

Abstract
CTCFL, a paralog of CTCF, also known as BORIS (brother of regulator of imprinted sites), is a testis-expressed gene whose function is largely unknown. Its product is a cancer testis antigen (CTA), and it is often expressed in tumor cells and also seen in two benign human vascular malformations, juvenile angiofibromas and infantile hemangiomas. To understand the function of Ctcfl, we created tetracycline-inducible Ctcfl transgenic mice. We show that Ctcfl expression during embryogenesis results in growth retardation, eye malformations, multiorgan pathologies, vascular defects, and neonatal death. This phenotype resembles prior mouse models that perturb the transforming growth factor β (TGFB) pathway. Embryonic stem (ES) cells with the Ctcfl transgene reproduce the phenotype in ES cell-tetraploid chimeras. Transcriptome sequencing of the Ctcfl ES cells revealed 14 genes deregulated by Ctcfl expression. Bioinformatic analysis revealed the TGFB pathway as most affected by embryonic Ctcfl expression. Understanding the consequence of Ctcfl expression in nontesticular cells and elucidating downstream targets of Ctcfl could explain the role of its product as a CTA and its involvement in two, if not more, human vascular malformations.
INTRODUCTION
CTCFL, also known as BORIS (brother of regulator of imprinted sites), a paralog of the ubiquitous zinc finger gene CTCF, is a testicular transcript (1) expressed in spermatogonia and preleptotene spermatocytes (2). CTCF and CTCFL share the conserved 11 central zinc fingers but differ in their amino and carboxy termini (3). CTCF is a major chromatin architecture protein (4) that is implicated in gene activation and repression (5–7), nucleosome positioning (8), genetic imprinting (9), X inactivation (10, 11), and telomere length (12). Mice homozygous null for Ctcf die early in development (13), specifically at embryonic day 4.5 (e4.5) to e5.5 (14), and embryos derived from oocytes depleted of Ctcf develop poorly to the blastocyst stage (15, 16). What role the paralogous Ctcfl gene plays during spermatogenesis or when reactivated in somatic cells is less certain. Ctcfl knockout mice are viable but subfertile, with reduced testicular weight (2, 17) and decreased Gal3st1 (cerebroside sulfotransferase) enzyme activity (17). Reduction in Gal3st1 activity likely contributes to their subfertility, as Gal3st1 null animals are completely sterile (18).
A major exception to male germ line only expression of CTCFL, however, is seen in a variety of human tumors, which qualifies the product of CTCFL as a cancer testis antigen (CTA) (19). For example, Vatolin et al. reported that CTCFL is expressed in most breast, prostate, and colon cancers and melanomas (20). Additionally, CTCFL is reported to reactivate in lung, ovarian, testicular, uterine, hepatocellular, and esophageal carcinomas (21–31). Finally, evidence exists showing that two benign human vascular malformations express CTCFL, i.e., juvenile angiofibromas (JAs) (32) and infantile hemangiomas (IHs) (B. Schultz, X. Yao, Y. Deng, M. Waner, C. Spock, L. Tom, J. Persing, and D. Narayan, submitted for publication). What etiologic role CTCFL might play in the development of these vascular malformations is unknown.
To investigate aberrant somatic cell Ctcfl expression, we created transgenic mice that expressed a Ctcfl cDNA during embryogenesis. We accomplished this by first creating transgenic mice that are inducible with doxycycline and conditional by choice of the promoter driving the gene for Cre recombinase. This strategy proved to be critical, as our data show that expression of the Ctcfl transgene is lethal on the first day of life and founder animals presumably would have died if the transgene had been ubiquitously expressed. By breeding transgenic males where Ctcfl expression was restricted to the testis, we were able to induce the expression of Ctcfl in their progeny and report that ubiquitous embryonic/fetal expression of Ctcfl results in fetal growth retardation, congenital eye anomalies, vascular malformations, visceral organ pathology, and early postnatal death. Comparison of our Ctcfl transgenic mice with known mouse models led us to conclude that, on the basis of phenotype alone, they resemble mice with an altered transforming growth factor β (TGFB) pathway. From our transgenic mice, we created Ctcfl transgenic embryonic stem (ES) cells and introduced them into wild-type tetraploid blastocysts so that the embryonic portion of the conceptus derives entirely from the ES cells. We observed that these Ctcfl transgenic ES cell-tetraploid chimeras replicate the phenotype of the original Ctcfl transgenic mice. Transcriptome sequencing (RNA-Seq) studies of Ctcfl transgenic ES cells revealed significant alteration of the expression of 14 genes in response to Ctcfl transgene induction. The genes affected included those for transcription factors, including a homeoprotein-encoding gene, a gene for a meiotic chromosome binding protein, genes for signaling pathway proteins (including TGFB and Jak2), and genes for proteins involved in cell adhesion and tight junctions. Not unexpectedly, pathway analysis revealed a perturbation of the TGFB pathway as the major consequence of somatic cell Ctcfl expression. An understanding of which genes are altered in response to Ctcfl expression and the phenotypic consequences that result will lead to a better understanding of the role CTCFL might play in spermatogenesis and why, when acting as a CTA, it is aberrantly expressed in normal or cancerous somatic cells.
(This work was a part of the Ph.D. thesis of Leyla Sati.)
MATERIALS AND METHODS
Creation of conditional/inducible Ctcfl transgenic mice.
All of our animal experiments were performed under a protocol approved by the Yale Institutional Animal Care and Use Committee. To create inducible Ctcfl transgenic mice, we obtained codon-optimized Ctcfl cDNA (Codon Devices) and subcloned the insert into the TET ON vector (Clontech Laboratories, Inc., Mountain View, CA). The cDNA insert was injected into C57BL/6J oocytes. Four B6.Cg-tg(Tet-CTCFL)Jmg founders were obtained. Positive founders and their offspring were subsequently bred to two additional transgenic strains. The first had a reverse tetracycline-controlled transactivator (rtTA) transgene embedded in the Rosa26 locus with a floxed stop signal [JAX.org stock no. 005670; B6.Cg-Gt(ROSA)26Sortm1(rtTA,EGFP)Nagy/J]. We created mice doubly heterozygous for the Ctcfl and rtTA transgenes that express Ctcfl in the presence of doxycycline only after the floxed stop signal is excised by Cre recombinase. The Cre recombinase was provided by testicle-specific promoter Cre transgenic strains [JAX.org stock no. 007252 and 003466; B6Ei.129S4-tg(Prm-cre)580J/EiJ and B6;D2-tg(Sycp1-cre)4Min/J, respectively]. The breeding strategy used is shown in Fig. 1. PCR genotyping (see Fig. S1 in the supplemental material) identified males that contained all three transgenes and expressed Ctcfl (data not shown) and green fluorescent protein (GFP) (see Fig. S2 in the supplemental material) only in the testis, as expected. When triple-transgenic males are bred with wild-type females receiving doxycycline (1.5 mg/ml with 3% sucrose in drinking water), 25% of the developing fetuses are doubly heterozygous for Ctcfl and rtTA and are expected to express Ctcfl in all embryonic tissues.
FIG 1.
