

Abstract
Pseudomonas aeruginosa is one of only a few Pseudomonas species that are able to use acyclic monoterpenoids, such as citronellol and citronellate, as carbon and energy sources. This is achieved by the acyclic terpene utilization pathway (Atu), which includes at least six enzymes (AtuA, AtuB, AtuCF, AtuD, AtuE, AtuG) and is coupled to a functional leucine-isovalerate utilization (Liu) pathway. Here, quantitative proteome analysis was performed to elucidate the terpene metabolism of P. aeruginosa. The proteomics survey identified 187 proteins, including AtuA to AtuG and LiuA to LiuE, which were increased in abundance in the presence of citronellate. In particular, two hydratases, AtuE and the PA4330 gene product, out of more than a dozen predicted in the P. aeruginosa proteome showed an increased abundance in the presence of citronellate. AtuE (isohexenyl-glutaconyl coenzyme A [CoA] hydratase; EC 4.2.1.57) most likely catalyzes the hydration of the unsaturated distal double bond in the isohexenyl-glutaconyl-CoA thioester to yield 3-hydroxy-3-isohexenyl-glutaryl-CoA. Determination of the crystal structure of AtuE at a 2.13-Å resolution revealed a fold similar to that found in the hydratase (crotonase) superfamily and provided insights into the nature of the active site. The AtuE active-site architecture showed a significantly broader cavity than other crotonase superfamily members, in agreement with the need to accommodate the branched isoprenoid unit of terpenes. Glu139 was identified to be a potential catalytic residue, while the backbone NH groups of Gly116 and Gly68 likely form an oxyanion hole. The present work deepens the understanding of terpene metabolism in Pseudomonas and may serve as a basis to develop new strategies for the biotechnological production of terpenoids.
INTRODUCTION
Terpenes are compounds responsible for the pleasant aroma of ginger, cloves, turmeric, cinnamon, and many other plants. They are also characterized by (many) important biological properties, including antimicrobial, antineoplastic, antifungal, antiviral, antihyperglycemic, anti-inflammatory, and antiparasitic activities. Terpenes consist of isoprene units combined in a cyclic or acyclic form and are produced in large quantities (∼105 tons/year) (1). Introduction of additional carbon-carbon double bonds in terpenes or incorporation of heteroatoms, such as oxygen, results in the formation of terpenoids. Terpenoids are used in large quantities in perfumes, as aroma compounds for food additives, or, in some cases, even as natural insect repellents (e.g., citronellol) (2). They can also be found as precursors and building blocks for the synthesis of complex chiral compounds in the chemical and pharmaceutical industries. However, only a few terpenoids are available in large quantities at reasonable costs. Consequently, the characterization of biocatalysts important for terpenoid metabolism and the development of processes for the biotransformation of abundant terpenoids to commercially interesting derivatives have attracted considerable attention in recent years. At the same time, knowledge of the catabolic pathways of terpenoids in biological organisms is still very poor.
Only a few Pseudomonas species, such as Pseudomonas citronellolis, P. aeruginosa, P. mendocina, and P. delhiensis, as well as a few strains of P. fluorescens, are able to use acyclic terpenes as a single source of carbon and energy (3). In particular, the acyclic terpene utilization (Atu) pathway in P. aeruginosa broadens the catabolic capabilities of the species to the utilization of acyclic monoterpenes (1, 4). The Atu pathway has been found to involve seven proteins (AtuA, AtuB, AtuCF, AtuD, AtuE, AtuG, AtuH) (5, 6). Biochemical studies have not yet verified the function of all Atu proteins. Exceptions are AtuC and AtuF, which constitute the two subunits of geranyl coenzyme A (CoA) carboxylase (3, 4, 7, 8). Recently, purified AtuD was found to possess citronellyl-CoA dehydrogenase activity and to be important for a functional Atu pathway (9). It has been suggested that the end products of the Atu pathway—after β oxidation—enter the leucine-isovalerate utilization (Liu) pathway and are finally converted to the central metabolite acetyl-CoA (10).
The Atu pathway includes a single hydratase step, namely, the hydration of isohexenyl-glutaconyl-CoA to 3-hydroxy-3-isohexenyl-glutaryl-CoA (Fig. 1), presumably catalyzed by the AtuE gene product. Knowledge of the hydratases acting on substrates with a branched carbon skeleton is currently poor. The P. aeruginosa genome (11) has more than two dozen putative hydratases, most of which are predicted to act on CoA-activated acyl compounds. Annotation of P. aeruginosa hydratases suggests that many of these are involved in, e.g., fatty acid degradation and that the different hydratases either have different substrate specificities or are simply redundant enzymes. The presence of a putative hydratase in the atu gene cluster suggests that AtuE could be a hydratase with specificity for isoprenoid (branched) substrates. Unfortunately, the substrate of the isohexenyl-glutaconyl-CoA hydratase reaction is not commercially available, and all attempts to synthesize isohexenyl-glutaconyl-CoA for use in enzyme assays failed, despite the use of four different approaches. We intend to provide insights into the putative function of AtuE by the combination of a proteomics approach and determination of the structure of the AtuE protein.
FIG 1.

Catalytic reaction of AtuE.
MATERIALS AND METHODS
Bacterial strains, plasmids, and culture conditions.
The bacterial strains and plasmids used in this study are shown in Table 1. Escherichia coli strain JM109 and E. coli strain S17-1 were used for cloning and as the donor strain in conjugation experiments, respectively. E. coli strains were grown on LB medium supplemented with the appropriate antibiotics at 37°C. P. aeruginosa was routinely grown on nutrient broth (NB; 0.8%, wt/vol) medium at 30°C or on mineral salts medium (12) with sodium octanoate (0.15%, wt/vol) or with the carbon sources indicated below.
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Relevant characteristic(s) | Reference or source |
|---|---|---|
| Strains | ||
| E. coli JM109 | Plasmid storage, cloning strain | |
| E. coli S17-1 | Conjugation strain | 37 |
| P. aeruginosa PAO1 | Growth on citronellol, citronellate, and related acyclic terpenoids | ATCC 15692 |
| Plasmids | ||
| pJoe4036.1 | Broad-host-range plasmid, rhaP pT7 lacPOZ′ rrnB bla, used for expression of proteins from rhamnose-dependent promoter | J. Altenbuchner (personal communication) |
| pJoe4036.1::atuE | atuE-hexahistidine under the control of an l-rhamnose promoter | 38 |
Culture conditions for proteome analysis.
