

Abstract
Currently, it is estimated that only 0.001% to 15% of bacteria in any given system can be cultured by use of commonly used techniques and media, yet culturing is critically important for investigations of bacterial function. Despite this situation, few studies have attempted to link culture-dependent and culture-independent data for a single system to better understand which members of the microbial community are readily cultured. In amphibians, some cutaneous bacterial symbionts can inhibit establishment and growth of the fungal pathogen Batrachochytrium dendrobatidis, and thus there is great interest in using these symbionts as probiotics for the conservation of amphibians threatened by B. dendrobatidis. The present study examined the portion of the culture-independent bacterial community (based on Illumina amplicon sequencing of the 16S rRNA gene) that was cultured with R2A low-nutrient agar and whether the cultured bacteria represented rare or dominant members of the community in the following four amphibian species: bullfrogs (Lithobates catesbeianus), eastern newts (Notophthalmus viridescens), spring peepers (Pseudacris crucifer), and American toads (Anaxyrus americanus). To determine which percentage of the community was cultured, we clustered Illumina sequences at 97% similarity, using the culture sequences as a reference database. For each amphibian species, we cultured, on average, 0.59% to 1.12% of each individual's bacterial community. However, the average percentage of bacteria that were culturable for each amphibian species was higher, with averages ranging from 2.81% to 7.47%. Furthermore, most of the dominant operational taxonomic units (OTUs), families, and phyla were represented in our cultures. These results open up new research avenues for understanding the functional roles of these dominant bacteria in host health.
INTRODUCTION
Microorganisms represent a large portion of global biodiversity and are responsible for many important ecological functions, yet the majority of microbes have not been cultured (1–3). It is estimated that only 0.001% to 15% of microbes can be cultured by use of commonly used techniques and media (2, 4), although some recent attempts at culturing the uncultured have been successful. For example, a prevalent and abundant human gut bacterium with potential biomedical importance was recently cultured by a gene-targeted approach (5). Still, only a third of known bacterial phyla have cultured representatives (6). Recent advances in culture-independent rRNA-based molecular approaches have allowed for a greater understanding of bacterial diversity (1, 7, 8), but in most cases, the only data we have about uncultured microbes are the DNA sequences obtained directly from the environment. Next-generation sequencing in some cases has led to the discovery of entire groups, such as the TM7 candidate division, often found in terrestrial, aquatic, and clinical habitats (9). This group is phylogenetically distinct and widespread yet has not been isolated in pure culture (9–12). There are also bacterial sequences observed in culture-independent data sets that represent uncultured isolates but appear to be closely related to commonly cultured bacteria, such as Pseudomonas spp. (13–15).
The advantages of next-generation molecular technologies for describing microbial communities should complement, not replace, an organismal approach to explaining the functional roles of microbes (16), which includes growing and studying bacteria in culture. The study of bacteria in culture has led to a greater understanding of their functions, physiology, and metabolic capabilities. For example, fermentation processes of mixed-culture bacterial consortia can be used to improve renewable energy production (17), and knowledge of metabolic pathways and products of microbes in culture has accelerated bioremediation efforts to decontaminate pollutants (18, 19). Additionally, research on spore formation by Bacillus anthracis has enhanced preparedness against biowarfare threats (20, 21), and culture-based bioassays have largely contributed to the successes of probiotic therapy and bioaugmentation in a number of systems (22–25). Experiments with microbial cultures are also invaluable for studying the fundamental bases of evolution (26, 27), community assembly (28), and biodiversity-ecosystem functional relationships (29). The evaluation and optimization of current culture techniques for organisms from diverse habitats are critical to understanding the basic and applied functions of microorganisms.
Microbial symbionts play important roles in the life processes of plants and animals (30), but few studies have assessed the culturability of these symbionts (31–36). As has been demonstrated for free-living bacteria (29), work with these symbiotic isolates can provide important information on bacterial function and can help to ground “-omics” data in organismal biology. In amphibians, there has been growing interest in the skin microbiota because some cutaneous bacterial symbionts can inhibit growth of the fungal pathogen Batrachochytrium dendrobatidis, which is responsible for global amphibian population declines and extinctions (25, 37–42). Many initial studies of the amphibian skin microbiota focused on culturing bacteria to discover individual isolates with antifungal properties (culture-dependent approaches), while more recent studies have used next-generation sequencing to characterize the structural and functional diversity of these systems (culture-independent approaches). Both approaches have shown that the skin bacterial communities of different host species are distinct (41, 43, 44). However, it is not clear how comparable the culture-dependent and culture-independent characterizations of community diversity are and, by extension, whether current sets of cultured isolates are representative of these complex communities and can thus serve as an adequate starting point for understanding the functions of the bacteria in these systems.
The objective of the present study was to determine what portion of the amphibian skin bacterial community is readily cultured by matching isolate sequence data to culture-independent sequence data obtained from the same individuals. In addition, we tested if culture-dependent patterns of bacterial diversity among amphibian species were consistent with culture-independent patterns.
MATERIALS AND METHODS
Field sampling.
We performed culture-dependent and culture-independent molecular characterizations of the bacteria associated with 64 individuals of the following four amphibian species near Blacksburg, VA: bullfrogs (Lithobates catesbeianus; n = 19), eastern newts (Notophthalmus viridescens; n = 18), spring peepers (Pseudacris crucifer; n = 12), and American toads (Anaxyrus americanus; n = 15). Bullfrogs and newts were sampled in 2010 from a single pond at Mountain Lake Biological Station (Giles County, VA). Toads and spring peepers were sampled in 2012 from the Jefferson National Forest (Montgomery County, VA) and the Town of Blacksburg's Heritage Community Park and Natural Area (Montgomery County, VA), respectively. All sites were located within 33 km of each other. Amphibians were collected in the field by hand or dip net. Each individual was handled with fresh gloves, rinsed twice with sterile water to remove environmental debris and transient microbes (45), and then swabbed twice, using two separate sterile rayon swabs (MW113; Medical Wire & Equipment). Each amphibian was swabbed by 20 strokes along the ventral side and 5 strokes along each thigh and hind foot to standardize sampling. Because of their different body shape, newts were swabbed by 10 strokes on the ventral side, 10 strokes along the dorsal/lateral sides, and 5 strokes on the hind feet. In each case, the first swab was used for culture-independent microbial community characterization by MiSeq Illumina sequencing, and the second swab was used for culture-dependent characterization by plating onto R2A medium (Difco; Becton, Dickinson and Company), prepared according to the manufacturer's instructions. Animal handling procedures were approved by Virginia Tech's Institutional Animal Care and Use Committee (protocols 08-042-BIOL, 10-029-BIOL, and 12-040-BIOL), and the study was conducted with permission from the Virginia Department of Game and Inland Fisheries (permits 38862 and 44303).
Culture-independent microbial community characterization.
The first swab taken from each individual was used for culture-independent analysis of microbial communities. The swab was placed into an empty 1.5-ml microcentrifuge tube and frozen until DNA extraction by use of a Qiagen DNeasy blood and tissue kit (Valencia, CA), with lysozyme pretreatment (44). The V4 region of the 16S rRNA gene was amplified using the primers 515F and barcoded 806R (46, 47). Triplicate reaction mixtures for each sample and PCR conditions followed the procedure described by Caporaso et al. (47). Controls without a template were run for each sample. Equimolar amplified samples were pooled, cleaned using a Qiagen QIAquick PCR Clean Up kit, and sequenced on a MiSeq Illumina platform, using a 250-bp paired-end approach (46), at the Dana-Farber Cancer Institute's Molecular Biology Core Facilities (Boston, MA).
Paired reads were assembled with Fastq-join (48; https://code.google.com/p/ea-utils/wiki/FastqJoin) and processed with the Quantitative Insights into Microbial Ecology pipeline (QIIME v. 1.8.0) (49). Sequences were demultiplexed and quality filtered following methods similar to those of Bokulich et al. (50). Specifically, sequences were discarded if (i) there were any ambiguous base calls, (ii) there were errors in the bar code, (iii) less than 50% of the read length had consecutive base calls with a phred quality score of >30, (iv) there were more than 10 consecutive low-quality base calls, or (v) the read length was not between 252 and 255 bp. After quality filtering, 6,206,929 reads were retained (number of sequences per sample, 27,693 to 173,588). Quality-filtered sequences were then clustered into operational taxonomic units (OTUs) at a sequence similarity threshold of 97% by the UCLUST method (51), and clusters with <0.001% of the total reads were discarded (50). Sequences were aligned against the Greengenes database (May 2013 release) (52), and those that did not match the database were clustered de novo at 97% sequence similarity. The most abundant sequence for a given cluster was assigned to be the representative sequence for that OTU. Taxonomy was assigned with RDP Classifier (53, 54) and the Greengenes database (52). Representative OTU sequences were aligned to the Greengenes database by use of PyNAST (55), and a 16S rRNA gene tree was constructed with FastTree (56). After the 0.001% OTU threshold was implemented, the number of sequences per sample ranged from 23,482 to 169,562. All samples were rarefied to 23,400 sequences to standardize sampling efforts. This analysis identified 4,351 Illumina OTUs across all samples. The relative abundances of the rarified reads aligned at the 97% similarity level to each of these OTUs for each sample provided the culture-independent data set that was used to examine patterns of alpha and beta diversity across the four amphibian species and to test for a correlation between the culture-dependent and -independent methods.