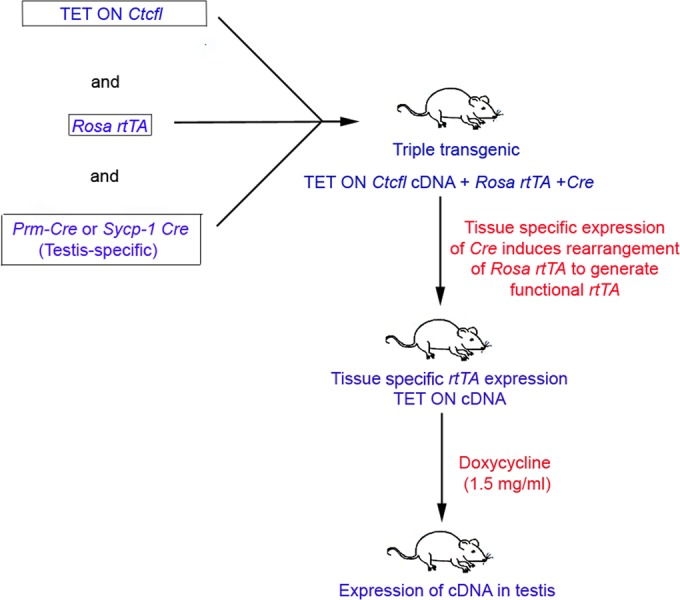
Transgenic strain breeding diagram. The breeding scheme shown illustrates the generation of testis-specific Ctcfl transgenic mice. In the presence of the gene for Cre recombinase, under the control of a testis-specific promoter, a DNA fragment with a stop signal is deleted to generate a functional rtTA transgene. This experimental design allows the specific expression of Ctcfl transgenes in any tissue, limited only by the availability of the tissue-specific Cre transgenic strain. In our case, we chose a testis-specific promoter to limit the expression of the Ctcfl transgene to the testis.
Genotyping of animals.
DNA was obtained from tissues with the Qiagen DNeasy kit according to the manufacturer's instructions. Genotyping was performed with the primers listed in Table S1 in the supplemental material. The cycling conditions for Ctcfl were as follows: 94°C for 2 min; 32 cycles of 94°C for 30 s, 56°C for 30 s, and 65°C for 3 min; and 68°C for 10 min. The cycling conditions for Cre were as follows: 94°C for 1.5 min; 35 cycles of 94°C for 30 s, 54°C for 1 min, and 72°C for 1 min; and 72°C for 5 min. The cycling conditions for rtTA were as follows: 94°C for 1.5 min; 35 cycles of 94°C for 30 s, 60°C for 40 s, and 72°C for 45 s; and 72°C for 10 min. The Ctcfl, Cre, and rtTA amplicons were 440, 520, and 560 bp, respectively, and were visualized on 1% agarose gels with ethidium bromide.
Creation of Ctcfl transgenic and control mice.
Transgenic males genotypically Ctcfl Cre rtTA were mated with C57B/6J females. Doxycycline water was given ad libitum at the time of mating and continued until after the females gave birth.
Creation of Ctcfl rtTA transgenic and control ES cells.
Transgenic males genotypically Ctcfl Cre rtTA were mated with C57B/6J females. Blastocysts were obtained on e3.5 (e0.5 is the day of vaginal plug detection). Blastocysts were cultured overnight in KSOMAA medium (33) at 37°C with 5% O2, 5% CO2, and 90% N2 in a modular incubator and transferred to 24-well dishes (Falcon) with inactivated mouse embryo fibroblast (MEF) feeder layers and ES medium containing Dulbecco's modified Eagle's medium with 20% ES-qualified serum (GIBCO), leukemia inhibitory factor (LIF) (1 × 103 U/ml; GIBCO BRL), 0.1 mM β-mercaptoethanol (American Bioanalytical), and the mitogen-activated protein kinase inhibitor PD 98059 (Calbiochem) at 50 μM (34). Cultures were observed for ES cell morphology, and when ES cells were present, they were trypsinized and expanded.
Creation of tetraploid-ES cell chimeras.
Ctcfl rtTA transgenic and wild-type ES cells were injected into e3.5 tetraploid blastocysts (35). Tetraploid embryos were generated by incubating two-cell-stage CD-1 (Charles River) embryos for 5 min at 37°C in electrofusion medium (0.3 M glucose, 0.1 mM CaCl2, 0.1 mM MgSO4, 0.3% bovine serum albumin, pH 7.2), performing electrofusion with a BTX ECM2001 pulse generator (Harvard Apparatus) with 2 V of AC for 10 s to orient the embryos, followed by two pulses of 50 V of DC for 35 μs and a postfusion 2 V of AC for 5 s. After electrofusion, embryos were washed in M2 medium and fused embryos were cultured in KSOMAA medium in an atmosphere of 5% O2, 5% CO2, and 90% N2 at 37°C and transferred to the uteri of e2.5 CD-1 pseudopregnant females on the day of injection.
Histology and immunohistochemistry analyses.
Tissues were fixed for 24 h in Bouin's fixative, cut along the sagittal and coronal planes, and submitted for routine paraffin embedding. Five-micrometer sections were used for hematoxylin-and-eosin (H&E) staining and immunostaining. Immunohistochemistry analysis was performed with monoclonal antibodies against CD34 (1:80; Abcam) and vascular endothelial growth factor (VEGF, 1:30; Dako) on a Dako Autostainer with Envision kits (Dako) according to the manufacturer's instructions. Sections were counterstained with hematoxylin.
RNA purification and qRT-PCR analysis.
Total RNA was extracted from tissues or cells with the Qiagen RNeasy minikit (Qiagen Inc., Valencia, CA, USA) by following the recommended protocol. Total RNA was treated with Qiagen RNase-free DNase. RNA samples were quantified with a NanoDrop ND-100 (Thermo Scientific, Wilmington, DE), and 600 ng of RNA was converted to cDNA with the Qiagen QuantiTect reverse transcription kit with genomic DNA wipeout buffer (Qiagen Inc., Valencia, CA). Quantitative real-time PCR (qRT-PCR) analysis was performed with the Bio-Rad Mini Opticon real-time PCR system. Ctcfl transgene and Ctcf or Ctcfl endogenous gene expression was determined with the primers listed in Table S2 in the supplemental material. PCR mixtures were prepared with 8.0 μl of cDNA (1:10 dilution) in SsoFast EvaGreen Supermix from Bio-Rad Laboratories, (Hercules, CA). All determinations were performed in triplicate and normalized to Gapdh gene expression by the comparative ΔΔCT method (36).
RNA-Seq studies.
ES cells for RNA-Seq studies were cultured in ES medium on MEFs and replated in 6-cm tissue culture dishes (Falcon) off MEFs for 48 h with or without doxycycline (2 μg/ml; Sigma, St. Louis, MO). Two biological replicates were performed for each of four samples (i.e., with and without the transgene and with and without doxycycline). Total RNA was isolated with TRIzol reagent (Invitrogen, Carlsbad, CA) and chloroform extraction. RNA was precipitated with isopropyl alcohol and washed with 75% ethanol, and total RNA was further purified with a Qiagen RNeasy minikit and DNase I digestion (Qiagen Inc., Valencia, CA). RNAs were sequenced on an Illumina HiSeq 2000 sequencing system generating 75-bp single-end strand-specific reads at the Yale Center for Genomic Analysis. The first six and last two nucleotides of each read were trimmed with the FASTX toolkit (http://hannonlab.cshl.edu/fastx_toolkit/index.html) to remove low-quality bases. Trimmed reads were mapped to the mouse reference genome (mm10) with a known transcriptome index (UCSC Known Gene annotation) by using TopHat v2.0.8 (37). Differential gene expression analysis was performed with Cufflinks v2.1.1 (38). Pathway analysis was performed with the DAVID program v6.7 (39, 40).