P. aeruginosa PAO1 was grown in a seed culture (100 ml) on mineral salts medium with sodium octanoate (0.15%, wt/vol) for 16 h at 30°C. For the main cultures, 200 ml mineral salts medium in a 1-liter Erlenmeyer flask was inoculated with the seed culture at an optical density of about 10 to 20 Klett units. Sodium octanoate (0.15%, wt/vol) as a control or 0.05% (wt/vol) octanoate plus 0.10% (wt/vol) sodium citronellate was used as the carbon source. Three biological replicates were used for each condition. The cells were incubated on a rotary shaker at 30°C. P. aeruginosa grew to 150 Klett units if only 0.05% octanoate was present as the sole carbon source. With additional carbon sources (citronellate or octanoate, each at 0.1%), the cultures grew to more than 250 Klett units. Cells were harvested by centrifugation when they had reached an optical density of ≈200 Klett units. Cells were stored at −80°C (or −20°C) until further use.
Sample preparation for proteome analysis.
Frozen cells were resuspended in 2 ml of 25 mM Tris-HCl, pH 7.5, containing 40% (vol/vol) glycerol per 1 g of cell pellet. Three hundred microliters DNase (1 mg/ml) and 300 μl RNase (1 mg/ml) were added before the cells were lysed by two subsequent passages through a French press. The cell extract was centrifuged twice at 35,000 rpm for 1 h at 4°C. The supernatant obtained after the second centrifugation step, from here on designated the soluble cell extract, was used for proteome analysis. The protein concentration of the soluble cell extract was determined by the Bradford method (13).
Protein preparation for LC-MS-based quantifications and in-gel digestion.
Soluble cell extracts were analyzed by label-free gel fractionation liquid chromatography (GeLC)–tandem mass spectrometry (MS/MS) approach as described recently (14). Briefly, 200 μg of each cell extract was applied to an SDS-10% polyacrylamide gel. The gel was stained with colloidal Coomassie G250 (Roth, Germany). Each sample lane was cut into 10 identical slices (fractions), resulting in 60 gel fractions in total. The gel slices were digested using trypsin (Roche, Penzberg, Germany) (15). After digestion, the gel slices were extracted with acetonitrile (ACN) for 10 min and the supernatant was vacuum dried. Samples were reconstituted in 15 μl 0.1% (vol/vol) formic acid (FA), and 5 μl was subjected to liquid chromatography (LC)-MS/MS analysis.
LC-MS/MS analysis.
Nano-LC-electrospray ionization-MS/MS experiments were performed on an Acquity nanoscale ultraperformance liquid chromatography system (Waters, USA) coupled to an LTQ-Orbitrap XL hybrid mass spectrometer (Thermo Fisher Scientific, Germany). Tryptic peptides were trapped and desalted on a precolumn (2 cm by 180 μm; particle size, 5 μm; Symmetry C18; Waters, USA) for 1 min at 15 μl/min using 0.1% FA. After trapping, the peptides were transferred to an analytical column (25 cm by 75 μm; particle size, 1.7 μm; BEH 130 C18; Waters, USA). The peptides were separated by the use of linear gradients ranging from 1% to 50% ACN in 0.1% FA within 60 min at a flow rate of 250 nl/min. The LTQ-Orbitrap mass spectrometer was operated under the control of XCalibur software (version 2.0.7). Survey spectra (m/z = 300 to 1,800) were detected in the LTQ-Orbitrap mass spectrometer at a resolution of 60,000 at m/z 400. Data-dependent tandem mass spectra for the seven most abundant peptide precursors in the linear ion trap were generated using a target value of 5,000 and a normalized collision energy of 35. For all measurements obtained using the LTQ-Orbitrap analyzer, internal calibration was performed using lock-mass ions from ambient air (16).
Data analysis.
For database searches, a specific P. aeruginosa PAO1 database was generated from the NCBInr database using Scaffold software (version 3.4.9; Proteome Software Inc., Portland, OR). In order to identify proteins that were not annotated in the P. aeruginosa-specific database, the bacterium subset of the NCBInr database (ftp://ftp.ncbi.nih.gov/blast/db/FASTA/nr.gz) was used as a second database. Both databases were searched simultaneously on an in-house Mascot server (version 2.3.02; Matrix Science, United Kingdom). Trypsin (allowing cleavage before proline) was specified as the cleavage enzyme, as it allows three missed cleavages in the Mascot database search algorithm. The carbamidomethylation of cysteine was set as fixed, and the oxidation of methionine was set as a variable modification. The mass tolerance was set to 5 ppm for the precursor and to 0.6 Da for fragment ions. To validate the Mascot search results, X! Tandem (version Cyclone [2010.12.01.1]; The Global Proteome Machine [thegpm.org]) was used as a second search engine, using the same parameters described above for the Mascot search.
Mascot and X! Tandem search results were imported into Scaffold software. Peptide identifications with a peptide probability of greater than 80.0%, as specified by the Peptide Prophet algorithm (17), were accepted. Proteins had to be identified by at least two peptides and a protein probability of at least 99% to be accepted. Protein probabilities were assigned by the Protein Prophet algorithm (18). Annotation of identified proteins was performed using the Pseudomonas genome database (19).
Label-free quantification.
Label-free quantification based on ion intensities was performed using the fractionation work flow of Progenesis LC-MS software (version 3.1; Nonlinear Dynamics, United Kingdom), as described by Simon et al. (14). Briefly, the 60 LC-MS runs of the GeLC-MS/MS experiment were aligned using the software's automatic alignment algorithm, and only peptides (features) with a charge state of 2+, 3+, or 4+ and at least two isotope signals were selected for further analysis. After normalization, biological replicates corresponding to the experimental design (e.g., octanoate versus octanoate plus citronellate) were grouped, and for each feature, a one-way analysis of variance (ANOVA) was performed and the fold change in expression was calculated. Protein identifications were validated by Scaffold and reimported into the Progenesis LC-MS software. Only proteins which were identified to have a minimum of 2 peptides and a protein probability of 99% were considered for quantification. Proteins which exhibited a fold change in expression of ≥3 and a P value of ≤0.05 were considered differentially abundant.
Construction of AtuE-His6.
The atuE gene was PCR amplified from the chromosomal DNA of P. aeruginosa using primers atuE-up and atuE-down (atuE-up, 5′-GGAATTCCATATGAGCCTGCCGCATTGGCAGAC-3′; atuE-down, 5′-CGGGATCCCTGGGCCCAGACCGGCTTG-3′; restriction sites for NdeI and BamHI in primers atuE-up and atuE-down, respectively, are underlined). The NdeI- and BamHI-digested PCR product was ligated into pJoe4036.1 that had been treated with the same restriction enzymes. The resulting product (pJoe4036.1::atuE, 5,417 bp) coded for a C-terminal hexahistidine-tagged AtuE protein under the control of an l-rhamnose-dependent promoter. The correct DNA sequence was verified by DNA sequencing. The atuE expression plasmid was transformed into E. coli JM109.
