

Abstract
Seagrass colonization changes the chemistry and biogeochemical cycles mediated by microbes in coastal sediments. In this study, we molecularly characterized the diazotrophic assemblages and entire bacterial community in surface sediments of a Zostera marina-colonized coastal lagoon in northern China. Higher nitrogenase gene (nifH) copy numbers were detected in the sediments from the vegetated region than in the sediments from the unvegetated region nearby. The nifH phylotypes detected were mostly affiliated with the Geobacteraceae, Desulfobulbus, Desulfocapsa, and Pseudomonas. Redundancy analysis based on terminal restriction fragment length polymorphism analysis showed that the distribution of nifH genotypes was mostly shaped by the ratio of total organic carbon to total organic nitrogen, the concentration of cadmium in the sediments, and the pH of the overlying water. High-throughput sequencing and phylogenetic analyses of bacterial 16S rRNA genes also indicated the presence of Geobacteraceae and Desulfobulbaceae phylotypes in these samples. A comparison of these results with those of previous studies suggests the prevalence and predominance of iron(III)-reducing Geobacteraceae and sulfate-reducing Desulfobulbaceae diazotrophs in coastal sedimentary environments. Although the entire bacterial community structure was not significantly different between these two niches, Desulfococcus (Deltaproteobacteria) and Anaerolineae (Chloroflexi) presented with much higher proportions in the vegetated sediments, and Flavobacteriaceae (Bacteroidetes) occurred more frequently in the bare sediments. These data suggest that the high bioavailability of organic matter (indicated by relatively lower carbon-to-nitrogen ratios) and the less-reducing anaerobic condition in vegetated sediments may favor Desulfococcus and Anaerolineae lineages, which are potentially important populations in benthic carbon and sulfur cycling in the highly productive seagrass ecosystem.
INTRODUCTION
Sediments colonized by seagrass in shallow estuarine and coastal environments are hot spots of microbial activities. Seagrass meadows enrich the sediment carbon matter by exuding dissolved organic carbon (DOC) through their roots and trapping organic particles from the overlying water (1). Due to the effects of seagrass on nutrient deposition, retention, and mineralization from organic matter, the nutrients in pore water are often richer in seagrass sediments than in sediments from unvegetated regions (1–3). Seagrass roots also release photosynthesis-produced O2 into sediments, which results in less-reducing conditions in seagrass meadows than unvegetated sediments and contributes to the prevention of the accumulation of sulfides, the toxic products of sulfate reduction in anaerobic sedimentary environments that could play a role in dieback events in seagrass meadows (1, 4). Higher bacterial populations and activities (particularly bacterium-mediated sulfate reduction) are usually found in seagrass-vegetated sites and not in unvegetated sediments (5–8).
Because the growth and the maintenance of the high productivity of seagrass meadows require the supply of substantial amounts of nitrogen (N), much effort has been made to reveal the activity of and contribution by N2-fixing bacteria (diazotrophs) in sediments (9). Although cultivation, tracing, and molybdate inhibition experiments have demonstrated that sulfate-reducing bacteria (SRB) are important contributors to nitrogen fixation in seagrass systems (9–11), the diversity and ecological studies of diazotrophs in seagrass meadow systems are still insufficient. Culturing methods have been employed in several studies to identify and enumerate seagrass diazotrophs, for example, the eelgrass Zostera marina in Kanagawa, Japan (12), and several other seagrass species in the Gulf of Mannar, India (13). However, only a few studies have used the nifH gene (a functional gene encoding the nitrogenase subunit NifH) as a molecular marker to explore the diazotrophic diversity of a limited species in seagrass systems, for example, a mixed meadow of Thalassia testudinum and Syringodium filiforme and the smooth cordgrass Spartina alterniflora (14, 15). The genetic diversity and abundance of diazotrophic populations in sediments of other seagrass species and/or in other regions largely remain unknown.
Because benthic diazotrophy is intimately linked to strictly anaerobic sulfate reduction, factors such as oxygen penetration into sediments, the quality and quantity of organic matter as electron donors, and nutrient (especially ammonium) levels may impact the distribution of diazotrophs in seagrass bed sediments (1, 4). It is therefore possible that different community compositions and sizes of diazotrophs exist in the seagrass-colonized and the unvegetated sediments. Furthermore, metal oxides, such as Fe(III) and Mn(IV) oxides, and pollutants can interact with sulfur and phosphate cycling in sediments (16, 17). However, the impact of these factors on the niche differentiation and distribution of benthic diazotrophs in seagrass systems remains poorly understood.
The overall bacterial communities in roots and/or in seagrass-vegetated sediments have been characterized by molecular approaches (8, 18, 19). To test the hypothesis that the input of organic matter from seagrass roots gives rise to different bacterial communities in vegetated and unvegetated sediments, James et al. (20) used double-gradient denaturing gradient gel electrophoresis, thereby demonstrating not only an effect of vegetation but also similarities between the bacterial communities in these sediment types on a seasonal scale. A relatively stable community composition of sulfate-reducing bacteria across these sediments was also noted (7). It was suggested that the differences in the communities likely stemmed from shifts in the abundance of some minor bacterial populations (20), but this has to be further investigated using more sensitive and quantitative approaches.
Zostera marina is an ecologically important seagrass species that is widespread on temperate coasts in both hemispheres. Studies on benthic microbial diversity and function are crucial to a better understanding of the global loss of seagrass habitats in coastal zones (1, 4, 7, 8). In this study, using a range of molecular tools, we characterized the diversity, quantity, and community composition and structure of diazotrophs and all bacteria in both vegetated and unvegetated sediments of a shallow Z. marina-colonized lagoon. The distribution of benthic diazotrophs, shifts in the relative abundance of several key bacterial lineages, and their involvement in C, N, S, and Fe cycles in seagrass bed systems are also discussed.
MATERIALS AND METHODS
Study area and sampling.
Swan Lake (also called the Yuehu Inlet) is a shallow lagoon located in the southwest part of Rongcheng Bay, Shandong Peninsula, northern China (see Fig. S1 in the supplemental material). The lagoon connects to the Yellow Sea with a narrow inlet, and it has an area of 4.8 km2 and an average water depth of less than 1.5 m. The annual mean water temperature is approximately 11.4°C, and the area represents a typical temperate habitat with a continental monsoon climate. The sediments are mainly sandy and rich in organic matter. Zostera marina meadows develop properly in this lagoon during both the spring and summer seasons and cover a significant area at the bottom.
A total of 10 sites in Swan Lake were sampled in May 2013. The five sites (bulk sediment) located within the seagrass meadow, here referred to as V1 to V5, were approximately 10 m away from each other. Another five sites (U1 to U5) from a barren region at a distance of approximately 20 to 40 m from the seagrass-covered region were selected. Sediment samples were collected with a custom-made corer (inner diameter, 7 cm) during the lower tide period, when the water depths were approximately 20 cm. The surface sediments of the top 5-cm layer were transported to the laboratory, sliced, placed on ice, and stored at −80°C.
Determination of environmental parameters.
The temperature, pH, salinity, and concentrations of dissolved oxygen (DO) and chlorophyll a (Chl-a) in the overlying water were measured at each site using an electronic probe (Hydrolab MS5; Hach, USA) (21). The particle size distribution was analyzed using a Malvern Mastersizer 2000F granulometer (Malvern, England), and the median diameter of the particles in each sediment sample was recorded. The concentrations of ammonium (NH4+-N), nitrate (NO3−-N), nitrite (NO2−-N), and soluble reactive phosphate (PO43−-P) in the sediment pore waters were determined with a nutrient AutoAnalyzer (Seal, Germany). The total organic carbon (TOC) and total organic nitrogen (TON) contents in the sediments were measured with a Vario Micro Cube elemental analyzer (Elementar, Germany). To determine the concentrations of metals, the sediments were pretreated with 1 M HCl, and the concentrations were then determined with an ELAN DRC II plasma mass spectrometer (an inductively coupled plasma mass spectrometer; PerkinElmer, Hong Kong) (22).
DNA extraction and clone library construction.
The DNA from approximately 0.5 g of sediment was extracted using a FastDNA spin kit for soil (MP Biomedical, USA) as specified by the manufacturer. The DNA concentrations were quantified using an ND-2000C spectrophotometer (NanoDrop, USA). The functional gene nifH, encoding one of the subunits of nitrogenase, was amplified by 35 PCR cycles using the primers PolF (5′-TGCGAYCCSAARGCBGACTC-3′) and PolR (5′-ATSGCCATCATYTCRCCGGA-3′) (23). The PCR products of the five samples from each region (vegetated and unvegetated) were pooled to generate two clone libraries. The amplicons were purified with a Tian Quick midipurification kit (Tiangen, China) and cloned into the pTZ57R/T vector (Thermo, USA). The resulting plasmids were transformed into Escherichia coli DH5α competent cells (Tiangen). The cloned plasmid inserts were amplified directly from the cells using M13 vector-specific primers (21). Approximately 100 clones in each library were randomly selected and sequenced (Sangon Biotech, China).
High-throughput sequencing and analysis.
Although the phylogenetic relationships among bacteria determined from the nifH sequences are largely congruent with those determined from the 16S rRNA gene sequences (24), the horizontal transfer of the nifH gene has been observed (e.g., see references 25 and 26). To check that the major nitrogen-fixing bacteria were present in our samples, high-throughput sequencing of bacterial 16S rRNA gene was carried out. For the DNA extracted from 6 samples (3 from vegetated sediments and 3 from bare sediments), we amplified the V3 hypervariable region using unique 12-bp bar codes and Ion Torrent adapter-modified core primers 338F (5′-ACTCCTACGGGAGGCAGCAG-3′) and 518R (5′-ATTACCGCGGCTGCTGG-3′) (27). The amplicons were gel purified and further purified with AMPure beads (Beckman Coulter, Brea, CA). Samples were pooled into equimolar proportions and sequenced on 318 chips with an Ion Torrent Personal Genome Machine according to the manufacturer's instructions (Life Technologies, Grand Island, NY).
