

Abstract
Bacillus thuringiensis produces chitinases, which are involved in its antifungal activity and facilitate its insecticidal activity. In our recent work, we found that a 16-bp sequence, drechiB (AGACTTCGTGATGTCT), downstream of the minimal promoter region of the chitinase B gene (chiB) was a critical site for the inducible expression of chiB in B. thuringiensis Bti75. In this work, we show that a GntR family transcriptional regulator (named YvoABt), which is homologous to YvoA of Bacillus subtilis, can specifically bind to the drechiB oligonucleotide sequences in vitro by using electrophoretic mobility shift assays (EMSAs) and isothermal titration calorimetry (ITC) assays. The results of quantitative real-time reverse transcription-PCR (qRT-PCR) and Western blotting indicated that deletion of yvoA caused an ∼7.5-fold increase in the expression level of chiB. Furthermore, binding of purified YvoABt to its target DNA could be abolished by glucosamine-6-phosphate (GlcN-6-P). We also confirmed, in the presence of the phosphoprotein Hpr-Ser45-P, that purified CcpABt bound specifically to the promoter of chiB, which contains the “crechiB” sequence (ATAAAGCGTTTACA). According to the results of qRT-PCR and Western blotting, deletion of ccpA resulted in a 39-fold increase in the chiB expression level, and glucose no longer influenced the expression of chiB. We confirm that chiB is negatively controlled by both CcpABt and YvoABt in Bti75.
INTRODUCTION
Chitinases (EC 3.2.1.14), which have the ability to digest chitin into N-acetylglucosamine, are produced by a wide range of organisms and can be potentially used in industry, medicines, scientific research, and agriculture (1–3). Bacillus thuringiensis (Bt) also produces chitinases (4–6). Several studies have reported that chitinases generated by Bt are involved in its antifungal activity and can enhance the insecticidal activity of Bt strains (7–10). Many studies have reported the expression and application of chitinases in microorganisms (11–15). However, reports on the regulation mechanism of chitinase genes are scarce.
The expression of the chitinase gene is controlled by the GntR family regulator DasR in Streptomyces coelicolor (16, 17). Actually, DasR is a global regulator that is involved in GlcNAc transport and metabolism, antibiotic synthesis (18), and morphological differentiation (16). Colson et al. previously identified a 16-bp consensus sequence (AGTGGTCTAGACCACT) in the promoters of chitin and GlcNAc metabolism-related genes in Streptomycetes, which was termed a DasR-responsive element (dre) (17). YvoA, the ortholog of DasR in Bacillus subtilis, is a bacterial repressor involved in GlcNAc transport and utilization; Titgemeyer and colleagues showed previously that YvoA binds specifically to a similar 16-bp consensus sequence (ATTGGTATAGACAACT) upstream of the nagAB and nagP genes (19, 20). In our recent work, we also found a 16-bp sequence, the drechiB sequence (AGACTTCGTGATGTCT), downstream of the minimal promoter region of the chitinase B gene (chiB) (Fig. 1A), which is a critical site for the inducible expression of chiB in B. thuringiensis Bti75 (21). Moreover, electrophoretic mobility shift assays (EMSAs) showed that some regulatory factors bind to the drechiB site in strain Bti75 cultured in the absence of the inducer. Thus, we hypothesized that YvoA of Bti75 is the regulator responsible for binding to drechiB to control the expression of chiB.
FIG 1.

(A) Map of the genetic elements of the chiB upstream region (altered according to Fig. 4 in reference 21). The transcription start site (TSS) is indicated as +1. The locations of the dre site and cre site are labeled. Gray regions in the minimal promoter represent the −10 box and the −35 box. SD, Shine-Dalgarno sequence. (B) Synteny of the yvoABt locus in Bacillus thuringiensis HD-789. Gene organization was deduced from the complete genome sequences retrieved from the NCBI database. Black rectangles upstream of the nagA ortholog represent identified dre-like sequences.
In addition, we also identified a 14-bp sequence that is similar to the catabolite response element (cre) consensus sequence inside the minimal promoter region. The cre consensus sequence is a 14-bp partially palindromic sequence that specifically interacts with CcpA (catabolite control protein A) in the metabolic process of carbon catabolite repression (CCR) (22). CCR is a general phenomenon whereby microbes adjust their expression of catabolic genes in response to the availability of rapidly metabolizable carbon sources (23–25). In low-GC Gram-positive bacteria such as B. subtilis, the key regulator of CCR is CcpA (a member of LacI/GalR family of bacterial regulatory proteins); with the help of Hpr phosphorylated at the Ser46 residue (Hpr-Ser46-P), CcpA can specifically bind to target promoters at cre sites (23, 26, 27). Chitinase can hydrolyze chitin into GlcNAc, which can be utilized by microorganisms as a carbon and nitrogen source; therefore, it is unsurprising that it is also regulated by CCR. Until now, there has been no detailed study of the mechanism of regulation of chitinases by CCR in Bt.
In the present study, we show that the upstream region of the Bti75 chiB gene contains drechiB and crechiB, which are similar to the cis-acting elements dre and cre in other species. In vitro experiments indicated that the transcriptional regulators YvoABt and CcpABt can specifically bind to drechiB and crechiB, respectively. The results of quantitative real-time reverse transcription-PCR (qRT-PCR) and Western blot analyses confirmed that chiB is negatively regulated by YvoABt and CcpABt in Bti75.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
Bti75, which is a highly efficiently inducible chitinase-producing strain, was previously isolated and maintained in our laboratory. Bti75 and its variants were cultured at 30°C with shaking at 200 rpm. Escherichia coli DH5α, for plasmid constructions, and E. coli BL21(DE3), for protein purification, were cultured at 37°C with shaking at 200 rpm. Luria-Bertani (LB) broth was used to grow E. coli as well as Bti75 and its mutants. For chitinase gene expression assays, S minimal medium [15 mM (NH4)2SO4, 80 mM K2HPO4, 44 mM KH2PO4, 3.4 mM Na-citrate, 1 mM MgSO4 · 7H2O (pH 7.4)] (28) supplemented with 0.5% yeast extract powder, G medium (S medium plus 0.5% glucose), and C medium (S medium plus 0.5% colloidal chitin) were used. Colloidal chitin was prepared according to a previously described method (9). For strains with antibiotic resistance, appropriate antibiotics were added to the culture medium at the following concentrations: 5 μg ml−1 chloramphenicol (Cam), 50 μg ml−1 erythromycin (Erm), 50 μg ml−1 ampicillin (Amp), and 50 μg ml−1 kanamycin (Kan). The primers used in this study are listed in Table 1. Plasmids and strains used in this study are listed in Table 2.
TABLE 1.
