

Abstract
The orphan nuclear receptor Nur77 plays critical roles in cardiovascular diseases, and its expression is markedly induced in the heart after beta-adrenergic receptor (β-AR) activation. However, the functional significance of Nur77 in β-AR signaling in the heart remains unclear. By using Northern blot, Western blot, and immunofluorescent staining assays, we showed that Nur77 expression was markedly upregulated in cardiomyocytes in response to multiple hypertrophic stimuli, including isoproterenol (ISO), phenylephrine (PE), and endothelin-1 (ET-1). In a time- and dose-dependent manner, ISO increases Nur77 expression in the nuclei of cardiomyocytes. Overexpression of Nur77 markedly inhibited ISO-induced cardiac hypertrophy by inducing nuclear translocation of Nur77 in cardiomyocytes. Furthermore, cardiac overexpression of Nur77 by intramyocardial injection of Ad-Nur77 substantially inhibited cardiac hypertrophy and ameliorated cardiac dysfunction after chronic infusion of ISO in mice. Mechanistically, we demonstrated that Nur77 functionally interacts with NFATc3 and GATA4 and inhibits their transcriptional activities, which are critical for the development of cardiac hypertrophy. These results demonstrate for the first time that Nur77 is a novel negative regulator for the β-AR-induced cardiac hypertrophy through inhibiting the NFATc3 and GATA4 transcriptional pathways. Targeting Nur77 may represent a potentially novel therapeutic strategy for preventing cardiac hypertrophy and heart failure.
INTRODUCTION
Cardiac hypertrophy is an adaptive process in response to various physiological or pathological stimuli associated with neurohumoral activation, elevated wall stress, and changes in volume load (1). Although initially adaptive, persistent hypertrophy induced by pathological conditions like myocardial infarction and hypertension has detrimental consequences on the heart and eventually progresses to heart failure, a major cause of death and disability in the industrialized world (1). At cellular and molecular levels, cardiac hypertrophy is characterized by increased myocyte size, sarcomerogenesis, and reexpression of a set of fetal genes, such as the atrial natriuretic factor, B-type natriuretic peptide, and β-myosin heavy chain genes, which are coordinately controlled by a subset of hypertrophy-responsive transcription factors, including myocyte enhancer factor 2 (MEF2), nuclear factor of activated T cells (NFAT), and GATA4 (2, 3) (4). Depending on the upstream hypertrophic stimuli, these transcriptional regulators can be directly phosphorylated or dephosphorylated by protein kinases, such as mitogen-activated protein (MAP) kinase and protein kinases A and C, as well as the calcium-activated phosphatase calcineurin, to facilitate hypertrophic gene expression (5–9). Disruption and/or attenuation of the activities of these transcription factors could serve as a potential therapeutic approach in terms of inhibiting the hypertrophic responses in the heart (10–12).
NR4A receptors are immediate early genes that are regulated by various physiological stimuli, including growth factors, hormones, and inflammatory signals, and are involved in a wide array of important biological processes, including cell apoptosis, brain development, glucose metabolism, and vascular remodeling (13). The NR4A subfamily consists of 3 well-conserved members, Nur77 (NR4A1), Nurr1 (NR4A2), and NOR-1 (NR4A3). Like other nuclear receptors, NR4A receptors consist of an N-terminal transactivation domain, a central 2-zinc-finger DNA-binding domain, and a C-terminal ligand-binding domain (13). So far, no ligands have been identified for these receptors, and therefore they are classified as orphan receptors. Recently, there has been much attention paid to the function of these receptors in cardiovascular system (14). In vascular smooth muscle cells, the expression of Nur77 and NOR-1 was significantly induced by atherogenic stimuli, such as platelet-derived growth factor BB, epidermal growth factor, and α-thrombin, and overexpression of Nur77 has been shown to inhibit cell proliferation and attenuate vascular injury-induced neointimal formation in vivo (15, 16). NR4A nuclear receptors are also induced in vascular endothelial cells (ECs) by several stimuli, such as hypoxia, tumor necrosis factor alpha (TNF-α), and vascular endothelial growth factor, and modulate EC growth, survival, and angiogenesis (17, 18). Most importantly, our recent study has implicated Nur77 as a potent negative regulator for the proinflammatory response in ECs via a selective inhibition of NF-κB activation (18). In cardiomyocytes, Nur77 expression can be induced by ischemia/reperfusion injury, and translocation of Nur77 to the mitochondria mediates cardiomyocyte apoptosis in response to oxidative stress (19). Interestingly, a recent report demonstrated that the expression of Nur77 in the heart was rapidly induced in vivo following β-adrenergic stimulation (20, 21). However, at this time, the exact functional significance of Nur77 in the heart remains unknown. Considering the significant importance of β-adrenergic signaling in cardiomyocytes, we aimed to determine whether Nur77 is induced by β-adrenergic stimulation in cardiomyocytes and whether Nur77 is involved in β-adrenergic cardiac hypertrophy and, if so, to determine the molecular mechanism(s) involved.
MATERIALS AND METHODS
Primary culture of NRVMs.
We obtained ventricles from 1-day-old Sprague-Dawley rats and isolated cardiac myocytes through digestion with trypsin-EDTA and type 2 collagenase as previously described (22). The protocol was approved by the Institutional Animal Care and Use Committee of the Thomas Jefferson University. Briefly, the tissues were cut into small pieces and digested with 0.25% trypsin at 4°C overnight. Collagenase (Worthington, Lakewood, NJ (1 mg/ml in Hanks balanced salt solution [HBSS]) was used to further digest tissues in a shaking bath at 37°C for 20 min. The cell suspension was centrifuged at 1,000 rpm for 5 min and resuspended in Dulbecco modified Eagle medium (DMEM) with 1 g/liter glucose and 10% fetal bovine serum (FBS) (GIBCO). Cells were cultured for 2 h to allow fibroblast cells to attach to the flask. Neonatal rat ventricular myocytes (NRVMs) were collected from the supernatants and cultured with DMEM containing 1 g/liter glucose plus 10% FBS and 1% penicillin-streptomycin (GIBCO).
Northern blot analysis.
Total RNA was isolated using TRIzol reagent (Invitrogen) according to the instructions of the manufacturer. Total RNA (20 μg per lane) was resolved on a 1% formaldehyde–agarose gel and transferred onto a positively charged nylon membrane (Ambion) in transfer buffer (1 mol/liter NaCl and 10 mmol/liter NaOH). The probes were labeled with [α-32P]dCTP (3,000 Ci/mmol; PerkinElmer) using a random-primed DNA labeling kit (Ambion) and purified with NucAway spin columns (Ambion). The blots were prehybridized and hybridized at 68°C in hybridization buffer (Sigma). After washing twice in 2× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate)–0.1% SDS at 68°C for 20 min and once in 0.1× SSC–0.1% SDS at 68°C for 30 min, the blots were exposed to autoradiography film at −80°C using 2 intensifier screens. Protein kinase A (PKA) inhibitor PKI (Santa Cruz), protein kinase C (PKC) inhibitor GF109203X (GFX) (Tocris), phospholipase C (PLC) inhibitor U-73122 (Tocris), calcium channel blocker nifedipine (Tocris), and beta-adrenergic receptor antagonist propranolol (Sigma) were used to define the molecular signaling pathways mediating isoproterenol (ISO)-induced Nur77 expression in cardiac cells.
Adenovirus construction.
Adenoviruses harboring wild-type Flag-tagged Nur77 (Ad-Nur77) and dominant negative Flag-tagged Nur77 lacking the transactivation domain (Ad-DN-Nur77) were made as previously described (18). The viruses were made and propagated in Ad293 cells and purified using CsCl2 banding.
Knockdown of Nur77 and luciferase assay.
Cardiomyocytes were seeded in 6-well plates, and knockdown of Nur77 was performed by using Nur77 small interfering RNAs (siRNAs) with AllStars negative siRNAs (Qiagen) as a control, as previously described (23). At 48 h after transfection, the cardiomyocytes were transfected with ANF-Luc reporter plasmid (100 ng) using FuGene 6 transfection reagent. Approximately 48 h after transfection, the myocytes were treated as indicated for an additional 24 h. To determine the effect of Nur77 on the NFAT transcriptional activity, myocytes were transduced with either Ad-lacZ or Ad-Nur77 (multiplicity of infection [MOI], 30). At 24 h after transduction, myocytes were then transduced with Ad-NFAT-Luc (MOI, 30) for another 24 h and then stimulated with ISO (10 μmol/liter) or ET-1 (200 nmol/liter) for 24 h. Cell lysates (20 μl) were assayed for luciferase activity with a dual-luciferase reporter assay system (Promega).