The fastq files were processed using the QIIME (v.1.8.0) work flow (28). Individual sequences were matched to their sample according to the bar codes and filtered to remove sequences that (i) failed to be longer than 70 bases, (ii) had quality scores of >20, (iii) had no ambiguous bases, and (iv) had homopolymer runs with <9 bases. Both primers were removed along with the bar code. Representative operational taxonomic units (OTUs) from each set were chosen at a minimum sequence identity of 97% with the UClust program (29), and their sequences were aligned against those in the Greengenes database (30) by use of the PyNAST program (31). Chimera check was performed with the ChimeraSlayer program (32) on the basis of the sequences in the Greengenes database. Putative chimeras and singletons (OTUs containing a single read across all samples) were discarded prior to further analysis. Taxonomy was assigned using the August 2013 release of Greengenes and a minimum confidence score of 0.9. Reads assigned to chloroplasts were not considered members of bacterial communities. For calculation of OTU numbers and Shannon indices, we rarefied an identical sampling effort of 1,813 reads per sample, which was the lowest number of quality reads among the samples. Sequences of potential diazotrophic taxa (i.e., Desulfobulbaceae and Geobacteraceae) were retrieved from bacterial reads using the QIIME filter_fasta.py work flow and then subjected to further phylogenetic analysis.
Phylogenetic analysis.
The newly obtained nifH sequences were checked for possible chimeric sequences through BLAST searches with partial sequences and then translated into amino acid sequences using BioEdit software (33). Subsequently, the translated versions of the nifH genes were analyzed using the mothur program (34) with a 95% similarity cutoff for grouping OTUs, as previously suggested (35). Rarefaction curves were depicted on the basis of the OTU assignments. The percent coverage (C) of the clone libraries was calculated as [1 − (n1/N)] × 100, where n1 is the number of unique sequences (i.e., sequences without a replicate) detected in a library and N is the total number of clones in the same library (36). To assess the statistically significant differences in the communities between the libraries of clones from vegetated and unvegetated areas, the Libshuff program in the mothur package was executed with 10,000 random shuffles (34). Reference sequences were downloaded from GenBank and aligned with representative amino acid sequences of each OTU using the ClustalW program (37). A maximum likelihood (ML) tree was built using the RaxML tool (v.8.0) (38) with the LG+G model, which was the best model suggested by the ProtTest program (39). The 16S rRNA gene sequences of Desulfobulbaceae and Geobacteraceae retrieved were aligned with reference sequences using BioEdit (33), and neighbor-joining (NJ) trees based on p-distances were constructed using the MEGA (v.6) program (40). A bootstrap analysis of 1,000 replications was applied in all phylogenetic analyses.
T-RFLP analysis.
For terminal restriction fragment length polymorphism (T-RFLP) analysis, the nifH gene was amplified by use of the same procedure used for library construction, with the exception that the forward primer was labeled with 5-carboxyfluorescein. The amplicons from triplicate PCRs for each sample were pooled and gel purified. DNA products of 200 ng were digested with the endonuclease HaeIII (Thermo) at 37°C for 1 h in the dark. The fluorescently labeled terminal restriction fragments (T-RFs) were analyzed using a 3130XL genetic analyzer and GeneScan (v.2.1) software (Applied Biosystems, USA). The baseline threshold for signal detection was set to 50 fluorescence intensity units to eliminate any background interference. Only the peaks with T-RF lengths ranging from 40 to 400 bp were included in the subsequent analysis. The relative abundance of each T-RF was calculated as the ratio of the peak area of that T-RF to the total peak area of all T-RFs detected for a given sample. Minor peaks with a relative abundance of <1% of the total were excluded, and the remaining peaks were presumed to represent phylotypes of diazotrophs.
qPCR assays.
The PolF and PolR primers were also used to quantify the copy numbers of the nifH genes in the samples (23). To assess the relative proportions of diazotrophs in the whole bacterial communities, we also quantified the bacteria through quantitative real-time PCR (qPCR) using the primers 341F and 518R, which are universal primers targeting a short fragment (ca. 171 bp) of the bacterial 16S rRNA genes (27). The qPCR assay was based on the fluorescence intensity of the SYBR green dye and performed as previously described (21). Briefly, the 20-μl reaction solution contained the reagents in the SYBR green PCR/carboxy-X-rhodamine qPCR kit (Thermo), 0.4 μM each primer, and 10 ng of template DNA. The PCR was performed using an ABI 7500 Fast real-time PCR system (Applied Biosystems) with the following program: an initial denaturation step of 95°C for 7 min, followed by 40 cycles of 30 s at 94°C, 30 s at the annealing temperature (60°C for 16S rRNA genes and 56°C for nifH), and an extension step of 30 s at 72°C. The data were retrieved at 72°C, and all of the reactions were finished with a melting curve from 60°C to 95°C using increases of 0.5°C.
A 10-fold serial dilution (10−1 to 10−8) of linear DNA fragments was used to generate standard curves. Using vector-targeted primers M13F/M13R, linear fragments were obtained from the PCR amplification of circular plasmids (pTZ57R/T vector; Fermentas) which contained inserts of the nifH (accession no. KT203777) and bacterial 16S rRNA gene fragments. The standard DNA was also quantified using a PicoGreen double-stranded DNA reagent kit (Invitrogen). The PCR efficiencies for amplifying the nifH and 16S rRNA genes were 97% and 102%, respectively, and were calculated as follows: E = (10−1/k −1) × 100, where E is the PCR efficiency and k is the slope. The copy numbers of the linear standard were calculated using the following formula: the number of molecules microliter−1 = a/(linear fragment length [in base pairs] × 660) × 6.022 × 1023, where a is the concentration of the standard (in grams microliter−1) and 6.022 × 1023 is the molar constant. The correlation coefficients (R2 values) for all of the assays were greater than 0.99. Controls without templates resulted in undetectable values for all of the samples.
Statistical analysis.
Student's t tests (two-tailed) were performed to test the differences in the environmental variables, the copy numbers of the nifH genes, and the relative abundance of a given bacterial taxon between the vegetated and unvegetated regions. The Pearson correlation coefficient (r) and Spearman's correlation coefficient (ρ) were calculated to explore the relationships between the log-transformed nifH abundances and environmental variables and between the proportions of dominant T-RFs and environmental variables (n = 10). These analyses were performed using SPSS (v.13.0) software for Windows (SPSS, Chicago, IL, USA). To visualize the diazotrophic community differences in all of the samples, nonmetric multidimensional scaling (NMDS) was conducted on the basis of a Bray-Curtis similarity matrix derived from the T-RFLP data using the PRIMER (v.6) software package (Primer-E, United Kingdom). For changes in the entire bacterial communities, principal coordinate analysis (PCoA) based on UniFrac distances was performed (41). Analysis of similarity (ANOSIM) was performed to statistically test the differences in the community structures of diazotrophs and all bacteria in the samples from vegetated and unvegetated regions. After detrended correspondence analysis to determine the length of the environmental gradient using the CANOCO (v.4.5) program (42), redundancy analysis (RDA) was selected to explore the environment-biota relationships. The statistical significance of the variable was tested using a Monte Carlo permutation test (999 permutations).
Nucleotide sequence accession numbers.
The nifH sequences obtained in this study were deposited in the GenBank database under accession numbers KR132012 to KR132191. Reads from the Ion Torrent sequencing of bacterial 16S rRNA genes are available under accession number SRR1985074.
RESULTS
Environmental setting.
The comparison of the vegetated and unvegetated regions revealed that the measured physicochemical parameters (DO, salinity, temperature, and pH) of the overlying waters appeared to be similar, with the exception of the Chl-a concentration, which was significantly higher in the former than the latter (P < 0.01; see Table S1 in the supplemental material). The analysis of the nutrients in pore waters showed that the dissolved inorganic nitrogen (DIN) concentrations were much lower in the seagrass sediments (P = 0.01), primarily because of the lower levels of NH4+ species in these samples (P < 0.01). The assessment of the sediment properties showed that the TOC and TON contents were not significantly different between the two types of sediments, but their ratio (TOC/TON) was significantly lower in the samples from the vegetated regions (on average, 6.6 versus 11.3 in samples from the unvegetated regions; P = 0.04). Relative to the properties of the unvegetated sediments, the seagrass sediments were much finer (P < 0.01), generally having higher concentrations of many of the metals measured in this study. Notably, iron (Fe) and manganese (Mn) were the richest in abundance, with mean concentrations of 4,400 mg/kg and 99 mg/kg in the vegetated sediments, respectively (see Table S1 in the supplemental material).
Gene copy numbers and correlations with environmental factors.
The abundances of the nifH gene were (2.7 ± 0.50) × 107 and (1.3 ± 0.33) × 108 copies g−1 (wet weight) of sediment in the vegetated and unvegetated samples, respectively. The bacterial 16S rRNA gene abundances were (1.1 ± 0.17) × 109 and (1.5 ± 0.25) × 109 copies g−1 (wet weight) of sediment. In comparison, the copy numbers of the bacterial 16S rRNA genes were not significantly different between the vegetated and the unvegetated sediments (P = 0.21, n = 5). However, the nifH gene was markedly more abundant in the vegetated sediments than in the unvegetated sediments (P = 0.03, n = 5).
The universal bacterial primers used in qPCR might also target chloroplast rRNA genes, which may affect our interpretation of bacterial abundance in these samples, especially in seagrass-covered sediments. Nevertheless, Ion Torrent sequencing targeting bacterial 16S rRNA genes indicated that the amplified chloroplast rRNA genes were present in minor amounts, accounting for 1.6 to 6.2% and 1.5 to 2.5% in the vegetated and unvegetated sediments, respectively (t test, P = 0.29), indicating that the chloroplast contamination could hardly affect our interpretation of the bacterial abundance assessed here.
Correlation analyses showed that the log-transformed nifH copy numbers were significantly and negatively correlated with the sediment grain size (r = −0.69, P = 0.03) and the concentration of NH4+-N in the sediment pore water (r = −0.69, P = 0.03) and positively correlated with the concentration of Chl-a in the overlying water (r = 0.75, P = 0.01) and the concentrations of the metals As, Cu, Cr, and Pb (r > 0.59, P < = 0.05) (Table 1).
TABLE 1.