Primers used in this study
| Primer | Sequence (5′→3′) | Function |
|---|---|---|
| SalI-erm-F | CTGGTCGACGCAAACTTAAGAGTGTGT | ccpA and yvoA deletion |
| XbaI-erm-R | CGCTCTAGAGACCTCTTTAGCTTCTTG | ccpA and yvoA deletion |
| KpnI-CcpAup-F | TCAGGTACCTTAAACGAAAGAGGTCGTCTCG | ccpA deletion |
| XbaI-CcpAup-R | CGCTCTAGATTCATCTCATCGCACACTCCTT | ccpA deletion |
| SalI-CcpAdown-F | TCTGTCGACCGTATCCAATTTAGAGATTCAACG | ccpA delection |
| PstI-CcpAdown-R | CCGCTGCAGAGGTAAGCTATATACTAGGGAGGATT | ccpA deletion |
| PstI-YvoAup-F | GTCTGCAGCAAGCTTTCCGAAATGTA | yvoA deletion |
| SalI-YvoAup-R | GCGGTCGACATTCTCAGATGGGATTTTAT | yvoA deletion |
| XbaI-YvoAdown-F | CGCTCTAGACAGTGGAAATGAAAATGGAT | yvoA deletion |
| KpnI-YvoAdown-R | AATGGTACCCCCTCGTACTTAAATAGCCT | yvoA deletion |
| NcoI-CcpA-F | TCGCCATGGTAATGAACGTAACAATCTATGATGTAG | ccpA cloning |
| XhoI-CcpA-R | CGCCTCGAGTTTCGTTGAATCTCTAAATTGGAT | ccpA cloning |
| NcoI-Hpr-F | CTGCCATGGTCATGGAAAAAATCTTTAAAGTAACT | hpr cloning |
| XhoI-Hpr-R | CATCTCGAGTTCTCCTAATCCTTCGTTTTTCAT | hpr cloning |
| NcoI-Hprk-F | CGGCCATGGGTATGAAATGTTTTTTTCTATT | hprK cloning |
| XhoI-Hprk-R | ATCTCGAGTATCTCCTGATTCCCTAACTCAATCGC | hprK cloning |
| NcoI-YvoA-F | GCCCCATGGTGATGAACATCGACAAG | yvoA cloning |
| XhoI-YvoA-R | CTGCTCGAGTTTGTTACGTGCAATATTC | yvoA cloning |
| EcoRI-ChiB-F | ATATGAATTCATGAGGTCTCAAAAATTCACACTG | chiB cloning |
| XhoI-ChiB-R | ATCTCGAGGTTTTCGCTAATGACGGCATT | chiB cloning |
| P16SrRNA-RT-F | GCCGTAAACGATGAGTGCTAAGTG | 16S rRNA RT-PCR |
| P16SrRNA-RT-R | TGAGTTTCAGTCTTGCGACCGTA | 16S rRNA RT-PCR |
| PchiB-RT-F | GCCGCTGATGAAAAGACAAGA | chiB RT-PCR |
| PchiB-RT-R | TTCCCAGTCTAAATCTACGCCA | chiB RT-PCR |
| PchiB-F | CCTTTCGTTTTCATATATAGTTTGT | PchiB cloning |
| PchiB-R | CTAGATAAAATGATCAGACATCACG | PchiB cloning |
| Pcre-F | TTTTTCAACTTAATAAAGCGTTTACACTAAATCTTACATT | Pcre-CcpA EMSA |
| Pcre-R | AATGTAAGATTTAGTGTAAACGCTTTATTAAGTTGAAAAA | Pcre-CcpA EMSA |
| Pcre-R(B) | AATGTAAGATTTAGTGTAAACGCTTTATTAAGTTGAAAAA(5′ biotin) | Pcre-CcpA EMSA |
| Pdre-F | GCTCCCTTGTATAGACTTCGTGATGTCTGATCATTTTATC | Pdre-YvoA EMSA |
| Pdre-R | GATAAAATGATCAGACATCACGAAGTCTATACAAGGGAGC | Pdre-YvoA EMSA |
| Pdre-R(B) | GATAAAATGATCAGACATCACGAAGTCTATACAAGGGAGC(5′ biotin) | Pdre-YvoA EMSA |
TABLE 2.
Bacterial strains and plasmids used in this study
| Plasmid or strain | Relevant characteristic(s)a | Source or reference |
|---|---|---|
| Plasmids | ||
| pET-28a(+) | Expression vector; Kanr; C/N-terminal His tag/thrombin/T7 tag, T7 lac promoter, T7 transcription start, F1 origin, lacI | Novagen |
| pET-CcpA | Kanr; ccpA gene cloned into pET-28a(+), His tag binding C terminus of CcpA | This study |
| pET-YvoA | Kanr; yvoA gene cloned into pET-28a(+), His tag binding C terminus of YvoA | This study |
| pET-Hpr | Kanr; hpr gene cloned into pET-28a(+), His tag binding C terminus of Hpr | This study |
| pET-HprK | Kanr; hprK gene cloned into pET-28a(+), His tag binding C terminus of HprK/P | This study |
| pET-ChiB | Kanr; chiB gene cloned into pET-28a(+), His tag binding C terminus of ChiB | This study |
| pKSV7 | Ampr Cmr; Bacillus-E. coli shuttle vector, temp sensitive | Laboratory collection |
| pKSV-e | Ampr Emr Cmr; erythromycin gene of pHT315 with its promoter cloned into the pKSV7 SalI/XbaI site | This study |
| pKSV7-ue-CcpA | Ampr Emr Cmr; 1,115-bp upstream fragment of ccpA cloned into the pKSV7-e KpnI/XbaI site | This study |
| pKSV7-ued-CcpA | Ampr Emr Cmr; 1,072-bp downstream fragment of ccpA cloned into the pKSV7-ue SalI/PstI site | This study |
| pKSV7-ue-YvoA | Ampr Emr Cmr; 1,042-bp upstream fragment of yvoA cloned into the pKSV7-e SphI/SalI site | This study |
| pKSV7-ued-YvoA | Ampr Emr Cmr; 1,015-bp downstream fragment of yvoA cloned into the pKSV7-ue XbaI/KpnI site | This study |
| pHT315 | Bacillus-E. coli shuttle vector; Ampr Emr | Pasteur Institute |
| Strains | ||
| E. coli DH5α | F− ϕ80dlacZΔM15 Δ(lacZYA-argF)U169 deoR recA1 endA1 hsdR17(rK− mK+) phoA supE44 λ− thi-1 gyrA96 relA1 | Laboratory collection |
| E. coli BL21(DE3) | Invitrogen | |
| Bti75 | Efficient chitinase-producing strain | Laboratory collection |
| Bti75ΔccpA | Bti75 ΔccpA | This study |
| Bti75ΔyvoA | Bti75 ΔyvoA | This study |
Ampr, ampicillin resistance; Emr, erythromycin resistance; Cmr, chloramphenicol resistance; Kanr, kanamycin resistance.
Construction of ccpA and yvoA deletion mutants of Bti75.
The plasmid used to generate strain Bti75ΔyvoA was constructed as follows: oligonucleotide primers SalI-erm-F and XbaI-erm-R were used to amplify the erythromycin resistance gene, erm, from pHT315. The PCR product was digested with SalI and XbaI and ligated into temperature-sensitive vector pKSV7 treated with the same enzymes to generate vector pKSV-e with erythromycin resistance. The upstream homologous fragment of yvoA was amplified by using primers PstI-YvoAup-F and SalI-YvoAup-R, and then ligated into pKSV-e after digestion with SphI and SalI, generating plasmid pKSV-ue-YvoA. Oligonucleotide primers XbaI-YvoAdown-F and KpnI-YvoAdown-R were used to amplify the downstream homologous fragment of yvoA, which was ligated into pKSV-ue-YvoA (digested by XbaI and KpnI), generating plasmid pKSV-ued-YvoA. Plasmid pKSV-ued-CcpA was constructed similarly to pKSV-ued-YvoA, using oligonucleotide primer pair KpnI-CcpAup-F and XbaI-CcpAup-R and primer pair SalI-CcpAdown-F, PstI-CcpAdown-R. The resultant plasmids were checked by restriction enzyme digestion and DNA sequencing.
Plasmids pKSV-ued-YvoA and pKSV-ued-CcpA were transformed into Bti75 by electroporation separately (29). When the correct transformant was obtained, we inoculated a single colony into LB medium containing Erm (50 μg ml−1) and cultivated it at 42°C for 36 h. The transformant was plated onto LB agar plates containing Erm or Cam and cultivated at 30°C overnight. Colonies with resistance to Erm but not to Cam were considered possible candidates for Bti75ΔyvoA or Bti75ΔccpA. The correct mutants were confirmed by PCR and DNA sequencing.
Protein expression and purification.
For the expression of YvoABt, the yvoA gene was amplified by PCR using oligonucleotide primers NcoI-YvoA-F and XhoI-YvoA-R. The fragment was digested with NcoI and XhoI and ligated into similarly digested pET28a(+) (Novagen, Germany), resulting in a His6 fusion at the C terminus of YvoA (plasmid pET-YvoA). The plasmids used to express CcpA (plasmid pET-CcpA), Hpr (histidine-containing phosphocarrier protein) (plasmid pET-Hpr), HprK/P (Hpr kinase/phosphorylase) (plasmid pET-HprK), and ChiB (plasmid pET-ChiB) were constructed similarly to plasmid pET-YvoA, using oligonucleotide primer pairs NcoI-CcpA-F and XhoI-CcpA-R, NcoI-Hpr-F and XhoI-Hpr-R, NcoI-Hprk-F and XhoI-Hprk-R, and EcoRI-ChiB-F and XhoI-ChiB-R, respectively.