Nucleotide sequence accession numbers.
Data were deposited in the Gene Expression Omnibus under accession no. GSE72178.
RESULTS
Creation and characterization of founder strains, birth weight, and percent survival of newborn Ctcfl transgenic mice.
Of 180 C57BL/6J zygotes injected with a codon-optimized Ctcfl cDNA, 4 founders/38 offspring were obtained (2 males and 2 females). All founders were fertile and bred to two additional transgenic strains, a testicle-specific Cre recombinase and a floxed-stop Rosa locus rtTA transgene as described in Materials and Methods. Twenty-five percent of the progeny of triple-transgenic males bred to wild-type females should be heterozygous for Ctcfl and rtTA and express the Ctcfl transgene ubiquitously during embryogenesis when pregnant females are maintained on doxycycline. Of 207 progeny from Ctcfl rtTA Cre × +/+ matings on doxycycline, 46 (22%) had unfused eyelids and a mean birth weight of 1.173 ± 0.019 g (Fig. 2A), while their phenotypically normal littermates weighed 1.392 ± 0.011 g. The unfused-eyelid progeny therefore had a 16% lower birth weight than their phenotypically normal littermates, which was statistically significant (P < 0.001; Mann-Whitney rank sum test). Most of the unfused-eyelid progeny died on the first day of life (P0), although a few survived to P1 and then died. In some cases, there was ocular or cranial hemorrhaging (Fig. 2B). Genotyping of pups revealed that the unfused-eyelid progeny possessed both the Ctcfl and rtTA transgenes, while the normal progeny possessed neither or one but not both. The proportion of unfused-eyelid progeny (22%) observed was not significantly different from the expected 25% (P > 0.05). Our observation of slightly less than the expected 25% of doubly heterozygous progeny could result from cannibalization of dead pups by the mother before investigator observation or by nonpenetrance of the unfused-eyelid phenotype in a small proportion of the offspring.
FIG 2.
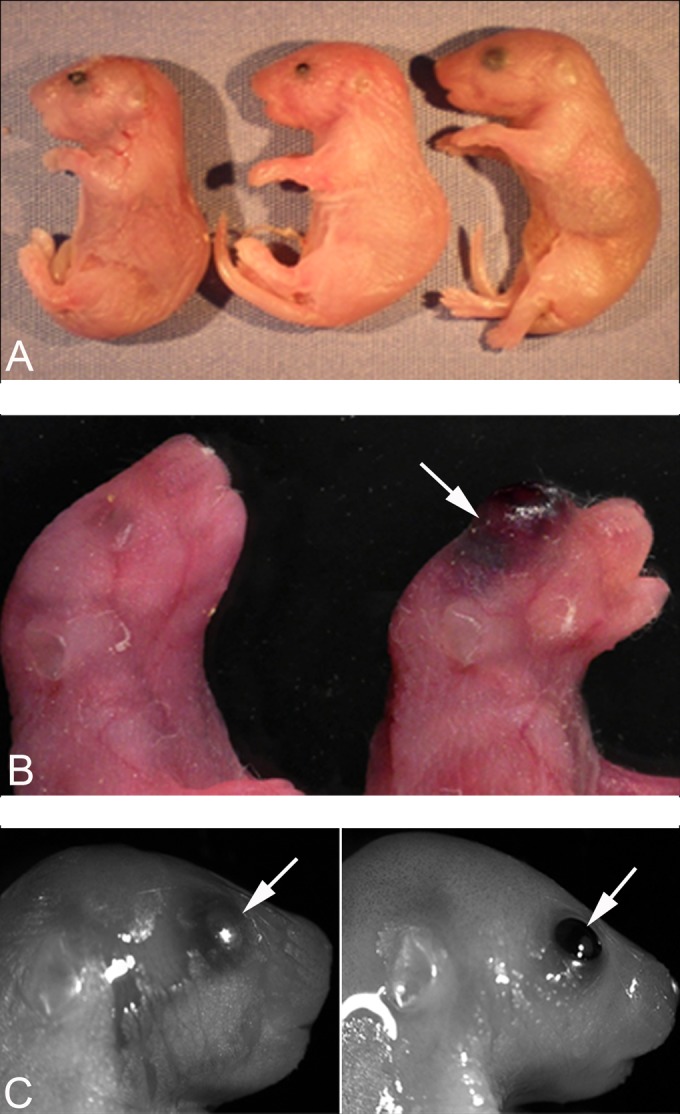
Abnormal pups observed after Ctcfl triple-transgenic mice were mated with wild-type females that were maintained on doxycycline during pregnancy. (A) Three P0 pups. The pups at the far left and center have unfused eyelids and are smaller. The rightmost pup is normal in size and with fused eyelids. (B) Normal P0 pup on the left and transgenic P0 pup with ocular hemorrhaging (arrow) on the right. (C) Control (left side) and Ctcfl transgenic (right side) ES cell-tetraploid chimeric e18.5 fetuses. Note the unfused eyelids in the transgenic fetus.
Histology of Ctcfl transgenic newborns.
Unfused-eyelid transgenic pups had an array of lens, vitreous, and anterior ocular abnormalities (Fig. 3). Microphthalmia was accompanied by microphakia, severe cataract, and in some cases, lens rupture. Multiple anterior segment abnormalities were present. Lenticular attachments to the posterior aspect of the cornea were accompanied by incomplete development of Descemet's membrane, excessive corneal vascularization, abnormal development of the iris, keratitis, and failure of eyelids to follow the normal developmental pattern of closure. Persistence of hyaloid vasculature was noted in the vitreous chamber and was variably accompanied by retinal folding or detachment.
FIG 3.