Expression and purification of AtuE.
E. coli JM109 cells harboring pJoe4036.1::atuE were grown in 2 × 400 ml of LB medium (30°C). l-Rhamnose (0.2%) was added when the optical density at 600 nm (OD600) had reached 0.3 to 0.5, and growth was allowed for another 5 to 6 h before the cells were harvested (4°C, 8,000 rpm, 30 min). The cell pellet was stored frozen at −20°C until use. Frozen cells were thawed on ice and resuspended in 1 ml of 0.1 M HEPES buffer, pH 7.5, per g (wet weight) of cells. A soluble cell extract was obtained by disruption with a French press (2 times) and subsequent ultracentrifugation (35,000 rpm; TFT rotor 65.13) for 60 min (4°C). The supernatant was applied to an Ni-Sepharose 6 Fast Flow column (bed volume, 5 ml; 1.6 by 2.5 cm; Macherey-Nagel, Düren, Germany) that had been equilibrated with equilibration buffer (20 mM sodium phosphate buffer containing 0.5 M NaCl and 20 mM imidazole, pH 7.4). After washing with equilibration buffer, AtuE was eluted by an imidazole gradient (0 to 500 mM imidazole in equilibration buffer) at ≈100 mM imidazole. AtuE-containing fractions were purified from salts and imidazole by a passage step over a Sephadex G25 size exclusion chromatography column (a HiPrep 26/10 column equilibrated with 50 mM Tris-HCl, pH 8.5). The yield of purified AtuE amounted to 5.7 mg per gram (wet weight) of E. coli cells. Purified AtuE (apparent molecular mass, ≈28 kDa) was homogeneous, as revealed by SDS-PAGE (see Data Set S1 and Fig. S1A in the supplemental material), and was stored at −20°C.
Crystallization.
For crystallization, AtuE was initially concentrated to ∼10 mg/ml with Amicon Ultra centrifugal filters (10,000-molecular-weight cutoff; Millipore) in 10 mM HEPES–NaOH, pH 7.0, buffer. Crystals were grown by the hanging-drop vapor diffusion method at 16°C using a well solution containing 0.2 M sodium iodide, 20% (vol/vol) polyethylene glycol 4000. The drop consisted of 2 μl of well solution mixed with an equal volume of protein solution. The crystals grew to their maximum size in a few days and formed clusters (see Data Set S1 and Fig. S1B in the supplemental material).
Data collection and processing.
X-ray diffraction data were collected on the X13 beamline at EMBL Hamburg, Hamburg, Germany (DESY), at cryogenic temperatures (100 K) in the presence of 20% (vol/vol) glycerol as a cryoprotectant. The data were processed with the XDS program package (20).
Structure determination and refinement.
Initial phases were obtained with molecular replacement using Phaser crystallographic software (21). Two search models were constructed (one with the structure with PDB accession number 3H81 [sequence identity, 37.0%] and one with the structure with PDB accession number 3I47 [sequence identity, 34.1%]). A solution with a Z score of 14.5 with an Rfree value (with 5% of the reflections excluded) of about 42% was obtained. Refinement was carried out with simulated annealing in a Phenix system (22) alternated with manual rebuilding in the Coot toolkit, which resulted in a drop of Rfree to ∼36%. At this stage, automatic rebuilding was employed using the Autobuild framework implemented in Phenix, resulting in a further drop of Rfree to ∼32%. Extensive manual rebuilding of the C terminus in each molecule and optimization of loops were carried out, resulting in a further drop of Rfree and improvement of the geometry. Manual rebuilding of the refined structures and visualization of the electron density maps were done with Coot (23). At the final stages of refinement, translation/liberation/screw (TLS) refinement was employed. The stereochemistry of the structure was validated with the MOLPROBITY program (24) and various tools in Coot. Structural superpositions were carried out with the PDBeFold tool (25). The data processing and final refinement statistics are shown in Table 2.
TABLE 2.
Data collection and refinement statistics for AtuE
| Parametera | Value(s) for AtuEb |
|---|---|
| Data processing statistics | |
| Beamline | EMBL Hamburg (DESY) X13 |
| Wavelength (Å) | 0.8123 |
| Space group | C2 |
| Unit cell dimensions | |
| a, b, c (Å) | 108.4, 134.0, 83.3 |
| β (°) | 139.5 |
| No. of molecules | 3 |
| Resolution range (Å) | 20.0–2.13 (2.25–2.13) |
| No. of reflections | 92,282 (13,394) |
| No. of unique reflections | 42,012 (6,310) |
| Completeness (%) | 95.8 (90.6) |
| Rmeas | 11.4 (98.1) |
| CC1/2 | 99.6 (39.0) |
| Wilson B factor (Å2) | 43.1 |
| Refinement statistics | |
| No. of reflections (working/test) | 39,014/2,045 |
| Rcryst/Rfree (%) | 19.8/25.7 |
| No. of: | |
| Protein atoms | 11,505 |
| Water molecules | 435 |
| Iodide ions | 14 |
| RMSD from ideal geometry | |
| Bond lengths (Å) | 0.015 |
| Bond angles (°) | 1.52 |
| Ramachandran plot (%) | |
| Residues in most favorable regions | 94.9 |
| Residues in additionally allowed regions | 4.7 |
| Avg B factors (Å2) | |
| Main chain | 34.4c |
| Side chain | 41.8d |
| Water molecules | 34.8 |
| Iodide | 56.0 |
I, intensity of a reflection; RMSD, root mean square deviation; Rmeas, redundancy-independent R factor as calculated by XDS (20); CC1/2, percentage of correlation between intensities from random half-data sets; Rcryst, the agreement between the observed and predicted structure factors.
Values for the outermost resolution shell are shown in parentheses.
The values for subunits A, B, and C are 32.0, 38.2, and 49.5 Å2, respectively.
The values for subunits A, B, and C are 34.0, 40.1, and 51.5 Å2, respectively.
Substrate docking.
