

Abstract
The eukaryotic Spt4-Spt5 heterodimer forms a higher-order complex with RNA polymerase II (and I) to regulate transcription elongation. Extensive genetic and functional data have revealed diverse roles of Spt4-Spt5 in coupling elongation with chromatin modification and RNA-processing pathways. A mechanistic understanding of the diverse functions of Spt4-Spt5 is hampered by challenges in resolving the distribution of functions among its structural domains, including the five KOW domains in Spt5, and a lack of their high-resolution structures. We present high-resolution crystallographic results demonstrating that distinct structures are formed by the first through third KOW domains (KOW1-Linker1 [K1L1] and KOW2-KOW3) of Saccharomyces cerevisiae Spt5. The structure reveals that K1L1 displays a positively charged patch (PCP) on its surface, which binds nucleic acids in vitro, as shown in biochemical assays, and is important for in vivo function, as shown in growth assays. Furthermore, assays in yeast have shown that the PCP has a function that partially overlaps that of Spt4. Synthesis of our results with previous evidence suggests a model in which Spt4 and the K1L1 domain of Spt5 form functionally overlapping interactions with nucleic acids upstream of the transcription bubble, and this mechanism may confer robustness on processes associated with transcription elongation.
INTRODUCTION
Transcription by RNA polymerase (RNAP) II is a regulated process that requires over 60 different accessory factors that interact dynamically with the polymerase as it proceeds through the three main stages of transcription: initiation, elongation, and termination (1). Each RNAP II complex is structurally and biochemically tuned to cope with a set of conditions (e.g., the chromatin state) and to accomplish specific functions (e.g., promoter-dependent initiation) associated with a particular stage of transcription. In eukaryotes, the processes of RNAP II elongation and termination interact closely with pathways of pre-mRNA processing, 3′-end formation, and RNA export (2–4) and thus form temporally and spatially coupled activities that together determine transcriptional responses to intrinsic and extrinsic signaling and regulate RNA metabolism. The heterodimeric complex of Spt4-Spt5 is one of several protein factors that participate in all of the steps that follow transcription initiation. Saccharomyces cerevisiae Spt4-Spt5 is known to coordinate transcription elongation with chromatin remodeling and histone modification; it functions as a general elongation factor for both RNAP II and RNAP I and coordinates with 5′ capping, splicing, and 3′-end processing of transcripts (reviewed by Hartzog and Fu [5]). The metazoan homolog of Spt4-Spt5, DSIF, partners with NELF and additional RNAP II-associated complexes (e.g., P-TEFb and Mediator) to impart a pause-and-release mechanism that regulates RNAP II activity during the initiation-to-elongation transition near promoter-proximal regions (6–8). While facilitative for transcription elongation, Spt4-Spt5 also plays roles in other nuclear transactions, including repression of transcription-coupled DNA repair (9–11), maintenance of chromosome integrity, and immunoglobulin somatic hypermutation and class switch recombination (12). Current data indicate that Spt4-Spt5 executes these functions as part of a stable complex with the elongating RNAP II (13–17); Spt4-Spt5 is not found in the preinitiation complex; rather, it first associates with RNAP II early in elongation (14, 18–20). It is conceivable that Spt4-Spt5 facilitates these functions by providing interfaces with other factors that bring about regulation of transcription and pre-mRNA processing. As such, Spt4-Spt5 may be generally considered the mediator of elongation and the complex it forms with an RNAP as the holo-elongation complex.
A large number of reported genetic and functional interactions suggest that Spt4-Spt5 may either directly or indirectly mediate interactions between an elongating RNAP II and a large set of its accessory factors (5). Consistent with this, Spt5 is a large protein with multiple domains that can present, along with Spt4, many potential interaction surfaces. Eukaryotic Spt5 contains an unfolded N-terminal acidic region interspersed with two short segments enriched in basic residues (see Fig. S1 in the supplemental material). Another unstructured region (the C-terminal repeated region [CTR]), which comprises a set of short repeats whose consensus motifs vary across species, is found at its C terminus (21–23). The CTR is subject to phosphorylation and participates in Spt5's mediation of nuclear activities, such as RNA capping to transcription (5). The central region of Spt5 begins with an NGN domain that is structurally similar to the N-terminal domain of bacterial elongation factor NusG (NGN). The NGN domain is structurally conserved throughout bacterial NusG and archaeal and eukaryotic Spt5 proteins (24). In eukaryotes and archaea, the NGN domain forms a heterodimer with Spt4, a small zinc finger protein with no counterpart in bacteria (see Fig. S2A in the supplemental material). The archaeal NGN binds to the Clamp domain of RNAP (25, 26), forming an arch over the extensive DNA/RNA binding cleft in RNAP; this suggests nucleic acid (NA) encirclement in the holo-elongation complex as a mechanism for promoting transcription processivity (27). This model is consistent with the NGN-based RNAP interaction model for RfaH (28), a paralog of NusG, and offers a mechanistic explanation for functional roles of the NusG family factors in promoting elongation (25, 29, 30). In eukaryotes, the remainder of the central region between the NGN and the CTR contains five KOW motif-containing domains (31). In contrast, the archaeal and bacterial proteins contain only a single C-terminal KOW domain without a CTR-like tail (see Fig. S2A in the supplemental material).
What are the roles of the multiple KOWs in Spt5? Deletions of yeast KOW2 and KOW4-KOW5 are associated with severe growth defects in yeast (10, 11, 17), indicating important functions of the KOW domains in vivo. Structural studies have shown that isolated KOW domains of NusG and both archaeal (30, 32–34) and human Spt5 (Protein Data Bank [PDB] IDs 2E6Z, 2E70, and 3H7H) all adopt the same fold as the β-barrel of Tudor domains. Tudor domains are structurally compact (∼50-amino-acid) modules that mediate protein-protein and protein-NA interactions in diverse molecular systems (35). It is logical to propose that the KOWs of Spt5 mediate interactions between transcription-coupled activities and RNAP, and they may also promote interactions with NAs. A recent protein cross-linking study identified interaction partners between certain modules of RNAP II and the KOWs that are important for transcription elongation and transcription-coupled DNA repair (11), which has begun to shed light on these roles. However, the extent of structural similarity among the KOW domains of eukaryotic Spt5, whether the KOWs mediate similar or distinct functions, and their functional relationship with other domains of Spt5 and Spt4 remain unknown.
Here, we present X-ray results demonstrating that Spt5's KOW domains form different structures. Surprisingly, the first KOW domain cofolds with the polypeptide (Linker1) that links the first and second KOW domains, and Linker1 assumes a novel fold in the KOW1-Linker1 (K1L1) rigid-body structure. The second and third KOWs adopt a tandem Tudor fold to form a rigid structure (K2K3). The high-resolution structure of K1L1 reveals a positively charged surface area that binds NAs in a non-sequence-specific manner in vitro. Binding site mutations that abolished NA interactions in vitro generated severe growth defects in the yeast strain lacking the SPT4 gene but no observable defects in cells with the wild type (WT). This suggests an iterated NA interaction function within the Spt4-Spt5 heterodimer. Synthesis with available structural data prompts the proposal that Spt4 and K1L1 of Spt5 form functionally overlapping interactions with NAs upstream of the transcription bubble and may thus impart robustness to transcription elongation and associated processes.
MATERIALS AND METHODS
Recombinant proteins.
For crystallographic studies, the various Spt5 domains were each subcloned into the pET-SUMO plasmid (Life Technology) after optimization of codons according to the Escherichia coli codon usage. Sequence-confirmed plasmids were each transformed into E. coli strain BL21(DE3) for protein expression. Cultures (1.0 liter) were grown for each expression construct to an optical density at 600 nm (OD600) of about 0.6 at 37°C and then induced with 0.5 mM isopropyl-β-d-thiogalactopyranoside (IPTG) at 16°C overnight. The harvested cells were lysed on ice with 0.2 mg/ml lysozyme and gentle sonication (200 W for 3 s 20 times) in the loading buffer (10 mM Tris-HCl, pH 8.0, 15% glycerol, 150 mM sodium chloride, 2.0 mM 2-mercaptoethanol, 1.0 mM phenylmethylsulfonyl fluoride [PMSF]). The purification entails Ni-nitrilotriacetic acid (NTA) metal-chelating chromatography via a His6 tag on the N-terminal SUMO moiety, Ulp1 cleavage of the SUMO tag (1 mg of Ulp1 at 4°C overnight), ionic-exchange chromatography, a second Ni-NTA step to remove the SUMO protein, and gel filtration chromatography. The K2K3 protein with a selenomethionine (Se-Met) substitution was expressed from a culture of E. coli strain B834 in M9 minimal medium with Se-Met (Novagen) and purified similarly.
For the in vitro nucleic acid binding assay, the various Spt5 domains were each subcloned into the plasmid pET24b(+) (Novagen) and transformed into E. coli BL21(DE3). Cultures (300 ml) were grown at 37°C to an OD600 of 0.6 to 0.7 and induced with 1 mM IPTG at 20°C overnight. The harvested cells were lysed by sonication and cleared by centrifugation. The recombinant proteins were purified over Ni-NTA resin via a C-terminal His6 tag. Linker2-KOW4 (L2K4) and Linker3-KOW5 (L3K5) were further purified with additional ionic-exchange (Mono-S) and gel filtration (Superose-6) chromatographic steps. K1L1 mutants were generated with QuikChange (Stratagene), using the pET24b expression construct as the starting template, and purified similarly.
Protein primary structure analysis.
Sequence alignments were performed using ClustalX (36) or ClustalW (37) and visualized with SEAVIEW (38). Initial domain annotation for the S. cerevisiae Spt5 sequence was done following the method of Hartzog and Fu (5) to delineate the NGN domain, five KOW domains, the N-terminal acidic region, and the CTR. Three linker regions of significant size separating the KOW domains were named Linker1 (between KOW1 and KOW2), Linker2 (between KOW3 and KOW4), and Linker3 (between KOW4 and KOW5). Figures showing the sequence alignment and annotation were created with Aline (39). The N-terminal acidic and linker regions were initially evaluated for potential folding through disorder predications using GlobPlot (40) and DisEMBL (41).
Consensus sequences of the KOW1 through KOW5 domains are shown with letters scaled according to the aligned sequences of Spt5 from S. cerevisiae, Schizosaccharomyces pombe, Caenorhabditis elegans, Drosophila melanogaster, Homo sapiens, Danio rerio, and Mus musculus, and the numbering follows that of S. cerevisiae. The scaled consensus sequence for bacterial KOW domains was calculated from the aligned NusG sequences of the eubacteria Aquifex aeolicus, E. coli, and Thermus thermophilus and the Spt5 sequences of the archaea Thermococcus gammatolerans, Pyrococcus furiosus, and Methanoculleus bourgensis, and the numbering follows that of A. aeolicus. Single-letter amino acid characters were scaled according to a method described previously (42). Secondary structures were predicted using the Jpred3 server (43).
Protein-folding prediction.