Plasmid pET-YvoA was then transformed into E. coli BL21(DE3) (Invitrogen, Carlsbad, CA, USA). When cells reached an optical density at 600 nm (OD600) of ∼0.6 to 0.9, they were induced by using 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside). After ultrasonic disruption (400 W) of E. coli BL21(DE3), native YvoABt was purified by using its His tag and a nickel column (GE Healthcare, Piscataway, NJ). The eluate was dialyzed in buffer containing 50 mM Tris-HCl (pH 8.0), 50 mM NaCl, 0.5 mM EDTA, and 5% glycerol for ∼18 h. The YvoA protein was stored at −20°C in the presence of 50% glycerol. A similar strategy was used to generate the CcpABt, Hpr, HprK/P, and ChiB proteins.
Phosphorylation of Hpr by HprK/P.
To check whether HprK/P purified in this study could phosphorylate Hpr or not, phosphorylation assays were performed in the presence of 20 μM Hpr and 1 μM HprK/P in phosphorylation buffer (10 mM Tris-HCl [pH 7.0], 50 mM NaCl, 1 mM MgCl2, 0.5 mM dithiothreitol, 5% glycerol, 2 mM ATP, 20 mM fructose-1,6-bisphosphate [FBP]). The reactions were carried out at 37°C for 30 min, and the mixtures were then incubated for 5 min at 70°C to stop the reaction. Phosphorylation of Hpr was detected by both nondenaturing 12% PAGE and SDS-PAGE (30) (Fig. 2).
FIG 2.
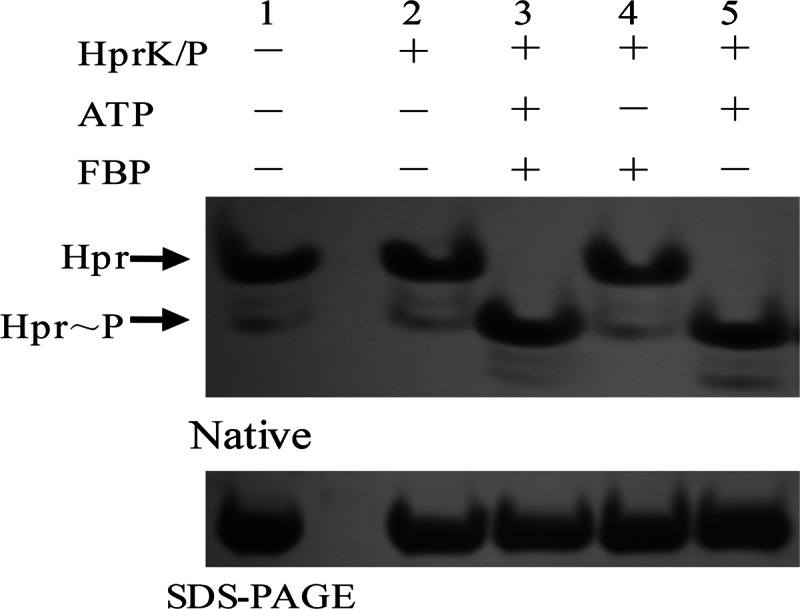
Hpr phosphorylation by HprK/P. Hpr-His6 (20 μM) was mixed with HprK/P-His6 (1 μM) in the presence of 2 mM ATP or 20 mM FBP. The reaction products were analyzed by both nondenaturing 12% PAGE and SDS-PAGE. Lane 1, Hpr; lane 2, Hpr and HprK/P; lane 3, Hpr, HprK/P, ATP, and FBP; lane 4, Hpr, HprK/P, and FBP; lane 5, Hpr, HprK/P, and ATP.
EMSAs.
Protein-DNA interactions were evaluated by an electrophoretic mobility shift assay. Long DNA probes were amplified by PCR using primers and the Bti75 genome as a template. Short DNA probes were generated by annealing primers in Tris-EDTA (TE) buffer. The reaction mixture was heated to 95°C for 5 min and then kept at room temperature for 40 min. The 5′ ends of several primers were labeled with biotin (listed in Table 1). The primers used to generate the probes were as follows: PchiB-F and PchiB-R were used to amplify a 158-bp fragment (named PchiB) of the chiB promoter that contained drechiB and crechiB, and Pdre-F and Pdre-R as well as Pcre-F and Pcre-R were used to generate 40-bp DNA fragments (Pdre and Pcre) that contained drechiB and crechiB, respectively.
The concentrations of probes and proteins used in this study are indicated in the corresponding figures and the legends to these figures. The reaction mixtures were incubated for 30 min at 37°C in reaction buffer containing 10 mM Tris-HCl (pH 7.0), 50 mM NaCl, 1 mM MgCl2, 0.5 mM dithiothreitol, 0.5 mM EDTA, and 5% glycerol. Nonspecific and specific competition assays were carried out in the presence of 0.5 μg ml−1 sheared salmon sperm DNA and 100-fold, 150-fold, and 300-fold excesses of unlabeled fragments, respectively. After the reaction, the mixtures were separated with an 8% nondenaturing polyacrylamide gel in Tris-borate-EDTA (TBE) buffer. In the competition reaction, the probes were transferred onto a nylon membrane from the gels in TBE buffer. Finally, the nylon membrane was stained by using a biotin chromogenic detection kit (Thermo Fisher Scientific Inc.) to show the location of the labeled probes.
RNA extraction and quantitative real-time reverse transcription-PCR.
Bacteria were grown at 30°C in culture medium for ∼9 h to the logarithmic phase (OD600 of ∼2.5), with shaking. Total RNA was extracted by using RNAiso Plus (TaKaRa, Dalian, China), according to the manufacturer's instructions. After the removal of genomic DNA by using RNase-free recombinant DNase I (TaKaRa), cDNA was synthesized from total RNA by using the PrimeScript RT reagent kit (Perfect Real Time; TaKaRa). Quantification of cDNA was carried out by using SYBR Premix Ex Taq (Perfect Real Time; TaKaRa), and real-time amplification of the PCR product was analyzed by using StepOne software (Applied Biosystems, Foster City, CA, USA), according to the supplier's instructions. The 16Sr RNA gene acted as the endogenous control. The relative amount of cDNA was calculated according to the 2−ΔΔCT method (31). The sequences of the primers for qRT-PCR are presented in Table 1.
Isothermal titration calorimetry.
Isothermal titration calorimetry (ITC) measurements were carried out as described previously by Wang et al. (32). The experiments were performed on a MicroCal iTC200 isothermal titration calorimetry instrument (GE Healthcare) with a solution containing 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, and 1 mM EDTA at 16°C. A 50 μM DNA probe (Pdre) was titrated into 10 μM YvoABt. Both DNA probe and protein solutions were degassed by spinning at 15,000 × g for 15 min. The titration consisted of an initial injection of 0.4 μl, followed by 26 injections of 1.5 μl every 120 s at 16°C. To determine the baseline, the DNA probe was titrated into the same buffer without the protein under the same conditions. The titration data and binding plot after baseline subtraction were analyzed by using MicroCal Origin software.
Western blot assays.
The purified ChiB protein was used for rabbit polyclonal antibody generation. Bti75 and Bti75ΔyvoA were grown in 100 ml S medium and C medium at 30°C for ∼9 h, respectively. The two bacterial strains were collected by centrifugation at 7,000 × g for 15 min at 4°C and resuspended in 5 ml lysis buffer (50 mM Tris-HCl [pH 8.0], 150 mM NaCl, and 1 mM PMSF [phenylmethanesulfonyl fluoride]). The same amount of bacteria was disrupted by sonication (400 W). Thirty-five microliters of the crude proteins was resolved by SDS-PAGE and transferred onto Immobilon-P membranes (Millipore, Billerica, MA). Anti-ChiB polyclonal antibodies and horseradish peroxidase-coupled goat anti-rabbit antibodies were used to detect the expression level of chiB. ChiB expressed from Bti75 and Bti75ΔccpA was assessed similarly.
Computational prediction of CcpA- and YvoA-responsive elements in Bt.
The PREDetector software program (33) was used to predict the positions of cre-like and dre-like sequences in B. thuringiensis subsp. israelensis strain HD-789 by using a list of cre targets in B. subtilis reported previously by Fujita (22) and three dre targets in Bti75 that we found in this work. The cutoff score was set at 8.0 to predict the cre-like sequence, and the score was set at 6.0 to predict the dre-like sequence, as fewer dre targets were used to generate the matrix.
