of Simpson's index of diversity. J Clin Microbiol 26:2465–2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goudet J. 1995. FSTAT (version 1.2): a computer program to calculate F-statistics. J Hered 86:485–486. [Google Scholar]
- 40.Jombart T, Devillard S, Balloux F. 2010. Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genet 11:94. doi: 10.1186/1471-2156-11-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Achtman M. 2008. Evolution, population structure, and phylogeography of genetically monomorphic bacterial pathogens. Annu Rev Microbiol 62:53–70. doi: 10.1146/annurev.micro.62.081307.162832. [DOI] [PubMed] [Google Scholar]
- 42.Zhou K, Aertsen A, Michiels CW. 2014. The role of variable DNA tandem repeats in bacterial adaptation. FEMS Microbiol Rev 38:119–141. doi: 10.1111/1574-6976.12036. [DOI] [PubMed] [Google Scholar]
- 43.Vernière C, Bui Thi Ngoc L, Jarne P, Ravigné V, Guérin F, Gagnevin L, Le Mai N, Chau NM, Pruvost O. 2014. Highly polymorphic markers reveal the establishment of an invasive lineage of the citrus bacterial pathogen Xanthomonas citri pv. citri in its area of origin. Environ Microbiol 16:2226–2237. doi: 10.1111/1462-2920.12369. [DOI] [PubMed] [Google Scholar]
- 44.Li X, Li J, Soejarto DD. 2009. Advances in the study of the systematics of Actinidia Lindley. Front Biol China 4:55–61. doi: 10.1007/s11515-008-0110-2. [DOI] [Google Scholar]
- 45.Feil EJ, Li BC, Aanensen DM, Hanage WP, Spratt BG. 2004. eBURST: inferring patterns of evolutionary descent among clusters of related bacterial genotypes from multilocus sequence typing data. J Bacteriol 186:1518–1530. doi: 10.1128/JB.186.5.1518-1530.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stefani E, Giovanardi D. 2012. Dissemination of Pseudomonas syringae pv. actinidiae through pollen and its epiphytic life on leaves and fruits. Phytopathologia Mediterranea 50:489–496. [Google Scholar]
- 47.Vanneste J, Yu J, Cornish D, Max S, Clark G. 2011. Presence of Pseudomonas syringae pv. actinidiae, the causal agent of bacterial canker of kiwifruit, on symptomatic and asymptomatic tissues of kiwifruit. New Zealand Plant Protection 64:241–245. [Google Scholar]
- 48.Spinelli F, Donati I, Vanneste JL, Costa M, Costa G. 2011. Real time monitoring of the interactions between Pseudomonas syringae pv. actinidiae and Actinidia species. Acta Hortic 913:461–465. doi: 10.17660/ActaHortic.2011.913.61. [DOI] [Google Scholar]
- 49.Serizawa S, Ichikawa T, Takikawa Y, Tsuyumu S, Goto M. 1989. Occurrence of bacterial canker of kiwifruit in Japan: description of symptoms, isolation of the pathogen and screening of bactericides. Ann Phytopathol Soc Jpn 55:427–436. doi: 10.3186/jjphytopath.55.427. [DOI] [Google Scholar]
- 50.Spadaro D, Amatulli MT, Garibaldi A, Gullino ML, Vittone G, Nari L, Pellegrino S, Morone C, Mason G, Ortalda E, Grosso S. 2010. È arrivato in Piemonte il cancro batterico del kiwi. L'Informatore Agrar 66(27):58–59. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.