Isohexenyl-glutaconyl-CoA, the substrate for AtuE, was prepared with the ChemBio3D program (version 13.0; PerkinElmer), a part of the ChemBioOffice 2012 suite. The LibDock docking protocol (26) from Discovery Studio (version 3.5.0.12158; Accelrys Software Inc., 2007) was utilized to dock the substrate into the putative active site of AtuE. AtuE and the ligand were prepared using the Prepare Protein and Prepare Ligand tools, respectively. The tentative binding site in AtuE was also identified using the From Receptor Cavities tool under the Define and Edit Binding Site tool, which searches for cavities in the receptor structure in the form of spheres. After setting the parameter values, the LibDock protocol was initiated.
Protein structure accession number.
The atomic coordinates and the structure factors have been deposited in the Protein Data Bank (PDB) under accession number 4ZU2.
RESULTS AND DISCUSSION
Proteome analysis of soluble P. aeruginosa proteins.
P. aeruginosa was grown in a mineral salts medium supplemented with octanoate or octanoate plus citronellate (3 biological replicates for each carbon source). Soluble cell extracts were prepared from each culture, separated by SDS-PAGE, and subjected to quantitative proteome analysis using a label-free GeLC-MS/MS approach, as described in Materials and Methods.
A total of 1,640 proteins with at least two unique peptides and a protein probability of >99%, corresponding to 29% of the predicted P. aeruginosa proteome, were identified in the GeLC-MS/MS experiment. One hundred eighty-seven proteins that showed at least a 3-fold increase in abundance in citronellate-grown cells were identified (see Data Set S2 in the supplemental material for a complete overview and Table 3 for a selection of proteins with a postulated function in the catabolism of acyclic terpenes). Remarkably, all products of the liu gene cluster (LiuA to LiuE) were identified and were upregulated 14- to 47-fold in the presence of citronellate compared to their levels in octanoate-grown cells. Most of the Atu peptides (AtuA to AtuG) were identified only in citronellate-grown cells and were also significantly increased in abundance (23- to 116-fold). Previous two-dimensional gel electrophoresis data had already shown the specific upregulation of several Liu and Atu proteins in citronellate-grown cells (6). Those Liu and Atu proteins that were not identified previously (AtuD, LiuE) were clearly identified in the present work and were strongly increased in abundance in citronellate-grown cells. Only AtuH, a predicted acyl-CoA synthetase (a putative citronellyl-CoA synthetase), was not identified in this study or in our previous study (6). The absence of AtuH is in agreement with the absence of a detectable growth defect of atuH insertion mutants on acyclic terpenes (citronellol, citronellate). Since the P. aeruginosa genome has many putative acyl-CoA synthetase genes, the function of a citronellyl-CoA synthetase can presumably be covered by at least one of those isoenzymes.
TABLE 3.
Proteins increased in abundance in citronellate-grown cells with postulated function in catabolism of acyclic terpenesa
| Locus | Gene | Function | Fold change in expression | P value by ANOVA | No. of peptides used for quantification | Mol wt |
|---|---|---|---|---|---|---|
| PA1535 | Citronellyl-CoA dehydrogenase | 39.9 | 2.13E−05 | 15 | 42,040.8 | |
| PA1982 | exaA | Quinoprotein ethanol dehydrogenase | 60.8 | 1.51E−02 | 36 | 68,122.9 |
| PA1984 | exaC | NAD+-dependent aldehyde dehydrogenase | 20.4 | 1.35E−03 | 32 | 54,892.3 |
| PA2011 | liuE | 3-Hydroxy-3-methylglutaryl-CoA lyase | 13.5 | 5.46E−04 | 11 | 31,837.1 |
| PA2012 | liuD | Methylcrotonyl-CoA carboxylase, alpha subunit | 17.2 | 2.22E−04 | 27 | 71,282 |
| PA2013 | liuC | 3-Methylglutaconyl-CoA hydratase | 40.5 | 3.76E−06 | 14 | 28,946.7 |
| PA2014 | liuB | Methylcrotonyl-CoA carboxylase, beta subunit | 46.7 | 1.31E−06 | 26 | 57,433.3 |
| PA2015 | liuA | Isovaleryl-CoA dehydrogenase | 24.2 | 2.68E−05 | 32 | 42,208.8 |
| PA2886 | atuA | Protein of citronellol catabolism | 92.8 | 2.40E−07 | 33 | 64,435.1 |
| PA2887 | atuB | Putative dehydrogenase | 115.6 | 5.71E−05 | 16 | 30,750.7 |
| PA2888 | atuC | Geranyl-CoA carboxylase, beta subunit | 50.3 | 1.40E−06 | 22 | 57,328 |
| PA2889 | atuD | Citronellyl-CoA dehydrogenase | 25.2 | 8.54E−05 | 32 | 42,714 |
| PA2890 | atuE | Isohexenylglutaconyl-CoA hydratase | 23.1 | 8.33E−05 | 12 | 27,717.4 |
| PA2891 | atuF | Geranyl-CoA carboxylase, alpha-subunit | 38.2 | 2.99E−05 | 34 | 71,747.2 |
| PA2892 | atuG | Short-chain dehydrogenase | 43.5 | 1.27E−06 | 17 | 29,630.6 |
| PA4330 | Probable enoyl-CoA hydratase/isomerase | 21.2 | 2.49E−04 | 13 | 28,152.2 |
Selection of proteins that were increased in abundance in citronellate-grown cells compared to their abundance in octanoate-grown cells and with a proposed function in the catabolism of citronellol. Proteins of the Atu and Liu gene clusters are in bold. For a complete overview of all proteins identified in our proteomics survey, see Data Set S2 in the supplemental material.
Additional proteins previously suggested to be involved in the pathway of acyclic terpene degradation were strongly increased in abundance in citronellate-grown cells. Namely, PA1535, which encodes an isoenzyme of citronellyl-CoA dehydrogenase (AtuD) with experimentally verified citronellyl-CoA dehydrogenase activity (9), and the gene encoding an unspecific pyrroloquinoline quinone (PQQ)-dependent alcohol dehydrogenase with verified terpene alcohol dehydrogenase activity (PA1982, exaA) were increased in abundance (27). Interestingly, the abundance of a gene encoding an NAD+-dependent aldehyde dehydrogenase (PA1984, exaC) belonging to the same gene cluster as exaA was likewise increased. Further experiments are, however, needed to clarify if ExaC is directly involved in terpene metabolism in P. aeruginosa.