Pearson correlations between nifH gene copy number and environmental factorsa
| Environmental variable | r | P |
|---|---|---|
| DO concn | −0.24 | 0.50 |
| Salinity | 0.01 | 1.00 |
| Temp | 0.17 | 0.65 |
| Chl-a concn | 0.75 | 0.01 |
| pH | 0.11 | 0.76 |
| Concn of: | ||
| NO3−-N | 0.15 | 0.69 |
| NO2−-N | −0.25 | 0.49 |
| NH4+-N | −0.69 | 0.03 |
| DIN | −0.24 | 0.51 |
| PO43−-P | 0.28 | 0.44 |
| N/P | −0.52 | 0.12 |
| Grain size | −0.69 | 0.03 |
| TOC content | −0.21 | 0.56 |
| TON content | 0.07 | 0.85 |
| TOC/TON | −0.56 | 0.09 |
| Concn of: | ||
| Pb | 0.60 | 0.07 |
| Cr | 0.63 | 0.05 |
| Mn | 0.48 | 0.16 |
| Fe | 0.61 | 0.06 |
| Co | 0.63 | 0.06 |
| Ni | 0.64 | 0.06 |
| Cu | 0.65 | 0.05 |
| Zn | 0.58 | 0.08 |
| As | 0.68 | 0.03 |
| Cd | 0.47 | 0.18 |
The gene copy number was log transformed. Significant differences (P < 0.05) are highlighted in bold.
Community composition of diazotrophs based on clone libraries.
The clone library analysis revealed a high degree of genetic diversity of diazotrophs in both vegetated and unvegetated sediments. A total of 180 nifH sequences were obtained from the two clone libraries: 85 from the vegetated sediment library and 95 from the unvegetated sediment library. These sequences showed 54.8 to 99.4% similarity at the nucleotide sequence level and 51.1 to 100% similarity at the amino acid sequence level. BLAST analysis of the NifH sequences from the clone libraries obtained in the present study against the sequences in GenBank showed that the sequences had 47.5 to 100% identities. The coverage of the vegetated sediment and unvegetated sediment libraries was 79.3% and 81.9%, respectively, indicating that major phylotypes within the diazotrophic communities had been recovered (for rarefaction curves, see Fig. S2A in the supplemental material). Among the 41 OTUs identified, 13 were shared between both sets of samples, accounting for 31.7% of all OTUs detected, whereas 13 OTUs were unique to the vegetated samples and 15 OTUs were unique to the unvegetated samples.
The ML tree showed that all of the newly obtained nifH sequences belonged to cluster III (104 sequences, 57.8%) or cluster I (76 sequences, 42.2%) (Fig. 1), according to the classification proposed by Zehr et al. (24). Overall, the nifH phylotypes affiliated with known sulfate-reducing deltaproteobacterial taxa (e.g., Desulfocapsa and Desulfobulbus) were highly represented, with proportions of 37.6% and 28.4% in the clone libraries of the vegetated and unvegetated sediments, respectively. The most abundant OTU of SRB, SL2, had a sequence identity of 98% to the NifH protein sequence of Desulfocapsa sulfexigens in the family Desulfobulbaceae translated from the nifH sequence and represented 17.6% and 12.6% of the sequences in the clone libraries of the vegetated and the unvegetated sediments, respectively. SL4 was the second most abundant OTU of SRB, with a 98% similarity to Desulfobulbus mediterraneus. The frequencies of occurrence of this OTU in the two habitats were markedly similar (vegetated sediment, 9.4%; unvegetated sediment, 10.5%). There were some OTUs affiliated with genera of SRB, such as Desulfuromonas, Desulfotomaculum, and Desulfatibacillum, with low similarities (<90%). Each of these OTUs occurred infrequently but collectively accounted for 10.6% and 5.3% of the OTUs in the samples from vegetated and unvegetated sediments, respectively.
FIG 1.

A maximum likelihood tree showing the phylogeny of the NifH amino acid sequences translated from the nifH sequences. Blue, newly obtained OTUs (named SL1 to SL42) from the lagoon of Swan Lake; green, sequences from tropical Thalassia testudinum and Syringodium filiforme mixed seagrass bed sediments; orange, sequences from the Spartina alterniflora rhizosphere. The numbers of clones of each OTU in the clone libraries are indicated and shown as black (the vegetated) and white (the unvegetated) bars on the scale. Bootstrap values lower than 50% are not shown. The scale bar indicates 0.2 amino acid substitution per site. Archaeal sequences are used as the outgroups. GenBank accession numbers are given in parentheses.
One of the most abundant OTUs (SL1) inferred from analysis of the clone library consisting of the NifH protein sequences translated from the nifH sequences exhibited 99% similarity to the NifH protein of the Fe(III)-reducing bacterium Geopsychrobacter electrodiphilus (Deltaproteobacteria), accounting for 21.2% and 29.5% of the OTUs in the vegetated and the unvegetated sediments, respectively. Another dominant OTU, SL3, was 97% identical to Pseudomonas stutzeri (Gammaproteobacteria) and presented a higher relative abundance in the vegetated sediments than the unvegetated sediments (17.6% versus 10.5%).
Phylotypes of the Epsilonproteobacteria were less represented (2.4%) and exclusively detected in the vegetated sediments. A single nifH sequence from Verrucomicrobia was exclusively detected in the library for unvegetated sediments. Diazotrophic phylotypes of Betaproteobacteria, Cyanobacteria, or Archaea were not recovered (Fig. 1). The Libshuff program indicated that there were no significant differences in community composition between these two libraries (x-y comparison, P = 0.46; y-x comparison, P = 0.35).
Recent studies of the diazotrophic diversity in bare coastal sediments revealed two main nifH groups which appeared to be phylogenetically related to Pelobacter and Desulfovibrio and were named NB3 and NB7, respectively (43, 44). To explore their phylogenetic relationships with our major OTUs (e.g., SL1, SL2, and SL4), we performed ML analyses of the NifH protein sequences of NB3 and NB7 translated from the nifH sequences together with our sequences. We found that the SL1 and NB3 sequences together formed a monophyletic group containing Geopsychrobacter electrodiphilus, which was supported with a 50% bootstrap support value (Fig. 2A; see Fig. S3 in the supplemental material); all SL2 and NB7 sequences were related to Desulfocapsa sulfexigens, forming another monophyletic group (bootstrap support, <50%; Fig. 2C; see also Fig. S4 in the supplemental material).
FIG 2.

Phylogenetic trees based on the NifH amino acid sequences translated from the nifH sequences and 16S rRNA gene sequences. (A, C) ML trees showing that the sequences of the major OTUs from this study (SL1 and SL2) translated from the nifH sequence cluster, respectively, with NB3 and NB7, two abundant clades identified in previous work (43, 44) (see Fig. S3 and S4 in the supplemental material). (B, D) The p-distance-based NJ trees were constructed using the high-throughput sequencing reads for Geobacteraceae (B) and Desulfobulbaceae (D), which show topologies similar to those of the nifH-based trees, respectively. Bootstrap values lower than 50% are not shown. The scale bar indicates 0.04 amino acid or nucleotide substitution per site. For details of the grouping of cable bacterium-like sequences retrieved from Ion Torrent sequencing, see Fig. S5 in the supplemental material. GenBank accession numbers are given in parentheses.
Geobacteraceae and Desulfobulbaceae 16S rRNA gene sequences.
In all of the 17,409 bacterial 16S rRNA reads that we obtained, 11 reads for Geobacteraceae and 481 reads for Desulfobulbaceae were retrieved, which accounted for 0.06% and 2.8% of the total, respectively. The phylogenetic trees based on the 16S rRNAs of these two families generally exhibited topologies similar to those of the trees based on the NifH protein sequences of SL1 (NB3), SL2 (NB7), and SL4, respectively, translated from the nifH sequence (Fig. 2). Bacteria of the Geobacteraceae formed a highly supported monophyly (bootstrap value, 97%) and clustered with Pelobacter, Geopsychrobacter, Desulfuromonas, and Desulfuromusa in the 16S rRNA tree (bootstrap value, 58%; Fig. 2B). In the 16S rRNA tree of Desulfobulbaceae (Fig. 2D), these phylotypes from our samples branched off the genus Desulfovibrio and grouped with Desulfocapsa and Desulfobulbus species. However, their interrelationship was not resolved (bootstrap value, <50%, Fig. 2D). In addition, 27 reads from our samples clustered well with previously published cable bacterial 16S rRNA sequences (45), which formed a highly supported monophyletic group (bootstrap value, 80%; Fig. 2D; see also Fig. S5 in the supplemental material).
Diazotrophic community variations based on T-RFLP of nifH genes and correlations with environmental factors.
The T-RFLP analysis of all 10 samples revealed a total of 34 distinct T-RFs. The number of nifH T-RFs in each sample varied from 7 to 21, with averages of 14 and 15 in the vegetated and unvegetated samples, respectively. In silico endonuclease site analysis showed that the four dominant (translated) NifH OTUs revealed by the clone libraries were also found in the T-RFLP profiles (Fig. 3). The T-RF of approximately 45 bp (primarily indicative of SL1, related to Geopsychrobacter electrodiphilus) was significantly negatively correlated with the sediment TOC/TON ratio (ρ = −0.88, P < 0.01) and N/P ratio (ρ = −0.62, P = 0.05) and positively correlated with the Chl-a concentration (ρ = 0.62, P = 0.05). The Desulfobulbaceae T-RF (151/152 bp) was negatively correlated with the sediment TOC/TON ratio (ρ = −0.68, P = 0.03). Another abundant R-TF, which was 61 to 63 bp in length but exhibited no corresponding sequences in the clone libraries, was positively correlated with the sediment TOC/TON (ρ = 0.67, P = 0.03). No significant correlations were found for the Pseudomonas-related T-RF (see Table S2 in the supplemental material).
FIG 3.

Variation in the dominant T-RFs of nifH genes in all of the samples. A T-RF was considered dominant when the cumulative abundance of the T-RF was higher than 70% in each sample from the Swan Lake lagoon. The legend shows the lengths of the T-RFs and the corresponding amino acid OTUs from clone library analysis.
In multivariate analyses, the NMDS ordination based on T-RFLP analysis of the nifH genes showed a result consistent with the findings from the clone library analysis; i.e., there was no significant difference in the diazotrophic community structure between the vegetated and unvegetated sediments (Fig. 4A; ANOSIM, P = 0.40). For the analysis of the biota-environment relationships, the RDA plot showed that changes in the benthic diazotrophic community structure significantly covaried with the variations in the sediment TOC/TON (P = 0.01), the concentration of Cd (P = 0.03), and the pH of the overlying water (P = 0.04) (Fig. 4B).
FIG 4.