Measurements of cardiac hypertrophy.
[3H]leucine uptake was measured as described previously (24). For staining of filamentous actin, NRVMs were fixed in 3.7% formaldehyde, treated with 0.1% Triton X-100 for 3 to 5 min, and then stained with Oregon Green 514 phalloidin (Molecular Probes) at 5 units/ml for 20 min. Expression of atrial natriuretic factor (ANF), brain natriuretic peptide (BNP), and beta-myosin heavy chain (β-MHC) was determined by quantitative reverse transcription-PCR (qRT-PCR) as described previously (11).
Immunofluorescence staining.
For immunostaining Nur77, cardiomyocytes were stimulated with 10 μmol/liter ISO for 3 h, and then fixed and sequentially incubated with anti–Nur77 polyclonal antibody (1:25 dilution; Santa Cruz) and fluorescein-5-isothiocyanate (FITC)-conjugated donkey anti-rabbit antibody (1:200 dilution, Invitrogen). For immunostaining of Flag-Nur77, cardiomyocytes were fixed and incubated with Cy3-conjugated anti-Flag antibody (1:100 dilution, Sigma). Images were visualized using an Olympus IX70 epifluorescence microscope.
Subcellular fractionation.
Subcellular fractions were prepared by differential centrifugation of cell homogenates as described previously (18). Briefly, cells were homogenized manually in hypotonic buffer (10 mmol/liter Tris-HCl [pH 7.5], 1 mmol/liter MgCl2, 1 mmol/liter EGTA, 1 mmol/liter EDTA, 1 mmol/liter phenylmethylsulfonyl fluoride, 5 μg/ml leupeptin, and 5 μg/ml aprotinin). The nuclear fraction was obtained from low-speed centrifugation (500 × g), followed by 3 washes with phosphate-buffered saline (PBS). For electrophoretic mobility shift assay (EMSA), the nuclear fraction was dissolved in hypertonic buffer (20 mmol/liter HEPES [pH 7.9], 420 mmol/liter NaCl, 1.5 mmol/liter MgCl2, 0.2 mmol/liter EDTA, 25% glycerol, and the above-mentioned protease inhibitors). After high-speed centrifugation at 12,800 × g for 10 min, the supernatants (nuclear extracts) were stored at −80°C until use.
Coimmunoprecipitation.
Neonatal rat cardiomyocytes (NRCMs) were treated with 10 μmol/liter ISO for 3 h. Cytoplasmic and nuclear fractions were isolated. Coimmunoprecipitation of Nur77 and GATA4 or NFATc3 was performed as described previously (25). To examine the interaction of DN-Nur77 with GATA4 and NFATc3, NRCMs were transduced with Ad-DN-Nur77 and then subjected to ISO stimulation for 3 h. The following antibodies and beads were used for detection and immunoprecipitation: anti-GATA4 (Santa Cruz), anti-NFATc3 (Santa Cruz), anti-Flag (Gene Script).
EMSA.
The oligonucleotides corresponding to the consensus sequence of GATA4 (5′-CAC TTG ATA ACA GAA AGT GAT AAC TCT-3′) was synthesized and labeled with IRDye 700 (IDT). Electrophoretic mobility shift assay (EMSA) were performed with Odyssey IRDye 700 infrared dye-labeled double-stranded oligonucleotides coupled with the EMSA buffer kit (Li-Cor Bioscience) according to the manufacturer's instructions. The specificity of the binding was examined using competition experiments and the gel supershift assay by adding GATA4 antibody (Santa Cruz) prior to the addition of the fluorescently labeled probe.
Myocardial gene delivery.
Myocardial gene delivery was performed as previously described with some minor changes (26). Briefly, C57BL/6 mice (20 to 25 g) were anesthetized with 2% isoflurane inhalation and not ventilated. A skin cut (1.2 cm) was made over the left chest, and the heart was smoothly and gently “popped out” through a small hole made at the fourth intercostal space. Twenty-five microliters of the gene of interest or vehicle control was then injected directly into left ventricle (LV) free wall with a syringe (Hamilton Co., Reno, NV) with a needle size of 30.5. After gene delivery, the heart was immediately placed back into the intrathoracic space, followed by manual evacuation of pneumothoraces and closure of muscle and the skin suture.
Implantation of Azlet mini-osmotic pumps.
At 24 h after intramyocardial injection with Ad-Nur77 or Ad-GFP, the mini-osmotic pump set to dispense 0.5 μl/h for 14 days was filled with PBS or ISO (60 mg/kg/day) and implanted subcutaneously in the back through a subscapular incision. Mice were monitored by echocardiogram prior to pump implantation and then weekly until the end of the 2-week period as described previously (26).
Echocardiography.
To measure global cardiac function, echocardiography was performed with the VisualSonics VeVo 2100 imaging system in anesthetized animals (1.5% [vol/vol] isoflurane). The internal diameter of the left ventricle was measured in the short-axis view from M-mode recordings in end diastole and end systole as described previously (27).
Cardiomyocyte cross-sectional area.
Axial cut tissue sections were stained with Masson's Trichrome and Alexa Fluor 594 conjugated with wheat germ agglutinin. For each treatment group, 3 hearts were imaged using a Nikon Eclipse Ti microscope with the 20× objective. Only myocytes cut in the short axis with a visible nucleus were measured (minimum of 200 cells per heart). Cell borders were planimetered manually by an operator who was blinded to treatment group. Nikon NIS Elements software was used to calculate 2-dimensional cross-sectional areas.
ChIP assay.
Chromatin immunoprecipitation assay (ChIP assays were performed as described previously (28). Briefly, NRCMs were cross-linked for 10 min by directly adding formaldehyde (Sigma) to the culture medium at a final concentration of 1%. The fixed cells were lysed with lysis buffer (EZ ChIP; Upstate) and sonicated two times for 7 s with output 5 (Virsonic 60; Virtis) with a 2-min refractory period. For immunoprecipitation, cell lysates were incubated with an antibody against GATA4 (5 μg; Santa Cruz Biotechnology) or NFATc3 (5 μg; Santa Cruz) overnight at 4°C. The immunoprecipitated and input DNAs were subjected to quantitative PCR with primers rANF (5′-GGTGTATGCCCCCTTCAAT-3′ and 5′-GCCAGGAAGCCAACATGATA-3′) and rBNP (5′-CCCTGTCATTTCCAATACGG-3′ and 5′-CCAGCCCTTCTCATGGAGT-3′) to amplify rat promoter regions containing NFATc3- and GATA4-binding sites.
Statistical analyses.
Data are expressed as means ± standard errors of the means (SEM). The statistical significance of differences was assessed by Student t tests or analysis of variance (ANOVA), as appropriate; a P value of <0.05 was considered statistically significant.
RESULTS
Upregulation of Nur77 by hypertrophic stimuli in NRVMs.
To investigate the role of NR4A nuclear receptors in cardiomyocyte biology, we examined the expression of Nur77 in NRVMs in response to a variety of hypertrophic stimuli. As shown in Fig. 1A, except for angiotensin II, all tested stimuli, including fetal bovine serum (FBS), phorbol 12-myristate 13-acetate (PMA), isoproterenol (ISO), phenylephrine (PE), and endothelin-1 (ET-1), markedly upregulated the expression of Nur77 in NRVMs. Because the critical importance of beta-adrenergic receptor (β-AR) signaling in cardiomyocyte hypertrophy, ISO was chosen to further investigate its effects on the expression of Nur77 in NRVMs. The time course of Nur77 expression, when incubated with 10 μmol/liter ISO, showed a maximal induction of Nur77 expression at 1 h after ISO stimulation (Fig. 1B). At a concentration of as low as 0.3 μmol/liter, ISO induced Nur77 mRNA expression by approximately 3-fold. The maximal induction of ISO on Nur77 mRNA expression was observed at the concentration of 1 μmol/liter, which increased Nur77 expression by approximately 20-fold (Fig. 1C). To further examine whether Nur77 expression is upregulated in vivo by β-AR stimulation, we collected the mouse hearts at 1 h, 3 h, 6 h, and 9 h after a single intraperitoneal injection of ISO (1 mg/kg). The mRNA expression of Nur77 in the heart was found to be substantially induced within 1 to 3 h and reached a peak at 1 h, with a maximal induction of approximately 25-fold (Fig. 1D).
