

Abstract
Methanogens are anaerobic archaea that grow by producing methane, a gas that is both an efficient renewable fuel and a potent greenhouse gas. We observed that overexpression of the cytoplasmic heterodisulfide reductase enzyme HdrABC increased the rate of methane production from methanol by 30% without affecting the growth rate relative to the parent strain. Hdr enzymes are essential in all known methane-producing archaea. They function as the terminal oxidases in the methanogen electron transport system by reducing the coenzyme M (2-mercaptoethane sulfonate) and coenzyme B (7-mercaptoheptanoylthreonine sulfonate) heterodisulfide, CoM-S-S-CoB, to regenerate the thiol-coenzymes for reuse. In Methanosarcina acetivorans, HdrABC expression caused an increased rate of methanogenesis and a decrease in metabolic efficiency on methylotrophic substrates. When acetate was the sole carbon and energy source, neither deletion nor overexpression of HdrABC had an effect on growth or methane production rates. These results suggest that in cells grown on methylated substrates, the cell compensates for energy losses due to expression of HdrABC with an increased rate of substrate turnover and that HdrABC lacks the appropriate electron donor in acetate-grown cells.
INTRODUCTION
Methane is a combustible gas fuel that can be used to produce heat and electricity. Methanogens are anaerobic archaea that naturally produce methane in soil, ocean sediment, and the gastrointestinal tracts of animals (1). In anaerobic environments, methanogens catalyze the terminal step in the carbon cycle by reducing C1 compounds (such as CO2, CO, formate, methanol, methylsulfides, and methylamines) and acetate to methane gas, which diffuses up to aerobic zones, where it can be oxidized by methanotrophic bacteria (2). Because of their intrinsic ability to produce methane, methanogens are harnessed in anaerobic digesters to turn municipal, agricultural, and industrial waste products into renewable fuel (biogas), heat, and electricity (3).
The model organism Methanosarcina acetivorans is one of a small group of methanogens capable of producing methane both from acetate and from methylated substrates, such as methanol, methylamines, and methylsulfides (4). In the methylotrophic methanogenesis pathway, electrons obtained from oxidizing one molecule of methanol to carbon dioxide are used to reduce three molecules of methanol to methane (5–7). The cell conserves energy by coupling generation of a transmembrane ion gradient (ΔΨ) to electron transport between reduced electron carriers and the terminal electron acceptor, a coenzyme M (2-mercaptoethane sulfonate) (CoM)-coenzyme B (7-mercaptoheptanoylthreonine sulfonate) (CoB) heterodisulfide molecule (CoM-S-S-CoB) that is produced in the last step of methanogenesis (6). The CoM-S-S-CoB heterodisulfide must be reduced to the CoM-SH and CoB-SH thiols to be reused for subsequent rounds of methanogenesis.
Reduction of CoM-S-S-CoB is performed by one of two heterodisulfide reductases: two-subunit membrane-bound HdrED (energy-conserving CoM-S-S-CoB reductase) and three-subunit cytosolic HdrABC (heterodisulfide reductase) (8). Genes encoding HdrED (MA0687-MA0688) are essential for growth on trimethylamine, methanol, acetate, and methanol plus acetate (8). HdrED oxidizes the membrane electron carrier methanophenazine (MP) and reduces the CoM-S-S-CoB heterodisulfide. In the process, HdrED conserves energy using a q-loop mechanism to contribute to the transmembrane ion gradient by translocating protons across the cell membrane (9). The flow of protons back into the cell via ATP synthase produces ATP, which is then used for other biosynthetic reactions (10). The hdrA1C1B1 (MA3126-MA3128) operon (here referred to as hdrABC) is expressed on methylotrophic substrates but is nonessential. In contrast to HdrED, the HdrABC enzyme is cytoplasmic and does not conserve energy. M. acetivorans also expresses genes encoding HdrD2 (MA0526), HdrA2 and polyferredoxin (MA2867-MA2868), and HdrC2B2 (MA4236-MA4237), all of which are constitutively expressed (8). The physiological roles of the hdrD2, hdrA2, and hdrC2B2 genes are not yet understood.
In hydrogenotrophic methanogens, such as Methanococcus, HdrABC accepts electrons from hydrogen via a bound hydrogenase, Vhu. An electron bifurcation mechanism is used to drive reduction of CO2 to formyl-methanofuran by the formyl-methanofuran dehydrogenase Fmd, coupling the reaction with the thermodynamically favorable reduction of CoM-S-S-CoB (11, 12). Though it does not itself conserve energy, HdrABC is essential for methanogenesis by Methanococcus because it provides the low-potential electrons necessary for the first step of the hydrogenotrophic methanogenesis pathway.
However, because functional hydrogenases are not expressed in M. acetivorans and some other Methanosarcinales species, Vhu or another hydrogenase cannot be the electron donor for HdrABC in these organisms. In light of this fact, it is possible that HdrABC may use one of three general mechanisms for catalysis: an electron confurcation mechanism (defined as when one electron each from two separate donors is transferred to one two-electron acceptor), where electrons from both reduced ferredoxin (FdxH2) and reduced F420 (8-hydroxy-5-deazaflavin [F420H2]) are used to reduce two molecules of CoM-S-S-CoB (Fig. 1A); a direct-reduction mechanism, where either FdxH2 or F420H2 could reduce CoM-S-S-CoB (Fig. 1B); or an electron bifurcation mechanism that couples oxidation of FdxH2 to reduction of both CoM-S-S-CoB and another electron carrier, such as F420 (Fig. 1C). Based on the lack of hydrogenase activity in M. acetivorans and because the reaction would be energetically favorable, HdrABC was thought to use FdxH2 as a substrate to directly reduce CoM-S-S-CoB (Fig. 1B) (8).
FIG 1.

Putative models for the role of HdrABC during methylotrophic growth. (A) In an electron confurcation mechanism, HdrABC uses electrons from both FdxH2 and F420H2 in a 1:1 ratio to reduce two molecules of CoM-S-S-CoB and bypasses Rnf, Fpo, and HdrED energy-conserving enzyme complexes. (B) In a direct-reduction mechanism, HdrABC bypasses either Rnf or Fpo ion translocation steps by using FdxH2 or F420H2 as an electron donor. (C) HdrABC uses a bifurcation mechanism to reduce CoM-S-S-CoB while interchanging electrons between FdxH2 and F420H2 electron carriers. The model in panel B most closely matches the experimental data. ka is the rate of HdrABC enzyme activity; kCH4 is the rate of methane production; tg is the generation time.
Whether HdrABC uses both or either ferredoxin and F420 as an electron carrier has predictable consequences for the cell. Ferredoxin is also thought to be the electron donor for the sodium-pumping ferredoxin-methanophenazine oxidoreductase, Rnf (named for homology to the Rhodobacter nitrogen fixation enzyme complex) (13–15). On methylotrophic growth in high-salt (HS) medium, Rnf pumps 0.04 sodium ion across the cell membrane per 2 electrons (16). F420H2 is the electron donor for the proton-pumping F420H2-methanophenazine Fpo complex. Fpo pumps two protons across the cell membrane per two electrons (17). The reduced methanophenazine (MPH2) produced by both Rnf and Fpo is oxidized by HdrED to translocate protons across the cell membrane. Therefore, if HdrABC confurcates electrons from both FdxH2 and F420H2 in a 1:1 stoichiometry, HdrABC will compete for electrons with both Rnf and Fpo complexes, decreasing electron flux through HdrED and resulting in lower ion motive force generated per mole substrate consumed (Fig. 1A). If HdrABC uses a direct mechanism using either FdxH2 or F420H2 to reduce CoM-S-S-CoB, then it will compete with either Rnf or Fpo for substrate and lower the electron flux through HdrED. However, because Rnf and Fpo have a 50-fold difference in transmembrane ion translocation activity, the effect of HdrABC enzyme activity on the ion motive force generated would depend on whether HdrABC competes with Rnf or Fpo (Fig. 1B). Finally, if HdrABC can bifurcate electrons by using FdxH2 to reduce both CoM-S-S-CoB and F420, then HdrABC may compete with Rnf for substrate but increase the electron flux through the more energy-efficient Fpo (Fig. 1C).