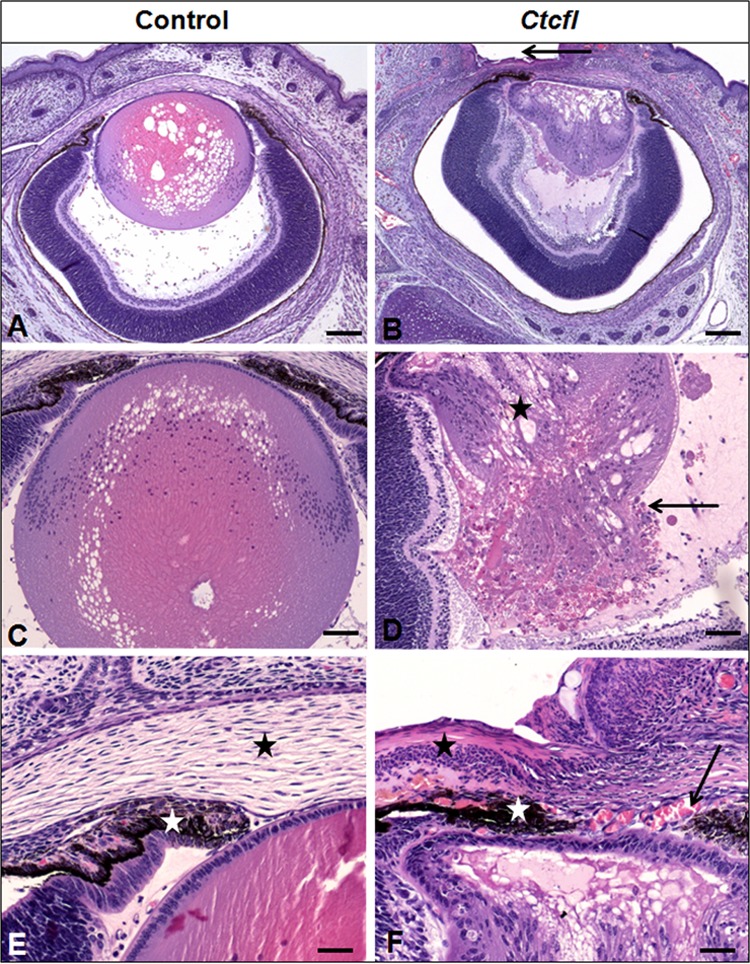
Ocular lesions in Ctcfl transgenic mice. Ctcfl mice have a smaller lens (B versus A) with cataract (star in panel D) and lens rupture (arrow in panel D). Eyelids have failed to fuse (arrow in panel B), contributing to keratitis (black stars in panels F and E). The anterior lens epithelium is adhered to the interior aspect of the cornea (B, F). This is accompanied by abnormal anterior segment vascularization (arrow in panel F) and impaired iridal development (white stars in panels F and E). Control lens morphology is shown in panel C (vacuolation results from a processing artifact), and normal stromal anatomy is shown in panel E (black star). H&E staining was used. Bars: 100 μm (A, B), 50 μm (C, D), and 20 μm (E, F).
Generalized developmental abnormalities were evident in Ctcfl transgenic mice (Fig. 4). Delayed alveolar development was characterized by alveolar interstitial thickening and accumulation of proteinaceous material within insufficiently inflated alveolar spaces. Fewer hematopoietic elements were present within the liver. Delayed cortical glomerulotubular development with increased mesenchyme and scattered tubular cysts were present in the kidney. Muscle fibers were narrow and periodically degenerate. In some newborns, hemorrhaging was grossly observed in the eye, face, and cranium (Fig. 2B). Histologic examination of the latter group revealed the presence of cerebral cavernous malformations, as well as midline brain and skull malformations (Fig. 5). Immunohistochemistry analysis of the vascular markers CD34 and VEGF revealed that vascular development of meninges and within the brain was poor, even in the most normal regions (Fig. 6). Ctcfl transgenics exhibited failure of midline ossification of the skull, meningocele, and focal excessive vascular proliferation of meninges. The latter was accompanied by increased VEGF expression (Fig. 6). Ctcfl transgenics exhibited reduced CD34 expression in the dermis and periadnexal regions (Fig. 7). Taken together, these results indicate that Ctcfl expression during embryogenesis results in multiorgan abnormalities that include eye, muscle, lung, liver, brain, renal, and vascular anomalies.
FIG 4.

Generalized developmental delay in Ctcfl transgenic P0 animal tissues (panels B, D, F, and H versus controls in panels A, C, E, and G) and ES cell-tetraploid e18.5 chimeric fetus tissues (panels J, L, N, and P versus controls in panels I, K, M, and O). Controls for P0 pups were nontransgenic littermates. Controls for e18.5 fetuses were from chimeras from pseudopregnant females that were not given doxycycline during pregnancy. Ctcfl mice display reduced alveolar maturation of lungs (B, J) and reduced glomerular maturation (D, L) accompanied by renal tubular cysts (D), reduced hematopoietic precursors in the liver (F), impaired pancreatic exocrine development (N), and skeletal muscle degeneration (H, P). H&E staining was used. Bars: 20 μm (A to F, I, J), 50 μm (K, L), and 10 μm (G, H, O, P).
FIG 5.

Sections at approximately the same coronal level from representative control (A to D) and Ctcfl transgenic (E to H) P0 newborns. Ctcfl transgenic mice develop cavernous malformations of the brain characterized by subcutaneous and cerebral hemorrhaging (F to H), midline malformations of the brain and skull (F, G), and meningocele with focal excessive vascular proliferation of meninges (F, G). H&E staining was used. Bar, 200 μm.
FIG 6.
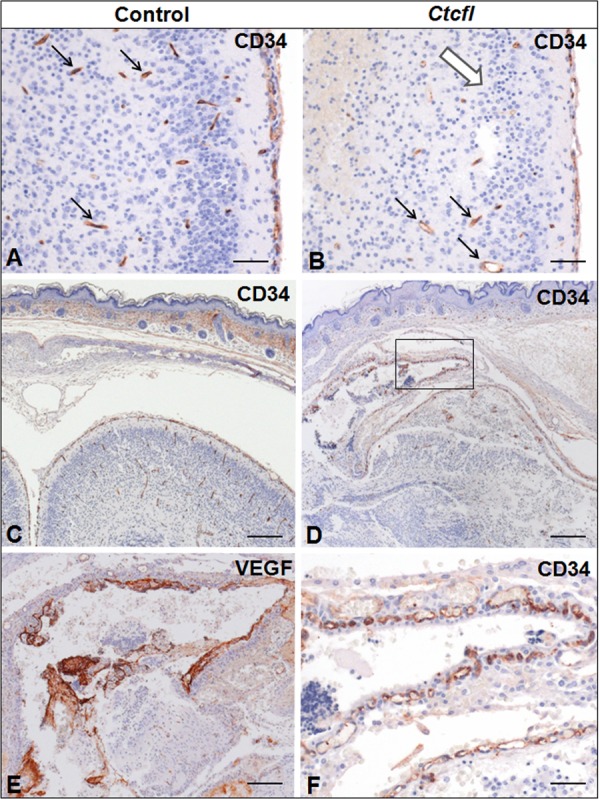
Immunohistochemistry analysis of CD34 and VEGF (brain and meninges) control and Ctcfl transgenic mice (P0). Fewer CD34-positive vascular profiles (black arrows) are present in the cortex of the Ctcfl transgenic animal (B versus A). This is accompanied by neuronal disorganization and nuclear pyknosis (white arrow). A focal region of meningeal proliferation and cortical dysplasia in the Ctcfl transgenic animal (D) contrasts with the comparable region of the control animal (C). The boxed area in panel D shows that meningeal proliferation is accompanied by abundant CD34-positive vascular profiles (F) and VEGF-positive proteinaceous material suggestive of vascular leakage (E). Bars: 50 μm (A, B), 100 μm (C, D), and 20 μm (E, F).
FIG 7.