FIG 1.

Induction of Nur77 expression by multiple hypertrophic stimuli. (A) NRVMs were treated with hypertrophic stimuli at the indicated concentrations for 1 h. Nur77 expression was determined by Northern blotting. (B) Time-dependent effect of ISO (10 μmol/liter) on Nur77 expression as determined by Northern blotting. (C) Dose-dependent effect of ISO on Nur77 expression as determined by Northern blotting 1 h after ISO stimulation. (D) Adult mice were treated with a single intraperitoneal injection of ISO (1 mg/kg), and their hearts were then harvested at the indicated time points for determining Nur77 expression by qRT-PCR. *, P < 0.05 versus untreated control or time at 0 h. The data represent 3 independent experiments. (E) Myocytes were pretreated with vehicle, various kinase inhibitors, or β-AR antagonist for 1 h and then treated with ISO (10 μmol/liter) for 1 h, and Nur77 expression was determined by Northern blotting.
To further determine the molecular signaling pathways involved in ISO-induced Nur77 expression, NRVMs were pretreated with various kinase inhibitors 1 h prior to the ISO stimulation. As shown in Fig. 1E, ISO-induced Nur77 expression was completely inhibited by PKA inhibitor PKI, the calcium blocker, and the β-AR antagonist, suggesting that both PKA and intracellular calcium pathways are involved in the expression of Nur77 induced by β-AR stimulation in cardiomyocytes.
Protein expression of Nur77 was confirmed by both Western blot and immunostaining analyses. As shown in Fig. 2A and B, Nur77 protein was upregulated in NRVMs, in a time- and dose-dependent fashion, by ISO treatment. Moreover, the immunoreactivity of Nur77 was increased predominantly in the nuclei of cardiomyocytes (Fig. 2C). Western blot analysis also indicated that ISO-induced Nur77 expression is localized primarily in the nuclear fraction, while ET-1-induced Nur77 is predominantly in cytoplasmic fraction (Fig. 2D). Chronic infusion of ISO into mice resulted in a marked Nur77 expression at 24 h which then declined to the baseline levels (Fig. 2E).
FIG 2.

ISO increases Nur77 protein expression in NRVMs. (A) Myocytes were treated with ISO (10 µmol/liter) for the indicated times. Nur77 expression was determined by Western blotting (n = 3). *, P < 0.05 versus time zero. (B) Myocytes were treated with indicated concentrations of ISO for 3 h. Nur77 expression was determined by Western blotting (n = 3). *, P < 0.05 versus ISO dose at time zero. (C) Myocytes were treated with ISO (10 µmol/liter) for 3 h and then fixed and sequentially incubated with anti-Nur77 polyclonal antibody and FITC-conjugated donkey anti-rabbit antibody. Images were visualized using an epifluorescence microscope. (D) Myocytes were treated with ISO (10 µmol/liter) or ET-1 (100 nmol/liter) for 3 h. Nuclear (Nucl) and cytoplasmic (Cyto) fractions were isolated and subjected to Western blot analysis to determine expression of Nur77. (E) Mice were administered ISO with a mini-osmotic pump (60 mg/kg/day). Hearts were harvested at the indicated time points, and Nur77 expression was determined by Western blotting (n = 3). *, P < 0.05 versus day 0.
Nur77 attenuates ISO-induced cardiac hypertrophy.
The induction of Nur77 levels in response to hypertrophic stimuli prompted us to speculate that Nur77 may play an important role in regulating cardiac hypertrophy. Since ISO-induced Nur77 expression is transient, we hypothesized that persistent expression of Nur77 may inhibit the development of cardiac hypertrophy. To test this hypothesis, we examined the effect of Nur77 overexpression on ISO induced cardiac hypertrophy. As shown in Fig. 3A, transduction of Ad-Nur77 (MOI = 50) markedly inhibited sarcomere organization induced by ISO. Furthermore, Nur77 overexpression substantially inhibited both basal and ISO-stimulated increases in cell size (Fig. 3B), protein synthesis (Fig. 3C), and ANF mRNA transcription (Fig. 3D). Interestingly, overexpression of dominant negative Nur77 (DN-Nur77), which is defective in its transactivation domain, demonstrated effects similar to those for wild-type Nur77 on ISO-induced cardiac hypertrophy, suggesting that a nongenomic effect of Nur77 might be responsible for its inhibitory effect on cardiac hypertrophy.
FIG 3.

Effect of Nur77 on ISO-induced cardiac hypertrophy. (A) Myocytes were transduced with either Ad-Nur77, Ad-DN-Nur77, or Ad-LacZ (MOI, 50) and stimulated with ISO (10 μmol/liter), PE (10 μmol/liter), or ET-1 (100 nmol/liter) for 48 h. The cells were stained with Oregon Green 514-phalloidin to visualize actin fibers. (B) The mean myocyte surface area (top) was obtained from 300 myocytes/well. (C) Cardiomyocytes cultured in 24-well plates were transduced with adenoviruses as indicated and cultured in serum-free medium for 24 h prior to stimulation with ISO (10 μmol/liter), PE (10 μmol/liter), or ET-1 (100 nmol/liter) for 24 h. [3H]leucine was added at 1 μCi per well, and incubation was continued for 6 h. The cells were washed and fixed with cold 5% trichloroacetic acid, and the radioactivity incorporated into the trichloroacetic acid-precipitable material was determined. The data are means ± standard errors (SE) from five independent studies. (D) Cardiomyocytes were transduced with the indicated adenoviruses (MOI, 50). At 24 h after transduction, myocytes were cultured in serum-free medium for 24 h prior to the stimulation with ISO (10 μmol/liter), PE (10 μmol/liter), or ET-1 (100 nmol/liter) for 48 h. The expression of ANF was determined by Northern blotting.
To test whether Nur77 has agonist-dependent specificity of action on cardiomyocytes, we examined the effect of Nur77 on PE- and ET-1-induced cardiac hypertrophy. Indeed, overexpression of both Nur77 and DN-Nur77 markedly inhibited the increases in cardiac cell size, protein synthesis, and ANF expression induced by PE (10 μmol/liter) but not by ET-1 (100 nmol/liter) (Fig. 3). These findings indicate that Nur77 functions as a negative-feedback regulator for both ISO- and PE-induced cardiac hypertrophy but not for ET-1 induced hypertrophy.
It was previously reported that the subcellular localization of Nur77 contributes to the diverse and sometimes even opposing biological activities of Nur77 in cancer cell lines (29, 30). To determine whether the subcellular localization of Nur77 accounts for the specificity of the Nur77 effects on cardiac hypertrophy induced by distinct agonists, we performed immunostaining to examine the subcellular localization of Nur77 in cardiomyocytes before and after ISO, PE, and ET-1 stimulation. Immunostaining using Cy3-conjugated anti-Flag M2 antibody yielded a profound perinuclear localization of Nur77 in cardiomyocytes infected with Ad-Nur77 but not in Ad-LacZ-transduced cells (data not shown). Intriguingly, both ISO and PE stimulation caused a rapid translocation of both Nur77 and DN-Nur77 from the cytoplasm into the nuclei of cardiomyocytes, whereas ET-1 had no effect on the Nur77 translocation (Fig. 4A). Furthermore, both ISO- and PE-induced Nur77 nuclear translocation was completely inhibited by PKA inhibitor H89 but not by a PKC inhibitor, suggesting that the PKA pathway may play a critical role in nuclear translocation of Nur77 induced by ISO and PE (Fig. 4B). Together, the results indicate that it is likely that the nuclear translocation of Nur77, as induced by ISO and PE stimulation but not by ET-1, may contribute significantly to its inhibitory effect on cardiac hypertrophy.
FIG 4.