RESULTS
YvoA and CcpA are presumed regulators of the expression of chiB.
From BLASTP analysis, we found a GntR family transcriptional regulator (GenBank accession no. AFQ27885.1) in B. thuringiensis HD-789 that has 45% sequence identity and 67% similarity to the B. subtilis regulator YvoA (YvoABs). Like YvoA in B. subtilis, this gene is located adjacent to nagB, which encodes the enzyme that catalyzes the deamination and isomerization of GlcNAc-6-P to Fru-6-P. Indeed, this gene is the third element of a tricistronic operon in which the first two positions are nagA (the GlcNAc-6-P deacetylase gene) and nagB (Fig. 1B). Thus, we named the gene yvoABt and speculated that its product may bind to the 16-bp locus upstream of chiB as a regulator.
At the same time, using the list of cre targets in B. subtilis reported by Fujita (22), we used the PREDetector software program (33) to identify an additional 14-bp sequence, crechiB (ATAAAGCGTTTACA), which is similar to the cis-acting element cre, in the promoter of chiB in the genomic sequence of B. thuringiensis HD-789 (see Data Set S1 in the supplemental material). The crechiB element overlaps the −35 box of the chiB promoter (Fig. 1A). The catabolite control protein CcpA is a pleiotropic regulator that mediates the global transcriptional response by binding cre elements site specifically. Thus, we hypothesized that chiB might also be regulated by CcpABt in Bti75.
YvoA can bind drechiB sequences in vitro.
In this study, YvoABt of Bti75 was heterologously expressed in E. coli. To confirm the interaction between YvoABt and drechiB, we used unlabeled and biotin-labeled DNA fragments containing the drechiB site for EMSAs with purified YvoABt. We incubated purified YvoABt with the 158-bp fragment of the chiB promoter PchiB (positions −106 to +52 relative to the transcription start site) and a 40-bp DNA fragment, Pdre (positions +9 to +48 relative to the transcription start site), which contains drechiB in the middle, for EMSAs. As shown in Fig. 3A, YvoABt retarded the migration of PchiB in a concentration-dependent manner.
FIG 3.

EMSAs to detect binding of CcpABt and YvoABt to their targets. DNA fragments were detected by staining. (The concentrations of CcpABt and YvoABt are indicated in the labels above each panel.) (A) Analysis of YvoABt binding to PchiB (158 bp). A total of 0.05 μM PchiB was mixed with various concentrations of YvoABt. (B) Analysis of CcpABt binding to PchiB. A total of 0.05 μM PchiB was mixed with various concentrations of CcpABt. (C to E) Analysis of CcpABt binding to Pcre (40 bp). (C) A total of 0.1 μM Pcre was mixed with various concentrations of CcpABt. (D) A total of 0.1 μM was Pcre was mixed with various concentrations of CcpABt in the presence of 4 μM Hpr. (E) A total of 0.1 μM Pcre was mixed with various concentrations of CcpABt in the presence of 4 μM Hpr-Ser45-P.
To further investigate the specific interaction between YvoABt and drechiB, 0.5 ng μl−1 salmon sperm DNA (nonspecific competition) and a 100-fold excess of unlabeled Pdre (specific competition) were added to the mixture for the reaction of YvoABt with Pdre (biotin labeled). As shown in Fig. 4, YvoABt also retarded the migration of Pdre, while salmon sperm DNA could not compete away YvoABt from Pdre (biotin labeled) (Fig. 4A). On the other hand, when a 100-fold excess of unlabeled probe was present, the retarded band of biotin-labeled Pdre disappeared completely (Fig. 4C). The results from these experiments suggested that YvoABt binds to the drechiB sequence specifically to regulate the expression of chiB.
FIG 4.

EMSAs to determine specific binding of YvoABt and CcpABt to drechiB and crechiB. DNA fragments were detected by using a biotin chromogenic reagent. (A and C) Nonspecific and specific competition assays with YvoABt (0.5 μM) and Pdre (0.1 μM) (40 bp). (A) Lane 1, Pdre (biotin-labeled DNA [Bio]); lane 2, Pdre (Bio) and YvoABt; lane 3, Pdre (Bio), YvoABt, and 0.5 ng μl−1 salmon sperm DNA. (C) Lane 1, Pdre (Bio); lane 2, Pdre (Bio) and YvoABt; lane 3, Pdre (Bio), YvoABt, and a 100-fold excess of unlabeled Pdre. (B and D) Nonspecific and specific competition assays with CcpABt (0.5 μM) and Pcre (0.1 μM). (B) Lane 1, Pcre (Bio); lane 2, Pcre (Bio) and CcpABt; lane 3, Pcre (Bio), CcpABt, and 0.5 ng μl−1 salmon sperm DNA; lanes 4 and 5, Pcre (Bio), CcpABt, and 0.5 ng μl−1 salmon sperm DNA plus 2 μM Hpr and 2 μM Hpr-Ser45-P, respectively. (D) Lane 1, Pcre (Bio); lanes 2 and 3, Pcre (Bio), CcpABt, and Hpr-Ser45-P plus 150- and 300-fold excesses of unlabeled Pcre, respectively; lanes 4 and 5, Pcre (Bio), CcpABt, and Hpr plus 150- and 300-fold excesses of unlabeled Pcre, respectively.
To study the interactions between YvoABt and Pdre quantitatively, we also used ITC to determine their binding affinity. As shown in Fig. 5, the binding of YvoABt to Pdre fitted well to a one-site binding model, with a calculated Kd (dissociation constant) value of ∼0.46 μM (Fig. 5B), which was significantly different from that of the negative control (Fig. 5A). Therefore, our results suggested that YvoABt binds to Pdre specifically.
FIG 5.

YvoABt DNA binding abilities measured by ITC. Shown are binding isotherms of 50 μM Pdre titrated with binding buffer (A) or binding buffer plus 10 μM YvoABt (B). For panel A, no binding was detected. For panel B, the data were fitted to a one-site binding model to give a Kd of ∼0.46 μM.
Hpr can be phosphorylated efficiently by Hprk/P.
The Hpr and HprK/P proteins were cloned and purified similarly to YvoABt. As shown in Fig. 2, we found that 1 μM HprK/P phosphorylated >20 μM Hpr at 37°C for 30 min in reaction buffer with ATP and FBP. Moreover, the same results were obtained by using reaction buffer with 2 mM ATP but without FBP (Fig. 2, lane 5), while HprK/P could not phosphorylate Hpr in reaction buffer without ATP (Fig. 2, lane 4).
CcpA can bind specifically to the chiB promoter with the assistance of Hpr-Ser45-P.
To confirm the interaction between CcpABt and crechiB, PchiB was tested with CcpABt alone in the reaction buffer. We found that CcpABt retarded the mobility of PchiB similarly to the interaction of YvoABt with PchiB (Fig. 3B). However, we observed a similar phenomenon (data not shown) when we used another DNA fragment (∼150 bp) without obvious cre sites at a CcpABt concentration of 0.1 μM. These results raised the possibility that the DNA retardation observed was the result of nonspecific binding of CcpABt because of its intrinsic DNA binding nature.
To further investigate the specific interaction between CcpABt and crechiB, we synthesized a 40-bp DNA fragment, Pcre (positions −53 to −14 relative to the transcription start site), of the chiB promoter, which contains crechiB in the middle. Only when its concentration reached 0.5 μM could CcpABt obviously retard the movement of Pcre alone (Fig. 3C). Thus, we hypothesized that CcpABt alone would have a higher affinity for the long DNA fragment than the short one because of its intrinsic DNA binding nature. We then tried to determine whether Hpr or Hpr-Ser45-P could further stimulate CcpABt binding to crechiB. Hpr and Hpr-Ser45-P (2 μM each) were mixed in the mixture for the reaction of CcpABt with Pcre. We found that Hpr did not increase the affinity of CcpABt for the DNA fragments (Fig. 3D). In contrast, Hpr-Ser45-P enhanced the affinity of CcpABt for the DNA fragments (Fig. 3E). We also further observed that even 10 nM CcpABt effectively retarded the movement of Pcre with the help of Hpr-Ser45-P (data not shown), which proved that Hpr-Ser45-P, rather than Hpr, significantly enhanced the affinity of CcpABt for Pcre. At the same time, we confirmed that Hpr-Ser45-P or HprK/P itself could not retard the movement of PchiB (data not show).