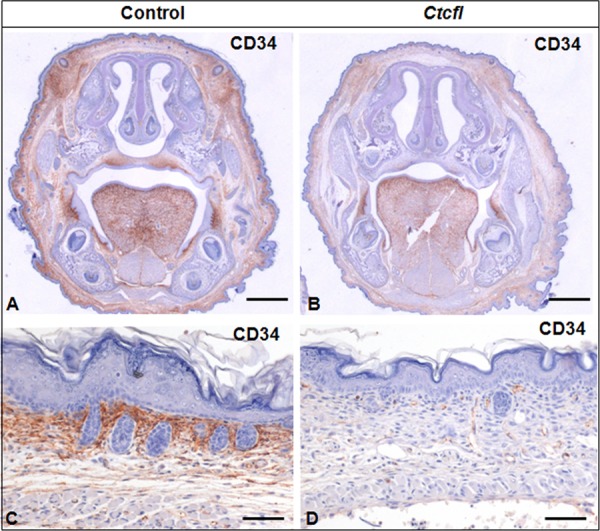
Lower CD34 expression is seen in the dermis and periadnexal regions of Ctcfl transgenic newborns (B, D) than in controls (A, C).
Expression of the Ctcfl transgene in P0 tissues.
In order to determine if Ctcfl transgene expression was present in newborn pups and to better quantitate the relative expression levels in different tissues, PCR primers that identify transgenic Ctcfl only were designed and qRT-PCR was performed with liver, lung, kidney, brain, eye, and skin tissues from control and Ctcfl transgenic progeny (Fig. 8). We did not see expression in wild-type littermates, as expected, since they do not possess the transgene. The transgene was expressed in transgenic pups at birth and the relative fold increase in Ctcfl was determined by setting expression in the brain as 1.0 and comparing its expression in other tissues with that in the brain. We observed similar levels of transgene expression in the brain, kidney, eye, and liver. Skin and lung expression levels were greater (10- and 40-fold, respectively). We did not observe a correlation between the level of expression and phenotypic abnormality, as the tissue with the second highest expression level, skin, was grossly and light microscopically normal.
FIG 8.
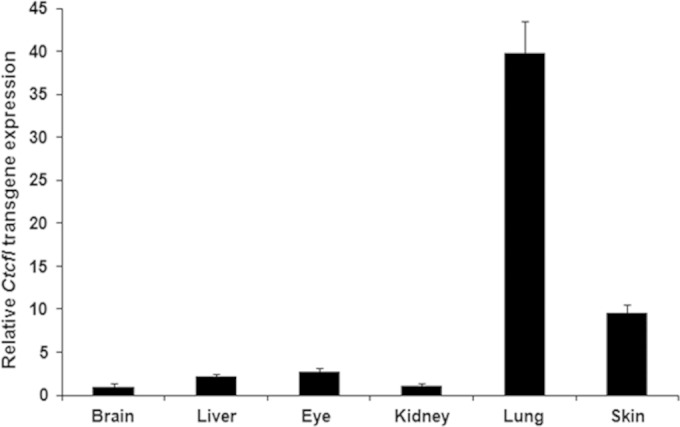
Relative expression of the Ctcfl transgene in different tissues as determined by qRT-PCR analysis.
Expression of Ctcfl in ES cells in vitro and the phenotype of tetraploid-Ctcfl ES cell chimeras in vivo.
ES cell lines were derived from our transgenic mice and genotyped, and Ctcfl rtTA and wild-type ES cells were characterized by (i) Ctcfl transgene expression in the presence or absence of doxycycline in vitro and (ii) the phenotype of Ctcfl ES-tetraploid chimeras when allowed to develop in pseudopregnant females in vivo. Our qRT-PCR results show that expression of the Ctcfl transgene in ES cells cultured in vitro increased 320-fold relative to that of Gapdh in the presence of doxycycline (data not shown). Expression of endogenous Ctcfl or Ctcf was not statistically significantly altered when the Ctcfl transgene was induced (data not shown). Having established that the Ctcfl transgene can be highly induced in our ES cells, we then tested these cells for the ability to replicate the mutant phenotype in vivo. The use of traditional chimeras in diploid host blastocysts is less than ideal, given the presence of wild-type cells that will diminish the phenotype of the transgenic cells. However, by using the ES cell-tetraploid chimera system, we can create embryos and newborns where the fetus originates solely from the ES cells while the placenta derives from the host tetraploid cells. Pseudopregnant females receiving Ctcfl ES cell-injected tetraploid blastocysts were given either doxycycline or regular water from the time of embryo transfer (e2.5), and resultant e18.5 fetuses or newborn pups were assessed for eye and internal pathology phenotypes. Our data show that Ctcfl transgenic ES cells always produced unfused-eyelid progeny that underwent perinatal death when pregnant mothers were given doxycycline and always produced normal-eyed progeny when doxycycline was withheld (Table 1 and Fig. 2C). When internal organs were surveyed by histology in the Ctcfl ES-derived animals on doxycycline, a phenotype of multiorgan pathology nearly identical to that initially observed in the transgenic animals was reproduced. Findings included ocular malformations, myodegeneration, and impaired pulmonary, pancreatic, hepatic, and renal development (Fig. 4), and these findings were not seen in the controls. Given the low number of fetuses that survive to e18.5 or birth in ES-tetraploid chimeras in general, we cannot comment on the presence or absence of growth retardation in this experimental series. However, the in vitro results showing a 320-fold increase in Ctcfl transgene expression in response to doxycycline and the in vivo results showing Ctcfl ES-tetraploid chimeras replicate the unfused-eyelid, lethal, internal organ pathology phenotype validate their use in our RNA-Seq studies.
TABLE 1.
Eyelid phenotypes of transgenic and control ES cell-tetraploid chimeras when pseudopregnant females were or were not given doxycycline
| ES cell line | Doxycycline | No. of embryos injected | No. of e18.5 fetuses/total | No. P0 pups/total | No. of pups with eyelids: |
|
|---|---|---|---|---|---|---|
| Fused | Unfused | |||||
| Ctcfl Tga | + | 189 | 9/159 | 1/30 | 0 | 10 |
| Ctcfl Tg | − | 117 | 4/62 | 2/55 | 6 | 0 |
| Not Tg | − | 70 | 4/58 | 1/12 | 5 | 0 |
Tg, transgenic.
RNA-Seq studies of Ctcfl transgenic ES cells.
Given that Ctcfl ES-tetraploid chimeras replicate the phenotype of transgenic mice, we grew transgenic and control cells with or without doxycycline for 48 h in vitro and performed RNA-Seq studies. On average, we obtained 21.12 million reads per sample, of which 96.8% were mappable. The data show 12 genes significantly upregulated and 2 significantly downregulated (P < 0.05, corrected for the false-detection rate) in response to Ctcfl transgene induction. These 14 gene products, their relative expression levels (in fragments per kilobase per million reads mapped), the fold changes in their expression, and the q values (P value corrected for the false-discovery rate) are listed in Table 2. Because our experimental design compared the same transgenic Ctcfl ES cell line with or without doxycycline, we were able to eliminate many false positives. For example, comparison of the Ctcfl ES cell line in the absence of doxycycline with a +/+ ES cell line, both derived from our Ctcfl transgenic mouse matings, revealed significant differences in the expression of 179 genes. Since doxycycline was not present to induce transgene expression, we conclude that these 179 genes represent the difference between two independently derived cell lines and not the effects of, in this case, the Ctcfl transgene. Only when a single transgene-inducible cell line is compared to itself, with or without doxycycline, are the differentially expressed genes truly revealed. Similarly, analysis of our nontransgenic ES cell line with and without doxycycline revealed no significant differences in the expression of any genes. These results, taken together, show that doxycycline itself, in the absence of the Ctcfl transgene, does not significantly alter gene expression by using the statistical parameters employed here and that the 14 deregulated genes we identified represent true downstream targets of Ctcfl induction.