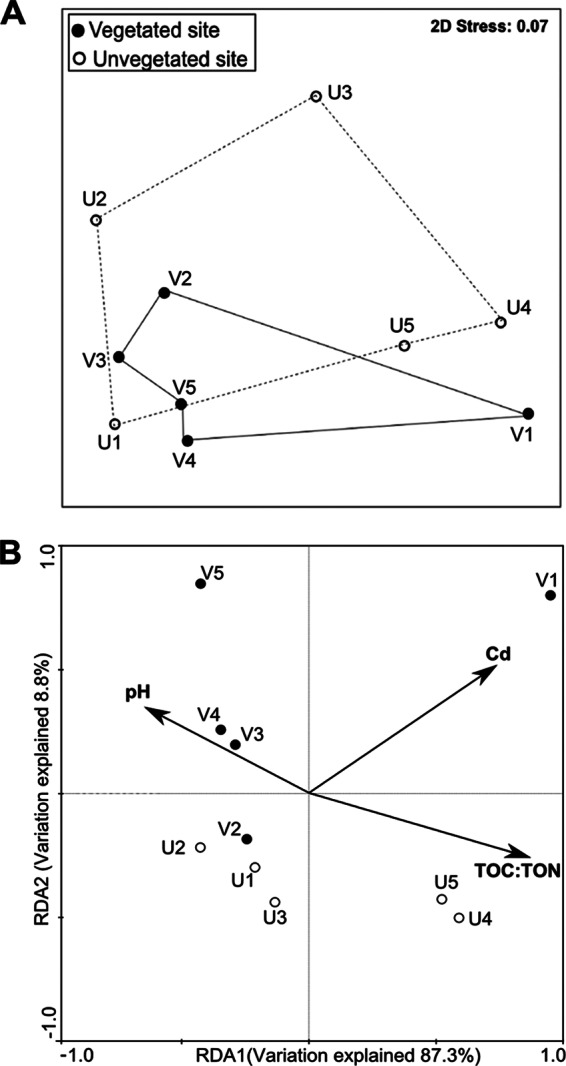
Nonmetric dimensional scaling (A) and RDA ordination diplots (B) for benthic diazotrophic communities on the basis of the T-RFLP profiles of the nifH genes. A two-dimensional (2D) stress value of 0.07 indicates a good ordination. Only the significantly correlated environmental factors are shown in panel B. The diazotrophic community was mostly influenced by the pH of the overlying water (P = 0.04), the sediment TOC/TON (P = 0.03), and the concentration of Cd (P = 0.04).
Shifts in bacterial community structure across seagrass-colonized and bare sediments.
Rarefaction curves of Ion Torrent sequencing of 16S rRNA genes indicated that a large portion of the bacterial diversity had been recovered in all samples, but alpha diversity estimators (OTU numbers, Shannon and Chao1 indices) were not much different between sediment types (t test, P > 0.70; Fig. S2B; see also Table S3 in the supplemental material). Through Greengenes classification, 36 bacterial phyla were identified in the vegetated and unvegetated sediment samples. In general, the most abundant taxa were the Deltaproteobacteria (relative abundance, 15.0 to 45.7%), Epsilonproteobacteria (8.7 to 25.3%), and Gammaproteobacteria (10.9 to 15.7%), followed by the Bacteroidetes (6.4 to 19.9%), Chloroflexi (3.5 to 11.2%), Actinobacteria (4.2 to 9.6%), and Alphaproteobacteria (3.3 to 9.3%) (Fig. 5A). The Desulfobacteraceae (15.1%) and Helicobacteraceae (14.6%), two families of the Deltaproteobacteria and Epsilonproteobacteria, respectively, were highly present in the community of vegetated sediments, accounting for approximately 1/3 of the bacterial 16S rRNA reads in these samples. Cyanobacteria appeared to be a minor group in all these samples (0.1 to 0.5%). In the PCoA plot, the bacterial community structure in vegetated samples clustered separately from that in bare sediment samples (Fig. 5B). However, the bacterial community differences between two sediment types were not statistically supported, regardless of the weighted (ANOSIM, P = 0.093) or unweighted (ANOSIM, P = 0.102) UniFrac metrics used.
FIG 5.

Relative proportions of major taxa (A) and a PCoA ordination diplot (B) for bacterial communities in vegetated (V1 to V3) and unvegetated (U1 to U3) sediment samples based on Ion Torrent sequencing of the 16S rRNA genes and Greengenes classification.
Although the overall bacterial communities were insignificantly differentiated, there were substantial changes in the relative abundance of some individual taxa between the two sediment types (Table 2). For example, bacteria of the phylum Chloroflexi occurred much more frequently in vegetated sediments (on average, 9.1%) than in bare sediments (on average, 4.0%) (P = 0.023), and these mainly consisted of the class Anaerolineae (6.8% in vegetated sediments versus 2.3% in bare sediments). Higher proportions of other taxa were also observed in seagrass sediments, including the gammaproteobacterial genus Desulfococcus (family Desulfobacteraceae; 12.5% in vegetated sediments versus 6.0% in bare sediments), the Syntrophobacteraceae (1.1 versus 0.3%), and the verrucomicrobial genus Luteolibacter (0.47 versus 0.17%). Bacteroidetes had much lower proportions in the vegetated sediments than in the bare ones (8.0 versus 18.5%) (P = 0.001), primarily due to the diminished amount of members of the family Flavobacteriaceae (4.1 versus 13.0%). Gammaproteobacterial Marinicellaceae (1.4 versus 2.8%) and actinobacterial Actinomycetales (0.2 versus 0.4%) exhibited lower rates of occurrence in the vegetated than in the bare sediments as well (P < 0.05; Table 2). The relative abundance of neither Geobacteraceae nor Desulfobulbaceae differed between these two sediment types (P > 0.70).
TABLE 2.
Shifts in relative abundance of major bacterial taxa in vegetated and unvegetated sedimentsa
| Taxonomy | % relative abundance (mean ± SE) in: |
P | |
|---|---|---|---|
| Vegetated sediment | Unvegetated sediment | ||
| Acidobacteria | 2.60 ± 0.32 | 2.43 ± 0.35 | 0.743 |
| BPC102, B110 | 0.40 ± 0.10 | 0.10 ± 0.00 | 0.040 |
| Actinobacteria | 5.00 ± 0.21 | 7.17 ± 1.58 | 0.246 |
| Acidimicrobiales | 4.50 ± 0.36 | 6.70 ± 2.82 | 0.251 |
| Actinomycetales | 0.17 ± 0.03 | 0.37 ± 0.03 | 0.013 |
| WCHB1-81, At425_EubF1 | 0.23 ± 0.03 | 0.10 ± 0.00 | 0.016 |
| Bacteroidetes | 8.03 ± 0.90 | 18.50 ± 0.95 | 0.001 |
| Flavobacteriales | 4.37 ± 0.81 | 13.97 ± 1.60 | 0.006 |
| Flavobacteriaceae | 4.13 ± 0.78 | 13.00 ± 1.46 | 0.006 |
| Lutimonas | 2.40 ± 0.59 | 5.27 ± 0.75 | 0.040 |
| Winogradskyella | 0.23 ± 0.09 | 1.53 ± 0.39 | 0.032 |
| Ulvibacter | 0.17 ± 0.03 | 0.87 ± 0.15 | 0.009 |
| [Rhodothermales], Rhodothermaceae | 0.00 ± 0.00 | 0.13 ± 0.03 | 0.016 |
| Chlorobi | 0.00 ± 0.00 | 0.10 ± 0.00 | <0.001 |
| Chloroflexi | 9.07 ± 1.37 | 4.03 ± 0.32 | 0.023 |
| Anaerolineae | 6.83 ± 1.13 | 2.33 ± 0.34 | 0.019 |
| GCA004 | 1.10 ± 0.12 | 0.43 ± 0.13 | 0.019 |
| OPB11 | 0.33 ± 0.07 | 0.03 ± 0.03 | 0.016 |
| SHA-20 | 2.27 ± 0.50 | 0.47 ± 0.03 | 0.023 |
| Dehalococcoidetes | 0.13 ± 0.03 | 0.00 ± 0.00 | 0.016 |
| Cyanobacteria | 0.20 ± 0.10 | 0.30 ± 0.12 | 0.548 |
| Firmicutes | 2.80 ± 0.79 | 2.20 ± 0.15 | 0.499 |
| Lentisphaerae | 0.07 ± 0.03 | 0.17 ± 0.07 | 0.251 |
| [Lentisphaeria], Z20 | 0.00 ± 0.00 | 0.13 ± 0.03 | 0.016 |
| Nitrospirae | 0.10 ± 0.00 | 0.00 ± 0.00 | <0.001 |
| [Thermodesulfovibrionaceae], LCP-6 | 0.10 ± 0.00 | 0.00 ± 0.00 | <0.001 |
| Planctomycetes | 1.80 ± 0.25 | 1.73 ± 0.22 | 0.851 |
| Verrucomicrobia | 2.63 ± 0.38 | 2.20 ± 0.26 | 0.406 |
| Verrucomicrobiaceae, Luteolibacter | 0.47 ± 0.09 | 0.17 ± 0.03 | 0.033 |
| Proteobacteria | 64.67 ± 2.80 | 57.93 ± 2.31 | 0.137 |
| Alphaproteobacteria | 5.00 ± 1.23 | 7.27 ± 1.02 | 0.230 |
| Rhizobiales | 2.20 ± 0.70 | 1.57 ± 0.40 | 0.246 |
| Rhodobacterales | 2.33 ± 1.31 | 5.33 ± 1.47 | 0.058 |
| Betaproteobacteria | 0.00 ± 0.00 | 0.07 ± 0.03 | 0.116 |
| Deltaproteobacteria | 33.50 ± 6.15 | 19.53 ± 2.82 | 0.108 |
| Desulfarculales | 0.33 ± 0.03 | 0.10 ± 0.06 | 0.025 |
| Desulfobacterales | 17.83 ± 1.67 | 9.97 ± 2.35 | 0.052 |
| Desulfobacteraceae | 15.10 ± 1.63 | 7.10 ± 2.25 | 0.045 |
| Desulfococcus | 12.47 ± 1.40 | 6.00 ± 1.68 | 0.042 |
| Desulfobulbaceae | 2.70 ± 0.12 | 2.87 ± 0.35 | 0.673 |
| Desulfuromonadales | 11.07 ± 5.49 | 6.27 ± 0.44 | 0.433 |
| Desulfuromonadaceae | 10.87 ± 5.50 | 6.17 ± 0.48 | 0.442 |
| Geobacteraceae | 0.07 ± 0.07 | 0.07 ± 0.07 | 1.000 |
| NB1-j | 0.47 ± 0.03 | 0.17 ± 0.03 | 0.003 |
| Syntrophobacterales | 1.17 ± 0.03 | 0.30 ± 0.06 | <0.001 |
| Syntrophobacteraceae | 1.07 ± 0.07 | 0.30 ± 0.06 | 0.001 |
| Epsilonproteobacteria | 14.67 ± 3.13 | 17.23 ± 4.04 | 0.642 |
| Campylobacterales | 14.67 ± 3.13 | 17.23 ± 4.04 | 0.642 |
| Helicobacteraceae | 14.63 ± 3.11 | 17.17 ± 4.03 | 0.645 |
| Gammaproteobacteria | 11.53 ± 0.45 | 13.83 ± 1.27 | 0.164 |
| Alteromonadales | 3.83 ± 0.39 | 5.37 ± 0.50 | 0.075 |
| OM60 | 3.70 ± 0.42 | 4.53 ± 0.54 | 0.287 |
| Alteromonadaceae | 0.00 ± 0.00 | 0.13 ± 0.03 | 0.016 |
| Chromatiales | 4.03 ± 0.24 | 2.83 ± 0.43 | 0.070 |
| [Marinicellales], [Marinicellaceae] | 1.40 ± 0.15 | 2.83 ± 0.32 | 0.015 |
| Thiotrichales | 1.27 ± 0.25 | 1.93 ± 0.40 | 0.072 |
| Others | 2.83 ± 1.64 | 3.20 ± 1.85 | 0.635 |
Data are based on Ion Torrent sequencing of bacterial 16S rRNA genes and Greengenes taxonomy. P values were derived from t tests (n = 3). Significant differences (P < 0.05) are highlighted in bold. Taxon names above the rank of genus in square brackets are names proposed by the Greengenes curators and will not be found in NCBI databases.