Culture-dependent microbial community characterization.
R2A is a low-nutrient medium originally designed for cultivation of bacteria from potable water (57). It was chosen because it is the most commonly used medium for culturing amphibian-associated bacteria (25, 41, 42, 45, 58–61) and because it supports the growth of slow-growing bacteria, which would quickly be suppressed by faster-growing species on a high-nutrient medium. The culture-dependent swabs obtained from bullfrogs and newts were plated directly onto R2A plates in the field by streaking each swab in a “W” pattern while simultaneously rotating the swab. The swabs from spring peepers and American toads were placed in 1.5-ml microcentrifuge tubes containing 100 μl TSYE-glycerol medium (2% Trypticase soy broth, 1% yeast extract, 20% glycerol) and incubated at room temperature for 45 min before storage at −80°C. These glycerol swabs were later thawed and vortexed to homogenize bacterial cells in the solution, and 30-μl samples were plated onto R2A. Because colony densities in the toad samples were too high, 1:10 dilutions of the glycerol solutions were plated to enable colony growth and isolation.
Culture plates were incubated at room temperature for 14 days, and during that time, morphologically distinct colonies were isolated into pure culture based on whole-colony color, form, margin, elevation, and substance (62). Reasoner and Geldreich (57) found that maximal bacterial counts were observed on R2A medium after 14 days of incubation at 20°C. Pure cultures were frozen at −80°C in TSYE-glycerol medium until DNA extraction. DNAs from pure bacterial cultures were extracted by using PrepMan Ultra sample preparation reagent (Applied Biosystems, Foster City, CA), an UltraClean microbial DNA isolation kit (Mo Bio Laboratories, Inc., Carlsbad, CA), or a DNeasy blood and tissue kit (Qiagen, Inc., Valencia, CA) and then amplified with the universal eubacterial primers 8F and 1492R, using the protocol described by Lauer et al. (60). Two-directional sequencing of the 16S rRNA gene was performed by Sanger DNA sequencing at either The University of Kentucky Advanced Genetic Technologies Center (Lexington, KY) or Beckman Coulter Genomics (Danvers, MA).
A total of 719 bacterial isolates were obtained from the 64 amphibians sampled. However, sequences could not be obtained for 41 of these isolates because they either could not be amplified or could not be sequenced well, despite multiple attempts at modifying PCR conditions with various cycle numbers, addition of bovine serum albumin (BSA), or use of diluted (1:10) DNA. Furthermore, 12 isolate sequences were removed from the analysis because they were not long enough to overlap the culture-independent sequences of the V4 region of the 16S rRNA gene. Quality sequences for the remaining 666 isolates were clustered into 259 OTUs at a sequence similarity threshold of 97% by using UCLUST (51) in QIIME (49), and 237 of these OTUs matched sequences in the Greengenes database. Using the same methods as those for the culture-independent sequence analysis, the most abundant sequence for a given cluster was assigned to be the representative sequence for that OTU. Taxonomy was assigned by use of RDP Classifier (53) and the Greengenes database (52). Representative sequences were aligned to the Greengenes database by use of PyNAST (55), and a 16S rRNA gene tree was constructed with FastTree (56). The resulting culture-dependent data set was used to examine patterns of alpha and beta diversity across the four amphibian species and to test for a correlation between the culture-dependent and -independent methods.
Analysis of patterns of alpha and beta diversity.
We assessed within-species diversity patterns by use of alpha diversity metrics (richness and phylogenetic diversity) computed with QIIME for each individual for both culture-independent and culture-dependent data sets. OTU richness was defined as the number of OTUs observed in a sample after rarefaction, and phylogenetic diversity was determined by Faith's phylogenetic diversity metric, which sums the branch lengths in the phylogenetic tree of each sample and is a way to quantify the phylogenetic scope of each individual's microbial community (63). To compare OTU richness levels across amphibian species, we used a generalized linear model with a Poisson error distribution and a log link function. To compare phylogenetic diversity levels across species, we used a linear model with a normal error distribution. For each of these analyses, pairwise comparisons were also conducted. To test for an overall correlation between culture-independent and culture-dependent OTU richness, a generalized linear model (GLM) with a Poisson error distribution and a log link function was used, while to test for a correlation between culture-independent and culture-dependent phylogenetic diversity, a simple linear regression model (LM) with a normal error distribution was used. The above analyses were all conducted using R, version 2.15.2 (64).
To compare patterns of diversity among amphibian host species and to determine whether both culture-dependent and -independent data sets led to similar conclusions about potential among-species differences, two indices of beta diversity were assessed, using Primer 6, version 6.1.15, and PERMANOVA+, version 1.0.5 (65). The incidence-based Sorensen index and the incidence-based unweighted UniFrac distance were calculated for both the culture-dependent and square-root-transformed culture-independent data. The Sorensen index is a measure of community similarity among samples, with an emphasis on the number of shared OTUs, while the UniFrac distance is a phylogenetically based measure of community similarity (66). For both distance matrices, differences in beta diversity across species were analyzed by permutational multivariate analysis of variance (PERMANOVA) and visualized with nonmetric multidimensional scaling (NMDS) (67). Pairwise tests between each pair of species were performed on both Sorensen and UniFrac matrices of the culture-independent and -dependent data.
Direct comparison of culture-independent and -dependent sequences.
The above-described analyses examined patterns of diversity based on the culture-dependent and -independent data sets separately. To calculate the percentage and abundance of culturable bacteria, we then performed a direct comparison of the culture-dependent and -independent sequences. To determine the fraction of the total community for each individual amphibian host that we cultured with R2A medium, the 259 representative sequences from the culture-dependent OTUs (Sanger sequences of isolates clustered at 97% similarity) were used as a reference database to cluster the quality-filtered Illumina sequences at 97% sequence similarity, using UCLUST (51) in QIIME (49). The Illumina sequences that did not cluster with the Sanger OTU reference sequences were then clustered de novo, also at 97% similarity. OTUs that did not contain at least 0.001% of the total reads were discarded (50), and samples were rarified to an even sampling depth of 24,900 sequences per sample (number of prerarefaction sequences/sample, 24,948 to 169,125). This combined analysis resulted in 3,779 short (252 to 255 bp), distinct OTUs clustered at 97% similarity. Of these, 179 OTUs represented both culture-dependent and culture-independent sequences, and 3,600 represented only culture-independent sequences. The rarified reads for each sample were aligned to this set of 3,779 combined OTUs to estimate the relative abundance of each of the culture-dependent and culture-independent OTUs on each amphibian individual and species.
To establish the proportion of bacteria culturable from each individual amphibian, we calculated percentages of the total numbers of OTUs that were “individually matched” and “species matched” to cultured OTUs for each individual. An OTU was considered “individually matched” if it was both cultured and detected by the culture-independent approach for the same individual. An OTU was considered “species matched” if it was detected on an individual by the culture-independent approach and not cultured from that specific individual but was cultured from a different individual of the same species. We then averaged the percentages for each amphibian species. Dominant bacterial phyla were defined as those with mean relative abundances of >0.5% for each species. To evaluate the portion of the dominant bacterial phyla that was culturable, relative abundances of cultured OTUs were divided by the relative abundances of all the combined OTUs within each phylum.
Lastly, to visualize the distribution and abundance of culturable OTUs among all the combined OTUs, a 16S rRNA gene tree for OTUs resulting from the direct comparison of culture-independent and culture-dependent sequences obtained from bullfrogs, newts, spring peepers, and toads was constructed based on the PyNAST (55) alignment of all combined OTU sequences by using FastTree (56) and was visualized by using the Interactive Tree of Life (iTOL) (68), with OTUs that were cultured on any species indicated. OTUs with ≥0.01% relative abundance across all amphibian species were included in this analysis.
Nucleotide sequence accession numbers.