AtuE, the predicted isohexenyl-glutaconyl-CoA hydratase, was 23-fold more abundant in citronellate-grown cells than in octanoate-grown cells. Remarkably, of the more than 2 dozen predicted hydratases in the P. aeruginosa genome, only one additional putative hydratase (the PA4330 gene product) was also increased in abundance in the presence of citronellate (21-fold). We assume that AtuE and possibly also the PA4330 gene product were responsible for the hydration of isohexenyl-glutaconyl-CoA. This might explain why inactivation of the atuE gene by insertion mutagenesis led only to a growth reduction and did not completely inhibit growth when acyclic terpenes were used as the substrates (28). In order to gain further insights into the function of AtuE at the molecular level, despite futile efforts to synthesize the physiological AtuE substrate, isohexenyl-glutaconyl-CoA, for biochemical studies, the crystal structure of AtuE was determined. To this end, AtuE was recombinantly expressed in E. coli as a His-tagged fusion protein, purified, and crystallized.
Overall description of AtuE structure.
AtuE was crystallized as a trimer in the asymmetric unit. Analysis with the PDBePISA tool (http://www.ebi.ac.uk/pdbe/pisa/) also revealed a trimer assembly for AtuE in solution. The shape of the trimer resembles a propeller with overall dimensions of approximately 76 by 79 by 55 Å (Fig. 2A). No significant differences between the three subunits were found, as shown by the root mean square deviation values after superposition on each other (0.39 Å, 0.47 Å, and 0.42 Å between subunits A and B, A and C, and B and C, respectively). Each subunit consists of 258 amino acid residues and folds into two separate and distinct domains, an N-terminal domain (residues Glu7 to Arg205) and a C-terminal domain (residues Arg206 to Gln297) (Fig. 2B). The N-terminal domain consists of 9 strands that form two β sheets. The first sheet consists of 6 almost parallel β strands, whereas the second sheet consists of 3 β strands which are oriented nearly perpendicular to the first β strand. The β sheets are flanked by 7 α helices and 5 310 helices. The C-terminal domain comprises about 60 amino acids arranged into four α helices and no β strands.
FIG 2.

(A) Ribbon representation of the AtuE trimer. Each subunit is shown in a different color. The proposed catalytic residue Glu139 is shown in each subunit in sphere representation. (B) Chain A of the trimer. The N- and C-terminal domains are shown in yellow and blue, respectively. Secondary structure elements are labeled. Glu139 is depicted as a stick. The figure was created with the Chimera (version 1.10) program (34).
Quality of the structure.
The crystal structure of P. aeruginosa AtuE was refined to a 2.13-Å resolution to an Rcryst (i.e., {[∑(|Fobs| − k|Fcalc|)]/∑|Fobs|} × 100, where Fobs and Fcalc are the observed and calculated structure factor amplitudes, respectively, and k is a weighting factor) and an Rfree of 19.8% and 25.7%, respectively. The final refined structure comprises 11,954 protein atoms and 435 water molecules. The average B factors calculated for protein atoms and water molecules are 36.4 Å2 and 24.8 Å2, respectively. The Ramachandran plot revealed that 96.6% of the total residues fall within the most favored combination of ϕ and ψ rotations, while 0.91% of the residues are outliers. In addition, the MOLPROBITY all atom contact score is 10.94, placing it in the 83rd percentile in the 19.7- to 2.13-Å resolution range of solved structures.
Comparison with other structures.
AtuE shows a close structural similarity with hydratase/isomerase enzymes from the crotonase superfamily (Fig. 3). The sequence identity of AtuE with members of this superfamily varies from 56% to as low as 15%. However, a significant similarity in terms of secondary structural elements (sse; in percent) ranging from 73% to as high as 95% was identified. It can therefore be inferred that several structures in the crotonase superfamily share a high level of conserved structural identity, despite having less than 50% amino acid sequence identity with the majority of the family members. This finding is also consistent with the findings of previous studies with enzymes of the crotonase superfamily, which displayed a low level of sequence identity/similarity but adopted similar three-dimensional architectures, thereby solving the mechanistic problem of catalysis by a common structural solution (29).
FIG 3.

Structure-based alignment of AtuE homologs. Only the top six structures are listed in descending order of the Q score (from 0.88 to 0.71). The residues corresponding to rat ECH catalytic glutamates are shown with a blue triangle. Details of the structures and their structure-based sequence identities to AtuE are as follows: the structure with PDB accession number 4K3W (Marinobacter aquaeolei enoyl-CoA hydratase), 56% sequence identity; the structure with PDB accession number 3MYB (Mycobacterium smegmatis enoyl-CoA hydratase), 29% sequence identity (Seattle Structural Genomics Center for Infectious Disease); the structure with PDB accession number 4KPK (Shewanella pealeana ATCC 700345), 32% sequence identity (New York Structural Genomics Research Consortium); the structure with PDB accession number 3KQF (Bacillus anthracis enoyl-CoA hydratase), 31% sequence identity (G. Minasov, A. Halavaty, Z. Wawrzak, T. Skarina, O. Onopriyenko, L. Papazisi, A. Savchenko, and W. F. Anderson, structure to be published); the structure with PDB accession number 2ZQR (human AU RNA binding protein/enoyl-coenzyme A hydratase), 27% sequence identity (35); and the structure with PDB accession number 2VX2 (human enoyl-CoA hydratase domain-containing protein 3), 21% sequence identity (W. W. Yue, K. Guo, G. Kochan, E. Pilka, J. W. Murray, E. Salah, R. Cocking, Z. Sun, A. K. Roos, A. C. W. Pike, P. Filippakopoulos, C. Arrowsmith, M. Wikstrom, A. Edwards, C. Bountra, and U. Oppermann, structure to be published). The homologous hydratases have a trimer composition according to the PDB. The figure was created with the ESPript program (version 3.0) (36). The structure-based alignment was carried out by PDBeFold. Conserved residues are indicated by white letters on a red background (strictly conserved) or red letters on a white background (global similarity score, >0.7) and framed in blue boxes.
Subunit interface.
The solvent-accessible surface area of the trimer is 27,710 Å2, and the buried surface area (the solvent-accessible surface area of monomeric units buried upon assembly formation) is 11,430 Å2. The average surface area is 13,049.6 Å2 for each molecule. The interface areas (calculated as the difference in the total accessible surface areas of isolated and interfacing structures divided by 2) are 1,932 Å2 (subunits B and C), 1,907 Å2 (subunits A and B), and 1,878 Å2 (subunits C and A). About 50 to 55 residues from each molecule from the regions from residues 70 to 99 and residues 137 to 171 are involved in interface formation. Comparison of the interface with other interfaces found in PDB showed similarities with the trimeric enzymes of the hydratase/isomerase class. The most striking interface similarity was found with the enoyl-CoA hydratase (ECH)/isomerase from Marinobacter aquaeolei (PDB accession number 4K3W; New York Structural Genomics Research Consortium; 56% sequence identity) and the enoyl-CoA hydratase from Mycobacterium smegmatis (PDB accession number 3MYB; 29% sequence identity; Seattle Structural Genomics Center for Infectious Disease). Currently, no biochemical data are available for these proteins.