Further prediction of folding was performed using Rosetta (44). Two sequences within the Linker1 region were selected: Spt5 residues 433 to 511, covering the entire Linker1, and a shorter sequence, 460 to 511, with a lower degree of disorder based on the disorder prediction. For each sequence, 30,000 low-energy conformations were generated and clustered, and the clusters were ranked by membership size following the standard Rosetta ab initio protocol. Control computations, conducted for the KOW/Tudor domains of Spt5, suggested that 30,000 conformations would be sufficient for structures of this size. The top-ranking structures for the Linker1 sequence were consistent with the top-ranking structures for the shorter sequence (root mean square [RMS], 2.826 Å), further suggesting that this region could correspond to a folded domain. Subsequent to the determination of the X-ray structure of K1L1, however, it was found that the predicted structures of Linker1 did not resemble the experimental structure. This might be due to the cross-domain region that was used in the Rosetta prediction—part of Linker1 plus the C-terminal part of the KOW1/Tudor domain—not an independently folded domain.
Crystallographic procedures.
Crystallization of the K1L1 domain was carried out in hanging drops at 20°C. The drops were formed by mixing 2.5 μl protein (3.5 mg/ml) and 1 μl mother liquor (0.1 M Tris-HCl, pH 8.0, 30% [wt/vol] polyethylene glycol [PEG] 3350). Crystals of K1L1 were cryoprotected in 0.1 M Tris-HCl, pH 8.0, 40% (wt/vol) PEG 3350, 150 mM sodium chloride, 5% glycerol and flash-frozen in liquid nitrogen. The K2K3 and Se-Met K2K3 proteins were concentrated to 20 mg/ml (in 10 mM Tris-HCl, pH 8.0, 5% glycerol, 0.5 mM EDTA, and 2.0 mM β-mercaptoethanol [β-ME]) and crystallized in hanging drops at 20°C with mother liquor consisting of 0.1 M sodium citrate, pH 5.0, 0.5 M ammonium sulfate, and 1.0 M lithium sulfate.
X-ray diffraction experiments were carried out at both the Shanghai Synchrotron Radiation Facility and the Australian Synchrotron. A native data set was collected from the K1L1 crystal to 1.09-Å resolution at the Shanghai facility, and a sulfa-anomalous data set (λ = 1.5484 Å) was collected at the Australian site to 1.55-Å resolution. Protein phases for K1L1 were calculated based on the single-wavelength anomalous diffraction (SAD) signal arising from sulfur atoms of the cystines in the protein, using standard programs (XDS [45] and SHLEX C/D/E and CCP4 [46]), and improved with PARROT. Model building was initiated with ARP/wARP and refined with Refmac5 and Coot (47). Final refinement of K1L1 was carried out against the native data set (1.09 Å) using Coot and PHENIX (48). Molprobity (49) was used to monitor stereochemical factors, such as bond length, clashes, and Ramachandran (main-chain torsion angle) plot. The K2K3 structure was determined based on the experimental electron density map that had been phased using the Se-Met SAD data set collected at the selenium absorption edge (λ = 0.9793 Å). The same data set was used, after merging Friedel pairs, for refining the K2K3 model.
Structure analysis.
Structural homologs of the yeast KOW/Tudor domains were identified by full-text search of the PDB (50) and manually removing unrelated results. For comparisons of electrostatic surface potentials, nonprotein components (water and other small molecules) were removed. For nuclear magnetic resonance (NMR) structures, all models but the first were removed. The models were parameterized by PDB 2PQR (51) using the CHARMM force field (52). The electrostatic potentials on the solvent-accessible surfaces of structural models were calculated using the APBS algorithm (53) and presented in kT units (k is the Boltzman constant; T is the absolute temperature) (54). The values of buried protein surface areas were calculated using the CNS package (http://cns-online.org/v1.3). Identification of hydrogen bond partners was aided by the program CONTACT within the CCP4 package. Secondary-structure topology diagrams were generated using the TopDraw procedure (55).
EMSA.
An electrophoretic mobility shift assay (EMSA) was used to assess protein-nucleic acid interactions. Recombinant proteins of the various KOW domains with and without a linker region were purified as described above. To measure binding with DNA, single-stranded deoxyoligonucleotides of 38 and 20 bases were chemically synthesized (ITD) with a 5′ 6-carboxytetramethylrhodamine (TAMRA) (N-hydroxysuccinimide [NHS] ester) for fluorescence detection. The sequences were randomly chosen. Double-stranded 20-mer probes were formed from annealing reactions with an unlabeled cDNA strand of the same length. For binding involving RNA, a single-stranded oligonucleotide was synthesized in the same sequence as that of the fluorescently labeled single-stranded DNA (ssDNA) 20-mer and also labeled with a 5′ TAMRA. Binding reactions were assembled in the gel shift buffer (10 mM Tris-HCl, 80 mM KCl, 2 mM MgCl2, 12% glycerol, 12 mM EDTA, pH 7.0) with 0.1 mg/ml bovine serum albumin (BSA) and proteins and nucleic acids at the concentrations indicated for the figures. EMSAs were performed with 8% (37.5:1 cross-linking) nondenaturing polyacrylamide gels strengthened with 0.2% agarose (using a hot stock of 1% agarose) and standard Tris-borate-EDTA (TBE) as the running buffer, and the gels were run at 150 V in a Protean II assembly (Bio-Rad) at room temperature for 2 h. The wet gels were imaged by fluorescence scanning on a Typhoon FLA 7000 laser scanner (GE Healthcare) using filters of 532 nm for excitation and 580 nm for detection. Quantification of fluorescent bands was carried out with the ImageQuant TL software (GE Healthcare). The apparent dissociation constant (KD) for a particular binding reaction was determined from the 50% saturation point of a binding curve; each data point was taken as a percentage of the bound amount of the input probe. For all the in vitro binding experiments, a percent bound value was measured from the disappearance of a free probe, which consistently appeared as a sharp band.
Plasmid shuffle and yeast growth.
Standard methods were followed in making yeast media and conducting growth assays (56). The strains GHY1066 (MATα his3Δ200 lys2-128δ leu2Δ1 ura3-52 trp1Δ63 spt5Δ3::HIS3 [pMS4]) and GHY1172 (MATa his3Δ200 lys2-128δ leu2Δ1 ura3-52 trp1Δ63 spt5Δ4::TRP1 spt4Δ2::HIS3 [pMS4]) were generated for this study using standard methods. spt4Δ2::HIS3 has the entire SPT4 coding sequence removed, and spt5Δ3::HIS3 has all but the first 4 and last 20 codons of SPT5 removed. Plasmid pMS4 (URA3 CEN SPT5) (21) was used to maintain viability of the strains in the absence of the chromosomal copy of SPT5. The two strains were each transformed with a derivative of pRS415 (LEU2 CEN) carrying either the wild-type SPT5 modified with a triplicate N-terminal Myc epitope (pRS415 3Myc-SPT5) (10) or a 3Myc-spt5 mutant allele (pRS415 3Myc-spt5_Q1 through -Q25). Transformants were selected from synthetic complete medium lacking Leu and Ura (SC−Leu−Ura) plates and then cultured in liquid SC−Leu medium to allow the loss of pMS4. Similar aliquots of each culture were applied to and streaked out on SC−Leu plus 5-fluoroorotic acid (5-FOA) plates to select for cells that had lost pMS4 and thus depended upon the pRS415 plasmid carrying an spt5 allele for growth. Growth rates of nonlethal strains isolated from the 5-FOA plates (5-FOA resistant) were monitored using a spot dilution assay, in which cells that had grown to stationary phase were adjusted to the same density (OD600 = 0.20; no morphological differences were observed among the strains), serially 10-fold diluted, and applied to solid medium plates (5.0 μl each). The compositions of the plates and incubation temperatures are specified in the text. For shuffle assays in the absence of TFIIS (ppr2Δ), the strain GHY2731 (MATa his3Δ200 lys2-128δ leu2Δ1 ura3-52 trp1Δ63 spt5Δ4::TRP1 ppr2Δ0::KAN [pMS4]) was used with the above-mentioned plasmids bearing the spt5 alleles Q1 to Q20.
Site-directed mutations in SPT5 were generated by the QuikChange (Stratagene) method using a plasmid carrying an NcoI and BglII fragment excised from pRS415 3Myc-SPT5. This fragment encompasses the K1L1 region of Spt5. Once verified by sequencing, a fragment carrying a mutation was subcloned back into pRS415 3Myc-SPT5. The specific amino acid changes caused by the mutations are described in the text, and the sequences of the oligonucleotides used to introduce the mutations are available upon request.
The spt4-IM interface mutation that disrupts the Spt4-Spt5 interface was generated as follows. First, the wild-type SPT4 open reading frame (ORF), along with 5′ and 3′ flanking regions, was amplified from genomic DNA by PCR and cloned into the plasmid pRS416 (URA3 CEN) to create pRS416 SPT4 using the SLIC technique (57). Next, the mutations corresponding to the amino acid changes that disrupt the Spt4-Spt5 interface were introduced by the QuikChange (Stratagene) method to create pRS416 spt4-IM. For N-terminally tagging the spt4 alleles, the wild-type SPT4 ORF was amplified from pRS416 SPT4 by PCR and cloned downstream of the 3× FLAG tag in the plasmid pFA6a-6xGLY-3xFLAG-hphMX4 (58) with SLIC to create a 3Flag-SPT4 cassette. The 3Flag-SPT4 cassette was then transferred into pRS416 SPT4 to replace the wild-type SPT4 ORF using SLIC, creating the plasmid pRS416 3Flag-SPT4. Finally, the mutations corresponding to the spt4-IM allele were introduced with QuikChange to create pRS416 3Flag-spt4-IM.
The pRS416 plasmids bearing these spt4 alleles were transformed into the 5-FOA-selected strains from the shuffle assay, which each carried an spt5 allele (pRS415 3Myc-SPT5 or its mutant derivatives, as described above). Transformants were selected on SC−Leu−Ura and analyzed by the spot dilution assay, as described above.
Overexpression of Spt5 variants.
As described in the text and consistent with the previous observation (10), the protein level of Spt5 in the spt4Δ strain (GHY1172) expressed from the pRS415 3Myc-SPT5 (CEN) plasmid is significantly reduced relative to that in the SPT4 strain (GHY1066) bearing the same CEN plasmid. Ding and coworkers overexpressed 3Flag-SPT5 under the control of the inducible GAL10 promoter from a multicopy 2μ plasmid to a level several times that of the chromosomal copy (10). The same pESC 3Flag-SPT5 plasmid (2μ URA3) used by Ding et al. was used to create positively charged patch (PCP) variants of Q1, Q6, Q8, Q20, and Q5: the 2μ plasmid was gapped by digestions with Bsu36I and BglII and then received a Bsu36I-BglII fragment encompassing a PCP mutation excised from the pRS415 3Myc-spt5 mutant plasmid (CEN LEU2) to produce a pESC 3Flag-spt5 PCP mutant plasmid (2μ URA3). To replace a pRS415 plasmid in the spt4Δ strain (GHY1172) with its pESC counterpart carrying a corresponding spt5 PCP allele, the cells were additionally transformed with a pESC 3Flag-spt5 PCP mutant plasmid. Transformants were cultured in liquid SC−Ura medium with raffinose (2%) plus galactose (2%) to allow loss of the pRS415 (CEN LEU2) plasmid and induction of an Sp5 variant. An aliquot of each overnight culture was streaked on solid SC−Ura plus galactose, followed by replica plating onto SC−Ura−Leu plus galactose and selection of Ura+ and Leu− colonies. The selected colonies were purified by repeating the selection process, starting from the liquid culture. The success of the plasmid swap was finally confirmed by verifying expression of the differently tagged Spt5 proteins with Western blotting using an anti-Flag antibody (Pierce/LifeTechnology) and an anti-Myc antibody (Santa Cruz Biotechnology) (data not shown). The growth phenotype of the spt5 PCP mutants driven by the GAL10 promoter from the 2μ plasmid were assessed using a spot assay on solid SC−Ura plus raffinose (2%) and galactose (2%). The protein levels of Spt5 variants were measured using Western blotting against Spt5, with anti-glucose-6-phosphate dehydrogenase (anti-G6PDH) (Sigma-Aldrich) blotting serving as a loading control.