Sequences of the 16S rRNA genes of cultured isolates were deposited in GenBank under accession numbers KM186980 to KM187640. Culture-independent Illumina sequences were deposited in NCBI's Sequence Read Archive (SRA) under study accession number SRP062395.
RESULTS
The values for OTU richness per individual ranged from 1 to 24 for the culture-dependent method and from 200 to 1,525 for the culture-independent method. Phylogenetic diversity values ranged from 0.15 to 1.7 for the culture-dependent method and from 19.3 to 100.7 for the culture-independent method. While the culture-independent method clearly detected OTU richness and phylogenetic diversity at levels that were orders of magnitude higher than those for the culture-dependent method, differences among skin bacterial communities of different host species were observed using both methods of characterization. Spring peepers and toads had higher OTU richness values than bullfrogs and newts by both the culture-independent method (Fig. 1a) (overall χ2 = 4,612; df = 3; overall P < 0.001; bullfrog-newt P = 0.19; spring peeper-toad P = 0.99; for all other pairwise comparisons, P < 0.001) and the culture-dependent method (Fig. 1c) (overall χ2 = 149.5; df = 3; overall P < 0.001; bullfrog-newt P = 0.06; spring peeper-toad P = 0.96; for all other pairwise comparisons, P < 0.001). In addition, spring peepers and toads had higher phylogenetic diversity values for skin bacteria than those of bullfrogs and newts (Fig. 1b and d). This pattern was observed for both methods (for the culture-independent method, overall F = 16.5, overall P < 0.001, spring peeper-toad P = 0.69, bullfrog-newt P = 0.06, and for all other pairwise comparisons, P < 0.04; for the culture-dependent method, overall F = 41.9, overall P < 0.001, spring peeper-toad P = 0.99, and for all other pairwise comparisons, P < 0.001). Both methods also suggested a difference in phylogenetic diversity between bullfrogs and newts (for the culture-independent method, P = 0.06; for the culture-dependent method, P < 0.001). Finally, there was a positive relationship between culture-dependent and -independent measures of OTU richness (Fig. 1e) (GLM; χ2 = 4,108.8; df = 1; P < 0.001) and phylogenetic diversity (Fig. 1f) (LM; t = 7.04; P < 0.001), such that individuals with higher culture-dependent OTU richness and phylogenetic diversity values also had higher culture-independent OTU richness and phylogenetic diversity values.
FIG 1.

Amphibian skin bacterial OTU richness (number of OTUs) and phylogenetic diversity values based on culture-independent (a and b) and culture-dependent (c and d) characterizations. Dashes represent the mean diversity for each amphibian species. Correlations between culture-dependent and -independent measures of OTU richness (e) and phylogenetic diversity (f) are shown for all amphibians combined. Lines in panels e and f represent GLM and LM predictions, respectively.
Culture-independent microbial community composition (beta diversity), as measured by the Sorensen index, differed among amphibian species (Fig. 2a) (PERMANOVA; overall pseudo-F = 16.14; overall P < 0.001; for all pairwise comparisons, P < 0.001). Although the host species do not cluster as clearly in the culture-dependent ordination, microbial community composition based on cultured isolates also differed significantly across species (Fig. 2b) (PERMANOVA; overall pseudo-F = 4.56; overall P < 0.001; for all pairwise comparisons, P < 0.005). The phylogenetics-based distance metric (unweighted UniFrac distance) also revealed host species-specific microbiota with both the culture-independent approach (Fig. 2c) (PERMANOVA; overall pseudo-F = 10.45; overall P < 0.001; for all pairwise comparisons, P < 0.001) and the culture-dependent approach (Fig. 2d) (PERMANOVA; overall pseudo-F = 9.75; P < 0.001; for all pairwise comparisons, P < 0.002).
FIG 2.

NMDS ordinations of Sorensen similarity (a and b) and unweighted UniFrac distance (c and d) matrices for culture-independent (a and c) and culture-dependent (b and d) microbial communities associated with four amphibian species.
Using the mapping of the Illumina reads to the combined culture-dependent and culture-independent OTUs, we estimated that we cultured, on average, 0.59%, 0.60%, 1.12%, and 0.95% of the OTUs for individual bullfrogs, newts, spring peepers, and toads, respectively (Fig. 3, “individually matched” analysis). However, the average percentages of bacteria that were culturable from these species (i.e., “species matched”) were higher: 5.26%, 2.81%, 7.02%, and 7.47% of the OTUs from bullfrogs, newts, spring peepers, and toads, respectively, were culturable (Fig. 3). Across all amphibian species, 179 of the 3,779 combined OTUs (4.7%) were culturable from at least one of the four species.
FIG 3.
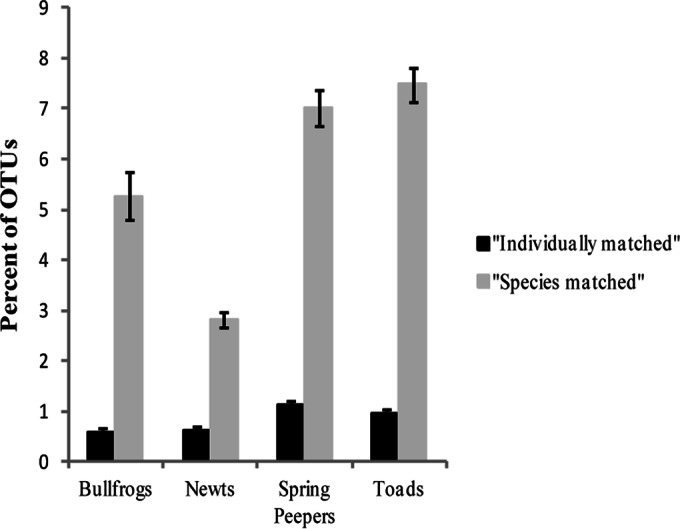
Mean percentages of total OTUs “individually matched” and “species matched” to cultured OTUs for each amphibian species. An OTU was considered individually matched if it was present in the Illumina data for the same individual from which it was cultured, and it was considered species matched if it was cultured from a different individual of the same amphibian species.
Many of the abundant (>1% mean relative abundance) culture-independent OTUs were culturable (Fig. 4; Table 1). For example, 10 of the 14 most abundant culture-independent OTUs on toads were cultured from toads (Table 1). For bullfrogs, 7 of the 13 most abundant culture-independent OTUs were cultured, and for spring peepers, 8 of the 17 most abundant culture-independent OTUs were cultured (Table 1). In contrast, for newts, only 2 of the top 16 culture-independent OTUs were cultured (Table 1). However, if OTUs that were cultured from other amphibian species are included, 11 of the top 16 culture-independent OTUs from newts were culturable; we just did not isolate them from newts in this study. Eight culture-independent OTUs were considered abundant (>1%) on more than one amphibian species (Table 1), and six of these were cultured. The two that were not cultured were a proteobacterium of the family Comamonadaceae and a proteobacterium of the genus Pseudoalteromonas. One culture-independent OTU in the family Cellulomonadaceae (phylum Actinobacteria) was abundant (>1%) on all four species sampled and was also culturable, but it was isolated only from toads (Table 1).
FIG 4.

16S rRNA gene tree of culture-independent OTUs, with the mean relative abundance of OTUs for each amphibian species (blue = bullfrogs, teal = newts, red = spring peepers, and green = toads). Only OTUs with mean relative abundances of ≥0.01% across all amphibian species were included in this analysis. Cultured OTUs are indicated with black bars, and bacterial phyla are indicated with colored branches. The phylum Proteobacteria was divided into classes (Alpha-, Beta-, Delta-, and Gammaproteobacteria).
TABLE 1.