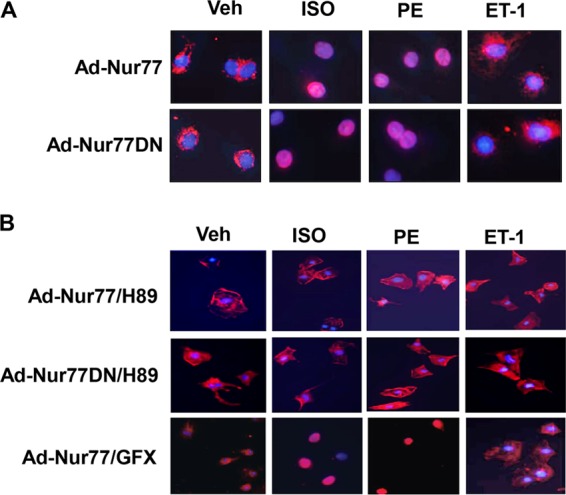
ISO induces Nur77 translocation in cardiomyocytes. (A) Myocytes were transduced with either Ad-Nur77 or Ad-DN-Nur77 (MOI, 50). At 24 h after transduction, myocytes were cultured in serum-free medium for 24 h prior to the stimulation with ISO (10 μmol/liter), PE (10 μmol/liter), or ET-1 (100 nmol/liter) for 3 h. Myocytes were then fixed and directly incubated with Cy3-conjugated anti-Flag antibody. Images were visualized using epifluorescence microscope. (B) Myocytes were transduced with either Ad-Nur77 or Ad-DN-Nur77 as indicated above and pretreated with H89 and GFX for 1 h prior to stimulation with ISO (10 μmol/liter), PE (10 μmol/liter), or ET-1 (100 nmol/liter) for 3 h. Myocytes were then fixed and directly incubated with Cy3-conjugated anti-Flag antibody.
Knockdown of Nur77 accelerates ISO-induced cardiac hypertrophy.
To further evaluate the functional role of endogenous Nur77 in cardiac hypertrophy, we performed loss-of-function studies. Transfection of myocytes with Nur77-specific siRNA (siNur77) markedly inhibited both ISO and PE induced Nur77 expression in myocytes as determined by qRT-PCR (Fig. 5A) and Western blotting (Fig. 5B). Furthermore, transfection of siNur77 significantly enhanced both ISO- and PE-induced increases in protein synthesis, whereas the basal protein synthesis in unstimulated cardiomyocytes was not affected (Fig. 5C). Likewise, knockdown of Nur77 barely affected the basal ANF transcription but significantly enhanced the ISO- and PE-induced ANF gene transcription, as determined by ANF promoter-driven luciferase activity (Fig. 5D). Taken together, these findings further suggest that Nur77 is a negative regulator of ISO-induced cardiac hypertrophy.
FIG 5.

Knockdown of Nur77 accelerates ISO induced cardiac hypertrophy. (A) Myocytes were transfected with Nur77 siRNA (siNur77) or control siRNA (siCTL). At 72 h after transduction, cells were cultured in serum-free medium for 24 h prior to stimulation with ISO (10 μmol/liter) or PE (10 μmol/liter) for 1 h. The expression of Nur77 was determined by qRT-PCR. The data represent 5 independent experiments. (B) Myocytes were transfected with siNur77) or siCTL. At 72 h after transduction, Nur77 expression was determined by Western blotting after stimulation with ISO (10 µmol/liter) or PE (10 µmol/liter) for 3 h. (C) Myocytes cultured in 24-well plates were transfected with siNur77 or siCTL. At 72 h after transduction, cells were cultured in serum-free medium for 24 h prior to stimulation with ISO (10 μmol/liter) or PE (10 μmol/liter) for 24 h. Leucine incorporation was determined (n = 5). (D) Myocytes cultured in 24-well plates were transfected with siNur77 or siCTL. At 48 h after transduction, myocytes were transfected with ANF-Luc and then stimulated with ISO (10 μmol/liter) or PE (10 μmol/liter) for 24 h. Luciferase assays were performed (n = 5).
Nur77 attenuates ISO-induced cardiac hypertrophy in vivo.
To further substantiate the in vivo functional significance of Nur77 in cardiac hypertrophy, we explored whether cardiac overexpression of Nur77 could inhibit cardiac hypertrophy in a mouse model of hypertrophy induced by the chronic infusion of ISO (7). To these ends, intramyocardial injection of Ad-Nur77 was applied to induce the expression of Nur77 in the heart. At 2 weeks after injection of Ad-Nur77, the expression of Nur77 was markedly increased in Ad-Nur77-injected hearts compared with the Ad-green fluorescent protein (Ad-GFP)-injected hearts (Fig. 6A), and increased Nur77 expression was localized predominantly in the nucleus as determined by immunoblotting on subcellular fractionations (Fig. 6B) and immunofluorescent staining (Fig. 6C). A single intramyocardial injection of Ad-Nur77 resulted in approximately 50% of cardiac cells expressing Nur77 (Fig. 6C). Furthermore, myocardial overexpression of Nur77 markedly inhibited ISO-induced cardiac hypertrophy, as demonstrated by heart weight/body weight ratio (Fig. 7A and B), cross-sectional areas (Fig. 7C and E) (Masson's trichrome staining), and cross-sectional diameter (Fig. 7D) (Alexa Fluor 594-conjugated wheat germ agglutinin staining). In addition, ISO-induced upregulation of hypertrophic markers, including ANF, BNP, and β-MHC, was attenuated in the hearts overexpressing Nur77 (Fig. 7F). Furthermore, we examined whether Nur77 overexpression can influence cardiac structure and function in vivo via echocardiography. Indeed, chronic administration of ISO significantly increased LV anterior wall thickness, which was significantly blunted when Nur77 was overexpressed (Fig. 8A and B). Overexpression of Nur77 also markedly attenuated the ISO-induced functional decompensation (Fig. 8C and D) but barely affected the ISO-induced increase in heart rate (Fig. 8E) and apoptosis as determined by terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) staining (data not shown). Together, these data suggest that upregulation of Nur77 in the heart inhibits ISO-induced cardiac hypertrophy and cardiac dysfunction in the animal model.
FIG 6.
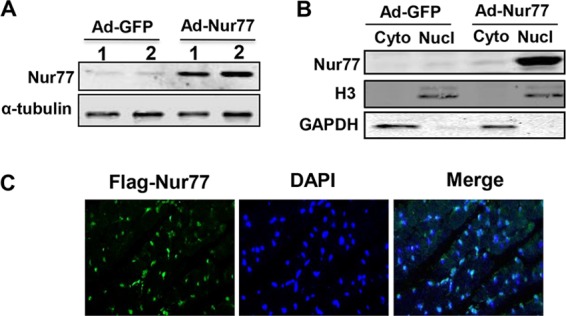
Expression of Nur77 in the heart after intramyocardial injection of Ad-Nur77. (A) At 2 weeks after intramyocardial injection of Ad-GFP or Ad-Nur77, Nur77 expression was determined by Western blotting. (B) At 14 days after chronic infusion of ISO, Nur77 expression in cytoplasmic and nuclear fractions in mouse hearts injected with either Ad-GFP or Ad-Nur77 was determined by Western blotting. (C) At 14 days after ISO infusion, Nur77 expression was determined in the heart by immunofluorescent staining using anti-Flag antibody.
FIG 7.

Cardiac Nur77 overexpression reduces ISO-induced myocardial hypertrophy. (A) Representative whole-heart picture taken at the end of a 2-week ISO pump infusion in Ad-GFP-PBS (a), Ad-GFP-ISO (b), Ad-Nur77-PBS (c), and Ad-Nur77-ISO (d) groups. (B) Ratio of heart weight (HW) to body weight (BW), *, P < 0.05 versus Ad-GFP-PBS; #, P < 0.05 versus Ad-GFP-ISO. (C) Representative heart tissue sections stained with Masson's trichrome. (D) Representative heart tissue sections stained with Alexa Fluor 594-conjugated wheat germ agglutinin. (E) Summary of cross-sectional area from panel C. *, P < 0.05 versus Ad-GFP-PBS group; #, P < 0.05 versus Ad-GFP-ISO (n = 5 for each group). (F) Expression of ANF, BNP, and β-MHC was examined by qRT-PCR. *, P < 0.05 versus Ad-GFP-PBS group; #, P < 0.05 versus Ad-GFP-ISO (n = 5 for each group).
FIG 8.