DISCUSSION
Comparison of environmental factors.
In this study, we characterized both the sediment properties and the diazotrophic diversity in eelgrass-colonized and adjacent surface sediments in a coastal lagoon (Swan Lake) of the Yellow Sea. Our results showed lower levels of NH4+-N in the vegetated sediments, suggesting that Z. marina seagrass roots take up NH4+-N as a favorable N source (2, 9, 46). The seagrass sediments were also characterized by finer particles and lower ratios of TOC/TON. These findings indicate that the seagrass affects water flow, leading to increased sedimentation, reduced sediment resuspension and erosion (47), and high rates of retention of organic nitrogen from seagrass debris (1). Because finer fractions carry most of the metals in natural sediments (48), we expected that markedly higher concentrations of many metals would be found in the vegetated than in the bare sediments. Compared with the metal concentrations in surface sediment samples collected from deeper sites (15 to 75 m) in the Bohai Sea and the Yellow Sea, where the sediment grain size was much finer (49), the concentrations of all metals, with the exception of Cd, were markedly lower in the present study, suggesting worse Cd pollution in Swan Lake.
Benthic diazotroph composition and abundance.
To the best of our knowledge, the present study provides the first investigation of the genetic diversity of the benthic diazotrophs in a Zostera marina ecosystem. We found that nifH phylotypes related to deltaproteobacterial SRB (Desulfobulbaceae) and gammaproteobacterial Geobacteraceae dominated the diazotrophic communities in both vegetated and unvegetated sediments. Diverse diazotrophs affiliated with Gammaproteobacteria and anaerobes (putatively SRB) were detected in the rhizosphere of Spartina alterniflora (14), and Alpha-, Gamma-, Betaproteobacteria and anaerobes were found in the sediments colonized by the seagrasses Thalassia testudinum and Syringodium filiforme (15). Compared with the findings of these former studies, we found some common diazotrophic groups with higher taxonomic ranks, e.g., Gammaproteobacteria and anaerobes that are most closely affiliated with deltaproteobacterial SRB. However, the major nifH phylotypes identified in this study are not closely related to those previously revealed for sediments of other seagrasses, suggesting that different seagrass species, different sampling seasons, geographic separation, and local environmental conditions may affect the diversity and distribution of N2-fixing bacteria in the seagrass-associated sedimentary environment.
Diazotrophic populations of cyanobacteria and methanogenic archaea might be present in the Swan Lake samples, but the nifH sequences of these two groups, classified within clusters I and III, were not detected in this study. This could be due to the PCR bias of the primer set PolF/PolR, which would likely preclude recovery of these two groups (50, 51). Furthermore, cyanobacterial 16S rRNA genes were found to be rare in our Ion Torrent sequencing data set, representing only 0.1 to 0.5% of the sequences in the sets of sequences representing the entire bacterial communities; most of these cyanobacteria were also affiliated with nondiazotrophic Synechococcus in our samples. In fact, cyanobacterial nifH sequences were not detected in the sediments of Narragansett Bay, even though primers whose sequences matched the nifH sequences of most cyanobacteria were applied for PCR amplification (44). These findings indicate that nitrogen-fixing cyanobacteria were very likely not present or were minor members of the diazotrophic communities in these coastal sediments.
The results of the qPCR assays with the nifH genes indicated the significantly higher genetic potential of diazotrophy in the seagrass meadows than in the adjacent bare sediments, a finding which supports previous measurements of N2 fixation activities by acetylene reduction (1, 4). However, the results of qPCR with 16S rRNA genes showed that the abundance of the whole bacterial community was not significantly different between the seagrass and bare sediments, suggesting that diazotrophs are selectively promoted in seagrass meadows. Furthermore, we showed that nifH genes affiliated with SRB accounted for more than 1/3 of the diazotrophs in the seagrass-colonized sediments. This finding provides molecular evidence that SRB are important players in N2 fixation in seagrass-associated sedimentary environments (1, 4, 52).
It has been reported that the nifH gene abundance ranges from 1.0 × 106 to 6.0 × 108 copies g−1 sediment in several marine benthic habitats (44, 53, 54). Our data on nifH gene abundance in the shallow seagrass system are consistent with those reported for a Brazilian mangrove (53), a subtropical coastal lagoon (54), and the Narragansett Bay and the southern coast of Rhode Island and Massachusetts (44), but the nifH gene abundance that we found is higher than that from deep-sea sites in the northern South China Sea (55), suggesting a spatial gradient of benthic N2 fixation potential from shallow, plant-dwelling coastal systems to deep oceans.
Fe(III)-reducing bacteria and sulfate-reducing bacteria as dominant diazotrophs in coastal sediments.
Our phylogenetic analyses of the NifH amino acid sequences translated from the nifH sequence and the bacterial 16S rRNA gene sequences indicated the presence of an N2-fixing bacterial group belonging to the family Geobacteraceae in the seagrass sediments. Members of the Geobacteraceae are known to be Fe(III)-reducing bacteria and electrogens that are directly involved in using a wide range of organic compounds (e.g., acetate, butyrate, propionate, and aromatic compounds) as electron donors and transferring electrons to reduce Fe(III) and Mn(IV) oxides or other electron acceptors (56, 57). In addition to supplying fixed nitrogen to meet the needs for the rapid growth of seagrass, the Fe(II) produced by these Fe(III)-reducing bacteria might relieve the toxicity of sulfide, an end product of sulfate reduction, by precipitation to pyrite.
Phylogenetic analyses of both the nifH and 16S rRNA genes indicated that some members of two Desulfobulbaceae genera (i.e., Desulfocapsa and Desulfobulbus) are likely involved in nitrogen fixation in the seagrass sediments. Indeed, the cultured species Desulfobulbus mediterraneus is known to use lactate and sugars as electron donors for the reduction of sulfate, sulfite, or thiosulfate, with acetate and CO2 being the major carbon products (58), and a nifH gene has been identified in the genome sequence of this organism.
Desulfocapsa-related nifH phylotypes represented another major diazotrophic group in the seagrass sediment. With reference to the existing physiological and genomic data for Desulfocapsa sulfoexigens (59, 60), members of the genus Desulfocapsa could fix nitrogen gas and grow by disproportionating elemental sulfur to sulfide and sulfate under anaerobic conditions using CO2 as their sole carbon source. In the presence of Fe(III) or other hydrogen sulfide-scavenging agents, the disproportionation of sulfur contributes to the maintenance of low sulfide concentrations, even if there is intense sulfide production through sulfate reduction (59). Furthermore, the type strain of Desulfocapsa was also isolated from the marine sediment covered by Zostera noltii (59). These findings suggest that, in addition to diazotrophy, sulfur-disproportionating Desulfocapsa might also contribute to the scavenging of hydrogen sulfide in iron-rich seagrass sediments.
Another major OTU detected from the Zostera marina sediments is closely related to Pseudomonas stutzeri, which is a versatile bacterium involved in multiple biogeochemical processes, such as nitrate-dependent Fe(II) oxidation (61) and the oxidation of thiosulfate to tetrathionate and to elemental sulfur (62). It is possible that the P. stutzeri-related diazotrophs interact with Geobacteraceae and Desulfobulbaceae by regenerating Fe(III) and supplying elemental sulfur.
Our study shows that the major nifH phylotypes in seagrass sediments, SL1 of Fe(III)-reducing bacteria and SL2 of Desulfobulbaceae SRB, actually represent the same groups called NB3 and NB7, respectively, elsewhere (43, 44). The NifH amino acid sequences of the Desulfobulbus-related SL4 translated from the nifH sequence form a lineage different from the NB10 group (data not shown), a minor group related to Desulfovibrio (44). This suggests that SL4 could represent an important and previously unrecognized diazotrophic group in coastal sediments. In terms of the dominance of NB3 and NB7 in marine benthic diazotrophs, our results are largely consistent with those of previous studies of nifH clone libraries for bare sediments (43, 44, 63). Therefore, the findings of our work on seagrass beds and these previous studies suggest the prevalence of iron(III)-reducing and sulfate-reducing bacteria in benthic diazotrophic communities across diverse coastal marine ecosystems.
Distribution of diazotrophs in seagrass-colonized and nearby bare sediments.