Immunoprecipitation.
Immunoprecipitation (IP) of Spt5 and the associated Spt4 variants was carried out using magnetic resins coupled with a specific antibody (Pierce/LifeTechnology) against the Myc tag engineered to the N terminus of Spt5. Whole-cell lysate (WCL) of a yeast strain was prepared by bead beating with glass beads (0.5-mm diameter) in a salt-ice-cooled metal chamber. Each WCL was clarified by running high-speed centrifugation at 40,000 rpm in a Ti-45 rotor (Beckman Coulter) for 40 min, and the total protein concentration was adjusted to 7.0 mg/ml afterward. The binding reaction mixture included 50 μl of the antibody resin and 1.7 ml of the WCL and was rotated for 1.0 h at 4°C. The resin was then washed 2,000-fold with a buffer (50 mM Tris-HCl, 1 mM EDTA, 60 mM NaCl, 50 mM KCl, 5 mM MgCl2, 0.1% NP-40) and eluted with 60 μl of the same buffer augmented with 5 mg/ml of c-Myc peptide (Genscript). About 20 μl eluent from an IP reaction was taken for Western blot analysis using antibodies against Spt5 and the Flag tag (N terminal to Spt4 proteins).
Protein structure accession numbers.
Coordinates and structure factors for the K1L1 and K2K3 structures have been deposited in the Protein Data Bank under accession numbers 4YTK and 4YTL, respectively.
RESULTS
Folding units of the KOW-encompassing region and crystallization.
The KOW domains of Spt5 are currently viewed as independently folded Tudor domains linked by flexible regions of similar lengths (∼52 amino acids [aa]), except for the second and third KOWs (KOW2-KOW3), which lack an obvious linker region (see Fig. S1 and S2A in the supplemental material). The central KOW-containing region of Spt5 participates in binding to RNAP II (15, 59). However, the functional specificities of individual KOW domains have not been defined, nor is it precisely known how they interact with an RNAP and other nuclear factors to mediate cotranscriptional activities. Using photoreactive protein cross-linking, Li and coworkers have shown recently that yeast KOW4 and the linker between KOW4 and KOW5 interact with the Rpb4-Rpb7 stalk module of RNAP II and that the KOW4-KOW5 region participates in Spt5-mediated repression of transcription-coupled DNA repair (11). It was also shown that the NGN and KOW3 domains interact with the two largest subunits of RNAP II, although the precise regions of the largest subunits providing the contact points were not defined.
To better understand the domain organization of Spt5, we sought to express and structurally characterize different segments of the protein, including the presumed flexible linker regions (diagramed in Fig. S2A in the supplemental material). The protein expression and purification results were consistent with the notion that four soluble domains, K1L1, K2K3, L2K4, and L3K5, behave as folding units of the Spt5 central region (see the supplemental material for details). We obtained crystals of the K1L1 and K2K3 proteins but not of K1 alone or of L2K4 or L3K5. The structures were determined to 1.09 Å and 1.6 Å for K1L1 and K2K3, respectively (see the supplemental material for more details).
Crystal structure of the KOW1-Linker1 domain.
Consistent with our ability to express and purify K1L1, the high-resolution data revealed that K1 folds together with L1 to form a rigid structure with two lobes. The N-terminal lobe consists of most of the K1 sequence, which forms a β-barrel structure resembling the Tudor domain fold, plus a C-terminal region of the L1 sequence that forms the final β-strand, β7 (Fig. 1A and B, green). Distinctively, the K1 Tudor barrel is packed against an α-helix (α2/C-helix) formed by residues in the very C terminus of L1. The C-terminal lobe is formed by a sequence insertion in the Tudor barrel. It begins at the end of the third strand (β3), where instead of forming a tight turn to join β7, the protein chain leads away from the barrel (Fig. 1A and B, cyan). This main-chain topology breaks the consensus-derived definition of the K1 C terminus: the C terminus of the previously defined K1 domain (residues 418 to 430) forms the N terminus of the C-terminal lobe, and the remainder is completed by the residues of L1. Thus, L1 is in fact a well-folded domain inserted between the last two β-strands (β3 and β7) of K1 (Fig. 1B), and the K1 Tudor domain is formed by sequences flanking this insertion. An updated consensus for K1 Tudor is presented in Fig. 1C, and the revised domain organization of Spt5 is shown in Fig. 1D. Structural database searches with the isolated L1 structure did not find any structural homolog, indicating that L1 assumes a novel protein fold.
FIG 1.

Structure of K1L1 and domain organization of Spt5. (A) The crystal structure of K1L1 comprises a Tudor domain barrel encompassing most of the KOW region and an insertion domain with a novel fold formed mainly from the Linker1 region. The N and C termini of K1L1 correspond to L382 and F508, respectively. A disordered region (I428 to R438) is found between the indicated residues E427 and R439. (B) Secondary-structure diagram drawn from the K1L1 structure and colored similarly to panel A. α-Helices are shown as cylinders, β-strands as block arrows, and loops as solid lines. (C) Updated KOW1 consensus sequence modified from the previous KOW1 consensus shown in Fig. S2B in the supplemental material. L1 is marked at its inserted position within K1. (D) Updated Spt5 domain organization with the K1L1 region delineated according to the crystal structure, modified from Fig. S2A in the supplemental material.
Additional analysis (see the supplemental material) indicated a highly conserved interface formed between K1 and L1. The interface is stabilized by a minimum of five hydrogen bonds and one cation-π electron interaction, in addition to van der Waals interactions (Fig. 2A), and these atomic interactions are formed between residues that are strongly conserved across eukaryotes (Fig. 2B). Therefore, the yeast structure determined here provides a universal model for all K1L1 domains in eukaryotic Spt5. There is a disordered region (I428 to R438) in the K1L1 structure that appears to be an inherent feature of all eukaryotic K1L1 domains (Fig. 2B). The high quality of the K1L1 electron density (Fig. 2C) indicates that the disorder is not due to crystallographic errors but rather is due to intrinsic flexibility of the region. This region is longer in S. cerevisiae and Drosophila than in other eukaryotes, suggesting possible organism-specific functions. We discuss its participation in NA interactions below.
FIG 2.
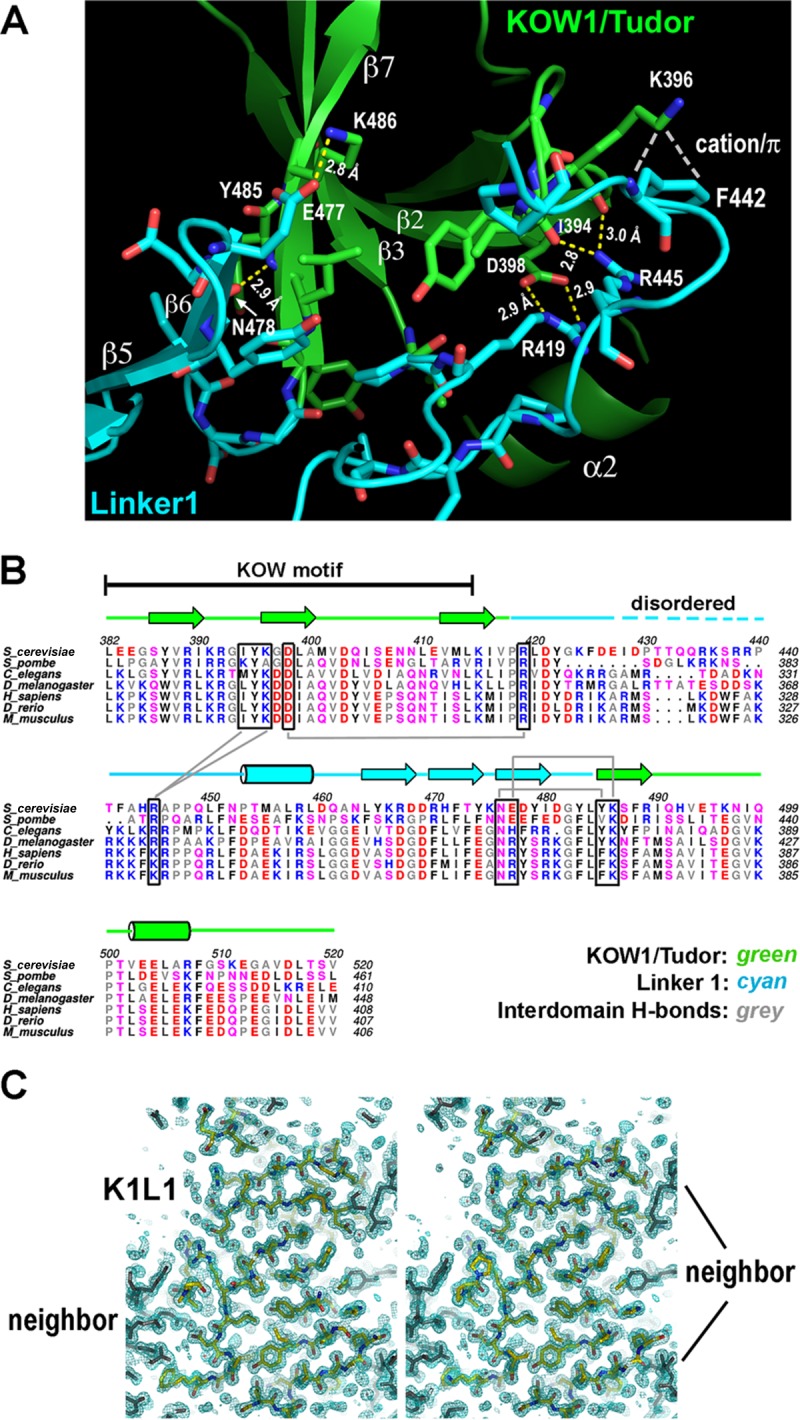
K1L1 forms a rigid-body structure with a flexible segment. (A) An extensive interface between the KOW1-Tudor and Linker1 lobes. Hydrogen bonds between K1 and L1 residues are shown with yellow dashed lines, a cation-π electron interaction is shown with white dashes, and van der Waals interactions are not shown. The interface buries a total of 1,145 Å2 of solvent-accessible surface. (B) Conservation of hydrogen bond-forming residues (boxed) in the interface. Secondary-structural elements are shown above the multiple aligned eukaryotic sequences, and the interdomain hydrogen bonds are indicated with gray lines. Basic, blue; acidic, red; polar, pink; hydrophobic, gray and black. (C) The K1L1 electron density at 1.09 Å (2Fo-Fc omit map contoured at 1.0 σ [green mesh; Fo, observed structure factor amplitudes; Fc, calculated structure factor amplitudes; omit map, model-omitted electron density map; σ, standard deviation of electron density]) is of high quality. Shown is a stereo plot of a slab through the middle of the structure. The K1L1 model is colored by atom type, and neighboring molecules in the crystal lattice are colored gray. Atomic features, such as phenol rings and carbonyl groups (red), are resolved.