Most abundant OTUs on each amphibian species (>1% relative abundance) and mean relative abundances and prevalences of these OTUsa
| Phylum | Family | OTU identificationb | Mean relative abundance (%) (prevalence [%])c |
|||
|---|---|---|---|---|---|---|
| Bullfrogs | Newts | Spring Peepers | Toads | |||
| Actinobacteria | Cellulomonadaceae | Cellulomonadaceae | 14.3 (100) | 7.9 (100) | 5.2 (100) | 3.3 (93) |
| Cellulomonas | 2.4 (100) | — | — | — | ||
| Nocardiaceae | Rhodococcus | 9.6 (100) | — | — | 1.8 (100) | |
| Bacteroidetes | Cytophagaceae | Flectobacillus | — | — | — | 5.4 (80) |
| Flavobacteriaceae | Flavobacterium | — | — | 3.1 (100) | — | |
| Flavobacterium | — | — | 2.6 (92) | — | ||
| Flavobacterium | — | — | 1.2 (92) | — | ||
| NA | Sphingobacteriales | — | — | 5.0 (92) | — | |
| Firmicutes | Bacillaceae | Bacillaceae | — | 2.8 (94) | — | — |
| Proteobacteria | Alcaligenaceae | Achromobacter | — | — | 11.8 (100) | — |
| Brucellaceae | Brucellaceae | 3.1 (100) | — | — | — | |
| Chromatiaceae | Rheinheimera | — | — | 1.3 (100) | — | |
| Comamonadaceae | Methylibium | 12.2 (100) | 3.7 (100) | — | — | |
| Comamonadaceae | 5.6 (95) | — | — | — | ||
| Roseateles depolymerans | 1.2 (89) | — | — | — | ||
| Comamonadaceae | — | 13.2 (100) | 1.3 (100) | — | ||
| Comamonadaceae | — | 1.9 (100) | — | — | ||
| Comamonadaceae | — | — | 4.7 (100) | — | ||
| Comamonadaceae | — | — | 1.7 (100) | — | ||
| Comamonadaceae | — | — | 1.5 (100) | — | ||
| Enterobacteriaceae | Enterobacteriaceae | — | 5.3 (78) | — | — | |
| Enterobacteriaceae | — | 1.7 (100) | — | — | ||
| Enterobacteriaceae | — | — | — | 1.1 (40) | ||
| Serratia | — | 4.9 (56) | — | — | ||
| Methylophilaceae | Methylophilaceae | — | 10.6 (100) | — | — | |
| Moraxellaceae | Acinetobacter | 4.6(95) | — | — | — | |
| Neisseriaceae | Neisseriaceae | 1.1 (95) | — | — | — | |
| Neisseriaceae | — | — | 1.9 (100) | — | ||
| Oxalobacteraceae | Oxalobacteraceae | 1.4 (95) | — | — | — | |
| Oxalobacteraceae | — | — | 2.2 (100) | — | ||
| Oxalobacteraceae | — | — | — | 2.1 (100) | ||
| Oxalobacteraceae | — | — | — | 1.7 (93) | ||
| Oxalobacteraceae | — | — | — | 1.6 (100) | ||
| Pseudoalteromonadaceae | Pseudoalteromonas | — | — | 3.6 (100) | 1.0 (100) | |
| Pseudomonadaceae | Pseudomonadaceae | 2.1 (100) | — | — | — | |
| Pseudomonas | — | 12.1 (100) | — | 2.2 (80) | ||
| Pseudomonas | — | 5.7 (100) | 2.8 (100) | 6.4 (93) | ||
| Pseudomonas | — | 3.4 (100) | — | — | ||
| Pseudomonas | — | — | 2.2 (100) | — | ||
| Pseudomonas | — | — | — | 2.0 (100) | ||
| Pseudomonas | — | — | — | 1.5 (93) | ||
| Rhodobacteraceae | Rhodobacter | — | — | 1.0 (100) | — | |
| Sphingomonadaceae | Sphingomonas | — | — | — | 2.0 (100) | |
| Xanthomonadaceae | Xanthomonadaceae | 1.4 (100) | 4.8 (100) | — | — | |
| Xanthomonadaceae | — | 3.5 (100) | — | — | ||
| Stenotrophomonas | — | 1.7 (94) | — | — | ||
| Xanthomonadaceae | — | — | — | 1.3 (93) | ||
| NA | Spirobacillales | 2.3 (63) | — | — | — | |
| Verrucomicrobia | “Chthoniobacteraceae” | “Candidatus Xiphinematobacter” | — | 4.6 (100) | — | — |
OTUs that were cultured from amphibian species are indicated in bold. NA, not available.
The lowest taxonomic resolution that could be defined for each OTU is listed.
Dashes indicate that an OTU either was not detected or was present at a mean relative abundance of <1% on an amphibian species.
In addition to many of the dominant OTUs being cultured, most of the prevalent OTUs, those found on all individuals of a species, were cultured. There were 13 OTUs that were detected on all the bullfrogs sampled (n = 19). Of these, 11 (85%) were cultured from at least one of the four amphibian species. Seventy-five percent (21 of 28 OTUs) of OTUs found on all the newts (n = 18) were cultured, 69% (50 of 72 OTUs) of OTUs found on all the spring peepers (n = 12) were cultured, and 77% (24 of 31 OTUs) of OTUs found on all the toads (n = 15) were cultured.
Four bacterial phyla (Proteobacteria, Actinobacteria, Bacteroidetes, and Firmicutes) were dominant across all four amphibian species, and all of the 179 culture OTUs belonged to these phyla (Fig. 5). Based on the culture-independent data, Proteobacteria was the most abundant phylum across all the amphibian species, representing, on average, 54% of all bacteria on bullfrogs, 80% of those on newts, 64% of those on peepers, and 60% of those on toads (Fig. 5). Proteobacteria was also the phylum with the largest portion of cultured bacteria, based on relative abundance (Fig. 5, black portion of bars). For example, in bullfrogs, 59% of the relative abundance of Proteobacteria was represented in the cultures. However, only 41 of the 1,016 Proteobacteria OTUs (4%) on bullfrogs contributed to this abundance (Fig. 5a). The portion of each phylum that was cultured varied, however, depending on the amphibian host species. In newts, only 12% of the Proteobacteria relative abundance was represented in the cultures, and 16 of the 764 Proteobacteria OTUs (2%) contributed to the abundance (Fig. 5b). Actinobacteria was another abundant phylum that varied in its culturability across amphibian hosts: 57% of this phylum's relative abundance was represented by cultures from toads, while less than 3.5% was represented by cultures from bullfrogs, newts, and spring peepers (Fig. 5).
FIG 5.
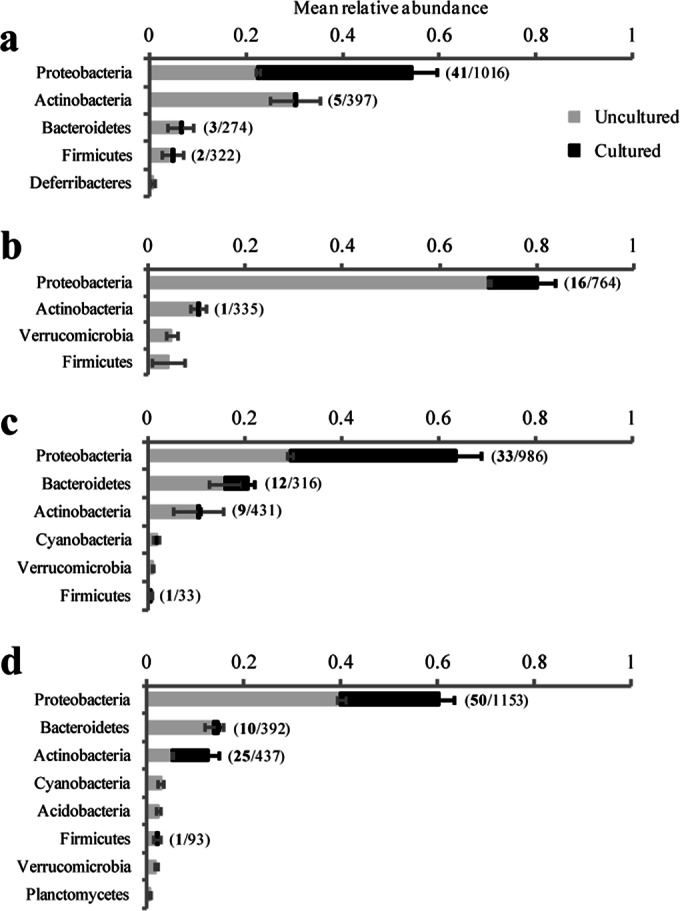
Relative abundances of culture-independent bacterial phyla associated with bullfrogs (a), newts (b), spring peepers (c), and toads (d). The complete bar represents the total mean relative abundance of each phylum, while the black portion of the bar represents the cultured portion of each phylum. The numbers in parentheses show the number of “species-matched” cultured OTUs (in bold) out of the total number of OTUs in the phylum for each amphibian species. Only phyla with mean relative abundances of >0.5% for each species are shown. Error bars represent standard errors.
Of the 32 most abundant bacterial families across bullfrogs, newts, spring peepers, and toads, 20 (63%) contained culturable OTUs. There were five bacterial families that were highly abundant (>1%) on all four amphibian species sampled. Three of these abundant bacterial families (Comamonadaceae, Oxalobacteraceae, and Pseudomonadaceae [all from the Proteobacteria phylum]) were cultured from all four amphibian species, while two (Cellulomonadaceae [phylum Actinobacteria] and Xanthomonadaceae [phylum Proteobacteria]) were cultured from only two of the four species.