This study of two types of sediments in the same season may allow us to detect the spatial patterns of diazotrophic communities and the environmental factors that drive these patterns. However, even though some physicochemical factors (e.g., grain size, the nitrogen nutrients found in pore water, and the concentrations of many metals) exhibited differences between the vegetated and unvegetated surface sediments, statistically supported differences between the two types of sediments according to either their amino acid-based clone libraries or the results of nifH gene-based T-RFLP analyses were found, providing little evidence that seagrass colonization is a primary factor structuring the diazotrophic community in these sediments. From an ecological point of view, a relatively stable functional potential of a benthic diazotrophic community is too important to lose, as nitrogen fixation fuels primary productivity and balances the nitrogen loss due to denitrification and anaerobic ammonia oxidation in anaerobic sediments. In the comparison of the two sediment types, the highly heterogeneous microenvironments inside seagrass meadows may partly account for the undifferentiated structuring of diazotrophic communities. For instance, site V2 was characterized by having a relatively lower TOC/TON ratio, pH, and concentration of Cd, which makes the environment and the diazotrophic community at this site more similar to those at unvegetated sites than to those at other vegetated sites (Fig. 4B). Although clone library and T-RFLP analyses yielded valuable information on the diazotrophic community structure and composition, these methods are not without limitations. Some rare or minor nifH genotypes presumably defining the difference between the niches could be undersampled or discarded, and some phylotypes may not be resolved. All these possibilities may make these methods not sensitive enough to distinguish diazotrophic communities with high levels of diversity. Nevertheless, it should be noted that the similar functional potential of nifH genes does not mean similar activities in these two sediment types, as the abundance and activity of these phylotypes are most likely increased in the vegetated sediments (1, 4; this study).
On the basis of the T-RFLP analysis of nifH genes, our RDA results reflect correlations between the structuring of nifH genotypes and environmental factor parameters across all samples on a local scale. This result, which differs from that of a comparison of the NifH protein sequences translated from the nifH sequences, shows another aspect of the diazotrophic genotype distribution in seagrass-associated sediments and can be explained by the much higher sequence divergence of nifH genes than their NifH protein products (64). Similarly, physicochemical parameters were found to correlate with differences among nifH gene pools in soils on a microscale (65).
The main sources for organic matter in seagrass sediments include seagrass root exudates, fallings, and phytoplankton debris, which exhibit different carbon-to-nitrogen ratios and decomposition rates (1). Our RDA plot demonstrates that changes in the diazotrophic community structure at the DNA level significantly covary with TOC/TON in sediments, suggesting an effect of carbon sources on the nifH genotype distribution in seagrass-associated sediments. The variation in the water pH in the shallow waters is known to be positively related to the photosynthetic activities, which influence the belowground release of DOC and O2 penetration via roots. This explains the link between the pH of the overlying water and the diazotrophic community (Fig. 4B). Additionally, the heavy metal Cd appears to be an important factor that influences the diazotrophic community structure, which could be due to the strong toxicity of Cd to diazotrophs and sulfate-reducing bacteria, as noted in previous studies (66, 67).
Selectively enriched bacterial populations in seagrass-colonized or bare sediments.
To our knowledge, our study is the first to investigate bacterial communities in seagrass-vegetated and unvegetated sediments using a high-throughput sequencing technology. Our results showed that Delta- and Gammaproteobacteria dominated the bacterial communities in Zostera-colonized marine sediments, as previously indicated (18, 19). In the comparison of the bacterial communities in these two sediment types, we did not detect statistically significant differences in abundances either in the entire bacterial community or in the dominating Deltaproteobacteria, which hosts most sulfate-reducing bacteria. This result is largely in line with previous conclusions (e.g., see references 7 and 20). With the power of high-throughput sequencing technology, we demonstrate that there are a number of major bacterial lineages with contrasting relative abundances in vegetated and unvegetated sediments (Table 2), a new finding indicating the stimulation of specific bacterial populations in either sediment type. Notable lineages are the genus Desulfococcus, the class Anaerolineae, and the family Flavobacteriaceae, of which the representative members are known to be heterotrophic bacteria involved in carbon cycling. Nevertheless, the TOC contents in these two sediments were determined to be similar in this study, indicating that the quantity of TOC hardly explains the selective enrichments of these lineages in either sediment type.
The pool of organic matter in the seagrass-colonized sediments consists of deposited phytoplankton-, root-, and rhizome-leached soluble organic matter that is readily used by bacteria and a relatively enduring particulate fraction (largely celluloses and hemicelluloses) that is retained in sediments and carried over from year to year (68). The unvegetated nearby sediments used to be dominated by seagrasses, so a large amount of particulate root material remains. Due to resuspension and transportation by waves and currents, phytoplankton deposition on the sediment surface may not be so different between the two sediment types in the shallow lagoon. A main difference in the source of organic matter between these two sediment types could be the input of root exudates (68). The composition of sedimentary organic matter was not determined in this study, but our measurements of TOC/TON ratios provide evidence for these suggestions. It is well-known that C/N ratios in algae typically range from 4 to 10, whereas vascular plants (e.g., seagrasses) have much higher C/N ratios (≥20) owing to the abundance of cellulose (69). Thus, the C/N ratios determined in this study (on average, 6.6 in vegetated sediments versus 11.3 in unvegetated sediments) support the notion that the organic matter pool in the bare sediments has a higher proportion of plant debris, whereas the sediments colonized by Zostera marina have more bioavailable organic matter, putatively root exudates.
The differences in the quality and source of organic matter might have caused selective enrichments of bacterial populations in the vegetated and unvegetated sediments. Desulfococcus SRB are known to be capable of the complete oxidation of a variety of electron donors to CO2 (70) and the degradation of short-chain hydrocarbons (e.g., 71). High cell numbers of Desulfococcus-related populations have also been found in coastal marine and hydrocarbon seep sediments (72–74). All these findings are consistent with our observation that Desulfococcus is one of the most abundant genera in the Ion Torrent sequencing data set. As mentioned above, the vegetated sediments might have diverse and more bioavailable organic matter, which favors the nutritionally versatile Desulfococcus populations. On the other hand, Flavobacteria are proficient in degrading the high-molecular-mass fraction of organic matter (e.g., cellulose and chitin) (75), which explains why Flavobacteria were more abundant in the bare sediments, which putatively possess larger amounts of particulate organic matter.
The exudates and oxygen released from roots in the vegetated sediments may stimulate Anaerolineae populations. Studies on cultured representatives of the Anaerolineae and metagenomic data for the Anaerolineae have indicated that these bacteria are capable of both aerobic sugar respiration and anaerobic fermentation of sugars and amino acids (76, 77). A high proportion of Anaerolineae populations was also found in rice field soil-based microbial fuel cells (78). These previous findings and our data suggest that the physiological flexibility of Anaerolineae also contributes to their persistence and competitiveness in alternating anaerobic/aerobic environments, such as the seagrass-colonized sediments.
Concluding remarks.
Using a range of molecular tools targeting the nifH and 16S rRNA genes, we detected highly diverse diazotrophs and bacterial phylogenetic groups in sediments of a Z. marina-colonized lagoon. Our study provides molecular evidence that sulfate-reducing bacteria are the most dominant diazotrophic populations in the sediments associated with seagrasses. A higher nifH gene abundance was found in the vegetated sites than the nearby sediments, confirming the results of previous studies on N2-fixing activities (1, 4–7). We also found that the Geobacteraceae (iron-reducing bacteria) and the Desulfobulbaceae (sulfate-reducing bacteria) could be dominant in diazotrophic communities of a seagrass system, a finding similar to that of previous work on other coastal habitats (43, 44). This indicates that diazotrophs of the Geobacteraceae and Desulfobulbaceae could be prevalent in coastal marine benthos and that they have multiple metabolic potentials in carbon, sulfur, and iron cycles in a sedimentary environment.
The high-throughput sequencing revealed that the overall bacterial communities in both vegetated and unvegetated sediments were dominated by Delta-, Epsilon-, and Gammaproteobacteria, none of which showed a statistically significantly different relative abundance between these two niches, indicating relatively stable bacterial communities in these sediments. Nevertheless, dramatic shifts in proportions across sediment types were recorded for some bacterial lineages (e.g., Desulfococcus, Anaerolineae, and Flavobacteriaceae), a new finding from this study indicating that the metabolically versatile and oxygen-tolerant anaerobic populations are selectively enriched in vegetated sediments, whereas the populations degrading the high-molecular-mass fraction of organic matter were stimulated in bare sediments. When these findings are taken together, our study highlights the impact of seagrasses on the benthic chemistry, microbial diversity, and biogeochemical potential in coastal marine ecosystems.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by a grant from the Natural Science Foundation of China (no. 41206155), awarded to X.Z.; the Strategic Priority Research Program of CAS (no. XDA11020702); the CAS Scientific Innovation Program-Interdisciplinary Field; the Natural Science Foundation for Distinguished Young Scholars of Shandong, China (no. JQ201210); a grant from the Science and Technology Development Program of Yantai, China (no. 2014ZH073), awarded to J.G.; and the NSFC project (no. 41371257), awarded to F.L. F.S. was supported by a joint studentship of YICZR.
We thank Bin Ma, Baoquan Li, and Qiuying Han for the sample collection.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.01382-15.