To determine whether the binding of CcpABt to crechiB was specific or not, EMSAs were carried out in the presence of Hpr-Ser45-P or Hpr with the addition of 0.5 ng μl−1 salmon sperm DNA (nonspecific competition) or 150- and 300-fold excesses of the same unlabeled DNA fragment of Pcre (specific competition). As shown in Fig. 4B, salmon sperm DNA easily competed away CcpABt from biotin-labeled Pcre without Hpr-Ser45-P (Fig. 4B, lane 3). In contrast, CcpABt bound strongly to the labeled probe in the presence of Hpr-Ser45-P mixed with a nonspecific fragment (Fig. 4B, lane 4). Moreover, Hpr could not assist CcpABt in binding to Pcre (Fig. 4B, lane 5). In the EMSA for specific competition, we found that when the amount of the specific fragment was increased 300-fold compared with the amount of the labeled probe, the retardation phenomenon in the presence of Hpr-Ser45-P disappeared (Fig. 4D, lane 3). However, a retarded band was observed in the lane with Hpr (Fig. 4D, lane 5). Thus, according to the results of assays for specific and nonspecific competition, we confirmed that CcpABt specifically binds to the cre site of the chiB promoter in Bti75 with the help of Hpr-Ser45-P, but Hpr did not have this function.
Both YvoA and CcpA can repress the expression of chiB in vivo.
To study the role of YvoABt and CcpABt in the expression of chiB in vivo, Bti75 and Bti75ΔyvoA were cultured in S minimal medium and C medium (with colloidal chitin), respectively. As shown in Fig. 6A, we observed that the relative expression level of chiB in Bti75ΔyvoA was elevated ∼7.5-fold compared to that in Bti75 in S medium. Moreover, the relative expression level of chiB in Bti75ΔyvoA in C medium was ∼8-fold higher than that in Bti75 in S medium. At the same time, the relative expression level of chiB in Bti75 in C medium was elevated by ∼4-fold compared with that in Bti75 in S medium but was also ∼2-fold lower than that in Bti75ΔyvoA in S medium.
FIG 6.

qRT-PCR analysis of the relative transcript levels of chiB genes of different strains in different media. (A) Relative transcript levels of the chiB genes of Bti75 and Bti75ΔyvoA in S medium (S minimal medium) and C medium (S medium plus 0.5% colloidal chitin). (B) Relative transcript levels of the chiB genes of Bti75 and Bti75ΔccpA in G medium (S medium plus 0.5% glucose) and S medium.
Strains Bti75 and Bti75ΔccpA were cultured in S medium and G medium, respectively. The qRT-PCR results (Fig. 6B) showed no obvious difference in the relative expression levels of chiB in Bti75 and Bti75ΔccpA in S medium. However, in G medium, the relative expression level of chiB in Bti75 was reduced by almost 39-fold compared with that in Bti75ΔccpA. This indicated that CcpABt could severely repress the expression of chiB when the medium contains rapidly metabolizable carbon sources such as glucose. Taken together, the results suggested that CcpABt and YvoABt act as negative regulators of chiB in Bti75.
Western blotting indicates that YvoA and CcpA repress the expression of chitinase B.
To detect the expression of chiB in Bti75 and its mutant strains at the protein level, Western blotting was performed. As shown in Fig. 7A, there was significantly more ChiB in Bti75ΔyvoA than in the parental strain in both C medium and S medium, while the amounts of ChiB in Bti75ΔyvoA in C and S media were similar (Fig. 7A, lanes 2 and 4). For Bti75, there was a clear increase in the level of ChiB in C medium compared to that in S medium (Fig. 7A, lanes 1 and 3). All these results are consistent with the qRT-PCR results for Bti75 and Bti75ΔyvoA in C and S media.
FIG 7.
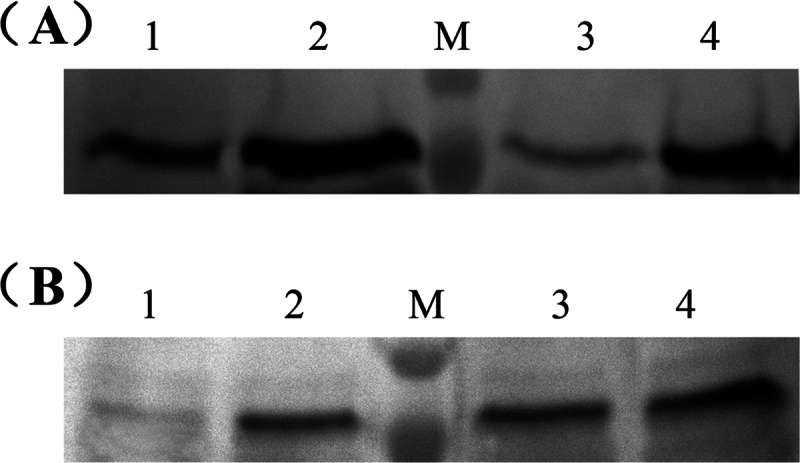
Western blot analysis to determine the expression levels of chiB in Bti75 and its mutants. (A) Expression levels of the chiB genes of Bti75 and Bti75ΔyvoA in C medium and S medium. Lanes 1 and 2, Bti75 and Bti75ΔyvoA in C medium, respectively; lanes 3 and 4, Bti75 and Bti75ΔyvoA in S medium, respectively. (B) Expression levels of the chiB genes of Bti75 and Bti75ΔccpA in G medium and S medium. Lanes 1 and 2, Bti75 and Bti75ΔccpA in G medium, respectively; lanes 3 and 4, Bti75 and Bti75ΔccpA in S medium, respectively. M is the molecular mass marker, which shows molecular masses of 70 kDa and 100 kDa.
On the other hand, as shown in Fig. 7B, there were roughly equal amounts of ChiB in Bti75 and Bti75ΔccpA in C medium without glucose (lanes 3 and 4); however, in G medium, the level of the ChiB protein in Bti75 was almost undetectable (lane 1), and the amount of ChiB in Bti75ΔccpA was similar to those in Bti75 and Bti75ΔccpA in C medium (lane 2). These results were also consistent with the qRT-PCR results for Bti75 and Bti75ΔccpA in G and C media. Thus, the Western blot results suggested that YvoABt and CcpABt repressed the expression of chiB in Bti75.
GlcN-6-P is an effector for YvoA.
To investigate the possible effectors for YvoABt, five different sugars (Glc, GlcNAc, Glc-6-P, GlcNAc-6-P, and GlcN-6-P) were incubated separately with Pdre and purified YvoABt for EMSAs; the final concentration of sugar was 100 mM. The EMSA results in Fig. 8 showed that only GlcN-6-P could abolish the DNA binding capability of YvoABt; the other sugars had no effect on the binding of YvoABt to Pdre. This result is in agreement with data from effector analyses of YvoA in B. subtilis (19) and DasR in S. coelicolor (16).
FIG 8.
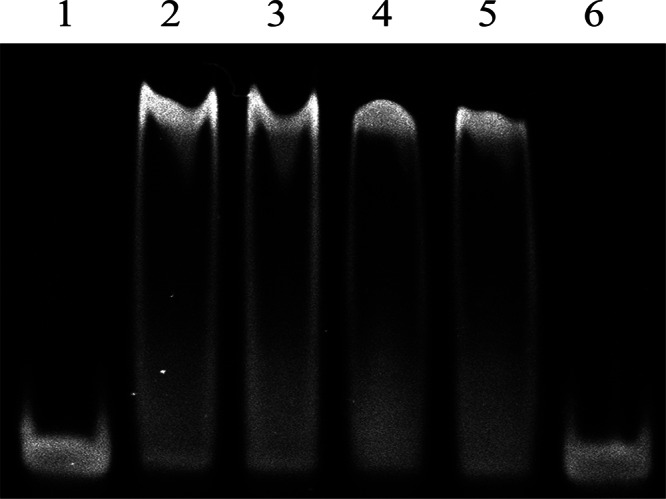
EMSAs to identify the inhibitor of binding of YvoABt to drechiB. Pdre (0.1 μM) was electrophoresed alone (lane 1) and after incubation with 0.4 μM YvoABt plus a final concentration of 100 mM Glc (lane 2), GlcNAc (lane 3), Glc-6-P (lane 4), GlcNAc-6-P (lane 5), or GlcN-6-P (lane 6).