Cardiac function was measured by echocardiography at 2 weeks after ISO pump infusion. (A) Representative echocardiograph from the various groups. (B and C) The left ventricle systolic anterior wall depth (LVAWDs) (B) and the LV internal diameters of diastole (LVIDd) (C) were measured at the view of the short axis from M mode. (D and E) LV ejection fraction (D) and heart rate (HR) (E) were calculated using VisualSonic analysis software (n = 5 animals/group). *, P < 0.05 versus Ad-GFP-ISO; #, P < 0.05 versus Ad-GFP-PBS.
Nur77 inhibits transcriptional activities of NFATc3 and GATA4 in NRVMs.
DN-Nur77, which lacks the transcriptional activation domain of Nur77, demonstrated as potent a antihypertrophic effect as wild-type Nur77, suggesting that the nongenomic effect of Nur77, such as protein-protein interactions in the nucleus, may be responsible for its antihypertrophic effects in cardiomyocytes. Furthermore, the aberrant activation of transcriptional pathways, such as the calcineurin/NFAT and GATA4 pathways, has been reported to play critical roles in ISO-induced cardiac hypertrophy (4, 10, 31). Therefore, to investigate the mechanism(s) responsible for the inhibitory effects of Nur77 on ISO-induced cardiac hypertrophy, we first examined whether there is a functional interaction of Nur77 with NFATc3, which is a key transcription factor for multiple cardiac hypertrophic signaling in the nuclei of cardiomyocytes. To this end, we performed coimmunoprecipitation with either anti-Nur77 or anti-NFATc3 antibody using lysates obtained from cardiomyocytes. As shown in Fig. 9A, Nur77 coprecipitated with the anti-NFATc3 antibody but not with the nonimmune control, indicating that the endogenous levels of Nur77 can specifically interact with NFATc3 in cardiomyocytes. To determine whether the interaction of Nur77 with NFATc3 affects the transcriptional activity of NFATc3, we transduced cardiomyocytes with adenovirus bearing an NFAT-luciferase reporter gene. As shown in Fig. 9B, ISO stimulation markedly increased the NFAT-luciferase activity in Ad-LacZ-transduced cardiomyocytes, which was inhibited in cardiomyocytes overexpressing Nur77. In addition, the ISO-stimulated binding of NFATc3 to the ANF promoter was significantly attenuated by Nur77 as determined by chromatin immunoprecipitation (ChIP) assay (Fig. 9C). In contrast, Nur77 overexpression had no effect on ET-1-induced NFAT-luciferase activity (Fig. 9B).
FIG 9.

Functional interaction of Nur77 with NFATc3. (A) Myocytes were treated with ISO (10 μmol/liter) for 3 h. The nuclear fraction of cardiomyocytes was isolated and then subjected to immunoprecipitation with either normal IgG, anti-NFATc3, or anti-Nur77 antibody. Immunocomplexes were separated by 10% SDS-PAGE and immunoblotted with either anti-Nur77 or NFATc3 antibody. (B) Myocytes were transduced with either Ad-LacZ or Ad-Nur77 (MOI, 30). At 24 h after transduction, myocytes were then transduced with Ad-NFAT-Luc (MOI, 30). At 24 h after transduction, myocytes were starved and stimulated with ISO (10 μmol/liter) or ET-1 (200 nmol/liter) for 24 h, and luciferase assays were performed (n = 5). *, P < 0.05 versus Ad-LacZ with Veh or Ad-Nur77 with ISO. (C) Nur77 inhibits the binding activity of NFATc3 to the ANF promoter. NRCMs were infected with Ad-Nur77 or Ad-LacZ for 48 h and then treated with vehicle or 10 μmol/liter ISO for 3 h. PCR analysis of sheared DNA from control and adenovirus-infected cells before immunoprecipitation (input) and after chromatin immunoprecipitation (ChIP) with antibody directed against NFATc3 and quantitative analysis of ChIP results from three independent experiments are shown. *, P < 0.05 versus Ad-LacZ without ISO; #, P < 0.05 versus Ad-LacZ with ISO.
GATA4 has been previously reported to regulate cardiac hypertrophy through a cooperative interaction with NFAT (31, 32). Similarly, Nur77 specifically interacts with GATA4 in neonatal rat cardiomyocytes as determined by coimmunoprecipitation (Fig. 10A). Similar to wild-type Nur77, DN-Nur77 also interacts with both GATA4 and NFATc3 in ISO-stimulated cardiomyocytes (Fig. 10B) but not in ET-1-stimulated cells (data not shown). To determine whether Nur77 affects the GATA4-mediated transcription pathway in cardiomyocytes, we performed EMSA with the nuclear extracts obtained from cardiomyocytes transduced with either Ad-lacZ or Ad-Nur77. As shown in Fig. 10C, in Ad-LacZ-infected cells, ISO stimulation led to a formation of a strong singular DNA-protein complex containing GATA4, which was verified by both competition and supershift studies. This GATA4 DNA binding, however, was markedly attenuated in the nuclear extracts from Nur77-overexpressing myocytes. Moreover, overexpression of Nur77 markedly attenuated the ISO-stimulated GATA4 binding to its consensus site in the BNP promoter as determined by ChIP assay (Fig. 10D). Together, these data suggest that Nur77 inhibits ISO-induced cardiac hypertrophy, at least in part, through the inhibition of the NFAT and GATA4 transcriptional pathways.
FIG 10.

Functional interaction of Nur77 with GATA4 in cardiomyocytes. (A) Myocytes were treated with ISO (10 μmol/liter) for 3 h. The nuclear fraction of cardiomyocytes was isolated and then subjected to immunoprecipitation with either normal IgG, anti-Nur77, or anti-GATA4 antibody. Immunocomplexes were immunoblotted with either anti-Nur77 or anti-GATA4 antibody. (B) Myocytes were transduced with either Ad-LacZ or Ad-DN-Nur77. At 48 h after transduction, cells were treated with ISO (10 μmol/liter) for 3 h. The nuclear fraction of cardiomyocytes was immunoprecipitated with either normal IgG or anti-Flag antibody and then immunoblotted with either anti-NFATc3, anti-GATA4, or anti-Flag antibody. (C) Myocytes were transduced with either Ad-LacZ or Ad-Nur77. At 48 h after transduction, nuclear extracts (NE) were isolated after ISO (10 µmol/liter) stimulation for 3 h and used for EMSA to detect the interaction of GATA4 with its conserved binding probe. A 100 times excess of unlabeled wild-type (WT) competitor was used to demonstrate the specificity of the shifted complex. To supershift the complex, 5 μl of GATA4 antibody was added to the binding reaction mixture. (D) Nur77 inhibits the binding of GATA4 to the BNP promoter. NRCMs were infected with Ad-Nur77 or Ad-LacZ for 48 h and then treated with vehicle or 10 μmol/liter ISO for 3 h. PCR analysis of sheared DNA from control and adenovirus-infected cells before immunoprecipitation (input) and after chromatin immunoprecipitation (ChIP) with antibody directed against GATA4 and quantitative analysis of ChIP results from 3 independent experiments are shown. *, P < 0.05 versus Ad-LacZ without ISO; #, P < 0.05 versus Ad-LacZ with ISO.
DISCUSSION
In this study, we have found that Nur77, an orphan nuclear receptor, has antihypertrophic properties in cardiomyocytes. We demonstrated that Nur77 expression is markedly upregulated in response to multiple hypertrophic stimuli, such as ISO and PE, and acts as a potent inhibitor for both ISO- and PE-induced hypertrophy. Our results show that ISO-induced expression of Nur77 is transient and that chronic stimulation of cardiac cells with ISO led to a decline of Nur77 expression to the baseline levels in mouse heart, which may inactivate the negative-feedback loop of Nur77 and lead to the development of cardiac hypertrophy. Therefore, we speculated that persistent expression of Nur77 would be a therapeutic strategy to block the ISO-induced cardiac hypertrophy. Consistent with this hypothesis, our results demonstrated that intramyocardial injection of Ad-Nur77 significantly ameliorated ISO-induced cardiac hypertrophy and preserved the cardiac dysfunction in the animal model after chronic infusion of ISO for 2 weeks. However, Nur77 overexpression barely affected the baseline expression of hypertrophic marker genes, which is different from what we observed in neonatal cardiomyocytes. Intriguingly, DN-Nur77, which is defective in its transactivation domain, also inhibited ISO-induced cardiac hypertrophy to the same extent as the wild-type Nur77 in cardiomyocytes, suggesting an involvement of the nongenomic effect of Nur77 in its antihypertrophic effect. Indeed, we found that in the nuclei of cardiomyocytes, Nur77 functionally interacts with at least two important prohypertrophic transcription factors, NFATc3 and GATA4, and suppresses their transcriptional activities. Therefore, we provide, for the first time, compelling evidence suggesting that the orphan nuclear receptor Nur77 functions as a novel negative inhibitor for ISO-induced cardiac hypertrophy, at least in part, through inhibiting the NFAT and GATA4 transcriptional pathways in cardiomyocytes.