We tested these hypotheses by adding a second copy of the methylotrophic HdrABC operon to the M. acetivorans chromosome and by using computational metabolic-flux modeling. Instead of observing a growth defect compared to the parental strain, as we had anticipated, we discovered that the mutant cells had growth kinetics and biomass identical to those of the parent while increasing the methane production rate by 30%. This suggests that an increased substrate uptake rate can compensate for decreased metabolic efficiency and that hdrA1B1C1 in M. acetivorans uses a direct mechanism (Fig. 1B) rather than an electron bifurcation mechanism to reduce CoM-S-S-CoB.
MATERIALS AND METHODS
Culture conditions.
Escherichia coli strains were grown at 35°C in lysis broth or 1.5% agar medium with 10 mM rhamnose and/or chloramphenicol (35 μg/ml or 8 μg/ml) when appropriate (18, 19). M. acetivorans strains were grown at 35°C in 18-mm by 150-mm anaerobic culture tubes with 10 ml HS mineral salt medium under a 5% H2–20% CO2-balance N2 atmosphere and strict anaerobic conditions (where “balance” indicates the remainder of the atmosphere) (20). HS mineral salt medium was supplemented with the carbon source 50 mM trimethylamine (TMA), 125 mM MeOH, 120 mM sodium acetate, or a mixture of 40 mM sodium acetate and 125 mM MeOH (21). M. acetivorans strains were plated on 1.4% HS agar, TMA (50 mM), and puromycin (2 mg/liter) and incubated at 35°C under a 0.1% H2S–20% CO2-balance N2 atmosphere.
Strain construction.
The primers and DNA sequences listed in Table S1 in the supplemental material were designed using Vector NTI software (Invitrogen). Genes, oligonucleotides, and multiple-cloning sites were synthesized commercially by IDT and Invitrogen. To overexpress hdrABC, the operon (MA3126-MA3128) was amplified from the M. acetivorans (strain NB34) chromosome using primers oNB68, oNB69, and oNB72 to -81 (see Table S1 in the supplemental material). Primers oNB72 to -81 introduce five point mutations to remove five NdeI restriction sites from the hdrABC operon. The resulting fused PCR product contained BamHI restriction sites at the 5′ and 3′ ends, as well as an NdeI site preceding the start codon of the hdrA gene. The PCR product was ligated into plasmid pNB708, creating pNB709, and verified by restriction digestion and sequencing. The mutated hdrABC* operon (Nicole R. Buan Murphy and Jennifer L. Catlett, U.S. provisional patent 61/980,656) was amplified from pNB709 using oligonucleotides oNB68 and oNB69 and ligated into pJK027A at the NdeI and BamHI restriction sites to create plasmid pJC1 (see Table S2 in the supplemental material). The hdrABC* operon is under the control of the PmcrB(tetO1) (also called Ptet) promoter. The Ptet promoter is constitutive when transformed into strains lacking the tetR repressor gene but can be controlled by addition of tetracycline in strains that express the TetR repressor. M. acetivorans was transformed with pJC1 as previously described (22). Briefly, 10 ml stationary-phase culture was pelleted by centrifugation and resuspended in 1 ml bicarbonate-buffered 0.85 M sucrose. Then, 10 μl plasmid DNA (0.2 μg/μl) was mixed with 30 μl DOTAP transfection reagent (1 μg/μl; Roche) and 70 μl bicarbonate-buffered 0.85 M sucrose and incubated at room temperature for 15 min in sterile glass test tubes. Cells were added to the liposome-encapsulated DNA and incubated for 2 to 4 h at room temperature. The transfected cells were incubated in 25 ml HS medium supplemented with 50 mM TMA overnight at 35°C. The cells were plated on HS TMA 0.5% agar plates with puromycin (2 mg/liter) and grown at 35°C under a 0.1% H2S–20% CO2-balance N2 atmosphere for 10 to 14 days. Colonies were streaked for isolation before being picked to liquid HS TMA medium. Transformants were verified via a PCR screen for wild-type hdrABC, the ΔhdrABC operon deletion, and Ptet hdrABC* (in pJC1) using primers listed in Table S1 in the supplemental material. Various PCR techniques were employed to create plasmids (listed in Table S2 in the supplemental material), including overlap extension and site-directed mutagenesis (23, 24).
For all PCRs, Phusion Flash PCR master mix (Thermo Scientific) was used as a source of proofreading DNA polymerase. DNA was purified using Wizard kits from Promega (Madison, WI). DNA fragments were joined using T4 DNA ligase (New England BioLabs) or GeneArt kits (Invitrogen). Restriction enzymes (AscI, BamHI, NdeI, NcoI, EcoRI, SphI, and XbaI) were purchased from New England BioLabs (Ipswich, MA). All plasmids were sequenced by Eurofins Operon MWG (Huntsville, AL).
Hdr enzyme assays.
Coenzyme M was purchased from Sigma-Aldrich (St. Louis, MO). Coenzyme B and CoM-S-S-CoB were synthesized in house (25, 26). Cells were grown to early stationary phase (optical density at 600 nm [OD600] = 1.0) in HS medium supplemented with 125 mM MeOH. Under strict anaerobic conditions, 30 ml of cell cultures was harvested, pelleted by centrifugation, and washed twice with 10 ml 0.1 M NaPO4, 0.4 M NaCl, pH 8.0. Cells were lysed osmotically by addition of 3 ml 0.1 M NaPO4, pH 8.0, without NaCl. Halt protease inhibitor cocktail (Thermo Pierce) was added, and the lysate was centrifuged at 22,000 × g for 1 h to pellet cell debris and membranes. The concentration of soluble cytoplasmic protein in the resulting extract was measured via Bradford assay against a 2-mg bovine serum albumin (BSA) standard (Thermo Pierce). Hdr assays according to the method of Welte and Deppenmeier (26) were modified for a 96-well plate format. Briefly, reaction mixtures were prepared with 170 μl 0.1 M NaPO4, pH 8.0, 28 μl extract, and 2 μl reduced methyl viologen (1 mM stock). The reaction was started by the addition of 30 μl CoM-S-S-CoB heterodisulfide (1.125 mM stock), and the increase in oxidized methyl viologen was followed at 578 nm in a Tecan Sunrise plate reader at 35°C under a 5% H2–20% CO2–75% N2 atmosphere and strict anaerobic conditions.
Growth curves.
M. acetivorans strains were grown in 10 ml TMA and then transferred to 125 mM MeOH, 120 mM acetate, or a mixture of 40 mM acetate and 125 mM MeOH. Growth was assessed by measuring the change in optical density at 600 nm using a Spec 20D spectrophotometer modified with an 18-mm tube adapter. Growth data were obtained immediately after inoculating from TMA into fresh medium containing a different carbon source, or the cultures were passaged for 25 generations before commencing growth measurements.
Methane production assays.
For methane endpoint assays, 10-ml cultures were grown in 125 mM methanol HS medium to stationary phase, and 100-μl headspace samples were transferred to empty crimped 2-ml serum vials using a gas-tight Hamilton syringe. For cell suspension assays, cells were grown with an appropriate carbon source to an OD600 between 0.3 and 0.5 (exponential growth) and put on ice. Under strict anaerobic conditions, cells from 10 ml of culture were harvested, pelleted by centrifugation, and washed twice in HS medium without a carbon source. The cells were resuspended in 0.5 ml HS medium with 50 μM mupirocin to halt protein synthesis and then aliquoted in replicates of 5 into 2-ml serum vials containing HS plus 125 mM MeOH or 120 mM acetate. The serum vials were sealed with aluminum crimps and warmed for 5 min at 35°C to start the assay. Methane in the headspace was measured via flame ionization on an Agilent 7890 gas chromatograph with a GS CarbonPlot column at 145°C using an autoinjector.