TABLE 2.
RNA-Seq data and affected gene productsa
| Gene product | Relative expression levelc |
Fold change | q valueb | |
|---|---|---|---|---|
| No doxycycline | With doxycycline | |||
| Upregulated | ||||
| Cited1 | 3.92 | 13.47 | +3.44 | 0.014 |
| Cdh3 | 14.08 | 25.17 | +1.79 | 0.034 |
| Cldn4 | 61.00 | 98.43 | +1.61 | 0.050 |
| Dsp | 9.27 | 17.07 | +1.84 | 0.008 |
| Jak2 | 2.53 | 5.30 | +2.09 | 0.008 |
| Krt18 | 96.30 | 166.89 | +1.73 | 0.008 |
| Krt8 | 111.76 | 184.18 | +1.65 | 0.020 |
| Prss50 | 3.83 | 12.20 | +3.19 | 0.008 |
| Rec8 | 5.70 | 14.10 | +2.47 | 0.008 |
| Six1 | 3.57 | 7.03 | +1.97 | 0.025 |
| TgfB1 | 12.69 | 23.60 | +1.86 | 0.040 |
| 1700019B21Rik | 0 | 0.660 | 0.008 | |
| Downregulated | ||||
| Id2 | 39.83 | 22.78 | −1.75 | 0.046 |
| P-Rex2 | 7.02 | 3.28 | −2.14 | 0.008 |
Twelve transcripts are significantly upregulated and two are significantly downregulated in response to Ctcfl induction by doxycycline in ES cells.
The q value is the P value corrected for the false-discovery rate for multiple samples.
Each value is the number of fragments per kilobase per million reads mapped.
Of the 12 genes upregulated, 2, 1700019B21Rik and Prss50 (also known as Tsp50), were previously identified as downregulated in testicular cells from Ctcfl knockout mice (17). Thus, control of these two genes by Ctcfl has now been shown to occur in two distinct cell types by two different methods, i.e., microarray analysis in a knockout model using testicular cells and RNA-Seq analysis in our transgenic model using ES cells. 1700019B21Rik is a noncoding mRNA cloned from adult mouse testis tissue (41). Of the two genes nearest to 1700019B21Rik, one is a 223-kb gene that encodes a hypothetical MAGE family (MAGE-11-like) centromeric protein. Therefore, expression of Ctcfl results in the upregulation of an X-linked noncoding RNA whose neighbor, also cloned from testis tissue, is a member of the MAGE family that, like Ctcfl, encodes a CTA. We report 12 additional genes, not previously identified, whose expression is significantly altered after Ctcfl induction. Among the latter, DNA binding genes (Rec8 and Id2), signal transduction genes (Cited1, Jak2, and TgfB1), and a homeobox-related gene (Six1) are represented. DAVID analysis of our RNA-Seq data revealed that the TGFB pathway is primarily affected by induced Ctcfl expression in ES cells.
DISCUSSION
Novel phenotype of Ctcfl transgenic mice and comparison to previous mouse models.
Our transgenic mice displayed a lethal malformation phenotype that was not expected on the basis of information obtained from prior knockout mouse studies and our knowledge of CTCFL as a human CTA. Indeed, we observed that Ctcfl rtTA transgenic mice, rather than exhibiting oncogenesis and promotion of growth, were approximately 16% smaller than their nontransgenic littermates, had multiple-organ pathology, and died in the first few hours of life. Especially striking was the presence of ocular malformations, including ocular hemorrhaging and unfused eyelids. During normal development, eyelid fusion occurs at e15.5 to e16.5 and the lids remain fused until P10 (42). Multiple specific keratin genes are expressed in a coordinated temporal sequence before, during, and after eyelid fusion (43). A review of the literature describing mice with unfused eyelids and death at birth reveals a number of alleles involved in the TGFB signal transduction pathway that exhibit these phenotypes. For example, a human integrin transgene controlled by a human involucrin promoter results in mice with unfused eyelids (44), as does homozygous knockout of the activin-βB-encoding gene (45). Homozygous activin-βA knockout mice die at birth (46), and when those authors created mice doubly null for activin-βA and activin-βB, they were shown to have unfused eyelids and undergo perinatal death. Similarly, Brown et al. reported on double mutants that combined a homozygous knock in of the activin-βB-encoding gene at the InhbA locus with a homozygous null at the InhbB locus that resulted in mice that were small at birth and had unfused eyelids (47). Conditional removal of Smad4 or addition of transgenic SMAD7, phosphorylation targets and effectors of TGFB signaling in the mouse eye, results in corneal defects, lens cataracts, adhesion of the retina to the lens, and unfused eyelids (48), a phenotype remarkably similar to what we observed in our Ctcfl transgenic mice. On the basis of the similarities between our Ctcfl transgenic mice and prior mouse models, we suspected that the TGFB signaling pathway was significantly altered by enforced expression of Ctcfl during embryogenesis.
Some of our newborns with unfused eyelids also had externally visible unilateral ocular hemorrhaging or craniofacial subcutaneous hemorrhaging. The histology of the brain in pups with gross hemorrhaging demonstrated cerebral cavernous malformations and, in some instances, focal excessive vascular proliferation of the meninges, resulting in meningocele. Because we did not perform cerebral histology analysis of most of our transgenic pups, the true incidence of cerebral vascular malformations cannot be determined from our data, although we never observed externally visible hemorrhaging in pups with fused eyelids. This abnormal cerebral vascular proliferation with cavernous malformations and hemorrhaging again implicates the TGFB pathway, as it has been shown that in mice with endothelial-tissue-specific CCM1 (cerebral cavernous malformation) deleted, upregulation of BMP6 and the TGFB pathway results in an endothelium-to-mesenchyme transition wherein endothelial cells acquire stem cell/mesenchymal characteristics that eventually lead to cerebral cavernous malformations (49). Perturbation of a mesenchyme-to-epithelium transition is also suspected from the renal pathology of our Ctcfl transgenic mice, as the kidneys showed an increase in mesenchymal elements and a corresponding decrease in epithelial structures such as glomeruli. Abnormal vessel formation mediated by dysregulated TGFB signaling may also provide a link between the cerebral vessel malformations and the ocular phenotype we observed, as double knockout of TgfB2 and TgfB3 results in a hypercellular/hypervascular posterior chamber of the eye (50), similar to what we observed in our Ctcfl transgenic mice.
Validation of transgenic ES cells in tetraploid-ES chimeras.
Although the phenotype and histology of our Ctcfl transgenic pups indirectly implicated the TGFB signaling pathway, a more direct molecular analysis of a single cell type was desired. We chose ES cells for this analysis, as (i) we could generate an immortal cell line from the transgenic mice used in our phenotype characterization and (ii) they allow us to assess whether the abnormal phenotype is reproduced in ES-tetraploid chimeras. When Ctcfl rtTA transgenic ES cells were injected into wild-type tetraploid blastocysts and transferred to pseudopregnant females on doxycycline, late-stage fetuses and P0 newborns exactly reproduced the lethal, unfused-eyelid, and aberrant internal pathology phenotype of the original transgenic mice, thus validating their use in gene expression experiments.
Comparison of altered gene expression in knockout versus transgenic Ctcfl studies.