Recently, the cardiovascular function of the NR4A receptor has received significant attention (14). Accumulating evidence suggests that NR4A receptors play essential roles in the development of atherosclerosis, restenosis, and angiogenesis (17, 18, 33). However, the functional role of the NR4A receptor in cardiomyocyte biology remains largely obscured, although several studies suggest that the NR4A receptors are highly expressed in the heart and that their expression is regulated by several external pathological stimuli, such as oxidative stress, pressure overload, and β-AR activation (19, 20, 34). In the present study, we sought to examine the role of Nur77 in cardiac hypertrophy and found that the expression of Nur77 is substantially increased in response to a wide range of hypertrophic stimuli. Furthermore, we demonstrated that stimulation of cardiomyocytes with the β-AR agonist ISO caused a rapid and robust induction of Nur77, mainly through the PKA- and intracellular calcium-dependent signaling pathways. Predominant nuclear localization of the induced Nur77 in cardiac cells prompted us to postulate that Nur77 may be a critical regulator of the β-AR-induced molecular signaling pathway in the nuclei of cardiomyocytes. Indeed, the activation of transcriptional factors in the nuclei of cardiomyocytes has been well established to contribute significantly to the development of cardiac hypertrophy. For instance, the activation of the transcriptional factor NFAT pathway has been demonstrated in a majority of models of pathological cardiac hypertrophy and plays an essential role in mediating β-AR hypertrophy and heart failure (32). Under basal and unstimulated conditions, NFAT is phosphorylated and resides in the cytoplasm. Stimulation of cardiac cells with hypertrophic stimuli such as ISO and PE can rapidly cause the activation of the calcium-dependent phosphatase calcineurin (PP2B), which in turn dephosphorylates NFAT, thus facilitating its nuclear translocation and transcriptional activation of target genes essential for cardiac hypertrophy (10, 35). In the current study, our data clearly indicated that Nur77 associates with NFATc3 after ISO stimulation, therefore suppressing its transcriptional activity, which is mainly responsible for the inhibitory effects of Nur77 on the β-AR-induced cardiomyocyte hypertrophy.
Our data also indicate that Nur77 can inhibit the transcriptional activity of GATA4, another critical transcriptional factor involved in cardiac hypertrophy. In cardiomyocytes, through a cooperative interaction with other transcription factors such as NFAT-3c and MEF-2, GATA4 has been shown to increase cell size, protein synthesis, and expression of prohypertrophic genes (31, 36). Direct interaction of GATA4 with NFAT has been shown to synergistically regulate the hypertrophic gene expression in the heart (31, 32). In addition, the transcriptional activity of GATA4 is regulated by posttranslational modifications, such as phosphorylation, acetylation, and methylation (4, 37, 38). Both MAP kinase and phosphatidylinositol (PI) 3-kinase/Akt/glycogen synthase kinase (GSK) pathways can phosphorylate GATA4 and regulate its transcriptional activity in the heart (4, 39). p300, a transcriptional coactivator, also directly interacts with GATA4 to synergistically activate the ANF and β-MHC promoters during myocardial hypertrophy (40, 41). In the present study, we demonstrated that Nur77 potently inhibits the GATA4 DNA binding and transcriptional activity through an inhibitory interaction with GATA4. However, whether Nur77 inhibits the GATA4 transcriptional pathway through its action on the posttranslational modification of GATA4 warrants further investigation.
In addition to inhibiting ISO-induced cardiac hypertrophy, Nur77 also inhibits PE-induced hypertrophy, but it has no effect on ET-1-induced cardiac hypertrophy. The differential effects of Nur77 on cardiac hypertrophy induced by different stimuli seem largely due to the intracellular localization of Nur77 in cardiomyocytes. In response to ISO and PE stimulation, Nur77 is rapidly translocated to the nuclei of cardiomyocytes, whereas ET-1 stimulation did not induce the nuclear translocation of Nur77, although ET-1 has been shown to induce the Nur77 expression to a similar extent as both ISO and PE did in cardiomyocytes. These results indicate a critical role of the nuclear localization of Nur77 in eliciting its antihypertrophic effects. Recently, Nur77 has been shown to be induced by 500 nmol/liter angiotensin II in NRVMs and to promote angiotensin II-induced cardiac hypertrophy through a cytoplasmic interaction with the TSC1/TSC2 complex (42). In our study, Nur77 expression was not significantly induced by angiotensin II at a concentration of 100 nmol/liter, which was much lower than the 500 nmol/liter used in the above-mentioned study (42). Indeed, multiple lines of evidence suggest that depending on the stimuli and its cellular localization, Nur77 can exert different or even opposite effects (29, 30). For instance, in response to certain apoptosis-inducing agents, Nur77 expression is induced in some cancer cells, and it subsequently translocates from the nucleus to mitochondria, where it causes the conformational change of Bcl-2 to promote apoptosis (30). While in the nucleus, Nur77 can function as a transcription factor by binding to its DNA response elements of target genes to promote cancer cell growth and survival (43, 44). Recently, Nur77 has been shown to function as a positive regulator of skeletal muscle growth in mice by transcriptional induction of insulin-like growth factor 1 (IGF1) as well as downregulation of growth-inhibitory genes (45). Our data suggest that such a genomic action of Nur77 is not likely involved in its antihypertrophic effect in cardiomyocytes, since DN-Nur77, which lacks the functional N-terminal AF-1 domain of Nur77, exerts the same antihypertrophic effects as wild-type Nur77, suggesting that the nongenomic action of Nur77, such as protein-protein interactions, may contribute to its antihypertrophic effect. At this point, the molecular mechanism mediating the translocation of Nur77 in cardiomyocytes remains elusive, but it may be dependent on the phosphorylation status of Nur77, as has been previously shown in some noncardiac cells lines. Several kinases, including PKA, PKC, Jun N-terminal protein kinase (JNK), p90 ribosomal S6 kinase (RSK), and Akt, have been reported to phosphorylate Nur77 in various in vitro and in vivo experiments (43, 46, 47). Phosphorylation of Nur77 by PKC, JNK, and RSK has been reported to target Nur77 to the mitochondria and induce cell apoptosis in T cells (29, 48) (48), whereas the Akt-mediated phosphorylation inhibits this process (43). In cardiomyocytes, our preliminary data indicate that inhibition of PKA but not PKC completely abolished the ISO- and PE-induced nuclear translocation, highlighting the importance of the PKA pathway in the translocation of Nur77 from the cytoplasm to the nuclei in cardiomyocytes. Indeed, PKA has been shown to phosphorylate Nur77 in an in vitro experiment (49); whether this phosphorylation mediates Nur77 translocation, however, is unknown. Thus, it would be interesting to investigate where Nur77 is a substrate of PKA and whether the PKA-mediated Nur77 phosphorylation accounts for its nuclear translocation in cardiomyocytes.
Overall, the data reported here provide evidence that the orphan nuclear receptor Nur77 is a novel negative-feedback regulator of cardiac hypertrophy in response to β-AR stimulation via inhibiting both NFAT and GATA4 transcriptional pathways. Chronic stimulation of the β-AR has been shown to contribute significantly to the development of progressive cardiac dysfunction and cardiac remodeling in patients (50). Understanding the molecular mechanism, particularly identifying the negative regulators of β-AR signaling in cardiomyocytes, will be of critical importance for a better treatment of patients with heart failure. In this regard, identification of Nur77 as a negative regulator of the β-AR signaling in cardiomyocytes may represent a potentially novel therapeutic strategy for preventing cardiac hypertrophy and heart failure.