Methanol consumption assays.
For each strain, 100-ml HS-MeOH medium (125 mM) cultures were grown to an OD600 of approximately 0.5. Under strict anaerobic conditions, cells were harvested by centrifugation, washed once in 10 ml plain HS medium, and resuspended in 10 ml HS-MeOH medium (125 mM). The cells were warmed to 35°C to start the assay. Every 15 min, 1-ml aliquots were withdrawn and filtered using a 0.2-μm PES syringe filter (Thermo Scientific) to remove cells. The MeOH content in the spent medium was measured via flame ionization on an Agilent 7890 gas chromatograph with a GS CarbonPlot column at 200°C using an autoinjector.
Biomass measurements.
For each strain, 10 cultures (10 ml each) were grown to stationary phase on HS medium containing 125 mM MeOH. The methane in the headspace was measured as described above, and cells were collected on preweighed 0.2-μm nylon filters by vacuum. The filters were dried at 95°C and weighed daily until the weights stabilized and remained consistent for 3 days. Uninoculated medium was used as a blank.
RESULTS
Overexpression of HdrABC in M. acetivorans.
To determine the effect of increased HdrABC enzyme activity on cell physiology, we created a plasmid to introduce a second hdrABC* operon on the M. acetivorans chromosome (Fig. 2A). The open reading frames corresponding to the methylotrophic hdrABC operon, MA3126-MA3128, were amplified from the M. acetivorans C2A chromosome and cloned into the pJK027A plasmid, resulting in plasmid pJC1. The hdrABC operon in pJC1 was designed to include five mutations to distinguish the inserted genes from the wild-type genes by removing NdeI restriction sites (see Table S1 in the supplemental material). Four of the mutations are silent mutations that do not change the translated protein sequence. One mutation necessitated a change to the translated protein primary sequence so that the pJC1 plasmid expressed HdrBM249V mutant protein. The HdrB M249V mutation is in a predicted disordered region within the second cysteine-rich motif at residues 166 to 257 (InterProScan) (27).
FIG 2.

Construction of plasmid pJC1 and strain verification. (A) Map of plasmid pJC1 containing the hdrABC* operon under the control of the Ptet promoter. The asterisks indicate the locations of point mutations in the hdrABC operon. (B) Agarose gel showing PCR identification of each strain.
The parent M. acetivorans strain contains a ϕC31 site-specific recombinase and a ϕC31 attP site at the hypoxanthine phosphoribosyltransferase (hpt) locus (see Table S2 in the supplemental material). The complementary attB sequence in the pJC1 plasmid (Fig. 2A) allows it to recombine into the host chromosome at the attP sequence (28, 29). Plasmid pJC1 was transformed into the parent strain, creating the att::hdrABC* strain, as well as into the ΔhdrABC deletion mutant strain, creating the ΔhdrABC att::hdrABC* mutant strain (see Fig. S1 in the supplemental material). We designed a PCR screen (Fig. 2B; see Fig. S1 in the supplemental material) to simultaneously detect the integrated hdrABC* operon (423-bp amplicon), the native MA3126-MA3128 operon (1,287-bp amplicon), and deletion of the MA3126-MA3128 operon (1,607-bp amplicon) to validate each transformed strain. This PCR screen was used as a quality control measure after all growth curves and methanogenesis assays to ensure that strains were not switched or contaminated.
Hdr enzyme activity in cell extracts of each strain was measured to check that the HdrB M249V point mutations did not abolish enzyme activity (Fig. 3D). Assaying Hdr enzyme activity in cell extracts is complicated by the fact that M. acetivorans expresses multiple hdr genes during growth on methanol. The hdrA1C1B1 (MA3126-MA3128) operon is methanol specific but nonessential, while genes encoding the membrane-bound HdrED (MA0687-MA0688) are essential. M. acetivorans also expresses hdrD2 (MA0526), hdrA2, polyferredoxin genes (MA2867-MA2868), and hdrC2B2 (MA4236-MA4237), which are constitutively expressed. We assayed CoM-S-S-CoB-dependent methyl viologen oxidation in clarified cell extracts to minimize HdrED protein levels, which could mask HdrABC activity. Methyl viologen was used as the electron donor because the specific electron donor for M. acetivorans HdrABC is unknown.
FIG 3.

Phenotypes of parent and hdrABC mutant strains. (A) Growth curve of methanol-adapted strains. Growth data were collected from triplicate biological replicates. (B and C) The rates of methane production by cell suspensions were measured from cells grown on methanol (B) and acetate (C). Methane production was measured in triplicate biological and triplicate technical replicates. *, P < 0.0001. (D) Heterodisulfide reductase activity in methanol-grown cell extracts. Assays were performed in triplicate. The error bars indicate the standard deviations and may not be visible behind the symbols.
The HdrABC enzyme activity correlates with each strain genotype (Fig. 3D). Hdr activity is 20% decreased in the ΔhdrABC mutant extract relative to the parental strain. This level of Hdr activity presumably results from incomplete removal of HdrED and from HdrA2C2B2 and/or HdrD2 protein. Extract from the att::hdrABC* overexpression strain has 14% increased activity versus the parental strain, indicating that HdrABC proteins expressed from the integrated pJC1 plasmid are enzymatically active. Seventy percent of the activity is lost after heat treatment, and CoM-S-S-CoB-dependent methyl viologen oxidation cannot be detected when protein is omitted from the assay. No CoM-S-S-CoM-dependent oxidation of methyl viologen was detected (data not shown). These results indicate that the HdrABC protein expressed from the integrated copy of the pJC1 plasmid is enzymatically active and that by integrating a second copy of the hdrABC* operon onto the chromosome it is possible to increase the level of HdrABC enzyme activity in cell extracts above wild-type levels.
pJC1 complements the growth and methanogenesis phenotypes of the ΔhdrABC deletion mutant on methylotrophic substrates.
Plasmid pJC1 was transformed into the ΔhdrABC deletion mutant to test whether the cloned hdrABC* operon on pJC1 functions in vivo and could complement the growth and methanogenesis phenotypes of the ΔhdrABC deletion mutant (Fig. 3A). When grown on methanol, the ΔhdrABC mutant displays an increased lag phase and a 20% decreased growth rate versus the parental strain. Integration of plasmid pJC1 in the ΔhdrABC strain results in growth rates and culture lag times similar to those of the original parent strain (Table 1). In cell suspension assays, the ΔhdrABC att::hdrABC* mutant had a 46% increased methane production rate versus the ΔhdrABC strain (Fig. 3B). These results indicate that the hdrABC* operon cloned into the pJC1 plasmid is capable of complementing the growth and methanogenesis phenotypes of a ΔhdrABC deletion mutant.
TABLE 1.
Culture doubling times for adapted cellsa
| Strain | MeOH |
MeOH + acetate |
Acetate |
||||||
|---|---|---|---|---|---|---|---|---|---|
| Doubling time (h) (±SD) | P vs parent | P vs ΔhdrABC | Doubling time (h) (±SD) | P vs parent | P vs ΔhdrABC | Doubling time (h) (±SD) | P vs parent | P vs ΔhdrABC | |
| Parent | 8.5 ± 0.17 | 1 | 0.0000 | 9.4 ± 0.28 | 1 | 0.0046 | 44.0 ± 2.80 | 1 | 0.0000 |
| att::hdrABC* | 8.4 ± 0.35 | NS | 0.0001 | 9.7 ± 0.16 | NS | NS | 47.6 ± 2.64 | NS | 0.0003 |
| ΔhdrABC att::hdrABC* | 9.1 ± 0.14 | 0.0012 | 0.0010 | 10.3 ± 0.48 | NS | NS | 58.0 ± 3.05 | 0.0002 | NS |
| ΔhdrABC | 9.9± 0.24 | 0.0000 | 1 | 10.1 ± 0.20 | 0.0046 | 1 | 61.8 ± 2.94 | 0.0000 | 1 |
Cells were adapted to methanol for 25 generations. Data were collected from five biological replicates (n = 5). Significance (P values) was determined by unpaired Student t tests. NS, not significant (P > 0.01).