The crystal structure of the KOW2-KOW3 tandem domains reveals a marked evolutionary conservation between yeast and human.
K2K3 consists of two Tudor domain β-barrel structures (49 residues each) separated by a single residue (I583) (Fig. 3A). The two domains share a significant contact area (541 Å2) that involves many side chain and main-chain groups and at least three hydrogen bonds (not shown). The result is a rigid tandem-domain structure in the middle region of Spt5 (Fig. 3B). This model is supported by its high-quality density map (Fig. 3C). The NMR structures of isolated human K2 (PDB ID 2e6z) and K3 (2do3) domains could be superimposed onto the crystal structure of the K2K3 tandem yeast domains with negligible deviations (main-chain root mean square deviations [RMSD], 0.99 Å for K2 and 0.66 Å for K3), and the ends from human K2 and K3 are poised to join in the same manner as is observed in the structure of yeast K2K3 (Fig. 4A). We conclude that the K2K3 tandem organization is conserved among eukaryotes, and this conclusion is corroborated by strong sequence conservation of residues at the K2K3 interface (Fig. 4B). The K2K3 structure is dramatically different from that of K1L1, since it lacks an L1 domain and a C helix. Rather, K2K3 bears a general similarity to the tandem Tudor structure of human KIN17 (main-chain RMSD, 3.09 Å) (see the supplemental material for additional descriptions).
FIG 3.

The KOW2 and KOW3 domains form a rigid-body structure of tandem Tudor barrels. (A) Ribbon models of K2 and K3, with the secondary-structural elements labeled in white. A single residue (I583) links the two domains. (B) Surface representation showing that K2 and K3 interact intimately to bury 541 Å2 of solvent-accessible surface. (C) Stereo plot of a central slab of the K2K3 electron density at 1.6 Å (2Fo-Fc omit map at 1.0 σ [green mesh]). The model is colored by atom type, and lattice-packing neighbors are shown in gray.
FIG 4.
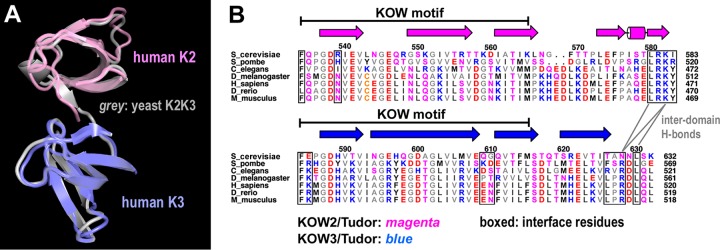
The tandem organization of K2K3 structure is conserved in eukaryotic Spt5. (A) Superposition of human K2 (PDB ID 2e6z) and K3 (2do3) with the yeast K2K3 model. (B) Sequence conservation of the hydrogen-bonded residues (boxed) between K2 and K3. The interdomain hydrogen bonds are indicated, and secondary elements are marked above the aligned sequences.
A positively charged patch on the K1L1 surface interacts with nucleic acids in vitro.
K1L1 and K2K3 present very different chemical properties on the surfaces of their structures. K1L1 has a dramatically segregated electrostatic surface with a highly positively charged patch (PCP), whereas K2K3 shows no significant bias in the distribution of charges on its surface. Interestingly, among Tudor domain proteins implicated in NA interactions, DNA repair factors 53BP1 and KIN17 (60–62) each possess a PCP that resembles that of K1L1 in terms of orientation and shape (see Fig. S3 and Table S2 in the supplemental material). To test the possibility that KOWs of Spt5 binds NA, we analyzed interactions of purified KOW domains with DNA fragments of a randomly chosen sequence using gel mobility shift assays (Fig. 5A and B) (see Materials and Methods). We observed binding of both single-stranded and double-stranded 38-mer DNAs by K1L1 but not by K1 alone, K2K3, L2K4, or K5 (Fig. 5C and D). Given the small dimensions of the K1L1 structure, gel shift experiments were repeated with defined fragments of 20 nucleotides (nt) (Fig. 6A). The results show that K1L1 is capable of binding with ssDNA, double-stranded DNA (dsDNA), ssRNA, and an RNA-DNA hybrid (Fig. 6B). While a discrete shifted band was not observed in the gels where L3K5 was analyzed (Fig. 5C and D and 6B), the disappearance of free probes suggests that L3K5 might be capable of NA interactions, which appeared to have a preference for ssRNA (Fig. 6B). The failure of L3K5 to produce an observable band of protein-NA complex may be attributable to oligomerization that precluded the bound species from entering the gel and loss of solution-bound materials from the gel wells, or to a very dynamic complex that dissociated during electrophoretic migration. However, we could not exclude the possibility that the L3K5 sample was contaminated with an RNase not present in the other proteins. Due to the scope, we focused on the K1L1 structure for functional analysis.
FIG 5.
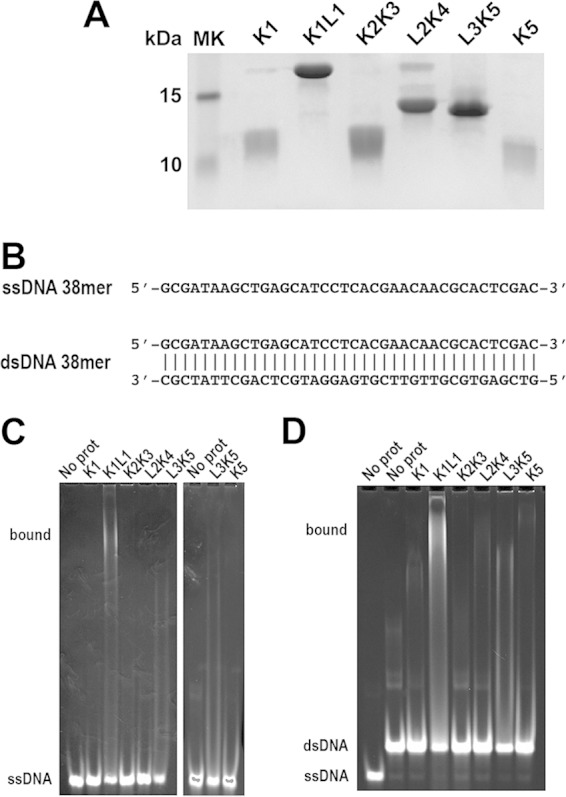
Differential DNA binding by the different KOW domains of Spt5. (A) SDS-PAGE analysis of purified recombinant KOW proteins (labeled above the lanes) with Coomassie blue staining. (B) ssDNA and dsDNA of a randomly chosen sequence used as a binding substrate for the Spt5 proteins. The DNAs were 5′ labeled with TAMRA (IDT). (C and D) Gel mobility shift assays to test the different Spt5 domains in binding ssDNA and dsDNA, respectively. Each binding reaction mixture contained 5 μM DNA probe, 50 μM the indicated Spt5 domain, and the buffer. The gels were stained with ethidium bromide and imaged with UV light. The positions of the free probe and bound complex are indicated on the left of the gels. In addition to binding by K1L1, note the disappearance of free probes due to L3K5 and the slower-migrating smear above the ssDNA and dsDNA bands in the L3K5 lanes, which may suggest low-affinity binding to ssDNA and dsDNA. Bands of the binding complex appeared persistently smeared, indicating heterogeneity in the binding stoichiometry due to the length of DNAs and/or partial dissociation of complexes during gel migration, and were also nonuniform due to adherence to the gel matrix (8% polyacrylamide with 0.2% agarose [see Materials and Methods]) of the loading wells.
FIG 6.

Interactions of recombinant Spt5 domains with various forms of NA. (A) Sequences of the single-stranded and double-stranded NAs. The 5′ end of a single strand and the top strand of a double strand were synthesized (IDT) with a TAMRA fluorescent group for detection. (B) Gel shift images from the binding assays. The reaction mixtures each contained 5 μM NA probe, 80 μM protein domain, and the buffer. A name of an NA form (bottom left side) indicates a free probe, while the label “bound” indicates a species of protein-NA complex. The protein domain used in a binding reaction is identified above the gels. (C) Direct comparison between two forms of NA for K1L1 binding in the same binding mixture. NA forms (5 μM) are indicated (bottom left side), and protein concentrations varied from 0 to 80 μM, as indicated. Binding curves for the NA forms are shown below the gels, with each data point averaged from multiple experiments (see Materials and Methods). The error bars indicate mean errors. (D) Binding competition of unlabeled dsDNAs of the same length but different sequences. Gel bands of the unbound 20-mer probe and bound species are indicated, and the probe and protein concentrations are given at the top.
The observation that K1L1, but not K1, binds DNA is consistent with the fact that the PCP is composed of basic residues from both the K1 and L1 lobes; K1L1 functions as one structural unit in this regard. In titration experiments (see Fig. S4 in the supplemental material), we estimated the NA-binding affinity of K1L1 to be ∼25 μM for ssDNA and RNA-DNA, ∼15 μM for dsDNA, and ∼12 μM for ssRNA. Consistent with the measured dissociation constants, K1L1 showed a preference for dsDNA over ssDNA in a direct-competition assay, while dsDNA was slightly outcompeted by ssRNA (Fig. 6C). An unlabeled dsDNA of the same length but with a sequence different than that of the fluorescently labeled probe was able to compete for binding (Fig. 6D), indicating sequence-independent interactions. The calculated dissociation constants resemble those previously observed for NA-interacting Tudor domains (see Table S2 in the supplemental material), whose weak affinities for NAs have been thought of as a fitting feature for their functions. Like Spt4-Spt5, these proteins normally are confined within larger molecular assemblies (63), such as the RNAP elongation complex, in which a binding substrate is often presented with spatial confinement to make the interaction tunable. High-strength NA affinities (e.g., in nanomolar ranges) are not compatible with dynamic on-and-off binding events that occur during the rapid process of translocation and polymerization by RNAP.
To identify residues responsible for the in vitro NA-binding activity of K1L1, we mutated the basic residues that line the PCP (Fig. 7A and B, blue) and found that single or double amino acid mutations resulted in a loss of dsDNA binding in vitro (Fig. 7C). In contrast, mutations of residues near the border of the PCP or a nonbasic residue did not significantly affect the binding (Fig. 7A to C, orange). This experiment also confirms that the PCP involves both the K1 and L1 moieties, including three basic residues (R435, K436, and R438) from the flexible loop I428 to R438 (Fig. 7A). As expected from this result, cassette mutations (Q21 to Q25) made from pairwise combination of the loss-of-binding PCP mutations caused complete loss of binding (Fig. 7B and D). The fact that all mutant K1L1 proteins could be expressed and purified at levels equivalent to those of the wild type indicated that these mutations did not induce adverse conformational alterations. Hence, the basic residues of the K1L1 PCP are required for the in vitro NA-binding activities.
FIG 7.