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health (HL103869 to J.S. and HL105414 to D.G.T.) and the Chinese Natural Science Foundation (no. 381170114 and no. 81370418 to J.S.).
REFERENCES
- 1.Frey N, Katus HA, Olson EN, Hill JA. 2004. Hypertrophy of the heart: a new therapeutic target? Circulation 109:1580–1589. doi: 10.1161/01.CIR.0000120390.68287.BB. [DOI] [PubMed] [Google Scholar]
- 2.Zhang CL, McKinsey TA, Chang S, Antos CL, Hill JA, Olson EN. 2002. Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell 110:479–488. doi: 10.1016/S0092-8674(02)00861-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Diedrichs H, Hagemeister J, Chi M, Boelck B, Muller-Ehmsen J, Schneider CA. 2007. Activation of the calcineurin/NFAT signalling cascade starts early in human hypertrophic myocardium. J Int Med Res 35:803–818. doi: 10.1177/147323000703500609. [DOI] [PubMed] [Google Scholar]
- 4.Suzuki YJ. 2011. Cell signaling pathways for the regulation of GATA4 transcription factor: Implications for cell growth and apoptosis. Cell Signal 23:1094–1099. doi: 10.1016/j.cellsig.2011.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sanna B, Bueno OF, Dai YS, Wilkins BJ, Molkentin JD. 2005. Direct and indirect interactions between calcineurin-NFAT and MEK1-extracellular signal-regulated kinase 1/2 signaling pathways regulate cardiac gene expression and cellular growth. Mol Cell Biol 25:865–878. doi: 10.1128/MCB.25.3.865-878.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liang Q, Bueno OF, Wilkins BJ, Kuan CY, Xia Y, Molkentin JD. 2003. c-Jun N-terminal kinases (JNK) antagonize cardiac growth through cross-talk with calcineurin-NFAT signaling. EMBO J 22:5079–5089. doi: 10.1093/emboj/cdg474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Braz JC, Bueno OF, Liang Q, Wilkins BJ, Dai YS, Parsons S, Braunwart J, Glascock BJ, Klevitsky R, Kimball TF, Hewett TE, Molkentin JD. 2003. Targeted inhibition of p38 MAPK promotes hypertrophic cardiomyopathy through upregulation of calcineurin-NFAT signaling. J Clin Invest 111:1475–1486. doi: 10.1172/JCI200317295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang J, Paradis P, Aries A, Komati H, Lefebvre C, Wang H, Nemer M. 2005. Convergence of protein kinase C and JAK-STAT signaling on transcription factor GATA-4. Mol Cell Biol 25:9829–9844. doi: 10.1128/MCB.25.22.9829-9844.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leenders JJ, Pinto YM, Creemers EE. 2011. Tapping the brake on cardiac growth-endogenous repressors of hypertrophic signaling. J Mol Cell Cardiol 51:156–167. doi: 10.1016/j.yjmcc.2011.04.017. [DOI] [PubMed] [Google Scholar]
- 10.Tokudome T, Horio T, Kishimoto I, Soeki T, Mori K, Kawano Y, Kohno M, Garbers DL, Nakao K, Kangawa K. 2005. Calcineurin-nuclear factor of activated T cells pathway-dependent cardiac remodeling in mice deficient in guanylyl cyclase A, a receptor for atrial and brain natriuretic peptides. Circulation 111:3095–3104. doi: 10.1161/CIRCULATIONAHA.104.510594. [DOI] [PubMed] [Google Scholar]
- 11.Fischer A, Klattig J, Kneitz B, Diez H, Maier M, Holtmann B, Englert C, Gessler M. 2005. Hey basic helix-loop-helix transcription factors are repressors of GATA4 and GATA6 and restrict expression of the GATA target gene ANF in fetal hearts. Mol Cell Biol 25:8960–8970. doi: 10.1128/MCB.25.20.8960-8970.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fisch S, Gray S, Heymans S, Haldar SM, Wang B, Pfister O, Cui L, Kumar A, Lin Z, Sen-Banerjee S, Das H, Petersen CA, Mende U, Burleigh BA, Zhu Y, Pinto YM, Liao R, Jain MK. 2007. Kruppel-like factor 15 is a regulator of cardiomyocyte hypertrophy. Proc Natl Acad Sci U S A 104:7074–7079. doi: 10.1073/pnas.0701981104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao Y, Bruemmer D. 2010. NR4A orphan nuclear receptors: transcriptional regulators of gene expression in metabolism and vascular biology. Arterioscler Thromb Vasc Biol 30:1535–1541. doi: 10.1161/ATVBAHA.109.191163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Tiel CM, de Vries CJ. 2012. NR4All in the vessel wall. J Steroid Biochem Mol Biol 130:186–193. doi: 10.1016/j.jsbmb.2011.01.010. [DOI] [PubMed] [Google Scholar]
- 15.Pires NM, Pols TW, de Vries MR, van Tiel CM, Bonta PI, Vos M, Arkenbout EK, Pannekoek H, Jukema JW, Quax PH, de Vries CJ. 2007. Activation of nuclear receptor Nur77 by 6-mercaptopurine protects against neointima formation. Circulation 115:493–500. doi: 10.1161/CIRCULATIONAHA.106.626838. [DOI] [PubMed] [Google Scholar]
- 16.Bonta PI, Matlung HL, Vos M, Peters SL, Pannekoek H, Bakker EN, de Vries CJ. 2010. Nuclear receptor Nur77 inhibits vascular outward remodelling and reduces macrophage accumulation and matrix metalloproteinase levels. Cardiovasc Res 87:561–568. doi: 10.1093/cvr/cvq064. [DOI] [PubMed] [Google Scholar]
- 17.Zeng H, Qin L, Zhao D, Tan X, Manseau EJ, Van Hoang M, Senger DR, Brown LF, Nagy JA, Dvorak HF. 2006. Orphan nuclear receptor TR3/Nur77 regulates VEGF-A-induced angiogenesis through its transcriptional activity. J Exp Med 203:719–729. doi: 10.1084/jem.20051523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.You B, Jiang YY, Chen S, Yan G, Sun J. 2009. The orphan nuclear receptor Nur77 suppresses endothelial cell activation through induction of IkappaBalpha expression. Circ Res 104:742–749. doi: 10.1161/CIRCRESAHA.108.192286. [DOI] [PubMed] [Google Scholar]
- 19.Cheng Z, Volkers M, Din S, Avitabile D, Khan M, Gude N, Mohsin S, Bo T, Truffa S, Alvarez R, Mason M, Fischer KM, Konstandin MH, Zhang XK, Heller Brown J, Sussman MA. 2011. Mitochondrial translocation of Nur77 mediates cardiomyocyte apoptosis. Eur Heart J 32:2179–2188. doi: 10.1093/eurheartj/ehq496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Myers SA, Eriksson N, Burow R, Wang SC, Muscat GE. 2009. Beta-adrenergic signaling regulates NR4A nuclear receptor and metabolic gene expression in multiple tissues. Mol Cell Endocrinol 309:101–108. doi: 10.1016/j.mce.2009.05.006. [DOI] [PubMed] [Google Scholar]
- 21.Grisanti LA, Talarico JA, Carter RL, Yu JE, Repas AA, Radcliffe SW, Tang HA, Makarewich CA, Houser SR, Tilley DG. 2014. β-Adrenergic receptor-mediated transactivation of epidermal growth factor receptor decreases cardiomyocyte apoptosis through differential subcellular activation of ERK1/2 and Akt. J Mol Cell Cardiol 72:39–51. doi: 10.1016/j.yjmcc.2014.