An increased rate of methane production is observed when the HdrABC expression level is increased.
After determining that the pJC1 plasmid can complement the ΔhdrABC mutant growth phenotype, we tested whether overexpression of HdrABC in the parent strain (att::hdrABC*) would have the opposite effect of a ΔhdrABC mutation and result in increased growth and methane production. Decreased expression of HdrABC (by deleting the hdrABC operon) resulted in a 50% decrease in methane production and a 20% decrease in the growth rate. However, instead of increasing growth and methane production, the att::hdrABC* mutant strain we created had a growth rate identical to that of the parent strain (Fig. 2A). When cells were switched from TMA to methanol, the parent had a doubling time of 10.1 ± 0.16 h versus 10.4 ± 0.21 for the att::hdrABC* mutant strain (see Table S3 in the supplemental material). When cells were preadapted to methanol for 25 generations, both the parent and the att::hdrABC* mutant had cell doubling times of 8.5 h (Table 1). When methane production was measured in resting cell suspensions, the att::hdrABC* mutant exhibited a 30% increase in the rate of methane production from methanol versus the parental strain (Fig. 3B). The methanol consumption rate was consistent with the methane production rates. The ΔhdrABC deletion mutant had a 15% decrease in the methanol consumption rate versus the parent strain, the ΔhdrABC att::hdrABC* complemented strain had a rate 16% higher than that of the parent strain, and the att::hdrABC* overexpression strain had a 16% increase in rate compared to the parent. The Hdr enzyme assays, growth phenotypes, and methane rate assays suggest that an increased HdrABC enzyme activity of 12% results in a 30% increase in conversion of methanol to methane without affecting the kinetics of cell growth. Our data show that the rate of methane production is dependent on the rate of Hdr enzyme activity by a direct linear correlation (Fig. 4A). As Hdr enzyme activity (kHdrABC) in the cell is increased, the rate of methane production (kCH4) increases according to equation 1:
| (1) |
FIG 4.

HdrABC uncouples methanogenesis from cell growth. (A) In cell suspension assays, the rate of methane production is dependent on the amount of HdrABC enzyme activity in the cell. (B) The growth rate and methane production rate are correlated by a second-order relationship. (C) Metabolic efficiency of each strain. The values are normalized to the ΔhdrABC mutant strain. (D) HdrABC enzyme activity has a strong negative correlation with metabolic efficiency and fits a direct-reduction mechanism model (R2 = 0.9981). The error bars indicate the standard deviations and may not be visible behind the symbols.
The correlation coefficient (R2) between HdrABC activity and the methane production rate is 1.00. This suggests that extra HdrABC may circumvent the production of a transmembrane ion gradient, leading to decreased ATP generation, and consequently may result in decreased cell growth from an equivalent amount of substrate. These results also suggest that, in the range of HdrABC expression we tested, the substrate (methanol), enzymes (methyltransferases, the oxidative and reductive branches of the methanogenesis pathway), and cofactor availability (coenzyme M) are not rate limiting.
In contrast, the rate of population growth (kg) depends on the rate of methane production (kCH4), which is described by equation 2:
| (2) |
The goodness of fit for the correlation is as follows: R2 = 0.9813 (Fig. 4B). This result suggests that other biosynthetic and metabolic reactions in the cell are constrained even when the rate of methane production increases. Presuming that the other biosynthetic and metabolic reactions are dependent on the turnover of reduced ferredoxin or F420H2 or ATP synthesis, these data are consistent with either the direct-reduction pathway shown in Fig. 1B or the bifurcation pathway shown in Fig. 1C. These results do not support the pathway shown in Fig. 1A, because if HdrABC confurcated electrons and competed equally with both Rnf and Fpo for electron donors, the growth rate would be inversely proportional to the rate of methane production.
HdrABC uncouples methanogenesis from ATP synthesis.
If overexpression of HdrABC increases the rate of methane production but does not affect the rate of biomass synthesis, this suggests that HdrABC can partially or completely uncouple methanogenesis from growth (Fig. 1B and C). The degree of uncoupling can be quantified by measuring the endpoint biomass or methane yield and by determining the metabolic efficiency of substrate turnover. We observed that the att::hdrABC* and ΔhdrABC att::hdrABC* strains had biomass yields equivalent to that of the parent strain but that the ΔhdrABC mutant cultures had a 16% increase in biomass compared to the parental strain (P = 0.00805) (Table 2). Therefore, when the hdrABC genes are deleted, cells are 16% more efficient at conserving energy, and in the parent strain, in the ΔhdrABC att::hdrABC* strain, and in the att::hdrABC* overexpression strain, HdrABC expression satisfies a lower-limit threshold for energy losses.
TABLE 2.
Growth yields and methane yieldsa
| Strain | Yieldb | SD | P vs parent |
|---|---|---|---|
| Biomass | |||
| Parent | 7.295 | 0.7378 | 1 |
| att::hdrABC* | 6.977 | 0.6565 | NS |
| ΔhdrABC att::hdrABC* | 7.020 | 0.7550 | NS |
| ΔhdrABC | 8.634 | 0.5642 | 0.0081 |
| Methanec | |||
| Parent | 95.0 | 3.66 | 1 |
| att::hdrABC* | 95.7 | 1.77 | NS |
| ΔhdrABC att::hdrABC* | 95.9 | 2.62 | NS |
| ΔhdrABC | 97.6 | 3.71 | NS |
Cells were adapted to methanol for 25 generations. Data were collected from 10 biological replicates (n = 10). Significance (P values) was determined by unpaired Student t tests. NS, not significant (P > 0.01).
Biomass yields are in grams per mole; methane yields are in millimoles per liter.
Theoretical methane yield, 93.75 mmol liter−1.
We measured the total methane yield from each strain grown on methanol. The total methane yields for all strains were equivalent (Table 2). These results indicate that when HdrABC is deleted, growth and methane production rates are constrained by a decreased CoM-S-S-CoB reduction rate but that substrate affinity is not affected. When HdrABC is overexpressed, cells take up and turn over substrate more quickly, but ATP and/or macromolecular biomass synthesis rates have reached a maximum.
If the cell always translocates the same number of ions across the membrane and, hence, attains the same ATP production rate, then any increase in the rate of methanogenesis (kCH4) should proportionally increase the growth rate (kg), assuming that the rate of substrate uptake is not rate limiting. However, if HdrABC uncouples ATP synthesis from methanogenesis, the ratio between kCH4 and kg will change in accordance with the expression level of HdrABC. The relationship between kCH4 and kg can be described by expressing the metabolic efficiency, e, of each strain using equation 3 (Fig. 4C) (30):
| (3) |
We observed a strong negative correlation between metabolic efficiency and HdrABC expression levels. As HdrABC levels increase, the rate of substrate conversion to methane increases at the expense of the growth rate. The relationship between HdrABC activity (kHdrABC) and e can be described by equation 4:
| (4) |
where the goodness of fit is as follows: R2 = 0.9838 (Fig. 4D). These results rule out the bifurcation pathway shown in Fig. 1C for the putative function of HdrABC. If HdrABC only partially bypassed energy-conserving steps in the methanogenesis pathway, then we would have expected a nonlinear correlation between metabolic efficiency and HdrABC activity. For electron bifurcation, the relationship between HdrABC enzyme activity and metabolic efficiency would follow equation 5:
| (5) |
This is because half of the electrons donated to HdrABC would be diverted to an energy-conserving ion translocation step. However, the linear relationship between metabolic efficiency and HdrABC activity does not fit the bifurcation model. Therefore, the experimental results are consistent with the idea that HdrABC uncouples methanogenesis from the generation of a transmembrane ion potential by increasing the rate of substrate turnover while bypassing HdrED and Rnf (Fig. 1B).