DISCUSSION
We confirmed that chiB is negatively controlled by YvoABt and CcpABt in Bti75. There have been some reports about cis-acting elements and regulatory mechanisms of the genes related to chitin metabolism in Streptomyces and Bacillus. Titgemeyer and collaborators demonstrated DasR, which is a pleiotropic transcription factor, regulates chitin uptake and GlcNAc utilization (17), antibiotic synthesis (18), and morphological differentiation (16) in S. coelicolor. However, Bertram et al. found only two genes (nagA and nagP) that were directly repressed by YvoA. Thus, those authors thought that YvoA might be a less-prominent regulator than DasR and may control only the uptake and subsequent utilization of GlcNAc in B. subtilis (19). Consequently, they suggested that YvoA be renamed NagR.
In this work, we showed that chiB was negatively controlled by YvoABt in Bti75, based on the combination of in silico, in vitro, and in vivo data. Our results also confirmed the speculation of Xie et al. that a sequence-specific DNA binding factor of strain Bti75 could bind to the dre sequence for the inducible expression of chiB (21). Moreover, we found that nagA and nagP of Bti75 contain a 16-bp dre-like sequence within their upstream regions that is also directly repressed by YvoABt (data not shown). To identify further possible YvoABt binding sites in the genome of B. thuringiensis HD-789, we used the PREDetector software program (33) to identify corresponding16-bp dre-like sequence of nagA, nagP, and chiB. The data are listed in Data Set S2 in the supplemental material. Unlike NagR in B. subtilis, the genes controlled by YvoABt are not limited to nagP and the nagAB-yvoA operon. Besides chiB, the chitin binding protein gene also contains a dre-like site (AGTTGGCTAGTCATCT) within its upstream region. Furthermore, the results of in vivo and in vitro experiments indicated that the chitin binding protein gene was also negatively controlled by YvoABt (data not shown). The chitin binding protein is believed to facilitate microbial attachment to chitin and act synergistically with chitinases for chitin degradation. Thus, we predicted that YvoABt might be a more prominent regulator than YvoABs. In addition to GlcNAc uptake and utilization, it also regulates genes involved in the chitin degradation pathway, such as the chitinase gene and the chitin binding protein gene. As to whether it can regulate other genes, further experiments are required.
Rigali et al. proved that GlcN-6-P was the inducing signal for DasR by using EMSAs (16). Resch et al. predicted that the effector of YvoA was GlcNAc-6-P, based on ITC data for B. subtilis (20). However, Bertram et al. incubated YvoA with drenagA in the presence of four different amino sugar compounds (GlcNAc, GlcNAc-6-P, GlcN-6-P, and GlcN) and found that only GlcN-6-P could abolish YvoA's binding to drenagA (19). Moreover, using DNase I footprinting, Gaugué et al. failed to detect any displacement of NagR from its dre binding sites by the amino sugar compounds that they used, whereas those authors state that under the same conditions, GlcN-6-P behaved as the inducing signal for the NagR homolog GamR (34). Recently, Fillenberg et al. produced crystallographic structures of NagR with the putative effector molecules GlcN-6-P and GlcNAc-6-P, implying that both of them are inducing signals for NagR in B. subtilis (35). However, we found that only GlcN-6-P could abolish the DNA binding capability of YvoABt among the five sugar compounds by EMSAs. Since YvoABt has ∼45% sequence identity to YvoABs, one could speculate that different inducing signals might be used. Besides this, the different methods and technologies that we used may yield inconsistent results. Elucidation of the inducing signal for YvoABt will require additional experiments.
Heravi et al. speculated that the chitinase gene (chiS) of Bacillus pumilus is under the control of CCR (36). Generally speaking, in low-GC Gram-positive bacteria such as B. subtilis, the key regulator for exerting CCR is CcpA. With the help of Hpr-Ser46-P, CcpA can specifically bind to target promoters at cre sites (23, 26). The cre consensus sequence is a 14-bp cis-activating, partially palindromic sequence, TGWAARCGYTWNCW, in B. subtilis (23), which is similar to the cre sequences in other microorganisms. Moreover, CcpA may act as either a repressor or an activator, depending on the relative positions of the cre sequences in the genes (37). Hpr is a small phosphocarrier protein (∼10 kDa), which is regarded as the central component of the PTS (phosphoenol pyruvate:carbohydrate phosphotransferase system), encoded by pstH (38). It can be phosphorylated on the His15 residue by enzyme I (EI) of the PTS during sugar uptake. On the other hand, Hpr-Ser46-P is produced by ATP-dependent Hpr kinase/phosphatase (HprK/P) in response to high intracellular concentrations of glycolytic intermediates (26). However, Khan et al. observed that the conserved His15 and Ser46 residues of Hpr were shifted by one amino acid to positions 14 and 45, respectively, in B. thuringiensis subsp. israelensis (38). In this study, we also found a similar amino acid sequence of Hpr in Bti75. In addition, Reizer et al. showed that neither Hpr-His15-P nor Hpr-(Ser46-P)-(His15-P) could bind CcpA to function in CCR (39). HprK/P is a bifunctional enzyme that presents kinase activity at high levels of ATP and FBP, whereas if the concentration of inorganic phosphate is high under starvation conditions, HprK/P will transform into a phosphorylase and dephosphorylate Hpr-Ser46-P into Hpr (40).
In the present study, we found that CcpABt could bind to nucleotide sequences with increasing concentrations and had a higher affinity for long nucleotide sequences than for short ones. We proved that CcpABt binds nonspecifically at high concentrations. The results confirmed that only with the help of Hpr-Ser45-P could CcpABt bind specifically to the cre site of the chiB promoter. Our findings are consistent with the generally accepted mode of binding of CcpA to cre sites. However, there are also several reports of specific DNA binding of CcpA to target sequences without cofactors (41–45), which is different from our data. Hammar et al. proposed that a transcription factor achieves specific binding to its targets by sliding along the DNA through nonspecific binding to specific binding sites (46). This hypothesis may explain the nonspecific binding of CcpA with the nucleotide sequence in some respects.
In addition, we found that HprK/P phosphorylated Hpr efficiently in reaction buffer with 2 mM ATP in the absence of FBP, which is slightly different from data in previous reports (22, 23, 30). Thus, we predicted that ATP might be a key element for HprK/P to function. When the concentration of ATP reaches a certain threshold, HprK/P could act on Hpr. FBP may act as an auxiliary factor to lower the threshold concentration of ATP for HprK/P to function; thus, HprK/P can phosphorylate Hpr at a relatively low concentration of ATP. Of course, this hypothesis requires further experimental support.
We propose the following more detailed model for the regulation of the inducible expression of chiB in Bti75. (i) When the strain is cultured in the presence of both rapidly metabolizable carbon (such as glucose) and chitin or chitooligosaccharides that can be degraded into the inducer of YvoA, YvoA is displaced from the dre site of chiB by the inducer, while CcpA still binds to the cre site of chiB and blocks transcriptional elongation with the help of Hpr-Ser45-P. (ii) When the culture contains rapidly metabolizable carbon sources but not the inducer, the expression of chiB is repressed by both CcpABt and YvoABt. (iii) Transcription of chiB progresses when the strain is cultured in the presence of the inducer but not rapidly metabolizable carbon sources that displace CcpABt and YvoABt from the promoter. (iv) When the culture lacks both rapidly metabolizable carbon sources and an inducer, CcpABt dissociates from the promoter, but YvoABt remains at the dre site and continues to repress the expression of chiB.