The in vitro NA-binding activity of K1L1 is mediated by its PCP. (A) (Left) The solvent-accessible surface of K1L1 is colored blue (+6 kT) to red (−6 kT) to show the electrostatic potential. (Right) The basic residues forming the PCP in K1L1 are shown in blue, and residues that are not in the center of the PCP (T441, F442, and R458) or are neutral (H492) are shown in orange. The disordered region is represented by dashes, with its sequence shown on the left. (B) Identification of the PCP and cassette mutations. (C and D) Gel shift assay showing losses of NA binding due to mutations of basic residues in the PCP. Each binding reaction mixture consisted of 5 μM probe, 22 μM a K1L1 protein variant, and the buffer. Free and bound NA species are indicated on the left, and K1L1 proteins are identified at the top with their mutation identifiers given at the bottom. The smeared appearance of bound complex bands is explained in the legend to Fig. 5.
The PCP of K1L1 is important for Spt5 function in vivo.
We next asked if the PCP is important for in vivo functions of Spt4-Spt5. A LEU2-marked CEN plasmid (pRS415) was used to express WT or mutant alleles of N-terminally 3Myc-tagged Spt5 as the sole source of Spt5 in yeast via plasmid shuffling (see Materials and Methods); each spt5 allele corresponded to a mutation from the in vitro NA-binding experiment (spt5-PCP and cassette mutants). When shuffled into an spt5Δ SPT4 strain (GHY1066), all the spt5-PCP alleles produced viable and robust colonies (Fig. 8A). This result and the fact that SPT5 is essential for yeast indicated that the mutations did not induce adverse misfolding of Spt5 in vivo. However, when introduced into an spt5Δ spt4Δ background (GHY1172), the plasmid-borne cassette mutations did not complement the spt5Δ mutation (Fig. 8B). As reasoned above, the lethality associated with Q21 to Q25 was not due to possible Spt5 protein misfolding in the cells, which was supported by circular dichroism (CD) spectra of purified cassette mutant proteins, which showed no significant conformational changes (Fig. 8C). These results indicate that the PCP of K1L1 mediates important physiological functions in the cell.
FIG 8.
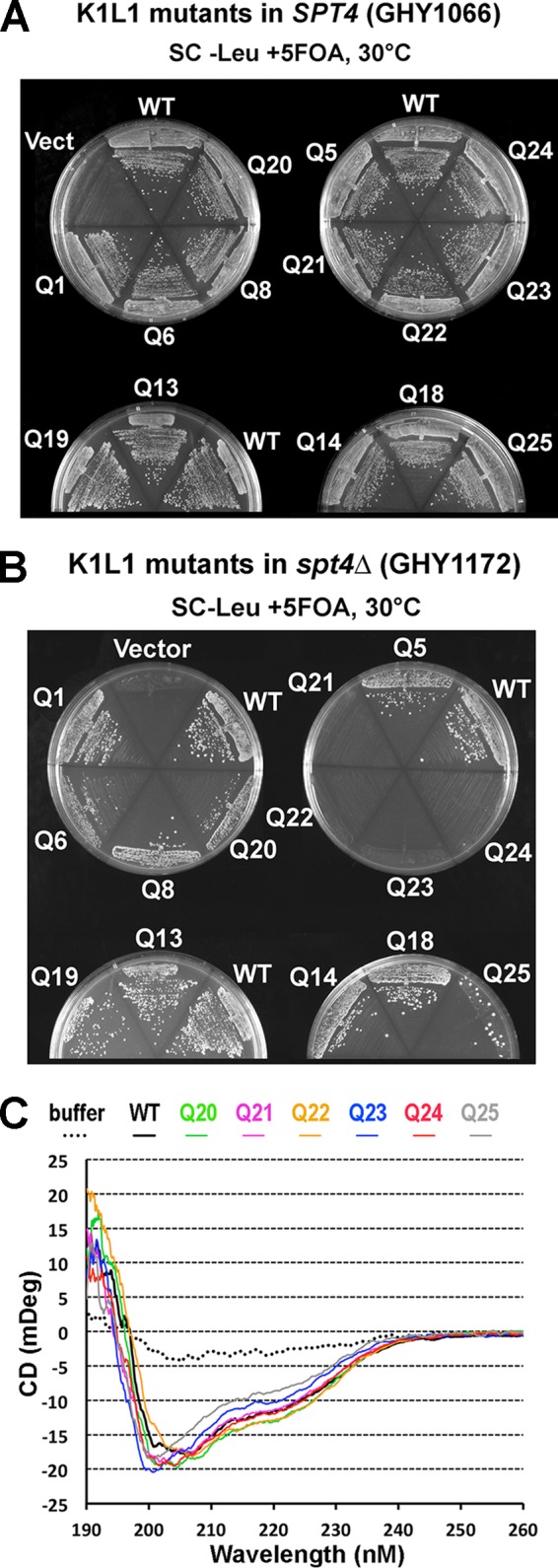
The PCP of Spt5 is important for in vivo function. (A) Growth of yeast strains following plasmid shuffle in the SPT4 background. (B) Growth in the spt4Δ background. Note that Q25 promoted the emergence of rare suppressor-like colonies. (C) Far-UV CD spectra taken from purified mutant K1L1 proteins of the WT and Q20 through Q25 (with the color code indicated).
The K1L1 PCP has a function that overlaps that of Spt4.
The spt5-PCP mutants that appeared nonlethal in the shuffle experiment (5-FOA-resistant colonies) were isolated for further analysis using a spot assay. The spt4Δ spt5-PCP mutant strains harboring mutations that maintained NA binding—Q14, Q18, Q19, and Q13—supported WT-like growth (Fig. 9A, spt4Δ, left). The spt5-PCP mutations that abolished NA binding—Q1, Q6, Q8, Q20, and Q5—conferred a slow-growth phenotype (at 30°C) (data not shown), which was exacerbated at the elevated temperature (37°C) (temperature sensitive [Ts]) and also displayed hyperosmotic-sensitive (1.0 M KCl) (Osm) growth defects (Fig. 9A, top left). However, in the SPT4 background, the spt5-PCP mutations, including the cassette mutations (Q21 to Q25) that were lethal in spt4Δ, showed no observable defects at 37°C or at high salt levels (Fig. 9A, right). Therefore, all the observed phenotypes—lethal, Ts, and Osm—associated with the spt5-PCP mutants are dependent on spt4Δ. We also assayed the SPT4 spt5-PCP and spt4Δ spt5-PCP strains for the Spt− phenotype (SC−Lys) and in the presence of mycophenolic acid (10 to 70 μg/ml), a condition commonly used for revealing defects associated with mutations in transcription elongation factors, but observed no additional defects (data not shown). Consistent with this absence of a phenotype, we did not observe any new growth defects when the spt5-PCP mutants were expressed in cells lacked the transcription elongation factor TFIIS (see Fig. S5 in the supplemental material). We observed the same phenotypes in growth assays using a different shuffling plasmid (pGH233) carrying untagged spt5 alleles (data not shown).
FIG 9.
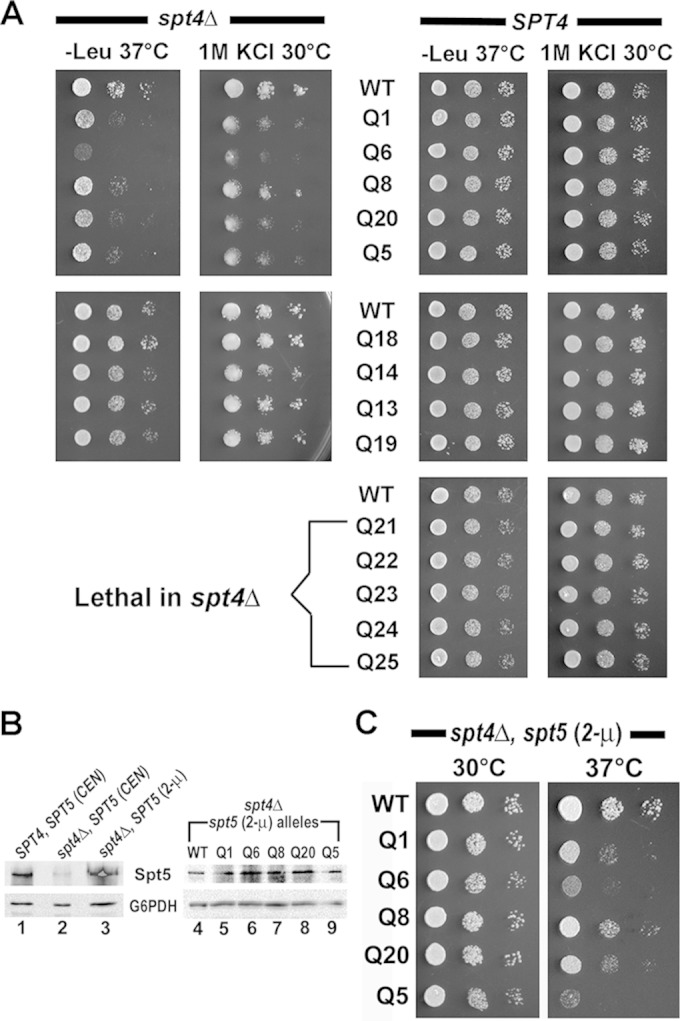
Functional overlap between Spt4 and the K1L1 domain of Spt5. (A) PCP mutations that cause losses of NA binding in vitro display Ts and Osm growth defects in the spt4Δ strain (left) but no defects in the SPT4 strain (right). Tenfold serial dilutions were spotted on SC−Leu plates and SC plates with 1.0 M KCl and incubated at the indicated temperatures. (B) Western blot showing cellular levels of Spt5 variants, with the anti-G6PDH blot serving as the loading control. Cell strains, the type of spt5-carrying plasmid (CEN or 2μ), and spt5 alleles are indicated at the top. (C) Overexpression of Spt5 does not affect the Ts defects associated with the PCP mutants. Growth of spt4Δ spt5-PCP (2μ) strains at 30° and 37°C on a solid medium (SC−Ura plus raffinose plus galactose) that is inductive for the spt5 alleles driven by the GAL10 promoter (see Materials and Methods).
The fact that the spt5-PCP mutations cause growth defects only when Spt4 is absent suggests that the PCP of K1L1 mediates important functions that overlap those of Spt4. To assess the possibility that the growth defects of spt4Δ spt5-PCP strains are due to reduction of the Spt5 protein level associated with SPT4 deletion (10), we overexpressed the Spt5 variants by swapping the pRS415 CEN plasmid with a multicopy (2μ) plasmid carrying the spt5 alleles under the control of the strong GAL10 promoter (see Materials and Methods). Consistent with the results of Ding et al. (10), the level of WT Spt5 was reduced in spt4Δ cells compared with that in SPT4 cells (Fig. 9B, cf. lanes 1 and 2), and so were levels of the PCP mutants (data not shown). The spt5 level dramatically increased in spt4Δ SPT5 (2μ) cells (Fig. 9B, lane 3) to several times that in the SPT4 cells. Although the levels of mutant Spt5 were comparable to that of WT Spt5 in the spt4Δ spt5 (2μ) strains (Fig. 9B, lanes 4 to 9), the PCP mutant alleles displayed Ts growth defects (Fig. 9C) similar to those observed in the spt4Δ spt5-PCP (CEN) strains. Therefore, the defects associated with spt5-PCP mutations are not due to alterations in the Spt5 protein level. Consistent with this conclusion, no growth defects were seen in the SPT4 spt5-PCP strains cultured on plates containing sublethal concentrations of cycloheximide (CHX) (0.1 to 1.0 μg/ml), which inhibits global protein synthesis (data not shown).