Hdr competes with Rnf for the same electron pool.
Determining the substrate of HdrABC is complicated by the fact that it contains two 4Fe-4S clusters and a bound flavin. Therefore, it is theoretically possible that HdrABC could directly accept electrons from any of the three electron carriers that shuttle electrons between the oxidative branch of the methylotrophic methanogenesis pathway and the electron transport system: a low-potential iron-sulfur cluster (via a ferredoxin or a protein-protein interaction), reduced F420H2, or the reduced CoM-SH and CoB-SH sulfhydryls (see Fig. S2 to S4 in the supplemental material). Therefore, there are seven possible mechanisms for HdrABC biochemistry: electron confurcation ([i] electrons from sulfhydryls and ferredoxin are used to reduce F420, or [ii] electrons from both ferredoxin and F420 are used to reduce CoM-S-S-CoB), electron bifurcation ([iii] electrons from ferredoxin are used to reduce F420 and CoM-S-S-CoB, or [iv] electrons from F420 are used to reduce both ferredoxin and CoM-S-S-CoB), direct reduction of CoM-S-S-CoB with either (v) ferredoxin or (vi) F420H2 (competitive with Rnf, competitive with Fpo, and noncompetitive with either Rnf or Fpo), and (vii) direct nonspecific oxidation of either ferredoxin or F420H2 to reduce CoM-S-S-CoB.
Electron confurcation using F420 and sulfhydryls as donors is not favorable with a positive change in Gibbs' free energy for the reaction under standard conditions (ΔG°′ = 38.59 kJ mol−1), but all other possible mechanisms are thermodynamically favorable, with ΔG°′ values ranging between −46.31 and −239.3 kJ mol−1 (see Table S4 in the supplemental material). Electron confurcation using either sulfhydryls and F420H2 or sulfhydryls and FdxH2 as electron donors, however, requires an additional 2 mol of electron input per mol substrate oxidized to balance the redox reactions and decreases the CH4/CO2 product ratio below the experimental 3:1. This is because methyl-CoM reductase (Mcr) requires CoB-SH as an electron donor. Hence, confurcation mechanisms would result in HdrABC competing with Mcr for electron donors, and increasing HdrABC activity would slow down methanogenesis. However, the ΔhdrABC mutant has a lower rate of methane production, and increased HdrABC activity results in an increased rate of methane production (Fig. 3B). For these reasons, HdrABC electron confurcation models using sulfhydryls as electron donors are excluded as possibilities in M. acetivorans growing on methanol. If HdrABC confurcates electrons from stoichiometric oxidation of both FdxH2 and F420H2 to reduce CoM-S-S-CoB (Fig. 1A), then HdrABC would compete equally with both Rnf and Fpo for electrons. Therefore, the ΔhdrABC mutant would have increased in biomass versus the parent strain because more electrons would flow through Rnf and Fpo and increased HdrABC activity would decrease the biomass. This mechanism is not supported by our growth rate or biomass yield results and can also be excluded (see Table S5 in the supplemental material).
Direct-reduction mechanisms can be distinguished by the physiological predictions they imply (Fig. 1C; see Table S5 in the supplemental material). If HdrABC accepts electrons only from F420, then high HdrABC activity would result in decreased growth. This is because, under the conditions we tested, Rnf only weakly couples electron transport with sodium ion translocation, and ATP synthesis is primarily dependent on proton pumping by Fpo and proton translocation by HdrED (16). This prediction does not match the experimental results. Conversely, if HdrABC can accept electrons only from reduced ferredoxin, then as HdrABC levels increase, the biomass would change negligibly because the electrons from ferredoxin are not used to conserve energy via Rnf and Mrp (31). If HdrABC can accept electrons nonspecifically from either FdxH2 or F420H2, HdrABC would probabilistically compete for substrate with Rnf (33%) and Fpo (66%) (see Table S5 in the supplemental material). However, because HdrABC is nonspecific, the decrease in biomass that results from bypassing Rnf and Fpo would follow a linear correlation with the amount of HdrABC activity in the cell. This prediction does not reflect the experimental data. Our growth data support a direct mechanism using only FdxH2 as the substrate because successive increases in HdrABC activity did not affect the growth rate or biomass yield (Tables 1 and 2).
If the model in Fig. 1C were correct and M. acetivorans HdrABC could bifurcate electrons from FdxH2, then half of the electrons from ferredoxin would be used to reduce F420. Electrons from F420H2 would still be used to conserve energy via Fpo and HdrED (Fig. 1C). As a result, at high HdrABC levels, there would be up to five protons pumped by Fpo per mole substrate instead of four, resulting in up to a 25% increase in biomass yield. If electrons from F420H2 were bifurcated to reduce FdxH2 and CoM-S-S-CoB, HdrABC would be competing with electrons from Fpo, and because Rnf translocates only 0.02 sodium ion per electron, overexpression of HdrABC would result in decreased biomass. In addition, if FdxH2 was a product of the reaction catalyzed by HdrABC, the only way to oxidize FdxH2 would be through Rnf or through production of acetyl-CoA and other biosynthetic reactions. This scenario would imply that a Δrnf mutant would be nonviable during methylotrophic growth, an implication that is at odds with the experimental data (13, 14, 16). Therefore, growth, biomass, and metabolic efficiency results support the conclusion that M. acetivorans HdrABC competes with Rnf for the same pool of electrons and does not use a bifurcation mechanism (Fig. 1B).
Metabolic-flux modeling of methylotrophic methanogenesis.
We next used computational modeling to quantitatively evaluate which of the 22 possible methylotrophic methanogenesis pathways most closely matches the experimental data (see Fig. S5 in the supplemental material). The 22 possible methanogenesis pathways (M1 to M22) compare roles of HdrABC and Fpo in methanogenesis: Fpo is an F420H2 oxidoreductase, Fpo uses ferredoxin (competes with Rnf), or Fpo obtains electrons from direct reduction by association with either methylene-tetrahydromethanopterin (H4MPT) reductase (Mer) or methylene-H4MPT dehydrogenase (Mtd) (see Fig. S2 to S4 in the supplemental material). Each of the 22 models was evaluated to predict whether HdrABC would be essential and what the relative biomass would be based on the magnitude of ion motive force that could be generated (see Table S4 in the supplemental material). The number of protons pumped per mole CH3OH (mH+) was calculated using equation 6:
| (6) |
where Ψ is the number of protons or sodium ions pumped across the membrane per electron. For Rnf, Ψ is equal to 0.02, and for Fpo and HdrED, Ψ is equal to 1. In the equation, n is the number of electrons accepted by each enzyme in each model pathway, and nHdrED is equal to the sum of nRnf and nFpo, because Fpo and Rnf reduce methanophenazine, which is the substrate for HdrED. Calculating the number of protons pumped in each pathway alone is insufficient to account for biochemical blockages that could occur if F420H2 pools accumulated without an oxidant.
To account for stoichiometric balance of electron donors and acceptors, steady-state electron flux was calculated for each model using equation 7:
| (7) |
where n is the number of electrons accepted by each electron carrier: Fdx, F420, or via an unknown direct protein-protein interaction (Fpo-Mer or Fpo-Mtd?). The term α is the mole fraction of electrons accepted by each enzyme (a combination of HdrABC, Fpo, and Rnf) in each model. For the purpose of simulation, we assumed that all enzymes (HdrABC, Fpo, and Rnf) could compete equally well for each electron donor and that flux through the oxidative branch of the methylotrophic methanogenesis pathway did not change. When input and output fluxes disagreed, a fault was declared, and the number of ions translocated was set to zero. Faults were manually checked for each model, and when theoretically biochemically possible, such as when two enzymes had the same substrate, accumulated electrons were cleared by increasing flux through the alternative enzyme(s).