In molecular studies, CTCFL expression was demonstrated to be complex, with three alternative promoters leading to the transcription of 5 mRNAs that differ in their 5′ untranslated regions (51) and 23 isoforms generated by alternate splicing (52). Genomic analysis has shown that there are many more CTCF (approximately 35,000) than CTCFL (5 to 6,000) binding sites, and although CTCF can bind to approximately 60% of CTCFL sites, CTCFL binds to only 10% of CTCF sites (2). In addition to reduced Gal3st1 expression as a result of Ctcfl knockout mentioned previously, Prss50 (also known as Tsp50), and the noncoding RNA 1700019B21Rik, are also significantly reduced in Ctcfl null testes (17). Prss50, like Ctcfl, was initially described as a testicular transcript, and it also encodes a CTA reactivated in cancer cells (53, 54). Ctcfl has been shown to directly bind to two distinct Prss50 promoter sites and upregulate it when those sites are not occupied by a nucleosome (2, 55). Here, we describe RNA-Seq analysis of Ctcfl transgenic ES cells and, despite major differences in the approach and methodology used (knockout versus transgenic, microarray versus RNA-Seq, testicular versus ES cells), we confirm the vital role of Ctcfl in the regulation of Prss50 and 170019B21Rik, as the expression of both of these genes was significantly upregulated in our transgenic ES cells (Table 2). Although we did not observe upregulation of Gal3st1 in our ES cells, this is not unexpected, as regulation of this gene in the testis results in a testis-specific Ctcfl isoform (17) that presumably is not produced in ES cells. Also, downregulation of Stra8 in knockout mice, not seen in our ES cells, is also not unexpected, as Stra8 is expressed only at certain testicular developmental time points and unlikely to be seen in ES cells (2). Sleutels and colleagues reported an additional 25 deregulated genes in Ctcfl-deregulated testicular cells, although none of them were as significantly altered as Gal3st1 or Prss50. Other than Prss50, there was no overlap between the deregulated genes they observed in testicular cells and the deregulated genes we observed in ES cells. Cell-type-specific expression, alternate methodologies, and quantitative differences in altered expression may underlie these differences. However, we understand that the gene expression profile we describe pertains only to ES cells and that a different profile could be observed in different cells or tissues.
Pathway analysis of Ctcfl transgene expression.
Our RNA-Seq results (Table 2) show significant alteration of 14 genes (12 upregulated, 2 downregulated) in response to Ctcfl induction (Fig. 9). Unpredicted from the Ctcfl knockout mouse data, seven of the affected genes encode signal transducers, transcription factors, and DNA binding proteins (or regulators of DNA binding) and one is a homeobox-related gene. DAVID analysis reveals the TGFB pathway to be the most affected by embryonic Ctcfl expression, as TGFB itself is significantly upregulated and five of the affected genes intersect directly or indirectly with TGFB. For example, we observed upregulation of CITED1, a transcriptional coactivator of the TFE3 transcription factor that, upon binding to TGFB response elements (TGFBRE), recruits SMAD3 and -4 to mediate the TGFB response (56). The KRT8 and KRT18 genes (contiguous and coordinately regulated) were also upregulated in our transgenic ES cells. Notably, mice transgenic for human KRT8 (57) phenocopy dominant negative TGFBRII mutant mice (58) and dominant negative TGFBRII mice upregulate Krt8 and Krt18 (57). Finally, we observed repression of the Id2 (inhibitor of DNA binding 2) gene by Ctcfl. TGFB is a known repressor of Id2 (59), so decreased Id2 expression in our Ctcfl transgenic ES cells may result from Ctcfl upregulation, leading to increased TGFB and consequent downregulation of Id2. Thus, pathway analysis and knowledge regarding 5 of the 14 Ctcfl-altered genes link Ctcfl expression to the TGFB pathway, confirming our phenotype-based suspicion that TGFB is involved in the ocular, abnormal vasculogenesis, and perturbed mesenchyme-to-epithelium transition phenotypes observed in our transgenic mice.
FIG 9.
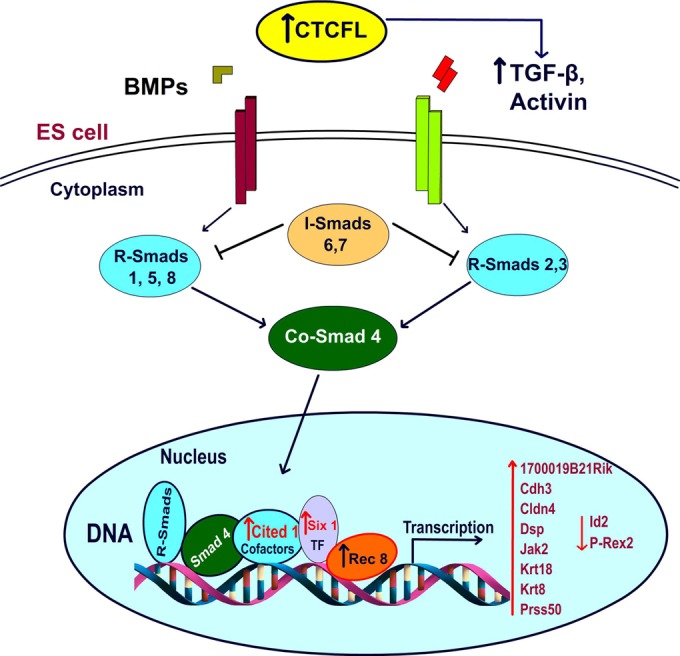
A hypothetical schema of the signal flow after application of CTCFL overexpression in ES cells is shown. RNA-Seq data revealed the TGFB pathway as most affected by embryonic Ctcfl expression.TF, transcription factor.
Of interest, one of the upregulated genes, Rec8, localizes to meiotic chromosomes and its product is part of the cohesin complex that joins sister chromatids at the site of chiasmata in spermatocytes and oocytes. When Rec8 is absent in mice, male and female sterility results (60). Immediately after fertilization, Rec8 is replaced by Scc1 (also known as Rad21) (61). Thus, a cohesin complex protein that is believed to function primarily in meiosis is upregulated in our Ctcfl transgenic ES cells and presumably in Ctcfl transgenic fetuses. We doubt, however, that expression of Rec8 in our transgenic animals explains the abnormal lethal phenotype, as Rec8 transgenic mice are viable, fertile, and seemingly normal (62). Finally, our data lead one to question whether the TGFB pathway may play a greater role in spermatogenesis than previously suspected. Dissection of TGFB signaling in gonad and germ cell development is made difficult by the functional redundancy of the multiple members of this superfamily. TGFB and its receptors are expressed in the developing testis, but elimination of any single ligand or receptor does not block testis development. Recently, however, use of the TGFB signaling antagonists ALK4, -5, and -7 was shown to block testis development, thus leading to a renewed interest in TGFB's role in male germ line development (63).
CTCFL in human vascular pathology.