Many Bt strains can generate more than one chitinase (4–7). In our previous work, we also found another chitinase gene (chiA) in Bti75 (47). We found that there is also a 16-bp sequence (ATACATCTAGACAACT) (drechiA), which is similar to dreBacillus downstream of the core promoter region of chiA. Also, disruption of this sequence resulted in the constitutive expression of chiA, and the site also appears in Data Set S2 in the supplemental material, so we speculate that YvoABt also participates in the regulation of chiA in Bti75. It is interesting to note that we did not find a cre-like sequence in the promoter of chiA according to Data Set S1 in the supplemental material. This suggests that different chitinase genes may have different regulatory mechanisms. At the same time, we also list other genes that we have proven or predicted to be regulated by YvoABt to show whether these genes have demonstrated or predicted cre sites according to Data Sets S1 and S2 in the supplemental material (Table 3). We found that some genes have both dre-like and cre-like sites in their promoters. This automatically raises the question of whether YvoABt and CcpABt are also involved in the regulation of other genes simultaneously. A definitive answer awaits additional experimental evidence.
TABLE 3.
Genes that have dre-like and cre-like sequences in their promoters in Bti75
| Gene(s) | dre-like sequence | cre-like sequencea |
|---|---|---|
| chiB | AGACATCACGAAGTCT | TGTAAACGCTTTAT |
| chiA | ATACATCTAGACAACT | − |
| nagP | ACACATCTATACAACT | AGAAAGCGTTTTCT |
| nagAB-yvoA | GCACGAGTAGTTGTCT | − |
| Chitin binding protein gene | AGTTGGCTAGTCATCT | − |
−, not found.
This work demonstrates that chiB of B. thuringiensis is regulated by YvoABt and CcpABt. Research on the regulatory mechanism of chitinase will permit better utilization of bacterial chitinase. Liu et al. demonstrated that the chitinase produced by B. thuringiensis improved its insecticidal activity (9). Furthermore, the roles of CcpA and YvoA have been studied in some detail in B. subtilis compared to B. thuringiensis. Therefore, the regulation mechanism of CcpABt and YvoABt in B. thuringiensis requires further investigation.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by grants from the Development Program of China (863 program) (grant no. 2011AA10A203), the National Natural Science Foundation of China (grant no. 31371979), and the Tianjin Natural Science Foundation (grant no. 15JCYBJC30200).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.01549-15.
REFERENCES
- 1.Khoushab F, Yamabhai M. 2010. Chitin research revisited. Mar Drugs 8:1988–2012. doi: 10.3390/md8071988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Felse PA, Panda T. 1999. Regulation and cloning of microbial chitinase genes. Appl Microbiol Biotechnol 51:141–151. doi: 10.1007/s002530051374. [DOI] [PubMed] [Google Scholar]
- 3.Flach J, Pilet PE, Jollès P. 1992. What's new in chitinase research? Experientia 48:701–716. doi: 10.1007/BF02124285. [DOI] [PubMed] [Google Scholar]
- 4.Thamthiankul S, Suan-Ngay S, Tantimavanich S, Panbangred W. 2001. Chitinase from Bacillus thuringiensis subsp. pakistani. Appl Microbiol Biotechnol 56:395–401. doi: 10.1007/s002530100630. [DOI] [PubMed] [Google Scholar]
- 5.Driss F, Kallassy-Awad M, Zouari N, Jaoua S. 2005. Molecular characterization of a novel chitinase from Bacillus thuringiensis subsp. kurstaki. J Appl Microbiol 99:945–953. doi: 10.1111/j.1365-2672.2005.02639.x. [DOI] [PubMed] [Google Scholar]
- 6.Barboza-Corona JE, Reyes-Rios DM, Salcedo-Hernández R, Bideshi DK. 2008. Molecular and biochemical characterization of an endochitinase (ChiA-HD73) from Bacillus thuringiensis subsp. kurstaki HD-73. Mol Biotechnol 39:29–37. doi: 10.1007/s12033-007-9025-4. [DOI] [PubMed] [Google Scholar]
- 7.Gomaa EZ. 2012. Chitinase production by Bacillus thuringiensis and Bacillus licheniformis: their potential in antifungal biocontrol. J Microbiol 50:103–111. doi: 10.1007/s12275-012-1343-y. [DOI] [PubMed] [Google Scholar]
- 8.Xiao L, Liu C, Xie CC, Cai J, Chen YH. 2012. The direct repeat sequence upstream of Bacillus chitinase genes is cis-acting elements that negatively regulate heterologous expression in E. coli. Enzyme Microb Technol 50:280–286. doi: 10.1016/j.enzmictec.2012.02.001. [DOI] [PubMed] [Google Scholar]
- 9.Liu D, Cai J, Xie C, Liu C, Chen Y. 2010. Purification and partial characterization of a 36-kDa chitinase from Bacillus thuringiensis subsp. colmeri, and its biocontrol potential. Enzyme Microb Technol 46:252–256. doi: 10.1016/j.enzmictec.2009.10.007. [DOI] [Google Scholar]
- 10.Hu SB, Liu P, Ding XZ, Yan L, Sun YJ, Zhang YM, Li WP, Xia LQ. 2009. Efficient constitutive expression of chitinase in the mother cell of Bacillus thuringiensis and its potential to enhance the toxicity of Cry1Ac protoxin. Appl Microbiol Biotechnol 82:1157–1167. doi: 10.1007/s00253-009-1910-2. [DOI] [PubMed] [Google Scholar]
- 11.Bhattacharya D, Nagpure A, Gupta RK. 2007. Bacterial chitinases: properties and potential. Crit Rev Biotechnol 27:21–28. doi: 10.1080/07388550601168223. [DOI] [PubMed] [Google Scholar]
- 12.Babashpour S, Aminzadeh S, Farrokhi N, Karkhane A, Haghbeen K. 2012. Characterization of a chitinase (Chit62) from Serratia marcescens B4A and its efficacy as a bioshield against plant fungal pathogens. Biochem Genet 50:722–735. doi: 10.1007/s10528-012-9515-3. [DOI] [PubMed] [Google Scholar]
- 13.Chandrasekaran R, Revathi K, Nisha S, Kirubakaran SA, Sathish-Narayanan S, Senthil-Nathan S. 2012. Physiological effect of chitinase purified from Bacillus subtilis against the tobacco cutworm Spodoptera litura Fab. Pestic Biochem Physiol 104:65–71. doi: 10.1016/j.pestbp.2012.07.002. [DOI] [Google Scholar]
- 14.Ozgen A, Sezen K, Demir I, Demirbag Z, Nalcacioglu R. 2013. Molecular characterization of chitinase genes from a local isolate of Serratia marcescens and their contribution to the insecticidal activity of Bacillus thuringiensis strains. Curr Microbiol 67:499–504. doi: 10.1007/s00284-013-0395-5. [DOI] [PubMed] [Google Scholar]
- 15.Hu S, Zhang X, Li Y, Ding X, Hu X, Yang Q, Xia L. 2013. Constructing Bacillus thuringiensis strain that co-expresses Cry2Aa and chitinase. Biotechnol Lett 35:1045–1051. doi: 10.1007/s10529-013-1171-0. [DOI] [PubMed] [Google Scholar]
- 16.Rigali S, Nothaft H, Noens EE, Schlicht M, Colson S, Müller M, Joris B, Koerten HK, Hopwood DA, Titgemeyer F, van Wezel GP. 2006. The sugar phosphotransferase system of Streptomyces coelicolor is regulated by the GntR-family regulator DasR and links N-acetylglucosamine metabolism to the control of development. Mol Microbiol 61:1237–1251. doi: 10.1111/j.1365-2958.2006.05319.x. [DOI] [PubMed] [Google Scholar]
- 17.Colson S, Stephan J, Hertrich T, Saito A, van Wezel GP, Titgemeyer F, Rigali S. 2007. Conserved cis-acting elements upstream of genes composing the chitinolytic system of streptomycetes are DasR-responsive elements. J Mol Microbiol Biotechnol 12:60–66. doi: 10.1159/000096460. [DOI] [PubMed] [Google Scholar]