The model simulations and flux calculations show that all but one model, M13 (see Table S5 and Fig. S4 in the supplemental material), are excluded by comparing predicted and actual biomass measurements. Model M13 is very similar to our physiological data in that a direct mechanism for CoM-S-S-CoB reduction by reduced ferredoxin is characterized by a 5 to 15% increase in biomass in the ΔhdrABC mutant relative to the parent strain (within the error of experimental ΔhdrABC biomass measurement), a 5 to 10% decrease in biomass in the Δrnf mutant (within the error of experimental Δrnf biomass measurement) (32), and equivalent biomasses between the parent and att::hdrABC* strains. The available data and model predictions suggest that when M. acetivorans is growing on methylotrophic substrates, the role of HdrABC is to bypass Rnf by using electrons from ferredoxin to reduce CoM-S-S-CoB (Fig. 5).
FIG 5.
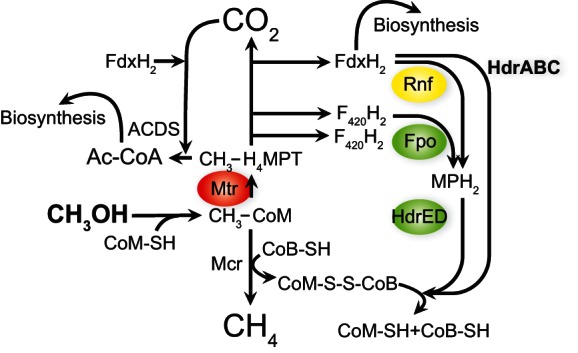
Methylotrophic methanogenesis pathway in M. acetivorans. The green ovals indicate energy-conserving steps. The red oval indicates an energy-consuming step. Yellow indicates an [Na+]-dependent energy conservation step. Ac-CoA, acetyl-coenzyme A; Fpo, proton-pumping F420-methanophenazine oxidoreductase; Mtr, methyl-coenzyme M methyltransferase.
Overexpression of HdrABC does not affect acetoclastic methanogenesis.
We also tested whether HdrABC could increase methanogenesis or affect growth when it is expressed during acetoclastic growth. Methanogens use a different methanogenesis pathway to grow on acetate as the sole carbon source (see Fig. S6 in the supplemental material). Expression of the hdrABC genes, like that of many of the methylotrophic methanogenesis genes, is greatly reduced when cells are growing acetoclastically (33, 34). Transcription from the PhdrABC promoter could not be detected on acetate, and deletion of the hdrABC operon did not affect growth or methane production from acetate versus the parental strain (8). HdrABC is thought to accept electrons from ferredoxin, the primary electron carrier during acetoclastic growth, which accepts two electrons per acetate molecule consumed (12, 35). Electrons from ferredoxin are donated to either the proposed sodium-pumping ferredoxin-methanophenazine oxidoreductase, Rnf, or potentially to HdrA2C2B2 to reduce CoM-S-S-CoB. Ferredoxin–CoM-S-S-CoB heterodisulfide oxidoreductase (Fho) activity has been detected in acetate-grown cells and is coupled with Rnf activity (16). Presumably, HdrA2C2B2 or HdrED may be the enzyme responsible for Fho activity during acetoclastic growth. If the methylotrophic HdrABC can substitute for Fho, or if it can compete with Rnf for electrons from ferredoxin, overexpression of HdrABC in the att::hdrABC* strain could potentially increase the rate of methanogenesis from acetate when expressed from a constitutive promoter on the integrated pJC1 plasmid.
However, integration of the plasmid pJC1 has no effect on either growth or methane production when acetate is used as the sole carbon source (Table 1). When cells are inoculated from trimethylamine to acetate, the parent strain has a doubling time of 51.2 ± 0.69 h versus 54.3 ± 0.86 h (P = 0.0012) for the att::hdrABC* mutant (see Table S3 in the supplemental material). When strains are preadapted to growth on acetate for 25 generations, the doubling times are 44.0 ± 2.8 h versus 47.6 ± 2.64 h, respectively (P = 0.1269) (Table 1). These results suggest the att::hdrABC* strain has a modest carbon source switching defect but that growth is not affected by overexpression of HdrABC once cells are adapted to acetate. Cell suspension assays show no change in the rate of methane production between the parent and the att::hdrABC* strains (Fig. 3C). Therefore, the HdrABC enzyme is specific for methylotrophic methanogenesis and overexpression does not increase methane production from acetate.
DISCUSSION
M. acetivorans has evolved multiple mechanisms for survival under a wide array of changing growth conditions. It can adjust to take advantage of changes in carbon and energy sources by altering gene expression of multiple substrate-specific methyltransferases to utilize methanol, methylamines, and methylsulfide (5, 34, 36–44). It can also adjust to changes in salt concentration by synthesizing glycine betaine as an osmoregulator; by upregulating phosphate and sodium transporters; by using a sodium proton antiporter, MrpA, to optimize the transmembrane proton gradient to maintain optimal ATPase function; and by using a promiscuous H+/Na+ ATP synthase (31, 45, 46). Our observations suggest Hdr enzymes have also evolved as a consequence of selective pressure to respond to fluctuations in substrate availability.
Phylogenomic analyses show that there are several classes of Hdr enzymes in methanogens. Obligate hydrogenotrophic methanogens generally have two sets of HdrABC enzymes (one encoding selenocysteines), while Methanosarcinales has two kinds of Hdr enzyme (HdrABC and HdrED), as well as multiple copies of Hdr subunit genes. Most Methanosarcinales genomes contain hdrABC genes that share similarity with methylotrophic and obligate acetoclastic Methanosarcinales hdrABC genes, while the other hdrA2 and hdrC2B2 genes are more similar to the obligate hydrogenotrophic methanogen hdrABC genes (8). Therefore, within the Methanosarcinales lineage, it appears as if there was a strong selective pressure to evolve increased HdrABC levels to uncouple methanogenesis and growth. Our physiological data suggest hdrABC genes allow methylotrophic methanogens to rapidly take up substrate and grow, albeit at submaximal efficiency. This scenario is consistent with M. acetivorans as a k-strategist at high substrate concentrations that turns over substrate faster but less efficiently. (Microbial k-strategists are characterized by an inability to adjust their growth rate in order to compete for high concentrations of substrate. Instead, k-strategists compete effectively at low substrate concentrations by having increased substrate affinity and uptake.) By converting substrate to methane faster and rapidly dropping the concentration of available substrate in the environment, over time, Methanosarcinales may outcompete other microbes for substrate as long as it is still able to generate ATP above a threshold maintenance level.
Multiple versions and copies of Hdr enzymes could have evolved to form specialized protein-protein interactions. Methanococcus HdrABC can use an electron bifurcation mechanism and forms specific protein-protein interactions with the hydrogenase Vhu; formyl-methanofuran dehydrogenase, Fmd; and formate dehydrogenase, Fdh (11, 12). HdrABC enzymes in M. acetivorans cannot interact with Vhu or another hydrogenase because none are expressed. However, Fmd or another protein partner(s) is possible. HdrED may also participate in protein-protein interactions in the cell. The HdrD subunit of HdrED interacts with acetyl-CoA decarbonylase/synthase (ACDS) and methylene-tetrahydromethanopterin reductase (Mer) (47). Further experiments are necessary to determine which proteins and enzymes interact with each other, how these relationships change as Methanosarcina switches between carbon and energy sources, and the degree of relevance to growth of the organism.