DISCUSSION
It is commonly thought that culturable microorganisms are not necessarily the dominant members in microbial communities but are isolated simply because they grow easily or rapidly in culture (69). For amphibian skin bacterial communities, however, we found that most of the highly prevalent and dominant bacterial phyla, families, and OTUs were cultured. This is encouraging, because these are the bacteria whose functions we are interested in, as dominant community members are often functionally important (70). In grassland ecosystems, for example, dominant grass species play an important role in community productivity and resistance to invasion (71, 72).
Bacteria that are relatively abundant in amphibian skin microbial communities are likely competitively dominant. Bacteria have evolved a wide range of competitive strategies (73), including specialized nutrient acquisition systems (74) and microbial defense systems that include the production of secondary metabolites (75). For example, some secondary metabolites that are produced by bacteria cultured from amphibian skin inhibit the growth of B. dendrobatidis and other amphibian fungal pathogens (25, 45, 60, 76–78). In addition, B. dendrobatidis zoospores exhibit chemotaxis away from several of these compounds (79). Therefore, dominant bacteria that produce secondary metabolites with antifungal properties may also play important roles in host defense, as demonstrated previously in a plant rhizosphere microbial community (80). Specifically, Costa et al. (80) demonstrated that the most dominant members exhibited antagonistic properties against a plant fungal pathogen.
Despite the fact that we found that only a small portion of the amphibian-associated microbiota was cultured, overall patterns of alpha and beta diversity were similar for the culture-independent and -dependent approaches. Spring peepers and toads had more diverse microbial communities (in terms of OTU richness and phylogenetic diversity) than bullfrogs and newts, and the microbial communities associated with bullfrogs, newts, spring peepers, and toads were different. This observation of host species-specific microbiota is consistent with other studies of amphibian skin microbiota (43, 44, 81). Furthermore, the culture-dependent OTU richness and phylogenetic diversity predicted the culture-independent OTU richness and phylogenetic diversity. Individuals with higher OTU richness or phylogenetic diversity values based on culturing had higher overall OTU richness and phylogenetic diversity values. This is encouraging because it allows for at least some basic comparisons among culture-dependent and -independent data sets, especially given the wealth of culture-based work that has been completed to date (e.g., see references 25, 41, 42, 45, 60, and 82 to 84).
Our culturing approach captured 0.59% to 1.1% of skin bacterial OTUs identified by culture-independent methods for individual bullfrogs, newts, spring peepers, and toads, which falls within the range observed in other systems (2). However, when bacteria that were cultured from other individuals of the same species were included, a larger portion of the community from a given host species (2.81 to 7.47%) was culturable. For any given individual, all of the culturable bacteria were not cultured consistently. This suggests that we are missing some culturable bacteria with our present sampling methods. To capture more diversity in our culture collections, the plating methodology needs to be refined. For example, serial dilution techniques (85, 86) could be used to more consistently recover all of the culturable bacteria from an individual. But, in general, to obtain a more complete profile of isolates for a species, it appears that sampling multiple individuals in a population greatly increases the chance of culturing the most dominant community members.
There are many factors that may influence the low culturability of amphibian skin bacteria. First, bacteria often need specific nutrient, pH, temperature, and oxygen conditions to grow (87), and laboratory and amphibian skin conditions are not identical. Second, cross-feeding and metabolic cooperation between bacterial species are common, and many bacteria require helper bacterial strains or their by-products for growth in vitro (87). For example, it has been found that a previously uncultured isolate related to Verrucomicrobia grows only in the presence of a siderophore growth factor produced by a particular neighbor (88). This group was among the dominant phyla associated with amphibians in our culture-independent data set; however, it was not culturable by our methods. Third, culture-independent methods capture DNAs from both active and inactive organisms, while culture-dependent methods capture only the viable and metabolically active cells. Approximately 20% to 80% of bacterial cells in various natural habitats are dormant, with environmental samples tending to have a larger inactive portion than host-associated samples, such as the human gut microbiota (89). There are likely numerous dormant, viable but nonculturable (VBNC) microbes (90) in our samples that further contribute to the small culturable portion of the community. In fact, the uncultured microbes in our samples are related to those that are known to enter this dormant state, such as Burkholderia, Enterobacter, Erwinia, Klebsiella, Legionella, Mycobacterium, Pseudomonas, Streptococcus, and Vibrio (90). Microbes may either be dormant on the amphibian skin or become VBNC once transferred to medium, which is an unfamiliar environment.
While it may be challenging to recreate the complex conditions of amphibian skin, certain aspects of this habitat, such as pH and mineral components, can be simulated in vitro to attempt to culture previously uncultured amphibian skin microbes. Indeed, the addition of specific minerals (e.g., CaCl2), sonication of the samples prior to plating, and an extended incubation time (e.g., 12 weeks) can enhance the culturability of bacteria, including groups also identified in our samples, such as Actinobacteria, Acidobacteria, Proteobacteria, and Verrucomicrobia (91, 92). Additionally, it may be possible to supplement media with amphibian skin washes, containing bacteria, their by-products, and amphibian-produced compounds, which may facilitate the growth of previously uncultured amphibian skin bacteria. Another method for increasing the culturability of microbial communities is to culture organisms in the actual habitat as opposed to in a laboratory setting (87, 93). This method has been used successfully in aquatic habitats (93–95), but it may not be realistic, or at least would be extremely challenging logistically, for living vertebrates, such as amphibians. Nonetheless, by incubating diffusion chambers in pond sediment, Bollmann et al. (94) successfully cultured rarely cultivated bacterial groups, such as Deltaproteobacteria, Verrucomicrobia, and Acidobacteria. These groups were among the more dominant phyla associated with amphibians in this study; however, only one OTU from these three taxa was culturable by our methods (Fig. 4 and 5). A similar approach may prove useful for exploring the amphibian skin system to target these groups. Lastly, rRNA-rRNA gene combined approaches can be used to establish the active and inactive (dormant or dead) portions of the community, which would inform the regulatory factors influencing the structure and function of these microbial communities (89, 96).
Recent advances in culture-independent molecular approaches have allowed for a greater assessment of microbial community structure (1, 7, 8) and, to some extent, function, through the use of metagenomics (97). However, culture collections are critical for advancing basic and applied microbial ecology, even in the age of sequencing. Cultures are useful for experimental work to understand microbiome functions, for identifying members of a community that produce key metabolites, and for applied purposes, such as probiotic therapy. In addition, microorganisms serve as useful models for testing basic ecological and evolutionary theories (28, 29). Given their importance, efforts should be made to expand culture collections, including those for the amphibian skin microbiome, even though most of the abundant, and likely functionally important, bacteria on amphibian skin were cultured with current methods. While it is clear that culture-independent approaches provide a more complete assessment of the microbial community composition, culture-dependent approaches may be adequate, and in some cases more valuable (98), for capturing patterns of community structural and functional diversity.
ACKNOWLEDGMENTS
This work was supported by the Morris Animal Foundation, The Fralin Life Sciences Institute at Virginia Tech, and the National Science Foundation (grant DEB-1136640).
We thank Mountain Lake Biological Station, the Jefferson National Forest, and the Town of Blacksburg for allowing us to perform our research on their lands. We also appreciate the Illumina sequencing provided by Zach Herbert and the Molecular Biology Core Facilities at Dana-Farber Cancer Institute and the statistical assistance provided by Skylar Hopkins.