REFERENCES
- 1.Duarte CM, Holmer M, Marbh N. 2005. Plant-microbe interactions in seagrass meadows, p 31–59. In Kristensen E, Haese RR, Kostka JE (ed), Interactions between macro- and microorganisms in marine sediments. Coastal and estuarine studies, vol 60 American Geophysical Union, New York, NY. [Google Scholar]
- 2.Caffrey JM, Kemp WM. 1990. Nitrogen cycling in sediments with estuarine populations of Potamogeton perfoliatus and Zostera marina. Mar Ecol Prog Ser 66:147–160. doi: 10.3354/meps066147. [DOI] [Google Scholar]
- 3.Miyajima T, Koike I, Yamano H, Iizumi H. 1998. Accumulation and transport of seagrass-derived organic matter in reef flat sediment of Green Island, Great Barrier Reef. Mar Ecol Prog Ser 175:251–259. doi: 10.3354/meps175251. [DOI] [Google Scholar]
- 4.Devereux R. 2005. Seagrass rhizosphere microbial communities, p 199–216. In Kristensen E, Haese RR, Kostka JE (ed), Interactions between macro- and microorganisms in marine sediments. Coastal and estuarine studies, vol 60 American Geophysical Union, New York, NY. [Google Scholar]
- 5.Moriarity DJW, Iverson RI, Pollard PC. 1986. Exudation of organic carbon by the seagrass Halodule wrightii Aschers, and its effect on bacterial growth in the sediment. J Exp Mar Biol Ecol 96:115–126. doi: 10.1016/0022-0981(86)90237-6. [DOI] [Google Scholar]
- 6.Holmer M, Andersen FØ, Nilesen SL, Boschker HTS. 2001. The importance of mineralization based on sulfate reduction for nutrient regeneration in tropical seagrass sediments. Aquat Bot 71:1–17. doi: 10.1016/S0304-3770(01)00170-X. [DOI] [Google Scholar]
- 7.Smith AC, Kostka JE, Devereux R, Yates DF. 2004. Seasonal composition and activity of sulfate reducing prokaryotic communities in seagrass bed sediment. Aquat Microb Ecol 37:183–195. doi: 10.3354/ame037183. [DOI] [Google Scholar]
- 8.Küsel K, Trinkwalter T, Drake HL, Devereux R. 2006. Comparative evaluation of anaerobic bacterial communities associated with roots of submerged macrophytes growing in marine or brackish water sediments. J Exp Mar Biol Ecol 337:49–58. doi: 10.1016/j.jembe.2006.06.004. [DOI] [Google Scholar]
- 9.Welsh DT. 2000. Nitrogen fixation in seagrass meadows: regulation, plant-bacteria interactions and significance to primary productivity. Ecol Lett 3:58–71. doi: 10.1046/j.1461-0248.2000.00111.x. [DOI] [Google Scholar]
- 10.Capone DG, Budin JM. 1982. Nitrogen-fixation associated with rinsed roots and rhizomes of the eelgrass Zostera marina. Plant Physiol 70:1601–1604. doi: 10.1104/pp.70.6.1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nielsen LB, Finster K, Welsh DT, Donelly A, Herbert RA, De Wit R, Lomstein BA. 2001. Sulphate reduction and nitrogen fixation rates associated with roots, rhizomes and sediments from Zostera noltii and Spartina maritima meadows. Environ Microbiol 3:63–71. doi: 10.1046/j.1462-2920.2001.00160.x. [DOI] [PubMed] [Google Scholar]
- 12.Shieh WY, Simidu U, Maruyama Y. 1989. Enumeration and characterization of nitrogen-fixing bacteria in an eelgrass (Zostera marina) bed. Microb Ecol 18:249–259. doi: 10.1007/BF02075812. [DOI] [PubMed] [Google Scholar]
- 13.Raja S, Thangaradjou T, Sivakumar K, Kannan L. 2012. Rhizobacterial population density and nitrogen fixation in seagrass community of Gulf of Mannar, India. J Environ Biol 33:1033–1037. [PubMed] [Google Scholar]
- 14.Lovell CR, Piceno YM, Quattro JM, Bagwell CE. 2000. Molecular analysis of diazotroph diversity in the rhizosphere of the smooth cordgrass, Spartina alterniflora. Appl Environ Microbiol 66:3814–3822. doi: 10.1128/AEM.66.9.3814-3822.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bagwell CE, La Rocque JR, Smith GW, Polson SW, Friez MJ, Longshore JW, Lovell CR. 2002. Molecular diversity of diazotrophs in oligotrophic tropical seagrass bed communities. FEMS Microbiol Ecol 39:113–119. doi: 10.1111/j.1574-6941.2002.tb00912.x. [DOI] [PubMed] [Google Scholar]
- 16.Thamdrup B. 2000. Bacterial manganese and iron reduction in aquatic sediments. Adv Microb Ecol 16:41–84. doi: 10.1007/978-1-4615-4187-5_2. [DOI] [Google Scholar]
- 17.Marbà N, Holmer M, Gacia E, Barrón C. 2006. Seagrass beds and coastal biogeochemistry, p 135–157. In Larkum AWD, Orth RJ, Duarte CM (ed), Seagrasses: biology, ecology and conservation. Springer, Amsterdam, Netherlands. [Google Scholar]
- 18.Cifuentes A, Antón J, Benlloch S, Donnelly A, Herbert RA, Rodrígues-Valera F. 2000. Prokaryotic diversity in Zostera noltii-colonized marine sediments. Appl Environ Microbiol 66:1715–1719. doi: 10.1128/AEM.66.4.1715-1719.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jensen SI, Kühl M, Priemé A. 2007. Different bacterial communities associated with the roots and bulk sediment of the seagrass Zostera marina. FEMS Microbiol Ecol 62:108–117. doi: 10.1111/j.1574-6941.2007.00373.x. [DOI] [PubMed] [Google Scholar]
- 20.James JB, Sherman TD, Devereux R. 2006. Analysis of bacterial communities in seagrass bed sediments by double-gradient denaturing gradient gel electrophoresis of PCR-amplified 16S rRNA genes. Microb Ecol 52:655–661. doi: 10.1007/s00248-006-9075-3. [DOI] [PubMed] [Google Scholar]
- 21.Zhang X, Song Y, Liu D, Keesing JK, Gong J. 18 December 2014. Macroalgal blooms favor heterotrophic diazotrophic bacteria in nitrogen-rich and phosphorus-limited coastal surface waters in the Yellow Sea. Estuar Coast Shelf Sci doi: 10.1016/j.ecss.2014.12.015. [DOI] [Google Scholar]
- 22.Zhang X, Agogué H, Dupuy C, Gong J. 2014. Relative abundance of ammonia oxidizers, denitrifiers, and anammox bacteria in sediments of hyper-nutrified estuarine tidal flats and in relation to environmental conditions. Clean Soil Air Water 42:815–823. doi: 10.1002/clen.201300013. [DOI] [Google Scholar]
- 23.Poly F, Monrozier LJ, Bally R. 2001. Improvement in the RFLP procedure for studying the diversity of nifH genes in communities of nitrogen fixers in soil. Res Microbiol 152:95–103. doi: 10.1016/S0923-2508(00)01172-4. [DOI] [PubMed] [Google Scholar]
- 24.Zehr JP, Jenkins BD, Short SM, Steward GF. 2003. Nitrogenase gene diversity and microbial community structure: a cross-system comparison. Environ Microbiol 5:539–554. doi: 10.1046/j.1462-2920.2003.00451.x. [DOI] [PubMed] [Google Scholar]
- 25.Raymond J, Siefert JL, Staples CR, Blankenship RE. 2004. The natural history of nitrogen fixation. Mol Biol Evol 21:541–554. [DOI] [PubMed] [Google Scholar]
- 26.Kechris KJ, Lin JC, Bickel PJ, Glazer AN. 2006. Quantitative exploration of the occurrence of lateral gene transfer by using nitrogen fixation genes as a case study. Proc Natl Acad Sci U S A 103:9584–9589. doi: 10.1073/pnas.0603534103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Muyzer G, De Waal EC, Uitterlinden AG. 1993. Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl Environ Microbiol 59:695–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Caporaso JG, Kuczynki J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. 2010. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Edgar RC. 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 30.McDonald D, Price MN, Goodrich J, Nawrocki EP, DeSantis TZ, Probst A, Andersen GL, Knight R, Hugenholtz P. 2012. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J 6:610–618. doi: 10.1038/ismej.2011.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Caporaso JG, Bittinger K, Bushman FD, DeSantis TZ, Andersen GL, Knight R. 2010. PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics 26:266–267. doi: 10.1093/bioinformatics/btp636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Haas BJ, Gevers D, Earl AM, Feldgarden M, Ward DV, Giannoukos G, Ciulla D, Tabbaa D, Highlander SK, Sodergren E, Methé B, DeSantis TZ, The Human Microbiome Consortium, Petrosino JF, Knight R, Birren BW. 2011. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res 21:494–504. doi: 10.1101/gr.112730.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hall TA. 1999. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser 41:95–98. [Google Scholar]
- 34.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF. 2009. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75:7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dang H, Luan X, Zhao J, Li J. 2009. Diverse and novel nifH and nifH-like gene sequences in the deep-sea methane seep sediments of the Okhotsk Sea. Appl Environ Microbiol 75:2238–2245. doi: 10.1128/AEM.02556-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Good IJ. 1953. The population frequencies of species and the estimation of population parameters. Biometrika 40:237–264. [Google Scholar]
- 37.Thompson JD, Higgins DG, Gibson TJ. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Darriba D, Taboada GL, Doallo R, Posada D. 2011. ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics 27:1164–1165. doi: 10.1093/bioinformatics/btr088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. 2013. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol 30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hamady M, Lozupone C, Knight R. 2010. Fast UniFrac: facilitating high-throughput phylogenetic analyses of microbial communities including analysis of pyrosequencing and PhyloChip data. ISME J 4:17–27. doi: 10.1038/ismej.2009.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.ter Braak CJF, Smilauer P. 2002. CANOCO reference manual and CanoDraw for Windows user's guide: software for canonical community ordination (version 4.5). Microcomputer Power, Ithaca, NY. [Google Scholar]
- 43.Fulweiler RW, Brown SM, Nixon SW, Jenkins BD. 2013. Evidence and a conceptual model for the co-occurrence of nitrogen fixation and denitrification in heterotrophic marine sediments. Mar Ecol Prog Ser 482:57–68. doi: 10.3354/meps10240. [DOI] [Google Scholar]