Hdr enzymes may also tailor electron donor specificity as cells switch from one methanogenesis pathway to another, for instance, between methylotrophic and acetoclastic pathways. Though HdrABC and Rnf compete for the same electron donor in methanol-grown cells, Rnf is upregulated 4- to 10-fold when cells are grown on acetate and genes necessary for the oxidative branch of the methanogenesis pathway are poorly expressed (15, 33, 34, 36). Transcriptional-fusion experiments suggest HdrA2, polyferredoxin, and HdrC2B2 genes in M. acetivorans are upregulated on acetate, and acetate-grown cells are known to have ferredoxin-heterodisulfide reductase activity (Fho), which is coupled with Rnf (16). If overexpressed HdrABC cannot kinetically compete with Fho, it may explain why overexpression of the methylotrophic HdrABC is not able to increase the rate of methanogenesis from acetate (8). If the methylotrophic HdrABC enzyme interacts directly with Fmd, or if it requires a ferredoxin that is not expressed on acetate, HdrABC would not be able to compete with Rnf for electrons when cells are grown on acetate. This idea is consistent with the fact that the acetate-induced HdrA2 gene is expressed as part of an operon that also contains a polyferredoxin. Regardless of substrate specificity details, our results suggest manipulating Hdr gene expression increases the rate of substrate turnover by bypassing rate-limiting redox reactions, further enhancing the k-strategist metabolism of M. acetivorans.
Supplementary Material
ACKNOWLEDGMENTS
This publication was made possible by National Science Foundation grant number IOS-1449525, by National Institutes of Health grant number P20 RR-17675 from the National Center for Research Resources, and by the Nebraska Tobacco Settlement Biomedical Research Development Funds.
Any opinions, findings, and conclusions or recommendations expressed in this material are ours and do not necessarily reflect the views of the institutions that provided funding.
We declare no competing interests.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.01162-15.
REFERENCES
- 1.Mah RA, Ward DM, Baresi L, Glass TL. 1977. Biogenesis of methane. Annu Rev Microbiol 31:309–341. doi: 10.1146/annurev.mi.31.100177.001521. [DOI] [PubMed] [Google Scholar]
- 2.Thauer RK, Kaster AK, Seedorf H, Buckel W, Hedderich R. 2008. Methanogenic archaea: ecologically relevant differences in energy conservation. Nat Rev Microbiol 6:579–591. doi: 10.1038/nrmicro1931. [DOI] [PubMed] [Google Scholar]
- 3.Weiland P. 2010. Biogas production: current state and perspectives. Appl Microbiol Biotechnol 85:849–860. doi: 10.1007/s00253-009-2246-7. [DOI] [PubMed] [Google Scholar]
- 4.Sowers KR, Baron SF, Ferry JG. 1984. Methanosarcina acetivorans sp. nov., an acetotrophic methane-producing bacterium isolated from marine sediments. Appl Environ Microbiol 47:971–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bose A, Pritchett MA, Metcalf WW. 2008. Genetic analysis of the methanol- and methylamine-specific methyltransferase 2 genes of Methanosarcina acetivorans C2A. J Bacteriol 190:4017–4026. doi: 10.1128/JB.00117-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deppenmeier U. 2004. The membrane-bound electron transport system of Methanosarcina species. J Bioenerg Biomembr 36:55–64. doi: 10.1023/B:JOBB.0000019598.64642.97. [DOI] [PubMed] [Google Scholar]
- 7.Deppenmeier U. 2002. The unique biochemistry of methanogenesis. Prog Nucleic Acid Res Mol Biol 71:223–283. doi: 10.1016/S0079-6603(02)71045-3. [DOI] [PubMed] [Google Scholar]
- 8.Buan NR, Metcalf WW. 2010. Methanogenesis by Methanosarcina acetivorans involves two structurally and functionally distinct classes of heterodisulfide reductase. Mol Microbiol 75:843–853. doi: 10.1111/j.1365-2958.2009.06990.x. [DOI] [PubMed] [Google Scholar]
- 9.Murakami E, Deppenmeier U, Ragsdale SW. 2001. Characterization of the intramolecular electron transfer pathway from 2-hydroxyphenazine to the heterodisulfide reductase from Methanosarcina thermophila. J Biol Chem 276:2432–2439. doi: 10.1074/jbc.M004809200. [DOI] [PubMed] [Google Scholar]
- 10.Abken HJ, Tietze M, Brodersen J, Baumer S, Beifuss U, Deppenmeier U. 1998. Isolation and characterization of methanophenazine and function of phenazines in membrane-bound electron transport of Methanosarcina mazei Gö1. J Bacteriol 180:2027–2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Costa KC, Lie TJ, Xia Q, Leigh JA. 2013. VhuD facilitates electron flow from H2 or formate to heterodisulfide reductase in Methanococcus maripaludis. J Bacteriol 195:5160–5165. doi: 10.1128/JB.00895-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Costa KC, Wong PM, Wang T, Lie TJ, Dodsworth JA, Swanson I, Burn JA, Hackett M, Leigh JA. 2010. Protein complexing in a methanogen suggests electron bifurcation and electron delivery from formate to heterodisulfide reductase. Proc Natl Acad Sci U S A 107:11050–11055. doi: 10.1073/pnas.1003653107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Suharti S, Wang M, de Vries S, Ferry JG. 2014. Characterization of the RnfB and RnfG subunits of the Rnf complex from the archaeon Methanosarcina acetivorans. PLoS One 9:e97966. doi: 10.1371/journal.pone.0097966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang M, Tomb JF, Ferry JG. 2011. Electron transport in acetate-grown Methanosarcina acetivorans. BMC Microbiol 11:165. doi: 10.1186/1471-2180-11-165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li Q, Li L, Rejtar T, Lessner DJ, Karger BL, Ferry JG. 2006. Electron transport in the pathway of acetate conversion to methane in the marine archaeon Methanosarcina acetivorans. J Bacteriol 188:702–710. doi: 10.1128/JB.188.2.702-710.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schlegel K, Welte C, Deppenmeier U, Muller V. 2012. Electron transport during aceticlastic methanogenesis by Methanosarcina acetivorans involves a sodium-translocating Rnf complex. FEBS J 279:4444–4452. doi: 10.1111/febs.12031. [DOI] [PubMed] [Google Scholar]
- 17.Welte C, Deppenmeier U. 2011. Re-evaluation of the function of the F420 dehydrogenase in electron transport of Methanosarcina mazei. FEBS J 278:1277–1287. doi: 10.1111/j.1742-4658.2011.08048.x. [DOI] [PubMed] [Google Scholar]
- 18.Uetake H, Luria SE, Burrous JW. 1958. Conversion of somatic antigens in Salmonella by phage infection leading to lysis or lysogeny. Virology 5:68–91. doi: 10.1016/0042-6822(58)90006-0. [DOI] [PubMed] [Google Scholar]
- 19.Metcalf WW, Jiang W, Daniels LL, Kim SK, Haldimann A, Wanner BL. 1996. Conditionally replicative and conjugative plasmids carrying lacZα for cloning, mutagenesis, and allele replacement in bacteria. Plasmid 35:1–13. doi: 10.1006/plas.1996.0001. [DOI] [PubMed] [Google Scholar]