Based on the altered NA-binding activities we observed (see above) for the Spt5-PCP proteins, we suggest that the function(s) shared between Spt4 and the Spt5 PCP includes an activity that is directed toward NA moieties in the elongation complex. A direct NA-binding assay for Spt4 was precluded by the insolubility of the Spt4 protein when expressed alone. Nonetheless, this NA interaction model is consistent with an extensive positively charged area on the Spt4-NGN surface (Fig. 10C) and the recently observed NA-binding activity in an Spt4-NGN complex (A. Vrielink, personal communication).
FIG 10.

Functional overlap between Spt4 and the K1L1 domain of Spt5 suggests iterated NA interactions located upstream of the transcription bubble. (A) The spt4-IM mutation (S58D_V60E) disrupts the recruitment of Spt4 to Spt5 in the cell. WCLs were made from the spt4Δ SPT5 strain transformed with pRS416-Flag-SPT4 or pRS416-Flag-spt4-IM. Protein levels (Input) were detected using Western blotting against the Myc tag on Spt5 and the Flag tag on Spt4, and anti-G6PDH served as a loading control. Spt5 was immunoprecipitated (IP) via the Myc tag borne by the pRS415 3Myc-SPT5 plasmid. (B) Masking of the spt5-PCP Ts phenotype by SPT4 requires an intact interface between Spt4 and the NGN domain of Spt5. The spt4Δ spt5-PCP mutant strains were transformed with pRS416 (URA3) alone (left), pRS416-Flag-SPT4 (middle), or pRS416-Flag-spt4-IM (right) and assayed for growth at 37°C on SC−Leu−Ura plates. (C) Mechanistic model showing interactions with the NAs (including DNA) upstream of the transcription bubble that are proposed to be shared between Spt4 and the K1L1 domain of Spt5. Electrostatic surfaces of the yeast K1L1 domain and the Spt4-NGN complex are shown relative to the NAs engaged in RNAP II. Blue, positively charged; red, negatively charged; black line, template DNA strand; thin gray line, nontemplate strand; thick gray line, RNA transcript. The arrows indicate the approximate locations of the proposed protein-DNA interactions. (D) Hypothetical three-dimensional model for the yeast holo-elongation complex, viewed from the back of panel C. The model highlights a potential location of the K1L1 domain relative to the RNAP (silver), the NGN domain, Spt4, and the upstream DNA (blue and red). A connector of 6 amino acids between NGN and K1L1 is indicated by the white dashes. The model was built on the basis of the semicrystallographic model of archaeal RNAP (26) and also incorporated the K1L1 placement suggested from the EM structure (34). Note that the location of NGN is the same as in the schematic model of Li et al. (11), and the K1L1 location is in a general agreement with that proposed in Li's model. CC, coiled-coil structural element of the Clamp module.
We next asked if the overlapping functions between Spt4 and the Spt5 PCP occur in the context of the Spt4-Spt5 complex. To test this, we constructed pRS416 (URA3 CEN)-derivative plasmids carrying Flag-SPT4 and a Flag-spt4 mutant (S58D_V60E; spt4-IM) predicted to disrupt the Spt4-Spt5 interface in the structure (64), and transformed spt4Δ spt5-PCP mutant strains with these plasmids. The level of WT Spt5 in the spt4-IM strain was reduced to a level similar to that observed in the spt4Δ strain, and the level of Spt4-IM was similar to that of WT Spt4 (Fig. 10A, Input). Immunoprecipitation, which captured similar amounts of Spt5 from whole-cell lysates of both strains, showed a dramatic loss of Spt4 binding to Spt5 in spt4-IM (Fig. 10A, IP). Growth assays showed that the plasmid-borne SPT4 complemented the Ts phenotype of spt4Δ spt5-PCP mutants, whereas neither the empty plasmid nor the spt4-IM mutant was able to overcome the defects (Fig. 10B). Therefore, the functional redundancy between Spt4 and the Spt5 PCP depends on Spt4 binding to Spt5.
In comparing the phenotypes of the related alleles, we noted that the spt4Δ spt5-PCP mutants and spt4Δ SPT5 (65) are both Ts, with the double mutants displaying enhanced growth defects. In contrast, the SPT4 spt5-PCP single mutants are WT-like. Thus, Spt5-PCP and Spt4 are unlikely to make distinct and sequential contributions to a common function. Rather, Spt4 may likely play a major role, with Spt5-PCP playing a minor role in a shared function (66). Altogether, our results indicate that Spt5 PCP has a function that partially overlaps that of Spt4, and this function shared with Spt4 likely involves interactions with NAs in the RNAP elongation complex.
Spt4 and the K1L1 of Spt5 are positioned to interact with NA upstream of the transcription bubble.
Is this shared NA interaction directed at the downstream DNA, transcription bubble, upstream NAs, or separated transcript emerging from the RNA pore (67)? Using UV cross-linking, Missra and Gilmour detected interaction of Drosophila Spt5 with RNA transcripts as short as 22 nt in an RNAP ternary complex (68), although they did not resolve the binding among specific Spt5 domains. Similarly, without resolving specific roles for the domains, Cheng and Price showed that human Spt4-Spt5 (also known as DSIF) interacted with the ternary complex and that longer transcripts facilitated the binding (69), while Viktorovskaya and coworkers observed direct RNA binding by the yeast heterodimer (17). It remains to be determined if RNA binding by Spt4-Spt5 is relevant to its in vivo functions and, if so, if RNA binding facilitates recruitment of Spt4-Spt5 or other functions. Structural and mutagenesis studies suggest, however, that in addition to RNA, Spt4-Spt5 may also interact with other NA moieties in the elongation complex. The electron microscopy (EM) structure of the archaeal Spt4-Spt5-RNAP complex (34) and modeling based on the crystal structure of an archaeal Spt4-NGN-Clamp module (26) showed that the Spt4-NGN subcomplex is located at the Clamp module, far away from the downstream DNA binding cleft, with the NGN poised near the transcription bubble (Fig. 10C). As such, it is unlikely that the in vitro RNA binding activity of Spt4-Spt5 can be explained solely by binding to the exiting transcript as it emerges from the RNA pore on RNAP (67).
Consistent with a model for their interactions with the transcription bubble, the NGN domains of bacterial NusG and its paralog RfaH contact the nontemplate DNA strand within the bubble (70–73). Moreover, it is obvious from the EM structure that both Spt4 and the archaeal KOW (organizationally equivalent to Spt5 K1) are distal to the downstream DNA or the bubble. Therefore, it can be suggested that the NA interaction that we have characterized as being shared by K1L1 and Spt4 reflects interactions with NAs upstream of the bubble. Consistent with this notion, K1L1 is expected to take up a flanking position near Spt4 in the elongation complex, since it is tethered to NGN by a 6-amino-acid linker (34) (see Fig. S1 in the supplemental material), seemingly allowing it to interact with NAs in the same region. A model synthesizing the results from the present and prior studies is presented (Fig. 10C and D), in which we hypothesize that Spt4 is recruited to the RNAP elongation complex via its binding to the NGN domain of Spt5 and thus is positioned to interact with the NAs immediately upstream of the transcription bubble, while the nearby K1L1 domain of Spt5 supplies a similar and compensatory NA interaction.
DISCUSSION
Despite well-supported genetic and functional studies that implicate Spt4-Spt5 in transcription-coupled processes ranging from chromatin modification and RNA processing (5) to 3′-end formation (74–76), not much is known about how Spt4-Spt5 interacts with the RNAP ternary complex to function in these nuclear processes. Mechanistic understanding is limited to an NA encirclement model based on the binding of the Spt5 NGN domain to the RNAP Clamp module (26, 27, 34) and a general understanding of the Spt5 KOW domains' involvement in binding RNAP (11, 17, 59). How biochemical and structural properties may differ across the individual KOW domains is not known, nor is it known how the domains constitute different facets of Spt4-Spt5 function. The present study addresses some of these questions at levels of high-resolution structure, biochemical assays for NA binding, and in vivo function.
First, the results of the protein expression experiment indicate that the central region of Spt5 is composed of four independent folding units, K1L1, K2K3, L2K4, and L3K5. The delineation of these domains, together with the dimeric Spt4-NGN domain and unfolded acidic and CTR domains, completes a framework for assigning specific functions of Spt5 in future analyses. The high-resolution structures demonstrate that the first two folding units, K1L1 and K2K3, assume very different tertiary structures. Based on this result and different consensus sequences of the individual KOW domains (see Fig. S2B in the supplemental material), we propose that L2K4 and L3K5 also assume unique structures and likely mediate distinct activities in the elongation complex. Our results redefine subdomain structures within K1L1, showing that the L1 region folds into a novel structure inserted N-terminally to the last β-strand of the K1 Tudor barrel. Moreover, the results demonstrate that the K2K3 domains adopt a twined Tudor fold that likely acts as a rigid structure. As such, a picture of the holo-elongation complex (RNAP II-Spt4/5) begins to emerge in which structurally distinct Spt5 domains distribute to different sites on the RNAP ternary complex. We hypothesize that these binding sites may directly or indirectly link RNA synthesis with cotranscriptional processes. Such a mechanism may be at work in the binding of the Spt4-NGN structure to the RNAP Clamp module, an event that could transmit a conformational effect to the RNAP catalytic center near the base of the Clamp. Consistent with this idea, RNAP II mutations in and near the catalytic center that suppress certain spt5 mutations have been isolated (G. Hartzog, unpublished results).
The newly discovered NA-binding surface on the K1L1 structure suggests an attractive role for the domain in interacting with upstream NAs. The NA-binding surface was identified from (i) the ultra-high-resolution structure of K1L1, where residues from both the K1 and L1 subdomains form a highly positively charged surface patch; (ii) an in vitro gel shift assay combined with site-directed mutagenesis targeting the PCP residues, which demonstrated non-sequence-specific NA binding with a weak preference for dsDNA and ssRNA; and (iii) growth defects of yeast strains carrying spt5-PCP mutations, which demonstrated that the PCP is important for in vivo functions even though it may not impact elongation per se. Interestingly, the phenotypes associated with PCP mutations are observed only in strains devoid of Spt4. These data suggest a functional redundancy between Spt4 and the K1L1 domain of Spt5 and the possibility that both are capable of interacting with NAs in the RNAP ternary complex. Interpretation of the phenotypes of spt4Δ SPT5, spt4Δ spt5-PCP, and SPT4 spt5-PCP strains further suggests that Spt4 and Spt5 K1L1 play nonequivalent roles, possibly with Spt4 as the main factor in this shared function and K1L1 providing a compensatory interaction.