REFERENCES
- 1.Pace NR. 1997. A molecular view of microbial diversity and the biosphere. Science 276:734–740. doi: 10.1126/science.276.5313.734. [DOI] [PubMed] [Google Scholar]
- 2.Amann RI, Ludwig W, Schleifer KH, Amann RI, Ludwig W. 1995. Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol Rev 59:143–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Van der Heijden MGA, Bardgett RD, van Straalen NM. 2008. The unseen majority: soil microbes as drivers of plant diversity and productivity in terrestrial ecosystems. Ecol Lett 11:296–310. doi: 10.1111/j.1461-0248.2007.01139.x. [DOI] [PubMed] [Google Scholar]
- 4.Schleifer K-H. 2004. Microbial diversity: facts, problems and prospects. Syst Appl Microbiol 27:3–9. doi: 10.1078/0723-2020-00245. [DOI] [PubMed] [Google Scholar]
- 5.Ma L, Kim J, Hatzenpichler R, Karymov MA, Hubert N, Hanan IM, Chang EB, Ismagilov RF. 2014. Gene-targeted microfluidic cultivation validated by isolation of a gut bacterium listed in Human Microbiome Project's Most Wanted taxa. Proc Natl Acad Sci U S A 111:9768–9773. doi: 10.1073/pnas.1404753111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Achtman M, Wagner M. 2008. Microbial diversity and the genetic nature of microbial species. Nat Rev Microbiol 6:431–440. doi: 10.1038/nrmicro1872. [DOI] [PubMed] [Google Scholar]
- 7.Shokralla S, Spall JL, Gibson JF, Hajibabaei M. 2012. Next-generation sequencing technologies for environmental DNA research. Mol Ecol 21:1794–1805. doi: 10.1111/j.1365-294X.2012.05538.x. [DOI] [PubMed] [Google Scholar]
- 8.Hugenholtz P, Goebel BM, Pace NR. 1998. Impact of culture-independent studies on the emerging phylogenetic view of bacterial diversity. J Bacteriol 180:4765–4774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hugenholtz P, Tyson GW, Webb RI, Wagner AM, Blackall LL. 2001. Investigation of candidate division TM7, a recently recognized major lineage of the domain Bacteria with no known pure-culture representatives. Appl Environ Microbiol 67:411–419. doi: 10.1128/AEM.67.1.411-419.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marcy Y, Ouverney C, Bik EM, Lösekann T, Ivanova N, Martin HG, Szeto E, Platt D, Hugenholtz P, Relman DA, Quake SR. 2007. Dissecting biological “dark matter” with single-cell genetic analysis of rare and uncultivated TM7 microbes from the human mouth. Proc Natl Acad Sci U S A 104:11889–11894. doi: 10.1073/pnas.0704662104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Podar M, Keller M, Hugenholtz P. 2009. Single cell whole genome amplification of uncultivated microorganisms, p 241–256. In Epstein SS. (ed), Uncultivated microorganisms. Springer Science & Business Media, New York, NY. doi: 10.1007/978-3-540-85465-4_10. [DOI] [Google Scholar]
- 12.Abrams M, Barton D, Vandaei E, Romero D, Caldwell A, Ouverney C. 2012. Genomic characteristics of an environmental microbial community harboring a novel human uncultured TM7 bacterium associated with oral diseases. Open Access Sci Rep 1:276. [Google Scholar]
- 13.Binnerup SJ, Jensen DF, Thordal-Christensen H, Sørensen J. 1993. Detection of viable, but non-culturable Pseudomonas fluorescens DF57 in soil using a microcolony epifluorescence technique. FEMS Microbiol Ecol 12:97–105. doi: 10.1111/j.1574-6941.1993.tb00021.x. [DOI] [Google Scholar]
- 14.Lloyd-Jones G, Laurie AD, Tizzard AC. 2005. Quantification of the Pseudomonas population in New Zealand soils by fluorogenic PCR assay and culturing techniques. J Microbiol Methods 60:217–224. doi: 10.1016/j.mimet.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 15.Costa R, Salles JF, Berg G, Smalla K. 2006. Cultivation-independent analysis of Pseudomonas species in soil and in the rhizosphere of field-grown Verticillium dahliae host plants. Environ Microbiol 8:2136–2149. doi: 10.1111/j.1462-2920.2006.01096.x. [DOI] [PubMed] [Google Scholar]
- 16.Lazcano A. 2011. Natural history, microbes and sequences: shouldn't we look back again to organisms? PLoS One 6:e21334. doi: 10.1371/journal.pone.0021334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hallenbeck PC, Ghosh D. 2009. Advances in fermentative biohydrogen production: the way forward? Trends Biotechnol 27:287–297. doi: 10.1016/j.tibtech.2009.02.004. [DOI] [PubMed] [Google Scholar]
- 18.Bachmann RT, Johnson AC, Edyvean RGJ. 2014. Biotechnology in the petroleum industry: an overview. Int Biodeterior Biodegradation 86:225–237. doi: 10.1016/j.ibiod.2013.09.011. [DOI] [Google Scholar]
- 19.Tyagi M, da Fonseca MMR, de Carvalho CCCR. 2011. Bioaugmentation and biostimulation strategies to improve the effectiveness of bioremediation processes. Biodegradation 22:231–241. doi: 10.1007/s10532-010-9394-4. [DOI] [PubMed] [Google Scholar]
- 20.Spencer RC. 2003. Bacillus anthracis. J Clin Pathol 56:182–187. doi: 10.1136/jcp.56.3.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hermanson G, Whitlow V, Parker S, Tonsky K, Rusalov D, Ferrari M, Lalor P, Komai M, Mere R, Bell M, Brenneman K, Mateczun A, Evans T, Kaslow D, Galloway D, Hobart P. 2004. A cationic lipid-formulated plasmid DNA vaccine confers sustained antibody-mediated protection against aerosolized anthrax spores. Proc Natl Acad Sci U S A 101:13601–13606. doi: 10.1073/pnas.0405557101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jankovic I, Sybesma W, Phothirath P, Ananta E, Mercenier A. 2010. Application of probiotics in food products—challenges and new approaches. Curr Opin Biotechnol 21:175–181. doi: 10.1016/j.copbio.2010.03.009. [DOI] [PubMed] [Google Scholar]
- 23.Jacobsen CN, Rosenfeldt Nielsen V, Hayford AE, Moller PL, Michaelsen KF, Parregaard A, Sandstrom B, Tvede M, Jakobsen M. 1999. Screening of probiotic activities of forty-seven strains of Lactobacillus spp. by in vitro techniques and evaluation of the colonization ability of five selected strains in humans. Appl Environ Microbiol 65:4949–4956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Verschuere L, Rombaut G, Sorgeloos P, Verstraete W. 2000. Probiotic bacteria as biological control agents in aquaculture. Microbiol Mol Biol Rev 64:655–671. doi: 10.1128/MMBR.64.4.655-671.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harris RN, James TY, Lauer A, Simon MA, Patel A. 2006. Amphibian pathogen Batrachochytrium dendrobatidis is inhibited by the cutaneous bacteria of amphibian species. Ecohealth 3:53–56. doi: 10.1007/s10393-005-0009-1. [DOI] [Google Scholar]
- 26.Barrick JE, Yu DS, Yoon SH, Jeong H, Oh TK, Schneider D, Lenski RE, Kim JF. 2009. Genome evolution and adaptation in a long-term experiment with Escherichia coli. Nature 461:1243–1247. doi: 10.1038/nature08480. [DOI] [PubMed] [Google Scholar]
- 27.Elena SF, Lenski RE. 2003. Evolution experiments with microorganisms: the dynamics and genetic bases of adaptation. Nat Rev Genet 4:457–469. [DOI] [PubMed] [Google Scholar]
- 28.Jessup CM, Kassen R, Forde SE, Kerr B, Buckling A, Rainey PB, Bohannan BJM. 2004. Big questions, small worlds: microbial model systems in ecology. Trends Ecol Evol 19:189–197. doi: 10.1016/j.tree.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 29.Krause S, Le Roux X, Niklaus PA, Van Bodegom PM, Lennon JT, Bertilsson S, Grossart H-P, Philippot L, Bodelier PLE. 2014. Trait-based approaches for understanding microbial biodiversity and ecosystem functioning. Front Microbiol 5:251. doi: 10.3389/fmicb.2014.00251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rosenberg E, Gophna U. 2011. Beneficial microorganisms in multicellular life forms, 1st ed Springer, Berlin, Germany. [Google Scholar]
- 31.Broderick NA, Raffa KF, Goodman RM, Handelsman J. 2004. Census of the bacterial community of the gypsy moth larval midgut by using culturing and culture-independent methods. Appl Environ Microbiol 70:293–300. doi: 10.1128/AEM.70.1.293-300.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rani A, Sharma A, Rajagopal R, Adak T, Bhatnagar RK. 2009. Bacterial diversity analysis of larvae and adult midgut microflora using culture-dependent and culture-independent methods in lab-reared and field-collected Anopheles stephensi—an Asian malarial vector. BMC Microbiol 9:96. doi: 10.1186/1471-2180-9-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bougoure DS, Cairney JWG. 2005. Fungi associated with hair roots of Rhododendron lochiae (Ericaceae) in an Australian tropical cloud forest revealed by culturing and culture-independent molecular methods. Environ Microbiol 7:1743–1754. doi: 10.1111/j.1462-2920.2005.00919.x. [DOI] [PubMed] [Google Scholar]