- 20.Sowers KR, Boone JE, Gunsalus RP. 1993. Disaggregation of Methanosarcina spp. and growth as single cells at elevated osmolarity. Appl Environ Microbiol 59:3832–3839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Metcalf WW, Zhang JK, Shi X, Wolfe RS. 1996. Molecular, genetic, and biochemical characterization of the serC gene of Methanosarcina barkeri Fusaro. J Bacteriol 178:5797–5802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guss AM, Mukhopadhyay B, Zhang JK, Metcalf WW. 2005. Genetic analysis of mch mutants in two Methanosarcina species demonstrates multiple roles for the methanopterin-dependent C-1 oxidation/reduction pathway and differences in H2 metabolism between closely related species. Mol Microbiol 55:1671–1680. doi: 10.1111/j.1365-2958.2005.04514.x. [DOI] [PubMed] [Google Scholar]
- 23.Horton RM, Hunt HD, Ho SN, Pullen JK, Pease LR. 1989. Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene 77:61–68. doi: 10.1016/0378-1119(89)90359-4. [DOI] [PubMed] [Google Scholar]
- 24.Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. 1989. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 25.Duin EC, Prakash D, Brungess C. 2011. Methyl-coenzyme M reductase from Methanothermobacter marburgensis. Methods Enzymol 494:159–187. doi: 10.1016/B978-0-12-385112-3.00009-3. [DOI] [PubMed] [Google Scholar]
- 26.Welte C, Deppenmeier U. 2011. Proton translocation in methanogens. Methods Enzymol 494:257–280. doi: 10.1016/B978-0-12-385112-3.00013-5. [DOI] [PubMed] [Google Scholar]
- 27.Jones P, Binns D, Chang HY, Fraser M, Li W, McAnulla C, McWilliam H, Maslen J, Mitchell A, Nuka G, Pesseat S, Quinn AF, Sangrador-Vegas A, Scheremetjew M, Yong SY, Lopez R, Hunter S. 2014. InterProScan 5: genome-scale protein function classification. Bioinformatics 30:1236–1240. doi: 10.1093/bioinformatics/btu031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guss AM, Rother M, Zhang JK, Kulkarni G, Metcalf WW. 2008. New methods for tightly regulated gene expression and highly efficient chromosomal integration of cloned genes for Methanosarcina species. Archaea 2:193–203. doi: 10.1155/2008/534081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Buan N, Kulkarni G, Metcalf W. 2011. Genetic methods for Methanosarcina species. Methods Enzymol 494:23–42. doi: 10.1016/B978-0-12-385112-3.00002-0. [DOI] [PubMed] [Google Scholar]
- 30.DeLong JP, Okie JG, Moses ME, Sibly RM, Brown JH. 2010. Shifts in metabolic scaling, production, and efficiency across major evolutionary transitions of life. Proc Natl Acad Sci U S A 107:12941–12945. doi: 10.1073/pnas.1007783107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jasso-Chavez R, Apolinario EE, Sowers KR, Ferry JG. 2013. MrpA functions in energy conversion during acetate-dependent growth of Methanosarcina acetivorans. J Bacteriol 195:3987–3994. doi: 10.1128/JB.00581-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Benedict MN, Gonnerman MC, Metcalf WW, Price ND. 2012. Genome-scale metabolic reconstruction and hypothesis testing in the methanogenic archaeon Methanosarcina acetivorans C2A. J Bacteriol 194:855–865. doi: 10.1128/JB.06040-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li Q, Li L, Rejtar T, Karger BL, Ferry JG. 2005. Proteome of Methanosarcina acetivorans. Part I: an expanded view of the biology of the cell. J Proteome Res 4:112–128. [DOI] [PubMed] [Google Scholar]
- 34.Li Q, Li L, Rejtar T, Karger BL, Ferry JG. 2005. Proteome of Methanosarcina acetivorans. Part II: comparison of protein levels in acetate- and methanol-grown cells. J Proteome Res 4:129–135. [DOI] [PubMed] [Google Scholar]
- 35.Kaster AK, Moll J, Parey K, Thauer RK. 2011. Coupling of ferredoxin and heterodisulfide reduction via electron bifurcation in hydrogenotrophic methanogenic archaea. Proc Natl Acad Sci U S A 108:2981–2986. doi: 10.1073/pnas.1016761108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li L, Li Q, Rohlin L, Kim U, Salmon K, Rejtar T, Gunsalus RP, Karger BL, Ferry JG. 2007. Quantitative proteomic and microarray analysis of the archaeon Methanosarcina acetivorans grown with acetate versus methanol. J Proteome Res 6:759–771. doi: 10.1021/pr060383l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rother M, Boccazzi P, Bose A, Pritchett MA, Metcalf WW. 2005. Methanol-dependent gene expression demonstrates that methyl-coenzyme M reductase is essential in Methanosarcina acetivorans C2A and allows isolation of mutants with defects in regulation of the methanol utilization pathway. J Bacteriol 187:5552–5559. doi: 10.1128/JB.187.16.5552-5559.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pritchett MA, Metcalf WW. 2005. Genetic, physiological and biochemical characterization of multiple methanol methyltransferase isozymes in Methanosarcina acetivorans C2A. Mol Microbiol 56:1183–1194. doi: 10.1111/j.1365-2958.2005.04616.x. [DOI] [PubMed] [Google Scholar]
- 39.Ding YH, Zhang SP, Tomb JF, Ferry JG. 2002. Genomic and proteomic analyses reveal multiple homologs of genes encoding enzymes of the methanol:coenzyme M methyltransferase system that are differentially expressed in methanol- and acetate-grown Methanosarcina thermophila. FEMS Microbiol Lett 215:127–132. doi: 10.1111/j.1574-6968.2002.tb11381.x. [DOI] [PubMed] [Google Scholar]
- 40.Sauer K, Harms U, Thauer RK. 1997. Methanol:coenzyme M methyltransferase from Methanosarcina barkeri. Purification, properties and encoding genes of the corrinoid protein MT1. Eur J Biochem 243:670–677. [DOI] [PubMed] [Google Scholar]
- 41.Krzycki JA, Kenealy WR, Deniro MJ, Zeikus JG. 1987. Stable carbon isotope fractionation by Methanosarcina barkeri during methanogenesis from acetate, methanol, or carbon dioxide-hydrogen. Appl Environ Microbiol 53:2597–2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van der Meijden P, Heythuysen HJ, Pouwels A, Houwen F, van der Drift C, Vogels GD. 1983. Methyltransferases involved in methanol conversion by Methanosarcina barkeri. Arch Microbiol 134:238–242. doi: 10.1007/BF00407765. [DOI] [PubMed] [Google Scholar]
- 43.Tallant TC, Paul L, Krzycki JA. 2001. The MtsA subunit of the methylthiol:coenzyme M methyltransferase of Methanosarcina barkeri catalyses both half-reactions of corrinoid-dependent dimethylsulfide: coenzyme M methyl transfer. J Biol Chem 276:4485–4493. doi: 10.1074/jbc.M007514200. [DOI] [PubMed] [Google Scholar]
- 44.Tallant TC, Krzycki JA. 1997. Methylthiol:coenzyme M methyltransferase from Methanosarcina barkeri, an enzyme of methanogenesis from dimethylsulfide and methylmercaptopropionate. J Bacteriol 179:6902–6911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pfluger K, Ehrenreich A, Salmon K, Gunsalus RP, Deppenmeier U, Gottschalk G, Muller V. 2007. Identification of genes involved in salt adaptation in the archaeon Methanosarcina mazei Gö1 using genome-wide gene expression profiling. FEMS Microbiol Lett 277:79–89. doi: 10.1111/j.1574-6968.2007.00941.x. [DOI] [PubMed] [Google Scholar]
- 46.Schlegel K, Leone V, Faraldo-Gomez JD, Muller V. 2012. Promiscuous archaeal ATP synthase concurrently coupled to Na+ and H+ translocation. Proc Natl Acad Sci U S A 109:947–952. doi: 10.1073/pnas.1115796109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lieber DJ, Catlett J, Madayiputhiya N, Nandakumar R, Lopez MM, Metcalf WW, Buan NR. 2014. A multienzyme complex channels substrates and electrons through acetyl-CoA and methane biosynthesis pathways in Methanosarcina. PLoS One 9:e107563. doi: 10.1371/journal.pone.0107563. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.