Active site of AtuE.
A comparative study of AtuE with the rat mitochondrial ECH (crotonase) in complex with the long-chain substrate octanoyl-CoA (PDB accession number 2DUB) (30) was performed for the identification of key residues involved in substrate binding. Crotonase is one of the first members of this superfamily and has been thoroughly studied (31). For comparison purposes, both enzymes were superimposed, and the cutaway of the active-site topology is shown in Fig. 4. Notably, the AtuE substrate (isohexenyl-glutaconyl-CoA) is 3 carbon atoms longer and more branched than the ECH (crotonase) substrate octenoyl-CoA. In addition, the active site of AtuE is partly formed by a well-ordered α helix found at the end of the tunnel, in sharp contrast to the presence of a disordered loop in crotonase in the same region. Therefore, a binding mode identical to that found in crotonase is unlikely. Analysis of the putative substrate binding site in AtuE shows that the binding tunnel starts at the CoA binding site, passes through the catalytic region, and opens onto the other end of the subunit mostly lined by hydrophobic residues (Fig. 5A). However, in the case of crotonase, this tunnel opens between the C-terminal α helix of the neighboring chain and its disordered loop to the intertrimer space. In fact, the modeling suggests that the longer branched chain of the substrate tail would clash with the neighboring subunit's C-terminal α helix if it were to adopt an extended conformation similar to that of octanoyl-CoA. Furthermore, the tunnel cavity broadens considerably toward the end to accommodate the branched isoprenoid unit of the substrate in AtuE. As a result, the binding pocket gets shielded from the solvent, which is not the case in crotonase. The expanded cavity might explain why AtuE is able to accommodate the bulky isoprenoid portion of the substrate. We therefore propose that AtuE has evolved to bind the CoA-activated branched isoprenoid substrate while maintaining the crotonase protein fold. On the basis of structural considerations, it is suggested that Glu139 could function as a catalytic residue. Glu139 is structurally equivalent to Glu164 in rat mitochondrial ECH, where it acts as the catalytic base. Moreover, the backbone amide nitrogen atoms of Gly116 and Gly68 could form an oxyanion hole via hydrogen bonds to the thioester carbonyl oxygen of the acyl substrate tail. The catalytic acid Glu144 of rat ECH is replaced by Gly119 in AtuE, a residue that cannot participate in catalysis, but it may assist in the correct positioning of the substrate. Thus, in the absence of a second catalytic residue in AtuE, Glu139 may play a dual role as a catalytic base and acid according to recent studies carried out on ECH mutants (32) (Fig. 5B). The presence of only one of the two catalytic Glu residues in members of the crotonase superfamily is not unusual. Variations in the active site have been previously reported, suggesting that the active-site residues are not strictly conserved within the crotonase superfamily (33).
FIG 4.

Comparison of the substrate-binding mode in AtuE (A) (following docking of isohexenyl-glutaconyl-CoA onto the active site, as described in Materials and Methods) and rat ECH (B) (the PDB accession number for the experimental structure is 2DUB). Colors correspond to the hydrophobicity of the active site, as shown in the scale (blue, less hydrophobic; brown, more hydrophobic). Isohexenyl-glutaconyl-CoA and octanoyl-CoA are shown as sticks with atoms indicated by colors: gray, carbon; red, oxygen; orange, phosphorus; yellow, sulfur; blue, nitrogen.
FIG 5.
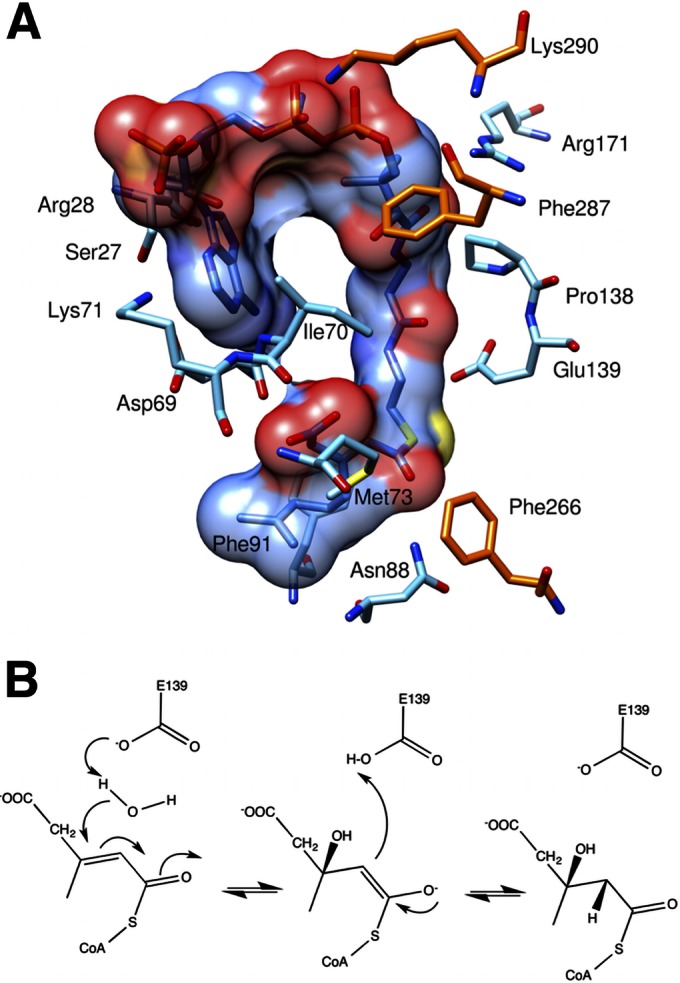
(A) Binding details of AtuE substrate. The residues involved in substrate binding and the substrate are shown in stick representation. The molecular surface of the substrate is depicted. Residues from the neighboring subunit in the trimer involved in substrate binding are shown in orange for carbon. The other atoms are colored as described in the legend to Fig. 4. (B) Proposed catalytic mechanism of AtuE.
Functional implications for Pseudomonas aeruginosa putative hydratases.