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun J, Yan G, Ren A, You B, Liao JK. 2006. FHL2/SLIM3 decreases cardiomyocyte survival by inhibitory interaction with sphingosine kinase-1. Circ Res 99:468–476. doi: 10.1161/01.RES.0000239410.65551.b3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li P, Liu Y, Yi B, Wang G, You X, Zhao X, Summer R, Qin Y, Sun J. 2013. MicroRNA-638 is highly expressed in human vascular smooth muscle cells and inhibits PDGF-BB-induced cell proliferation and migration through targeting orphan nuclear receptor NOR1. Cardiovasc Res 99:185–193. doi: 10.1093/cvr/cvt082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takemoto M, Node K, Nakagami H, Liao Y, Grimm M, Takemoto Y, Kitakaze M, Liao JK. 2001. Statins as antioxidant therapy for preventing cardiac myocyte hypertrophy. J Clin Invest 108:1429–1437. doi: 10.1172/JCI13350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sun J, Liao JK. 2002. Functional interaction of endothelial nitric oxide synthase with a voltage-dependent anion channel. Proc Natl Acad Sci U S A 99:13108–13113. doi: 10.1073/pnas.202260999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gao E, Lei YH, Shang X, Huang ZM, Zuo L, Boucher M, Fan Q, Chuprun JK, Ma XL, Koch WJ. 2010. A novel and efficient model of coronary artery ligation and myocardial infarction in the mouse. Circ Res 107:1445–1453. doi: 10.1161/CIRCRESAHA.110.223925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hullmann JE, Grisanti LA, Makarewich CA, Gao E, Gold JI, Chuprun JK, Tilley DG, Houser SR, Koch WJ. 2014. GRK5-mediated exacerbation of pathological cardiac hypertrophy involves facilitation of nuclear NFAT activity. Circ Res 115:976–985. doi: 10.1161/CIRCRESAHA.116.304475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qin Q, Chen M, Yi B, You X, Yang P, Sun J. 2014. Orphan nuclear receptor Nur77 is a novel negative regulator of endothelin-1 expression in vascular endothelial cells. J Mol Cell Cardiol 77:20–28. doi: 10.1016/j.yjmcc.2014.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kolluri SK, Bruey-Sedano N, Cao X, Lin B, Lin F, Han YH, Dawson MI, Zhang XK. 2003. Mitogenic effect of orphan receptor TR3 and its regulation by MEKK1 in lung cancer cells. Mol Cell Biol 23:8651–8667. doi: 10.1128/MCB.23.23.8651-8667.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin B, Kolluri SK, Lin F, Liu W, Han YH, Cao X, Dawson MI, Reed JC, Zhang XK. 2004. Conversion of Bcl-2 from protector to killer by interaction with nuclear orphan receptor Nur77/TR3. Cell 116:527–540. doi: 10.1016/S0092-8674(04)00162-X. [DOI] [PubMed] [Google Scholar]
- 31.Molkentin JD, Lu JR, Antos CL, Markham B, Richardson J, Robbins J, Grant SR, Olson EN. 1998. A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell 93:215–228. doi: 10.1016/S0092-8674(00)81573-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vega RB, Bassel-Duby R, Olson EN. 2003. Control of cardiac growth and function by calcineurin signaling. J Biol Chem 278:36981–36984. doi: 10.1074/jbc.R300023200. [DOI] [PubMed] [Google Scholar]
- 33.Zhao D, Qin L, Bourbon PM, James L, Dvorak HF, Zeng H. 2011. Orphan nuclear transcription factor TR3/Nur77 regulates microvessel permeability by targeting endothelial nitric oxide synthase and destabilizing endothelial junctions. Proc Natl Acad Sci U S A 108:12066–12071. doi: 10.1073/pnas.1018438108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grisanti LA, Repas AA, Talarico JA, Gold JI, Carter RL, Koch WJ, Tilley DG. 2015. Temporal and gefitinib-sensitive regulation of cardiac cytokine expression via chronic beta-adrenergic receptor stimulation. Am J Physiol Heart Circ Physiol 308:H316-330. doi: 10.1152/ajpheart.00635.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vega RB, Rothermel BA, Weinheimer CJ, Kovacs A, Naseem RH, Bassel-Duby R, Williams RS, Olson EN. 2003. Dual roles of modulatory calcineurin-interacting protein 1 in cardiac hypertrophy. Proc Natl Acad Sci U S A 100:669–674. doi: 10.1073/pnas.0237225100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liang Q, De Windt LJ, Witt SA, Kimball TR, Markham BE, Molkentin JD. 2001. The transcription factors GATA4 and GATA6 regulate cardiomyocyte hypertrophy in vitro and in vivo. J Biol Chem 276:30245–30253. doi: 10.1074/jbc.M102174200. [DOI] [PubMed] [Google Scholar]
- 37.He A, Shen X, Ma Q, Cao J, von Gise A, Zhou P, Wang G, Marquez VE, Orkin SH, Pu WT. 2012. PRC2 directly methylates GATA4 and represses its transcriptional activity. Genes Dev 26:37–42. doi: 10.1101/gad.173930.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen M, Yi B, Sun J. 2014. Inhibition of cardiomyocyte hypertrophy by protein arginine methyltransferase 5. J Biol Chem 289:24325–24335. doi: 10.1074/jbc.M114.577494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.van Berlo JH, Elrod JW, Aronow BJ, Pu WT, Molkentin JD. 2011. Serine 105 phosphorylation of transcription factor GATA4 is necessary for stress-induced cardiac hypertrophy in vivo. Proc Natl Acad Sci U S A 108:12331–12336. doi: 10.1073/pnas.1104499108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dai YS, Markham BE. 2001. p300 functions as a coactivator of transcription factor GATA-4. J Biol Chem 276:37178–37185. doi: 10.1074/jbc.M103731200. [DOI] [PubMed] [Google Scholar]
- 41.Takaya T, Kawamura T, Morimoto T, Ono K, Kita T, Shimatsu A, Hasegawa K. 2008. Identification of p300-targeted acetylated residues in GATA4 during hypertrophic responses in cardiac myocytes. J Biol Chem 283:9828–9835. doi: 10.1074/jbc.M707391200. [DOI] [PubMed] [Google Scholar]
- 42.Wang RH, He JP, Su ML, Luo J, Xu M, Du XD, Chen HZ, Wang WJ, Wang Y, Zhang N, Zhao BX, Zhao WX, Shan ZG, Han J, Chang C, Wu Q. 29 November 2012. The orphan receptor TR3 participates in angiotensin II-induced cardiac hypertrophy by controlling mTOR signalling. EMBO Mol Med doi: 10.1002/emmm.201201369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen HZ, Zhao BX, Zhao WX, Li L, Zhang B, Wu Q. 2008. Akt phosphorylates the TR3 orphan receptor and blocks its targeting to the mitochondria. Carcinogenesis 29:2078–2088. doi: 10.1093/carcin/bgn197. [DOI] [PubMed] [Google Scholar]
- 44.Jacobs CM, Boldingh KA, Slagsvold HH, Thoresen GH, Paulsen RE. 2004. ERK2 prohibits apoptosis-induced subcellular translocation of orphan nuclear receptor NGFI-B/TR3. J Biol Chem 279:50097–50101. doi: 10.1074/jbc.M409145200. [DOI] [PubMed] [Google Scholar]
- 45.Tontonoz P, Cortez-Toledo O, Wroblewski K, Hong C, Lim L, Carranza R, Conneely O, Metzger D, Chao LC. 2015. The orphan nuclear receptor Nur77 is a determinant of myofiber size and muscle mass in mice. Mol Cell Biol 35:1125–1138. doi: 10.1128/MCB.00715-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Davis IJ, Hazel TG, Chen RH, Blenis J, Lau LF. 1993. Functional domains and phosphorylation of the orphan receptor Nur77. Mol Endocrinol 7:953–964. [DOI] [PubMed] [Google Scholar]
- 47.Wingate AD, Campbell DG, Peggie M, Arthur JS. 2006. Nur77 is phosphorylated in cells by RSK in response to mitogenic stimulation. Biochem J 393:715–724. doi: 10.1042/BJ20050967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thompson J, Burger ML, Whang H, Winoto A. 2010. Protein kinase C regulates mitochondrial targeting of Nur77 and its family member Nor-1 in thymocytes undergoing apoptosis. Eur J Immunol 40:2041–2049. doi: 10.1002/eji.200940231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Katagiri Y, Hirata Y, Milbrandt J, Guroff G. 1997. Differential regulation of the transcriptional activity of the orphan nuclear receptor NGFI-B by membrane depolarization and nerve growth factor. J Biol Chem 272:31278–31284. doi: 10.1074/jbc.272.50.31278. [DOI] [PubMed] [Google Scholar]
- 50.Bristow MR. 2000. Beta-adrenergic receptor blockade in chronic heart failure. Circulation 101:558–569. doi: 10.1161/01.CIR.101.5.558. [DOI] [PubMed] [Google Scholar]