The relatively weak affinities of K1L1 for various forms of NA (12 to 15 μM) seem comparable to those of other NA-interacting Tudor domains and fit with its roles within the context of the remaining Spt5 domains and the RNAP elongation complex. A precedent for the hypothesis that Spt4-Spt5 interacts with NAs in elongation complexes is found in the NusG paralog RfaH, whose NGN domain is recruited to elongation complexes via its recognition of the ops sequence element (72). For rapid translocation during RNA chain synthesis, the RNAP of an elongation complex must make and break numerous contacts with the encompassed NA strands at very high frequencies, and similarly, the NGN and K1L1 domains may undergo dynamic interactions with the transcription bubble and NAs upstream of the bubble, respectively. Moreover, interactions of K1L1 and Spt4-NGN with upstream NAs could in themselves be an important component of the mechanism that facilitates reannealing of the DNA strands and limits the upstream edge of the bubble; overextension of transcriptional RNA-DNA hybrids is a known defect that impairs elongation- and transcription-associated processes (77, 78). We have not ruled out the possibility that the PCP has a non-NA interaction partner that participates in upstream DNA-dependent processes. It should be noted that TFIIS is not a candidate partner, since it binds the polymerase distal to the upstream DNA site and shows no functional interaction with the PCP mutations in the growth assay.
Taken together, our results suggest mechanistic aspects of the holo-elongation complex: (i) Spt4 is recruited to the RNAP elongation complex via its binding to the NGN domain of Spt5 and possibly contacts the upstream NAs; (ii) the K1L1 domain of Spt5 is capable of dynamic NA interactions in the same or a nearby region; and (iii) Spt4 performs an essential function in the cell possibly mediated by its interaction with the upstream NAs, and in spt4Δ cells, this function is rescued by Spt5. The last property possibly reflects the existence in the holo-elongation complex of a buffering mechanism against internal and/or external alterations, e.g., a loss of SPT4. We present the main features of this mechanism in a speculative model of the holo-elongation complex (Fig. 10C and D). A premise of this model is that interactions with the upstream NAs within the RNAP ternary complex is an essential function in vivo and that the loss-of-binding spt5-PCP mutations in spt4Δ cells would produce ill-formed upstream DNAs—most likely during the DNA-reannealing process—that are improperly constrained and/or topologically defective. It is conceivable that defects like these may result in transcriptional failures due to accumulation of topological DNA entanglement, misalignment of upstream DNA in the ternary complex, or failed upstream DNA-dependent recruitment of factors that are necessary for the completion of elongation and termination in vivo. It appears to us that the RNAP holo-elongation complex is endowed with redundant or compensatory functions distributed among its substructures, e.g., between Spt4 and the K1L1 domain of Spt5, which may be important to the events that occur in the wake of RNAP elongation.
Supplementary Material
ACKNOWLEDGMENTS
We thank Shisheng Li for advice on molecular cloning and yeast methods, Joseph Reese and David Gilmour for comments and suggestions, and Samantha Kohn and Tevin Hughley for assistance.
This research was supported by Public Health Service grant GM100997 (J.F. and G.A.H.) from the National Institutes of Health and by National Science Foundation grant MCB-1157688 (Y.T.). S.L. was supported in part by research funds from ShanghaiTech University, Shanghai, China.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00520-15.
REFERENCES
- 1.Thomas MC, Chiang CM. 2006. The general transcription machinery and general cofactors. Crit Rev Biochem Mol Biol 41:105–178. doi: 10.1080/10409230600648736. [DOI] [PubMed] [Google Scholar]
- 2.Perales R, Bentley D. 2009. “Cotranscriptionality”: the transcription elongation complex as a nexus for nuclear transactions. Mol Cell 36:178–191. doi: 10.1016/j.molcel.2009.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hsin JP, Manley JL. 2012. The RNA polymerase II CTD coordinates transcription and RNA processing. Genes Dev 26:2119–2137. doi: 10.1101/gad.200303.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rondon AG, Jimeno S, Aguilera A. 2010. The interface between transcription and mRNP export: from THO to THSC/TREX-2. Biochim Biophys Acta 1799:533–538. doi: 10.1016/j.bbagrm.2010.06.002. [DOI] [PubMed] [Google Scholar]
- 5.Hartzog GA, Fu J. 2013. The Spt4-Spt5 complex: a multi-faceted regulator of transcription elongation. Biochim Biophys Acta 1829:105–115. doi: 10.1016/j.bbagrm.2012.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou Q, Li T, Price DH. 2012. RNA polymerase II elongation control. Annu Rev Biochem 81:119–143. doi: 10.1146/annurev-biochem-052610-095910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yamaguchi Y, Shibata H, Handa H. 2013. Transcription elongation factors DSIF and NELF: promoter-proximal pausing and beyond. Biochim Biophys Acta 1829:98–104. doi: 10.1016/j.bbagrm.2012.11.007. [DOI] [PubMed] [Google Scholar]
- 8.Conaway RC, Conaway JW. 2013. The Mediator complex and transcription elongation. Biochim Biophys Acta 1829:69–75. doi: 10.1016/j.bbagrm.2012.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jansen LE, den Dulk H, Brouns RM, de Ruijter M, Brandsma JA, Brouwer J. 2000. Spt4 modulates Rad26 requirement in transcription-coupled nucleotide excision repair. EMBO J 19:6498–6507. doi: 10.1093/emboj/19.23.6498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ding B, LeJeune D, Li S. 2010. The C-terminal repeat domain of Spt5 plays an important role in suppression of Rad26-independent transcription coupled repair. J Biol Chem 285:5317–5326. doi: 10.1074/jbc.M109.082818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li W, Giles C, Li S. 2014. Insights into how Spt5 functions in transcription elongation and repressing transcription coupled DNA repair. Nucleic Acids Res 42:7069–7083. doi: 10.1093/nar/gku333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pavri R, Gazumyan A, Jankovic M, Di Virgilio M, Klein I, Ansarah-Sobrinho C, Resch W, Yamane A, Reina San-Martin B, Barreto V, Nieland TJ, Root DE, Casellas R, Nussenzweig MC. 2010. Activation-induced cytidine deaminase targets DNA at sites of RNA polymerase II stalling by interaction with Spt5. Cell 143:122–133. doi: 10.1016/j.cell.2010.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lindstrom DL, Squazzo SL, Muster N, Burckin TA, Wachter KC, Emigh CA, McCleery JA, Yates JR III, Hartzog GA. 2003. Dual roles for Spt5 in pre-mRNA processing and transcription elongation revealed by identification of Spt5-associated proteins. Mol Cell Biol 23:1368–1378. doi: 10.1128/MCB.23.4.1368-1378.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tardiff DF, Abruzzi KC, Rosbash M. 2007. Protein characterization of Saccharomyces cerevisiae RNA polymerase II after in vivo cross-linking. Proc Natl Acad Sci U S A 104:19948–19953. doi: 10.1073/pnas.0710179104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yamaguchi Y, Wada T, Watanabe D, Takagi T, Hasegawa J, Handa H. 1999. Structure and function of the human transcription elongation factor DSIF. J Biol Chem 274:8085–8092. doi: 10.1074/jbc.274.12.8085. [DOI] [PubMed] [Google Scholar]
- 16.Zhang Z, Wu CH, Gilmour DS. 2004. Analysis of polymerase II elongation complexes by native gel electrophoresis: evidence for a novel carboxyl-terminal domain-mediated termination mechanism. J Biol Chem 279:23223–23228. doi: 10.1074/jbc.M402956200. [DOI] [PubMed] [Google Scholar]
- 17.Viktorovskaya OV, Appling FD, Schneider DA. 2011. Yeast transcription elongation factor Spt5 associates with RNA polymerase I and RNA polymerase II directly. J Biol Chem 286:18825–18833. doi: 10.1074/jbc.M110.202119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ping YH, Rana TM. 2001. DSIF and NELF interact with RNA polymerase II elongation complex and HIV-1 Tat stimulates P-TEFb-mediated phosphorylation of RNA polymerase II and DSIF during transcription elongation. J Biol Chem 276:12951–12958. doi: 10.1074/jbc.M006130200. [DOI] [PubMed] [Google Scholar]
- 19.Bourgeois CF, Kim YK, Churcher MJ, West MJ, Karn J. 2002. Spt5 cooperates with human immunodeficiency virus type 1 Tat by preventing premature RNA release at terminator sequences. Mol Cell Biol 22:1079–1093. doi: 10.1128/MCB.22.4.1079-1093.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu Y, Warfield L, Zhang C, Luo J, Allen J, Lang WH, Ranish J, Shokat KM, Hahn S. 2009. Phosphorylation of the transcription elongation factor Spt5 by yeast Bur1 kinase stimulates recruitment of the PAF complex. Mol Cell Biol 29:4852–4863. doi: 10.1128/MCB.00609-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Swanson MS, Malone EA, Winston F. 1991. SPT5, an essential gene important for normal transcription in Saccharomyces cerevisiae, encodes an acidic nuclear protein with a carboxy-terminal repeat. Mol Cell Biol 11:3009–3019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pei Y, Shuman S. 2003. Characterization of the Schizosaccharomyces pombe Cdk9/Pch1 protein kinase: Spt5 phosphorylation, autophosphorylation, and mutational analysis. J Biol Chem 278:43346–43356. doi: 10.1074/jbc.M307319200. [DOI] [PubMed] [Google Scholar]
- 23.Yamada T, Yamaguchi Y, Inukai N, Okamoto S, Mura T, Handa H. 2006. P-TEFb-mediated phosphorylation of hSpt5 C-terminal repeats is critical for processive transcription elongation. Mol Cell 21:227–237. doi: 10.1016/j.molcel.2005.11.024. [DOI] [PubMed] [Google Scholar]
- 24.Werner F. 2012. A nexus for gene expression-molecular mechanisms of Spt5 and NusG in the three domains of life. J Mol Biol 417:13–27. doi: 10.1016/j.jmb.2012.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hirtreiter A, Damsma GE, Cheung AC, Klose D, Grohmann D, Vojnic E, Martin AC, Cramer P, Werner F. 2010. Spt4/5 stimulates transcription elongation through the RNA polymerase clamp coiled-coil motif. Nucleic Acids Res 38:4040–4051. doi: 10.1093/nar/gkq135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martinez-Rucobo FW, Sainsbury S, Cheung AC, Cramer P. 2011. Architecture of the RNA polymerase-Spt4/5 complex and basis of universal transcription processivity. EMBO J 30:1302–1310. doi: 10.1038/emboj.2011.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hartzog GA, Kaplan CD. 2011. Competing for the clamp: promoting RNA polymerase processivity and managing the transition from initiation to elongation. Mol Cell 43:161–163. doi: 10.1016/j.molcel.2011.07.002. [DOI] [PubMed] [Google Scholar]
- 28.Belogurov GA, Vassylyeva MN, Svetlov V, Klyuyev S, Grishin NV, Vassylyev DG, Artsimovitch I. 2007. Structural basis for converting a general transcription factor into an operon-specific virulence regulator. Mol Cell 26:117–129. doi: 10.1016/j.molcel.2007.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Burns CM, Richardson LV, Richardson JP. 1998. Combinatorial effects of NusA and NusG on transcription elongation and Rho-dependent termination in Escherichia coli. J Mol Biol 278:307–316. doi: 10.1006/jmbi.1998.1691. [DOI] [PubMed] [Google Scholar]