Several putative hydratases have been identified in the genome of Pseudomonas aeruginosa (see Data Set S1 and Fig. S2 in the supplemental material). Sequence alignment shows a sequence identity in the range of 24% to 30% compared with the sequence of AtuE (PA2890 gene product). The suggested catalytic residue Glu139 of AtuE is rather conserved in most of them, apart from a few exceptions where it is replaced by Phe or Tyr. Notably, most putative hydratases in P. aeruginosa also possess the second catalytic Glu residue, similar to the findings for rat ECH. Since no increase in abundance was detected for these enzymes, it can be postulated that they may be involved in other catalytic activities or are redundant enzymes. Apart from AtuE, the PA4330 gene product was the only putative hydrolase found to be increased in abundance in citronellate-grown cells. The sequence of the PA4330 gene product shows 44.3% similarity to the sequence of an enoyl-CoA hydratase/carnithine racemase from Magnetospirillum magneticum AMB-1 (New York Structural Genomics Research Consortium) and 24% similarity to the sequence of AtuE. It is therefore expected to share a similar fold with the crotonase superfamily. However, it lacks both glutamate residues and has a Thr residue structurally equivalent to AtuE Glu139. Therefore, the PA4330 gene product may have a catalytic mechanism different from that of AtuE.
Conclusion.
Proteome analysis identified 187 proteins in P. aeruginosa that were increased in abundance in the presence of citronellate. In particular, proteins belonging to the Atu and Liu pathways were increased in abundance, thus confirming the importance of these proteins and of both pathways for terpenoid catabolism in P. aeruginosa. Among the proteins that showed an increased abundance, two hydratases (AtuE and the PA4330 gene product) were identified. The structure of AtuE was subsequently determined at a 2.13-Å resolution, and AtuE was found to be a member of the crotonase superfamily. Substrate docking and comparative studies of AtuE with previously crystallized enzymes of the crotonase family in the presence of substrate revealed similar binding modes for CoA and the pantothenate moiety of the substrate. However, a much wider active-site topology suitable for the branched substrate of AtuE was identified. The structure of AtuE provides the first example of an enoyl-CoA hydrolase able to bind a branched acyl moiety of the substrate.
Supplementary Material
ACKNOWLEDGMENTS
We thank the Academy of Finland and the Deutsche Forschungsgemeinschaft for financial support (grant no. 121278 to A.C.P. and grant no. JE 152/11-2 to D.J., respectively). Access to EMBL Hamburg (DESY) was provided by the European Community's Seventh Framework Programme (FP7/2007-2012) under grant agreement no. 226716 and Biostruct-X (grant agreement no. 283570). Infrastructure support from Biocenter Finland is also acknowledged.
The docking procedure was carried out by remote servers at the CSC-IT Center for Science Ltd. (Espoo, Finland). We thank Nadine Randel for construction of the atuE expression plasmid and technical assistance in some experiments, and we thank J. Altenbuchner (University of Stuttgart) for providing pJoe4036.1.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.01686-15.
REFERENCES
- 1.Förster-Fromme K, Jendrossek D. 2010. Catabolism of citronellol and related acyclic terpenoids in pseudomonads. Appl Microbiol Biotechnol 87:859–869. doi: 10.1007/s00253-010-2644-x. [DOI] [PubMed] [Google Scholar]
- 2.Hierro I, Valero A, Pérez P, González P, Cabo MM, Montilla MP, Navarro MC. 2004. Action of different monoterpenic compounds against Anisakis simplex s.l. L3 larvae. Phytomedicine 11:77–82. doi: 10.1078/0944-7113-00375. [DOI] [PubMed] [Google Scholar]
- 3.Höschle B, Gnau V, Jendrossek D. 2005. Methylcrotonyl-CoA and geranyl-CoA carboxylases are involved in leucine/isovalerate utilization (Liu) and acyclic terpene utilization (Atu), and are encoded by liuB/liuD and atuC/atuF, in Pseudomonas aeruginosa. Microbiology 151:3649–3656. doi: 10.1099/mic.0.28260-0. [DOI] [PubMed] [Google Scholar]
- 4.Aguilar JA, Zavala AN, Díaz-Pérez C, Cervantes C, Díaz-Pérez AL, Campos-García J. 2006. The atu and liu clusters are involved in the catabolic pathways for acyclic monoterpenes and leucine in Pseudomonas aeruginosa. Appl Environ Microbiol 72:2070–2079. doi: 10.1128/AEM.72.3.2070-2079.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Höschle B, Jendrossek D. 2005. Utilization of geraniol is dependent on molybdenum in Pseudomonas aeruginosa: evidence for different metabolic routes for oxidation of geraniol and citronellol. Microbiology 151:2277–2283. doi: 10.1099/mic.0.27957-0. [DOI] [PubMed] [Google Scholar]
- 6.Förster-Fromme K, Jendrossek D. 2006. Identification and characterization of the acyclic terpene utilization gene cluster of Pseudomonas citronellolis. FEMS Microbiol Lett 264:220–225. doi: 10.1111/j.1574-6968.2006.00454.x. [DOI] [PubMed] [Google Scholar]
- 7.Aguilar JA, Díaz-Pérez C, Díaz-Pérez AL, Rodríguez-Zavala JS, Nikolau BJ, Campos-García J. 2008. Substrate specificity of the 3-methylcrotonyl coenzyme A (CoA) and geranyl-CoA carboxylases from Pseudomonas aeruginosa. J Bacteriol 190:4888–4893. doi: 10.1128/JB.00454-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Díaz-Pérez C, Rodríguez-Zavala JS, Díaz-Pérez AL, Campos-García J. 2012. Co-expression of α and β subunits of the 3-methylcrotonyl-coenzyme A carboxylase from Pseudomonas aeruginosa. World J Microbiol Biotechnol 28:1185–1191. doi: 10.1007/s11274-011-0921-1. [DOI] [PubMed] [Google Scholar]
- 9.Förster-Fromme K, Chattopadhyay A, Jendrossek D. 2008. Biochemical characterization of AtuD from Pseudomonas aeruginosa, the first member of a new subgroup of acyl-CoA dehydrogenases with specificity for citronellyl-CoA. Microbiology 154:789–796. doi: 10.1099/mic.0.2007/014530-0. [DOI] [PubMed] [Google Scholar]
- 10.Seubert W, Fass E. 1964. Studies on the bacterial degradation of isoprenoids. V. The mechanism of isoprenoid degradation. Biochem Z 341:35–44. [PubMed] [Google Scholar]