- 44.Brown SM, Jenkins BD. 2014. Profiling gene expression to distinguish the likely active diazotrophs from a sea of genetic potential in marine sediments. Environ Microbiol 16:3128–3142. doi: 10.1111/1462-2920.12403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pfeffer C, Larsen S, Song J, Dong M, Besenbacher F, Meyer RL, Kjeldsen KU, Schreiber L, Gorby YA, El-Naggar MY, Leung KM, Schramm A, Risgaard-Petersen N, Nielsen LP. 2012. Filamentous bacteria transport electrons over centimetre distances. Nature 491:218–221. doi: 10.1038/nature11586. [DOI] [PubMed] [Google Scholar]
- 46.Welsh DT, Bourgues S, de Wit R, Herbert RA. 1996. Seasonal variations in nitrogen-fixation (acetylene reduction) and sulphate-reduction rates in the rhizosphere of Zostera noltii: nitrogen fixation by sulphate-reducing bacteria. Mar Biol 125:619–628. doi: 10.1007/BF00349243. [DOI] [Google Scholar]
- 47.Gacia E, Duane CM. 2001. Sediment retention by a Mediterranean Posidonia oceanica meadow: the balance between deposition and resuspension. Estuar Coast Shelf Sci 52:505–514. doi: 10.1006/ecss.2000.0753. [DOI] [Google Scholar]
- 48.Maslennikova S, Larina N, Larin S. 2012. The effect of sediment grain size on heavy metal content. Lakes Reserv Ponds 6:43–54. [Google Scholar]
- 49.Gong J, Shi F, Ma B, Dong J, Pachiadaki M, Zhang X, Edgcomb VP. 11 January 2015. Depth shapes α- and β-diversities of microbial eukaryotes in surficial sediments of coastal ecosystems. Environ Microbiol doi: 10.1111/1462-2920.12763. [DOI] [PubMed] [Google Scholar]
- 50.Bauer K, Díez B, Lugomela C, Seppälä S, Borg AJ, Bergman B. 2008. Variability in benthic diazotrophy and cyanobacterial diversity in a tropical intertidal lagoon. FEMS Microbiol Ecol 63:205–221. doi: 10.1111/j.1574-6941.2007.00423.x. [DOI] [PubMed] [Google Scholar]
- 51.Gaby JC, Buckley DH. 2012. A comprehensive evaluation of PCR primers to amplify the nifH gene of nitrogenase. PLoS One 7:e42149. doi: 10.1371/journal.pone.0042149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Donnelly AP, Herbert RA. 1999. Bacterial interactions in the rhizosphere of seagrass communities in shallow coastal lagoons. J Appl Microbiol Symp Suppl 85:151S–160S. [DOI] [PubMed] [Google Scholar]
- 53.Dias ACF, Pereira e Silva MC, Cotta SR, Dini-Andreote F, Soares FL, Salles JF, Azevedo JL, van Elsas JD, Andreote FD. 2012. Abundance and genetic diversity of nifH gene sequences in anthropogenically affected Brazilian mangrove sediments. Appl Environ Microbiol 78:7960–7967. doi: 10.1128/AEM.02273-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bernard RJ, Mortazavi B, Wang L, Ortmann AC, MacIntyre H, Burnett WC. 2014. Benthic nutrient fluxes and limited denitrification in a sub-tropical groundwater-influenced coastal lagoon. Mar Ecol Prog Ser 504:13–26. doi: 10.3354/meps10783. [DOI] [Google Scholar]
- 55.Dang H, Yang J, Li J, Luan X, Zhang Y, Gu G, Xue R, Zong M, Klotz MG. 2013. Environment-dependent distribution of the sediment nifH-harboring microbiota in the northern South China Sea. Appl Environ Microbiol 79:121–132. doi: 10.1128/AEM.01889-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Holmes DE, Finneran KT, O'Neil RA, Lovley DR. 2002. Enrichment of members of the family Geobacteraceae associated with stimulation of dissimilatory metal reduction in uranium-contaminated aquifer sediments. Appl Environ Microbiol 68:2300–2306. doi: 10.1128/AEM.68.5.2300-2306.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Holmes DE, Nicoll JS, Bond DR, Lovley DR. 2004. Potential role of a novel psychrotolerant member of the family Geobacteraceae, Geopsychrobacter electrodiphilus gen. nov., sp. nov., in electricity production by a marine sediment fuel cell. Appl Environ Microbiol 70:6023–6030. doi: 10.1128/AEM.70.10.6023-6030.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sass A, Rütters H, Cypionka H, Sass H. 2002. Desulfobulbus mediterraneus sp. nov., a sulfate-reducing bacterium growing on mono- and disaccharides. Arch Microbiol 177:468–474. doi: 10.1007/s00203-002-0415-5. [DOI] [PubMed] [Google Scholar]
- 59.Finster K, Liesack W, Thamdrup B. 1998. Elemental sulfur and thiosulfate disproportionation by Desulfocapsa sulfoexigens sp. nov., a new anaerobic bacterium isolated from marine surface sediment. Appl Environ Microbiol 64:119–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Finster KW, Kjeldsen KU, Kube M, Reinhardt R, Mussmann M, Amann R, Schreiber L. 2013. Complete genome sequence of Desulfocapsa sulfexigens, a marine deltaproteobacterium specialized in disproportionating inorganic sulfur compounds. Stand Genomic Sci 8:58–68. doi: 10.4056/sigs.3777412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Muehe EM, Gerhardt S, Schink B, Kappler A. 2009. Ecophysiology and the energetic benefit of mixotrophic Fe(II) oxidation by various strains of nitrate-reducing bacteria. FEMS Microbiol Ecol 70:335–343. doi: 10.1111/j.1574-6941.2009.00755.x. [DOI] [PubMed] [Google Scholar]
- 62.Podgorsek L, Imhoff JF. 1999. Tetrathionate production by sulfur oxidizing bacteria and the role of tetrathionate in the sulfur cycle of Baltic Sea sediments. Aquat Microb Ecol 17:255–265. doi: 10.3354/ame017255. [DOI] [Google Scholar]
- 63.Bertics VJ, Sohm JA, Treude T, Chow CET, Capone DG, Fuhrman JA, Ziebis W. 2010. Burrowing deeper into benthic nitrogen cycling: the impact of bioturbation on nitrogen fixation coupled to sulfate reduction. Mar Ecol Prog Ser 409:1–15. doi: 10.3354/meps08639. [DOI] [Google Scholar]
- 64.Rosado AS, Duarte GF, Seldin L, van Elsas JD. 1998. Genetic diversity of nifH gene sequences in Paenibacillus azotofixans strains and soil samples analyzed by denaturing gradient gel electrophoresis of PCR-amplified gene fragments. Appl Environ Microbiol 64:2770–2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Poly F, Ranjard L, Nazaret S, Gourbère F, Monrozier LJ. 2001. Comparison of nifH gene pools in soils and soil microenvironments with contrasting properties. Appl Environ Microbiol 67:2255–2262. doi: 10.1128/AEM.67.5.2255-2262.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Loka Bharathi PA, Sathe V, Chandramohan D. 1990. Effect of lead, mercury and cadmium on a sulphate-reducing bacterium. Environ Pollut 67:361–374. doi: 10.1016/0269-7491(90)90072-K. [DOI] [PubMed] [Google Scholar]
- 67.Singh S, Shrivastava AK, Singh VK. 2014. Arsenic and cadmium are inhibitors of cyanobacterial dinitrogenase reductase (nifH1) gene. Funct Integr Genomics 14:571–580. doi: 10.1007/s10142-014-0375-2. [DOI] [PubMed] [Google Scholar]
- 68.Kenworthy WJ, Thayer GW. 1984. Production and decomposition of the roots and rhizomes of seagrasses, Zostera marina and Thalassia testudinum, in temperate and subtropical marine ecosystems. Bull Mar Sci 35:364–379. [Google Scholar]
- 69.Meyers PA. 1994. Preservation of elemental and isotopic source identification of sedimentary organic matter. Chem Geol 114:289–302. doi: 10.1016/0009-2541(94)90059-0. [DOI] [Google Scholar]
- 70.Widdel F, Bak F. 1992. Gram-negative mesophilic sulfate-reducing bacteria, p 3352–3378. In Balows A, Trüper HG, Dworkin M, Harder W, Schleifer K-H (ed), The prokaryotes, 2nd ed, vol 1 Springer-Verlag, New York, NY. [Google Scholar]
- 71.Kniemeyer O, Musat F, Sievert SM, Knittel K, Wilkes H, Blumenberg M, Michaelis W, Classen A, Bolm C, Joye SB, Widdel F. 2007. Anaerobic oxidation of short-chain hydrocarbons by marine sulphate-reducing bacteria. Nature 449:898–902. doi: 10.1038/nature06200. [DOI] [PubMed] [Google Scholar]
- 72.Ravenschlag K, Sahm K, Knoblauch C, Jørgensen BB, Amann R. 2000. Community structure, cellular rRNA content, and activity of sulfate-reducing bacteria in marine Arctic sediments. Appl Environ Microbiol 66:3592–3602. doi: 10.1128/AEM.66.8.3592-3602.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Llobet-Brossa E, Rabus R, Böttcher ME, Könneke M, Finke N, Schramm A, Meyer RL, Grötzschel S, Rosselló-Mora R, Amann R. 2002. Community structure and activity of sulfate-reducing bacteria in an intertidal surface sediment: a multi-method approach. Aquat Microb Ecol 29:211–226. doi: 10.3354/ame029211. [DOI] [Google Scholar]
- 74.Kleindienst S, Ramette A, Amann R, Knittel K. 2012. Distribution and in situ abundance of sulfate-reducing bacteria in diverse marine hydrocarbon seep sediments. Environ Microbiol 14:2689–2710. doi: 10.1111/j.1462-2920.2012.02832.x. [DOI] [PubMed] [Google Scholar]
- 75.Kirchman DL. 2002. The ecology of Cytophaga-Flavobacteria in aquatic environments. FEMS Microbiol Ecol 39:91–100. doi: 10.1111/j.1574-6941.2002.tb00910.x. [DOI] [PubMed] [Google Scholar]
- 76.Yamada T, Sekiguchi Y. 2009. Cultivation of uncultured Chloroflexi subphyla: significance and ecophysiology of formerly uncultured Chloroflexi ‘subphylum I’ with natural and biotechnological relevance. Microbes Environ 24:205–216. doi: 10.1264/jsme2.ME09151S. [DOI] [PubMed] [Google Scholar]
- 77.Hug LA, Castelle CJ, Wrighton KC, Thomas BC, Sharon I, Frischkorn KR, Williams KH, Tringe SG, Banfield JF. 2013. Community genomic analyses constrain the distribution of metabolic traits across the Chloroflexi phylum and indicate roles in sediment carbon cycling. Microbiome 1:22. doi: 10.1186/2049-2618-1-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cabezas A, Pommerenke B, Boon N, Friedrich MW. 2015. Geobacter, Anaeromyxobacter and Anaerolineae populations are enriched on anodes of root exudate-driven microbial fuel cells in rice field soil. Environ Microbiol Rep 7:489–497. doi: 10.1111/1758-2229.12277. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.