- 34.Lindh JM, Terenius O, Faye I. 2005. 16S rRNA gene-based identification of midgut bacteria from field-caught Anopheles gambiae sensu lato and A. funestus mosquitoes reveals new species related to known insect symbionts. Appl Environ Microbiol 71:7217–7223. doi: 10.1128/AEM.71.11.7217-7223.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hayashi H, Sakamoto M, Benno Y. 2002. Phylogenetic analysis of the human gut microbiota using 16S rDNA clone libraries and strictly anaerobic culture-based methods. Microbiol Immunol 46:535–548. doi: 10.1111/j.1348-0421.2002.tb02731.x. [DOI] [PubMed] [Google Scholar]
- 36.Webster NS, Wilson KJ, Blackall LL, Hill RT. 2001. Phylogenetic diversity of bacteria associated with the marine sponge Rhopaloeides odorabile. Appl Environ Microbiol 67:434–444. doi: 10.1128/AEM.67.1.434-444.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Harris RN, Brucker RM, Walke JB, Becker MH, Schwantes CR, Flaherty DC, Lam BA, Woodhams DC, Briggs CJ, Vredenburg VT, Minbiole KPC. 2009. Skin microbes on frogs prevent morbidity and mortality caused by a lethal skin fungus. ISME J 3:818–824. doi: 10.1038/ismej.2009.27. [DOI] [PubMed] [Google Scholar]
- 38.Becker MH, Brucker RM, Schwantes CR, Harris RN, Minbiole KPC. 2009. The bacterially produced metabolite violacein is associated with survival of amphibians infected with a lethal fungus. Appl Environ Microbiol 75:6635–6638. doi: 10.1128/AEM.01294-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Becker MH, Harris RN. 2010. Cutaneous bacteria of the redback salamander prevent morbidity associated with a lethal disease. PLoS One 5:e10957. doi: 10.1371/journal.pone.0010957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Myers JM, Ramsey JP, Blackman AL, Nichols AE, Minbiole KPC, Harris RN. 2012. Synergistic inhibition of the lethal fungal pathogen Batrachochytrium dendrobatidis: the combined effect of symbiotic bacterial metabolites and antimicrobial peptides of the frog Rana muscosa. J Chem Ecol 38:958–965. doi: 10.1007/s10886-012-0170-2. [DOI] [PubMed] [Google Scholar]
- 41.Flechas SV, Sarmiento C, Cárdenas ME, Medina EM, Restrepo S, Amézquita A. 2012. Surviving chytridiomycosis: differential anti-Batrachochytrium dendrobatidis activity in bacterial isolates from three lowland species of Atelopus. PLoS One 7:e44832. doi: 10.1371/journal.pone.0044832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Woodhams DC, Vredenburg VT, Simon M-A, Billheimer D, Shakhtour B, Shyr Y, Briggs CJ, Rollins-Smith LA, Harris RN. 2007. Symbiotic bacteria contribute to innate immune defenses of the threatened mountain yellow-legged frog, Rana muscosa. Biol Conserv 138:390–398. doi: 10.1016/j.biocon.2007.05.004. [DOI] [Google Scholar]
- 43.McKenzie VJ, Bowers RM, Fierer N, Knight R, Lauber CL. 2012. Co-habiting amphibian species harbor unique skin bacterial communities in wild populations. ISME J 6:588–596. doi: 10.1038/ismej.2011.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Walke JB, Becker MH, Loftus SC, House LL, Cormier G, Jensen RV, Belden LK. 2014. Amphibian skin may select for rare environmental microbes. ISME J 8:2207–2217. doi: 10.1038/ismej.2014.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lauer A, Simon MA, Banning JL, André E, Duncan K, Harris RN. 2007. Common cutaneous bacteria from the eastern red-backed salamander can inhibit pathogenic fungi. Copeia 2007:630–640. doi: 10.1643/0045-8511(2007)2007[630:CCBFTE]2.0.CO;2. [DOI] [Google Scholar]
- 46.Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, Owens SM, Betley J, Fraser L, Bauer M, Gormley N, Gilbert JA, Smith G, Knight R. 2012. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J 6:1621–1624. doi: 10.1038/ismej.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ, Fierer N, Knight R. 2011. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci U S A 108:4516–4522. doi: 10.1073/pnas.1000080107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Aronesty E. 2013. Comparison of sequencing utility programs. Open Bioinform J 7:1–8. doi: 10.2174/1875036201307010001. [DOI] [Google Scholar]
- 49.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. 2010. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bokulich NA, Subramanian S, Faith JJ, Gevers D, Gordon JI, Knight R, Mills DA, Caporaso JG. 2013. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat Methods 10:57–59. doi: 10.1038/nmeth.2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Edgar RC. 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 52.DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL. 2006. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 72:5069–5072. doi: 10.1128/AEM.03006-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang Q, Garrity GM, Tiedje JM, Cole JR. 2007. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73:5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Claesson MJ, O'Sullivan O, Wang Q, Nikkilä J, Marchesi JR, Smidt H, de Vos WM, Ross RP, O'Toole PW. 2009. Comparative analysis of pyrosequencing and a phylogenetic microarray for exploring microbial community structures in the human distal intestine. PLoS One 4:e6669. doi: 10.1371/journal.pone.0006669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Caporaso JG, Bittinger K, Bushman FD, DeSantis TZ, Andersen GL, Knight R. 2010. PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics 26:266–267. doi: 10.1093/bioinformatics/btp636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Price MN, Dehal PS, Arkin AP. 2010. FastTree 2—approximately maximum-likelihood trees for large alignments. PLoS One 5:e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Reasoner DJ, Geldreich EE. 1985. A new medium for the enumeration and subculture of bacteria from potable water. Appl Environ Microbiol 49:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Antwis RE, Haworth RL, Engelmoer DJP, Ogilvy V, Fidgett AL, Preziosi RF. 2014. Ex situ diet influences the bacterial community associated with the skin of red-eyed tree frogs (Agalychnis callidryas). PLoS One 9:e85563. doi: 10.1371/journal.pone.0085563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Walke JB, Harris RN, Reinert LK, Rollins-Smith LA, Woodhams DC. 2011. Social immunity in amphibians: evidence for vertical transmission of innate defenses. Biotropica 43:396–400. doi: 10.1111/j.1744-7429.2011.00787.x. [DOI] [Google Scholar]
- 60.Lauer A, Simon MA, Banning JL, Lam BA, Harris RN. 2008. Diversity of cutaneous bacteria with antifungal activity isolated from female four-toed salamanders. ISME J 2:145–157. doi: 10.1038/ismej.2007.110. [DOI] [PubMed] [Google Scholar]
- 61.Roth T, Foley J, Worth J, Piovia-Scott J, Pope K, Lawler S. 2013. Bacterial flora on Cascades frogs in the Klamath mountains of California. Comp Immunol Microbiol Infect Dis 36:591–598. doi: 10.1016/j.cimid.2013.07.002. [DOI] [PubMed] [Google Scholar]
- 62.Salle A. 1973. Laboratory manual on fundamental principles of bacteriology. McGraw-Hill, New York, NY. [Google Scholar]
- 63.Faith DP. 1992. Conservation evaluation and phylogenetic diversity. Biol Conserv 61:1–10. doi: 10.1016/0006-3207(92)91201-3. [DOI] [Google Scholar]
- 64.R Core Team. 2012. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- 65.Clarke K, Gorley R. 2006. PRIMER v6: user manual/tutorial. PRIMER-E, Plymouth, United Kingdom. [Google Scholar]
- 66.Lozupone C, Lladser ME, Knights D, Stombaugh J, Knight R. 2011. UniFrac: an effective distance metric for microbial community comparison. ISME J 5:169–172. doi: 10.1038/ismej.2010.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kruskal JB. 1964. Multidimensional scaling by optimizing goodness of fit to a nonmetric hypothesis. Psychometrika 29:1–27. doi: 10.1007/BF02289565. [DOI] [Google Scholar]