- 30.Mooney RA, Schweimer K, Rosch P, Gottesman M, Landick R. 2009. Two structurally independent domains of E. coli NusG create regulatory plasticity via distinct interactions with RNA polymerase and regulators. J Mol Biol 391:341–358. doi: 10.1016/j.jmb.2009.05.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kyrpides NC, Woese CR, Ouzounis CA. 1996. KOW: a novel motif linking a bacterial transcription factor with ribosomal proteins. Trends Biochem Sci 21:425–426. doi: 10.1016/S0968-0004(96)30036-4. [DOI] [PubMed] [Google Scholar]
- 32.Steiner T, Kaiser JT, Marinkovic S, Huber R, Wahl MC. 2002. Crystal structures of transcription factor NusG in light of its nucleic acid- and protein-binding activities. EMBO J 21:4641–4653. doi: 10.1093/emboj/cdf455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reay P, Yamasaki K, Terada T, Kuramitsu S, Shirouzu M, Yokoyama S. 2004. Structural and sequence comparisons arising from the solution structure of the transcription elongation factor NusG from Thermus thermophilus. Proteins 56:40–51. doi: 10.1002/prot.20054. [DOI] [PubMed] [Google Scholar]
- 34.Klein BJ, Bose D, Baker KJ, Yusoff ZM, Zhang X, Murakami KS. 2011. RNA polymerase and transcription elongation factor Spt4/5 complex structure. Proc Natl Acad Sci U S A 108:546–550. doi: 10.1073/pnas.1013828108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lasko P. 2010. Tudor domain. Curr Biol 20:R666–R667. doi: 10.1016/j.cub.2010.05.056. [DOI] [PubMed] [Google Scholar]
- 36.Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. 1997. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thompson JD, Higgins DG, Gibson TJ. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Galtier N, Gouy M, Gautier C. 1996. SEAVIEW and PHYLO_WIN: two graphic tools for sequence alignment and molecular phylogeny. Comput Appl Biosci 12:543–548. [DOI] [PubMed] [Google Scholar]
- 39.Bond CS, Schuttelkopf AW. 2009. ALINE: a WYSIWYG protein-sequence alignment editor for publication-quality alignments. Acta Crystallogr D Biol Crystallogr 65:510–512. doi: 10.1107/S0907444909007835. [DOI] [PubMed] [Google Scholar]
- 40.Linding R, Russell RB, Neduva V, Gibson TJ. 2003. GlobPlot: exploring protein sequences for globularity and disorder. Nucleic Acids Res 31:3701–3708. doi: 10.1093/nar/gkg519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Linding R, Jensen LJ, Diella F, Bork P, Gibson TJ, Russell RB. 2003. Protein disorder prediction: implications for structural proteomics. Structure 11:1453–1459. doi: 10.1016/j.str.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 42.Crooks GE, Hon G, Chandonia JM, Brenner SE. 2004. WebLogo: a sequence logo generator. Genome Res 14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cole C, Barber JD, Barton GJ. 2008. The Jpred 3 secondary structure prediction server. Nucleic Acids Res 36:W197–W201. doi: 10.1093/nar/gkn238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Leaver-Fay A, Tyka M, Lewis SM, Lange OF, Thompson J, Jacak R, Kaufman K, Renfrew PD, Smith CA, Sheffler W, Davis IW, Cooper S, Treuille A, Mandell DJ, Richter F, Ban YE, Fleishman SJ, Corn JE, Kim DE, Lyskov S, Berrondo M, Mentzer S, Popovic Z, Havranek JJ, Karanicolas J, Das R, Meiler J, Kortemme T, Gray JJ, Kuhlman B, Baker D, Bradley P. 2011. ROSETTA3: an object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol 487:545–574. doi: 10.1016/B978-0-12-381270-4.00019-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kabsch W. 2010. Xds. Acta Crystallogr D Biol Crystallogr 66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Collaborative Computational Project Number 4. 1994. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D 50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 47.Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 48.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB III, Snoeyink J, Richardson JS, Richardson DC. 2007. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res 35:W375–W383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. 2000. The Protein Data Bank. Nucleic Acids Res 28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dolinsky TJ, Czodrowski P, Li H, Nielsen JE, Jensen JH, Klebe G, Baker NA. 2007. PDB2PQR: expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res 35:W522–W525. doi: 10.1093/nar/gkm276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FT, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiorkiewicz-Kuczera J, Yin D, Karplus M. 1998. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B 102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 53.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. 2001. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci U S A 98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Honig B, Nicholls A. 1995. Classical electrostatics in biology and chemistry. Science 268:1144–1149. doi: 10.1126/science.7761829. [DOI] [PubMed] [Google Scholar]
- 55.Bond CS. 2003. TopDraw: a sketchpad for protein structure topology cartoons. Bioinformatics 19:311–312. doi: 10.1093/bioinformatics/19.2.311. [DOI] [PubMed] [Google Scholar]
- 56.Rose MD, Winston F, Hieter P. 1990. Methods in yeast genetics: a laboratory course manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 57.Li MZ, Elledge SJ. 2007. Harnessing homologous recombination in vitro to generate recombinant DNA via SLIC. Nat Methods 4:251–256. doi: 10.1038/nmeth1010. [DOI] [PubMed] [Google Scholar]
- 58.Funakoshi M, Hochstrasser M. 2009. Small epitope-linker modules for PCR-based C-terminal tagging in Saccharomyces cerevisiae. Yeast 26:185–192. doi: 10.1002/yea.1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ivanov D, Kwak YT, Guo J, Gaynor RB. 2000. Domains in the SPT5 protein that modulate its transcriptional regulatory properties. Mol Cell Biol 20:2970–2983. doi: 10.1128/MCB.20.9.2970-2983.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Iwabuchi K, Basu BP, Kysela B, Kurihara T, Shibata M, Guan D, Cao Y, Hamada T, Imamura K, Jeggo PA, Date T, Doherty AJ. 2003. Potential role for 53BP1 in DNA end-joining repair through direct interaction with DNA. J Biol Chem 278:36487–36495. doi: 10.1074/jbc.M304066200. [DOI] [PubMed] [Google Scholar]
- 61.Charier G, Couprie J, Alpha-Bazin B, Meyer V, Quemeneur E, Guerois R, Callebaut I, Gilquin B, Zinn-Justin S. 2004. The Tudor tandem of 53BP1: a new structural motif involved in DNA and RG-rich peptide binding. Structure 12:1551–1562. doi: 10.1016/j.str.2004.06.014. [DOI] [PubMed] [Google Scholar]
- 62.le Maire A, Schiltz M, Stura EA, Pinon-Lataillade G, Couprie J, Moutiez M, Gondry M, Angulo JF, Zinn-Justin S. 2006. A tandem of SH3-like domains participates in RNA binding in KIN17, a human protein activated in response to genotoxics. J Mol Biol 364:764–776. doi: 10.1016/j.jmb.2006.09.033. [DOI] [PubMed] [Google Scholar]
- 63.Gong W, Wang J, Perrett S, Feng Y. 2014. Retinoblastoma-binding protein 1 has an interdigitated double Tudor domain with DNA binding activity. J Biol Chem 289:4882–4895. doi: 10.1074/jbc.M113.501940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Guo M, Xu F, Yamada J, Egelhofer T, Gao Y, Hartzog GA, Teng M, Niu L. 2008. Core structure of the yeast spt4-spt5 complex: a conserved module for regulation of transcription elongation. Structure 16:1649–1658. doi: 10.1016/j.str.2008.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Basrai MA, Kingsbury J, Koshland D, Spencer F, Hieter P. 1996. Faithful chromosome transmission requires Spt4p, a putative regulator of chromatin structure in Saccharomyces cerevisiae. Mol Cell Biol 16:2838–2847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zheng J, Benschop JJ, Shales M, Kemmeren P, Greenblatt J, Cagney G, Holstege F, Li H, Krogan NJ. 2010. Epistatic relationships reveal the functional organization of yeast transcription factors. Mol Syst Biol 6:420. doi: 10.1038/msb.2010.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Westover KD, Bushnell DA, Kornberg RD. 2004. Structural basis of transcription: separation of RNA from DNA by RNA polymerase II. Science 303:1014–1016. doi: 10.1126/science.1090839. [DOI] [PubMed] [Google Scholar]
- 68.Missra A, Gilmour DS. 2010. Interactions between DSIF (DRB sensitivity inducing factor), NELF (negative elongation factor), and the Drosophila RNA polymerase II transcription elongation complex. Proc Natl Acad Sci U S A 107:11301–11306. doi: 10.1073/pnas.1000681107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cheng B, Price DH. 2008. Analysis of factor interactions with RNA polymerase II elongation complexes using a new electrophoretic mobility shift assay. Nucleic Acids Res 36:e135. doi: 10.1093/nar/gkn630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yakhnin AV, Yakhnin H, Babitzke P. 2008. Function of the Bacillus subtilis transcription elongation factor NusG in hairpin-dependent RNA polymerase pausing in the trp leader. Proc Natl Acad Sci U S A 105:16131–16136. doi: 10.1073/pnas.0808842105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yakhnin AV, Babitzke P. 2014. NusG/Spt5: are there common functions of this ubiquitous transcription elongation factor? Curr Opin Microbiol 18:68–71. doi: 10.1016/j.mib.2014.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Belogurov GA, Sevostyanova A, Svetlov V, Artsimovitch I. 2010. Functional regions of the N-terminal domain of the antiterminator RfaH. Mol Microbiol 76:286–301. doi: 10.1111/j.1365-2958.2010.07056.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Artsimovitch I, Landick R. 2002. The transcriptional regulator RfaH stimulates RNA chain synthesis after recruitment to elongation complexes by the exposed nontemplate DNA strand. Cell 109:193–203. doi: 10.1016/S0092-8674(02)00724-9. [DOI] [PubMed] [Google Scholar]
- 74.Cui Y, Denis CL. 2003. In vivo evidence that defects in the transcriptional elongation factors RPB2, TFIIS, and SPT5 enhance upstream poly(A) site utilization. Mol Cell Biol 23:7887–7901. doi: 10.1128/MCB.23.21.7887-7901.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kaplan CD, Holland MJ, Winston F. 2005. Interaction between transcription elongation factors and mRNA 3′-end formation at the Saccharomyces cerevisiae GAL10-GAL7 locus. J Biol Chem 280:913–922. doi: 10.1074/jbc.M411108200. [DOI] [PubMed] [Google Scholar]
- 76.Mayer A, Schreieck A, Lidschreiber M, Leike K, Martin DE, Cramer P. 2012. The spt5 C-terminal region recruits yeast 3′ RNA cleavage factor I. Mol Cell Biol 32:1321–1331. doi: 10.1128/MCB.06310-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kireeva ML, Komissarova N, Kashlev M. 2000. Overextended RNA:DNA hybrid as a negative regulator of RNA polymerase II processivity. J Mol Biol 299:325–335. doi: 10.1006/jmbi.2000.3755. [DOI] [PubMed] [Google Scholar]
- 78.Huertas P, Aguilera A. 2003. Cotranscriptionally formed DNA:RNA hybrids mediate transcription elongation impairment and transcription-associated recombination. Mol Cell 12:711–721. doi: 10.1016/j.molcel.2003.08.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.