- 18.Rigali S, Titgemeyer F, Barends S, Mulder S, Thomae AW, Hopwood DA, van Wezel GP. 2008. Feast or famine: the global regulator DasR links nutrient stress to antibiotic production by Streptomyces. EMBO Rep 9:670–675. doi: 10.1038/embor.2008.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bertram R, Rigali S, Wood N, Lulko AT, Kuipers OP, Titgemeyer F. 2011. Regulon of the N-acetylglucosamine utilization regulator NagR in Bacillus subtilis. J Bacteriol 193:3525–3536. doi: 10.1128/JB.00264-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Resch M, Schiltz E, Titgemeyer F, Muller YA. 2010. Insight into the induction mechanism of the GntR/HutC bacterial transcription regulator YvoA. Nucleic Acids Res 38:2485–2497. doi: 10.1093/nar/gkp1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xie CC, Shi J, Jia HY, Li PF, Luo Y, Cai J, Chen YH. 2015. Characterization of regulatory regions involved in the inducible expression of chiB in Bacillus thuringiensis. Arch Microbiol 197:53–63. doi: 10.1007/s00203-014-1054-3. [DOI] [PubMed] [Google Scholar]
- 22.Fujita Y. 2009. Carbon catabolite control of the metabolic network in Bacillus subtilis. Biosci Biotechnol Biochem 73:245–259. doi: 10.1271/bbb.80479. [DOI] [PubMed] [Google Scholar]
- 23.Stüelke J, Hillen W. 2000. Regulation of carbon catabolism in Bacillus species. Annu Rev Microbiol 54:849–880. doi: 10.1146/annurev.micro.54.1.849. [DOI] [PubMed] [Google Scholar]
- 24.Brückner R, Titgemeyer F. 2002. Carbon catabolite repression in bacteria: choice of the carbon source and autoregulatory limitation of sugar utilization. FEMS Microbiol Lett 209:141–148. doi: 10.1111/j.1574-6968.2002.tb11123.x. [DOI] [PubMed] [Google Scholar]
- 25.Göerke B, Stuelke J. 2008. Carbon catabolite repression in bacteria: many ways to make the most out of nutrients. Nat Rev Microbiol 6:613–624. doi: 10.1038/nrmicro1932. [DOI] [PubMed] [Google Scholar]
- 26.Deutscher J, Francke C, Postma PW. 2006. How phosphotransferase system-related protein phosphorylation regulates carbohydrate metabolism in bacteria. Microbiol Mol Biol Rev 70:939–1031. doi: 10.1128/MMBR.00024-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schumacher MA, Allen GS, Diel M, Seidel G, Hillen W, Brennan RG. 2004. Structural basis for allosteric control of the transcription regulator CcpA by the phosphoprotein HPr-Ser46-P. Cell 118:731–741. doi: 10.1016/j.cell.2004.08.027. [DOI] [PubMed] [Google Scholar]
- 28.Kraus A, Hueck C, Gärtner D, Hillen W. 1994. Catabolite repression of the Bacillus subtilis xyl operon involves a cis element functional in the context of an unrelated sequence, and glucose exerts additional xylR-dependent repression. J Bacteriol 176:1738–1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lecadet MM, Chaufaux J, Ribier J, Lereclus D. 1992. Construction of novel Bacillus thuringiensis strains with different insecticidal activities by transduction and transformation. Appl Environ Microbiol 58:840–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kravanja M, Engelmann R, Dossonnet V, Blüggel M, Meyer HE, Frank R, Galinier A, Deutscher J, Schnell N, Hengstenberg G. 1999. The hprK gene of Enterococcus faecalis encodes a novel bifunctional enzyme: the HPr kinase/phosphatase. Mol Microbiol 31:59–66. doi: 10.1046/j.1365-2958.1999.01146.x. [DOI] [PubMed] [Google Scholar]
- 31.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 32.Wang Z, Yang X, Chu X, Zhang J, Zhou H, Shen Y, Long J. 2012. The structural basis for the oligomerization of the N-terminal domain of SATB1. Nucleic Acids Res 40:4193–4202. doi: 10.1093/nar/gkr1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hiard S, Marée R, Colson S, Hoskisson PA, Titgemeyer F, van Wezel GP, Joris B, Wehenkel L, Rigali S. 2007. PREDetector: a new tool to identify regulatory elements in bacterial genomes. Biochem Biophys Res Commun 357:861–864. doi: 10.1016/j.bbrc.2007.03.180. [DOI] [PubMed] [Google Scholar]
- 34.Gaugué I, Oberto J, Plumbridge J. 2014. Regulation of amino sugar utilization in Bacillus subtilis by the GntR family regulators, NagR and GamR. Mol Microbiol 92:100–115. doi: 10.1111/mmi.12544. [DOI] [PubMed] [Google Scholar]
- 35.Fillenberg SB, Grau FC, Seidel G, Muller YA. 2015. Structural insight into operator dre-sites recognition and effector binding in the GntR/HutC transcription regulator NagR. Nucleic Acids Res 43:1283–1296. doi: 10.1093/nar/gku1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Heravi KM, Shali A, Naghibzadeh N, Ahmadian G. 2014. Characterization of cis-acting elements residing in the chitinase promoter of Bacillus pumilus SG2. World J Microbiol Biotechnol 30:1491–1499. doi: 10.1007/s11274-013-1569-9. [DOI] [PubMed] [Google Scholar]
- 37.Sonenshein AL. 2007. Control of key metabolic intersections in Bacillus subtilis. Nat Rev Microbiol 5:917–927. doi: 10.1038/nrmicro1772. [DOI] [PubMed] [Google Scholar]
- 38.Khan SR, Deutscher J, Vishwakarma RA, Monedero V, Bhatnagar NB. 2001. The ptsH gene from Bacillus thuringiensis israelensis—characterization of a new phosphorylation site on the protein HPr. Eur J Biochem 268:521–530. doi: 10.1046/j.1432-1327.2001.01878.x. [DOI] [PubMed] [Google Scholar]
- 39.Reizer J, Bergstedt U, Galinier A, Küester E, Saier MH Jr, Hillen W, Steinmetz M, Deutscher J. 1996. Catabolite repression resistance of gnt operon expression in Bacillus subtilis conferred by mutation of His-15, the site of phosphoenolpyruvate-dependent phosphorylation of the phosphocarrier protein HPr. J Bacteriol 178:5480–5486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dossonnet V, Monedero V, Zagorec M, Galinier A, Pérez-Martínez G, Deutscher J. 2000. Phosphorylation of HPr by the bifunctional HPr kinase/P-Ser-HPr phosphatase from Lactobacillus casei controls catabolite repression and inducer exclusion but not inducer expulsion. J Bacteriol 182:2582–2590. doi: 10.1128/JB.182.9.2582-2590.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moir-Blais TR, Grundy FJ, Henkin TM. 2001. Transcriptional activation of the Bacillus subtilis ackA promoter requires sequences upstream of the CcpA binding site. J Bacteriol 183:2389–2393. doi: 10.1128/JB.183.7.2389-2393.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim HJ, Roux A, Sonenshein AL. 2002. Direct and indirect roles of CcpA in regulation of Bacillus subtilis Krebs cycle genes. Mol Microbiol 45:179–190. doi: 10.1046/j.1365-2958.2002.03003.x. [DOI] [PubMed] [Google Scholar]
- 43.Puri-Taneja A, Schau M, Chen Y, Hulett FM. 2007. Regulators of the Bacillus subtilis cydABCD operon: identification of a negative regulator, CcpA, and a positive regulator, ResD. J Bacteriol 189:3348–3358. doi: 10.1128/JB.00050-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Puri-Taneja A, Paul S, Chen Y, Hulett FM. 2006. CcpA causes repression of the phoPR promoter through a novel transcription start site, P(A6). J Bacteriol 188:1266–1278. doi: 10.1128/JB.188.4.1266-1278.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ishii H, Tanaka T, Ogura M. 2013. The Bacillus subtilis response regulator gene degU is positively regulated by CcpA and by catabolite-repressed synthesis of ClpC. J Bacteriol 195:193–201. doi: 10.1128/JB.01881-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hammar P, Leroy P, Mahmutovic A, Marklund EG, Berg OG, Elf J. 2012. The lac repressor displays facilitated diffusion in living cells. Science 336:1595–1598. doi: 10.1126/science.1221648. [DOI] [PubMed] [Google Scholar]
- 47.Xie CC, Luo Y, Chen YH, Cai J. 2012. Construction of a promoter-probe vector for Bacillus thuringiensis: the identification of cis-acting elements of the chiA locus. Curr Microbiol 64:492–500. doi: 10.1007/s00284-012-0100-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.