- 68.Letunic I, Bork P. 2011. Interactive Tree Of Life v2: online annotation and display of phylogenetic trees made easy. Nucleic Acids Res 39:W475–W478. doi: 10.1093/nar/gkr201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hugenholtz P. 2002. Exploring prokaryotic diversity in the genomic era. Genome Biol 3:REVIEWS0003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Winfree R, Fox JW, Williams NM, Reilly JR, Cariveau DP. 2015. Abundance of common species, not species richness, drives delivery of a real-world ecosystem service. Ecol Lett 18:626–635. doi: 10.1111/ele.12424. [DOI] [PubMed] [Google Scholar]
- 71.Smith MD, Wilcox JC, Kelly T, Knapp AK. 2004. Dominance not richness determines invasibility of tallgrass prairie. Oikos 106:253–262. doi: 10.1111/j.0030-1299.2004.13057.x. [DOI] [Google Scholar]
- 72.Smith MD, Knapp AK. 2003. Dominant species maintain ecosystem function with non-random species loss. Ecol Lett 6:509–517. doi: 10.1046/j.1461-0248.2003.00454.x. [DOI] [Google Scholar]
- 73.Hibbing ME, Fuqua C, Parsek MR, Peterson SB. 2010. Bacterial competition: surviving and thriving in the microbial jungle. Nat Rev Microbiol 8:15–25. doi: 10.1038/nrmicro2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zubkov MV, Fuchs BM, Tarran GA, Burkill PH, Amann R. 2003. High rate of uptake of organic nitrogen compounds by Prochlorococcus cyanobacteria as a key to their dominance in oligotrophic oceanic waters. Appl Environ Microbiol 69:1299–1304. doi: 10.1128/AEM.69.2.1299-1304.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Riley MA, Wertz JE. 2002. Bacteriocins: evolution, ecology, and application. Annu Rev Microbiol 56:117–137. doi: 10.1146/annurev.micro.56.012302.161024. [DOI] [PubMed] [Google Scholar]
- 76.Brucker RM, Harris RN, Schwantes CR, Gallaher TN, Flaherty DC, Lam BA, Minbiole KPC. 2008. Amphibian chemical defense: antifungal metabolites of the microsymbiont Janthinobacterium lividum on the salamander Plethodon cinereus. J Chem Ecol 34:1422–1429. doi: 10.1007/s10886-008-9555-7. [DOI] [PubMed] [Google Scholar]
- 77.Brucker RM, Baylor CM, Walters RL, Lauer A, Harris RN, Minbiole KPC. 2008. The identification of 2,4-diacetylphloroglucinol as an antifungal metabolite produced by cutaneous bacteria of the salamander Plethodon cinereus. J Chem Ecol 34:39–43. doi: 10.1007/s10886-007-9352-8. [DOI] [PubMed] [Google Scholar]
- 78.Banning JL, Weddle AL, Wahl GW, Simon MA, Lauer A, Walters RL, Harris RN. 2008. Antifungal skin bacteria, embryonic survival, and communal nesting in four-toed salamanders, Hemidactylium scutatum. Oecologia 156:423–429. doi: 10.1007/s00442-008-1002-5. [DOI] [PubMed] [Google Scholar]
- 79.Lam BA, Walton DB, Harris RN. 2011. Motile zoospores of Batrachochytrium dendrobatidis move away from antifungal metabolites produced by amphibian skin bacteria. Ecohealth 8:36–45. doi: 10.1007/s10393-011-0689-7. [DOI] [PubMed] [Google Scholar]
- 80.Costa R, Gomes NCM, Krögerrecklenfort E, Opelt K, Berg G, Smalla K. 2007. Pseudomonas community structure and antagonistic potential in the rhizosphere: insights gained by combining phylogenetic and functional gene-based analyses. Environ Microbiol 9:2260–2273. doi: 10.1111/j.1462-2920.2007.01340.x. [DOI] [PubMed] [Google Scholar]
- 81.Kueneman JG, Parfrey LW, Woodhams DC, Archer HM, Knight R, McKenzie VJ. 2014. The amphibian skin-associated microbiome across species, space and life history stages. Mol Ecol 23:1238–1250. doi: 10.1111/mec.12510. [DOI] [PubMed] [Google Scholar]
- 82.Becker MH, Walke JB, Murrill L, Woodhams DC, Reinert LK, Rollins-Smith LA, Burzynski EA, Umile TP, Minbiole KPC, Belden LK. 2015. Phylogenetic distribution of symbiotic bacteria from Panamanian amphibians that inhibit growth of the lethal fungal pathogen Batrachochytrium dendrobatidis. Mol Ecol 24:1628–1641. doi: 10.1111/mec.13135. [DOI] [PubMed] [Google Scholar]
- 83.Woodhams DC, Alford RA, Antwis RE, Archer H, Becker MH, Belden LK, Bell SC, Bletz M, Daskin JH, Davis LR, Flechas SV, Lauer A, Gonzalez A, Harris RN, Holden WM, Hughey MC, Ibáñez R, Knight R, Kueneman J, Rabemananjara F, Reinert LK, Rollins-Smith LA, Roman-Rodriguez F, Shaw SD, Walke JB, McKenzie V. 2015. Antifungal isolates database of amphibian skin-associated bacteria and function against emerging fungal pathogens. Ecology 96:595. doi: 10.1890/14-1837.1. [DOI] [Google Scholar]
- 84.Antwis RE, Preziosi RF, Harrison XA, Garner TWJ. 2015. Amphibian symbiotic bacteria do not show a universal ability to inhibit growth of the global panzootic lineage of Batrachochytrium dendrobatidis. Appl Environ Microbiol 81:3706–3711. doi: 10.1128/AEM.00010-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Garland JL, Lehman RM. 1999. Dilution/extinction of community phenotypic characters to estimate relative structural diversity in mixed communities. FEMS Microbiol Ecol 30:333–343. doi: 10.1111/j.1574-6941.1999.tb00661.x. [DOI] [PubMed] [Google Scholar]
- 86.Connon SA, Giovannoni SJ. 2002. High-throughput methods for culturing microorganisms in very-low-nutrient media yield diverse new marine isolates. Appl Environ Microbiol 68:3878–3885. doi: 10.1128/AEM.68.8.3878-3885.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Vartoukian SR, Palmer RM, Wade WG. 2010. Strategies for culture of “unculturable” bacteria. FEMS Microbiol Lett 309:1–7. doi: 10.1111/j.1574-6968.2010.02000.x. [DOI] [PubMed] [Google Scholar]
- 88.D'Onofrio A, Crawford JM, Stewart EJ, Witt K, Gavrish E, Epstein S, Clardy J, Lewis K. 2010. Siderophores from neighboring organisms promote the growth of uncultured bacteria. Chem Biol 17:254–264. doi: 10.1016/j.chembiol.2010.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lennon JT, Jones SE. 2011. Microbial seed banks: the ecological and evolutionary implications of dormancy. Nat Rev Microbiol 9:119–130. doi: 10.1038/nrmicro2504. [DOI] [PubMed] [Google Scholar]
- 90.Oliver JD. 2010. Recent findings on the viable but nonculturable state in pathogenic bacteria. FEMS Microbiol Rev 34:415–425. doi: 10.1111/j.1574-6976.2009.00200.x. [DOI] [PubMed] [Google Scholar]
- 91.Janssen PH, Yates PS, Grinton BE, Taylor PM, Sait M. 2002. Improved culturability of soil bacteria and isolation in pure culture of novel members of the divisions Acidobacteria, Actinobacteria, Proteobacteria, and Verrucomicrobia. Appl Environ Microbiol 68:2391–2396. doi: 10.1128/AEM.68.5.2391-2396.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Taylor CB. 1951. Nature of the factor in soil-extract responsible for bacterial growth-stimulation. Nature 168:115–116. doi: 10.1038/168115a0. [DOI] [PubMed] [Google Scholar]
- 93.Kaeberlein T, Lewis K, Epstein SS. 2002. Isolating “uncultivable” microorganisms in pure culture in a simulated natural environment. Science 296:1127–1129. doi: 10.1126/science.1070633. [DOI] [PubMed] [Google Scholar]
- 94.Bollmann A, Lewis K, Epstein SS. 2007. Incubation of environmental samples in a diffusion chamber increases the diversity of recovered isolates. Appl Environ Microbiol 73:6386–6390. doi: 10.1128/AEM.01309-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Nichols D, Lewis K, Orjala J, Mo S, Ortenberg R, O'Connor P, Zhao C, Vouros P, Kaeberlein T, Epstein SS. 2008. Short peptide induces an “uncultivable” microorganism to grow in vitro. Appl Environ Microbiol 74:4889–4897. doi: 10.1128/AEM.00393-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Jones SE, Lennon JT. 2010. Dormancy contributes to the maintenance of microbial diversity. Proc Natl Acad Sci U S A 107:5881–5886. doi: 10.1073/pnas.0912765107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Knight R, Jansson J, Field D, Fierer N, Desai N, Fuhrman JA, Hugenholtz P, van der Lelie D, Meyer F, Stevens R, Bailey MJ, Gordon JI, Kowalchuk GA, Gilbert JA. 2012. Unlocking the potential of metagenomics through replicated experimental design. Nat Biotechnol 30:513–520. doi: 10.1038/nbt.2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ellis RJ, Morgan P, Weightman AJ, Fry JC. 2003. Cultivation-dependent and -independent approaches for determining bacterial diversity in heavy-metal-contaminated soil. Appl Environ Microbiol 69:3223–3230. doi: 10.1128/AEM.69.6.3223-3230.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]