- 11.Stover CK, Pham XQ, Erwin AL, Mizoguchi SD, Warrener P, Hickey MJ, Brinkman FSL, Hufnagle WO, Kowalik DJ, Lagrou M, Garber RL, Goltry L, Tolentino E, Westbrock-Wadman S, Yuan Y, Brody LL, Coulter SN, Folger KR, Kas A, Larbig K, Lim R, Smith K, Spencer D, Wong GKS, Wu Z, Paulsen IT, Reizer J, Saier MH, Hancock REW, Lory S, Olson MV. 2000. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 406:959–964. doi: 10.1038/35023079. [DOI] [PubMed] [Google Scholar]
- 12.Schlegel HG, Gottschalk G, von Bartha R. 1961. Formation and utilization of poly-beta-hydroxybutyric acid by Knallgas bacteria (Hydrogenomonas). Nature 191:463–465. doi: 10.1038/191463a0. [DOI] [PubMed] [Google Scholar]
- 13.Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 14.Simon O, Klaiber I, Huber A, Pfannstiel J. 2014. Comprehensive proteome analysis of the response of Pseudomonas putida KT2440 to the flavor compound vanillin. J Proteomics 109:212–227. doi: 10.1016/j.jprot.2014.07.006. [DOI] [PubMed] [Google Scholar]
- 15.Shevchenko A, Wilm M, Vorm O, Mann M. 1996. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal Chem 68:850–858. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- 16.Olsen JV, de Godoy LMF, Li G, Macek B, Mortensen P, Pesch R, Makarov A, Lange O, Horning S, Mann M. 2005. Parts per million mass accuracy on an Orbitrap mass spectrometer via lock mass injection into a C-trap. Mol Cell Proteomics 4:2010–2021. doi: 10.1074/mcp.T500030-MCP200. [DOI] [PubMed] [Google Scholar]
- 17.Keller A, Nesvizhskii AI, Kolker E, Aebersold R. 2002. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal Chem 74:5383–5392. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- 18.Nesvizhskii AI, Keller A, Kolker E, Aebersold R. 2003. A statistical model for identifying proteins by tandem mass spectrometry. Anal Chem 75:4646–4658. doi: 10.1021/ac0341261. [DOI] [PubMed] [Google Scholar]
- 19.Winsor GL, Lam DKW, Fleming L, Lo R, Whiteside MD, Yu NY, Hancock REW, Brinkman FSL. 2011. Pseudomonas Genome Database: improved comparative analysis and population genomics capability for Pseudomonas genomes. Nucleic Acids Res 39:D596–D600. doi: 10.1093/nar/gkq869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kabsch W. 2010. XDS. Acta Crystallogr D Biol Crystallogr 66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. 2007. Phaser crystallographic software. J Appl Crystallogr 40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L-W, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 24.Lovell S, Davis I, Arendall W, de Bakker P, Word J, Prisant M, Richardson J, Richardson D. 2003. Structure validation by Cα geometry: ϕ, ψ, and Cβ deviation. Proteins 50:437–450. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- 25.Krissinel E, Henrick K. 2004. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr D Biol Crystallogr 60:2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- 26.Diller DJ, Merz KM Jr. 2001. High throughput docking for library design and library prioritization. Proteins 43:113–124. doi:. [DOI] [PubMed] [Google Scholar]
- 27.Chattopadhyay A, Förster-Fromme K, Jendrossek D. 2010. PQQ-dependent alcohol dehydrogenase (QEDH) of Pseudomonas aeruginosa is involved in catabolism of acyclic terpenes. J Basic Microbiol 50:119–124. doi: 10.1002/jobm.200900178. [DOI] [PubMed] [Google Scholar]
- 28.Förster-Fromme K, Höschle B, Mack C, Bott M, Armbruster W, Jendrossek D. 2006. Identification of genes and proteins necessary for catabolism of acyclic terpenes and leucine/isovalerate in Pseudomonas aeruginosa. Appl Environ Microbiol 72:4819–4828. doi: 10.1128/AEM.00853-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Holden HM, Benning MM, Haller T, Gerlt JA. 2001. The crotonase superfamily: divergently related enzymes that catalyze different reactions involving acyl coenzyme A thioesters. Acc Chem Res 34:145–157. doi: 10.1021/ar000053l. [DOI] [PubMed] [Google Scholar]
- 30.Engel CK, Kiema TR, Hiltunen JK, Wierenga RK. 1998. The crystal structure of enoyl-CoA hydratase complexed with octanoyl-CoA reveals the structural adaptations required for binding of a long-chain fatty acid-CoA molecule. J Mol Biol 275:847–859. doi: 10.1006/jmbi.1997.1491. [DOI] [PubMed] [Google Scholar]
- 31.Engel CK, Mathieu M, Zeelen JP, Hiltunen JK, Wierenga RK. 1996. Crystal structure of enoyl-coenzyme A (CoA) hydratase at 2.5 angstroms resolution: a spiral fold defines the CoA-binding pocket. EMBO J 15:5135–5145. [PMC free article] [PubMed] [Google Scholar]
- 32.Hofstein HA, Feng Y, Anderson VE, Tonge PJ. 1999. Role of glutamate 144 and glutamate 164 in the catalytic mechanism of enoyl-CoA hydratase. Biochemistry 38:9508–9516. doi: 10.1021/bi990506y. [DOI] [PubMed] [Google Scholar]
- 33.Gerlt JA, Babbitt PC. 2001. Divergent evolution of enzymatic function: mechanistically diverse superfamilies and functionally distinct suprafamilies. Annu Rev Biochem 70:209–246. doi: 10.1146/annurev.biochem.70.1.209. [DOI] [PubMed] [Google Scholar]
- 34.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. 2004. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 35.Kurimoto K, Kuwasako K, Sandercock AM, Unzai S, Robinson CV, Muto Y, Yokoyama S. 2009. AU-rich RNA-binding induces changes in the quaternary structure of AUH. Proteins 75:360–372. doi: 10.1002/prot.22246. [DOI] [PubMed] [Google Scholar]
- 36.Robert X, Gouet P. 2014. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res 42:W320–W324. doi: 10.1093/nar/gku316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Simon R, Priefer U, Pühler A. 1983. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in Gram negative bacteria. Nat Biotechnol 1:784–791. doi: 10.1038/nbt1183-784. [DOI] [Google Scholar]
- 38.Randel N. 2012. Biologische Untersuchungen zum “lower acyclic terpene utilisation (atu) pathway” in Pseudomonas aeruginosa und chemische Synthese wichtiger Intermediate dieses Stoffwechselweges. Universitätsbibliothek der Universität Stuttgart, Stuttgart, Germany. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.