Two benign human vascular malformations unexpectedly express CTCFL, JAs (32) and IHs (Schultz et al., submitted). JAs are fibrovascular tumors that arise in the nasal cavity in adolescent males. In some cases, there is CTCFL gene duplication (64). JAs exhibit increased levels of VEGF, insulin-like growth factor 2 (IGF2), TGFB, and other growth factors (65). In IHs, a vascular proliferation seen predominantly in females, CTCFL is again overexpressed (Schultz et al., submitted). Like JAs, IHs are also known to overexpress IGF2 (66). However, Yu et al. did not observe loss of imprinting of IGF2 in IHs, despite CTCF's role as a chromatin insulator element at IGF2 differentially methylated region I (DMRI) (67, 68), making it unlikely that CTCFL directly alters methylation and CTCF binding at this site. Our results offer an alternative explanation wherein CTCFL may deregulate the TGFB pathway, which, combined with IGF2 overexpression, leads to IH vascular proliferation. Our results, obtained with a Ctcfl mouse transgenic model, combined with two known instances of human CTCFL overexpression in benign vascular malformations, warrants the consideration of CTCFL somatic expression in additional human vascular malformations.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the Yale Comprehensive Cancer Center, New Haven, CT, with a grant to J.M., by the Scientific and Technological Research Council of Turkey (TUBITAK) (SBAG 110S383 and SBAG 213S109), and by Akdeniz University, Scientific Research Projects Coordination Unit (2008.03.0122.004).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00381-15.
REFERENCES
- 1.Loukinov DI, Pugacheva E, Vatolin S, Pack SD, Moon H, Chernukhin I, Mannan P, Larsson E, Kanduri C, Vostrov AA, Cui H, Niemitz EL, Rasko JE, Docquier FM, Kistler M, Breen JJ, Zhuang Z, Quitschke WW, Renkawitz R, Klenova EM, Feinberg AP, Ohlsson R, Morse HC III, Lobanenkov VV. 2002. BORIS, a novel male germ-line-specific protein associated with epigenetic reprogramming events, shares the same 11-zinc-finger domain with CTCF, the insulator protein involved in reading imprinting marks in the soma. Proc Natl Acad Sci U S A 99:6806–6811. doi: 10.1073/pnas.092123699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sleutels F, Soochit W, Bartkuhn M, Heath H, Dienstbach S, Bergmaier P, Franke V, Rosa-Garrido M, van de Nobelen S, Caesar L, van der Reijden M, Bryne JC, van Ijcken W, Grootegoed JA, Delgado MD, Lenhard B, Renkawitz R, Grosveld F, Galjart N. 2012. The male germ cell gene regulator CTCFL is functionally different from CTCF and binds CTCF-like consensus sites in a nucleosome composition-dependent manner. Epigenetics Chromatin 5:8. doi: 10.1186/1756-8935-5-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Campbell AE, Martinez SR, Miranda JJ. 2010. Molecular architecture of CTCFL. Biochem Biophys Res Commun 396:648–650. doi: 10.1016/j.bbrc.2010.04.146. [DOI] [PubMed] [Google Scholar]
- 4.Ong CT, Corces VG. 2014. CTCF: an architectural protein bridging genome topology and function. Nat Rev Genet 15:234–246. doi: 10.1038/nrg3663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baniahmad A, Steiner C, Kohne AC, Renkawitz R. 1990. Modular structure of a chicken lysozyme silencer: involvement of an unusual thyroid hormone receptor binding site. Cell 61:505–514. doi: 10.1016/0092-8674(90)90532-J. [DOI] [PubMed] [Google Scholar]
- 6.Lobanenkov VV, Nicolas RH, Adler VV, Paterson H, Klenova EM, Polotskaja AV, Goodwin GH. 1990. A novel sequence-specific DNA binding protein which interacts with three regularly spaced direct repeats of the CCCTC-motif in the 5′-flanking sequence of the chicken c-myc gene. Oncogene 5:1743–1753. [PubMed] [Google Scholar]
- 7.Filippova GN, Fagerlie S, Klenova EM, Myers C, Dehner Y, Goodwin G, Neiman PE, Collins SJ, Lobanenkov VV. 1996. An exceptionally conserved transcriptional repressor, CTCF, employs different combinations of zinc fingers to bind diverged promoter sequences of avian and mammalian c-myc oncogenes. Mol Cell Biol 16:2802–2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fu Y, Sinha M, Peterson CL, Weng Z. 2008. The insulator binding protein CTCF positions 20 nucleosomes around its binding sites across the human genome. PLoS Genet 4:e1000138. doi: 10.1371/journal.pgen.1000138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Filippova GN. 2008. Genetics and epigenetics of the multifunctional protein CTCF. Curr Top Dev Biol 80:337–360. [DOI] [PubMed] [Google Scholar]
- 10.Chao W, Huynh KD, Spencer RJ, Davidow LS, Lee JT. 2002. CTCF, a candidate trans-acting factor for X-inactivation choice. Science 295:345–347. doi: 10.1126/science.1065982. [DOI] [PubMed] [Google Scholar]
- 11.Donohoe ME, Zhang LF, Xu N, Shi Y, Lee JT. 2007. Identification of a Ctcf cofactor, Yy1, for the X chromosome binary switch. Mol Cell 25:43–56. doi: 10.1016/j.molcel.2006.11.017. [DOI] [PubMed] [Google Scholar]
- 12.Deng Z, Wang Z, Stong N, Plasschaert R, Moczan A, Chen HS, Hu S, Wikramasinghe P, Davuluri RV, Bartolomei MS, Riethman H, Lieberman PM. 2012. A role for CTCF and cohesin in subtelomere chromatin organization, TERRA transcription, and telomere end protection. EMBO J 31:4165–4178. doi: 10.1038/emboj.2012.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heath H, Ribeiro de Almeida C, Sleutels F, Dingjan G, van de Nobelen S, Jonkers I, Ling KW, Gribnau J, Renkawitz R, Grosveld F, Hendriks RW, Galjart N. 2008. CTCF regulates cell cycle progression of alphabeta T cells in the thymus. EMBO J 27:2839–2850. doi: 10.1038/emboj.2008.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moore JM, Rabaia NA, Smith LE, Fagerlie S, Gurley K, Loukinov D, Disteche CM, Collins SJ, Kemp CJ, Lobanenkov VV, Filippova GN. 2012. Loss of maternal CTCF is associated with peri-implantation lethality of Ctcf null embryos. PLoS One 7:e34915. doi: 10.1371/journal.pone.0034915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fedoriw AM, Stein P, Svoboda P, Schultz RM, Bartolomei MS. 2004. Transgenic RNAi reveals essential function for CTCF in H19 gene imprinting. Science 303:238–240. doi: 10.1126/science.1090934. [DOI] [PubMed] [Google Scholar]
- 16.Wan LB, Pan H, Hannenhalli S, Cheng Y, Ma J, Fedoriw A, Lobanenkov V, Latham KE, Schultz RM, Bartolomei MS. 2008. Maternal depletion of CTCF reveals multiple functions during oocyte and preimplantation embryo development. Development 135:2729–2738. doi: 10.1242/dev.024539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Suzuki T, Kosaka-Suzuki N, Pack S, Shin DM, Yoon J, Abdullaev Z, Pugacheva E, Morse HC III, Loukinov D, Lobanenkov V. 2010. Expression of a testis-specific form of Gal3st1 (CST), a gene essential for spermatogenesis, is regulated by the CTCF paralogous gene BORIS. Mol Cell Biol 30:2473–2484. doi: 10.1128/MCB.01093-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Honke K, Hirahara Y, Dupree J, Suzuki K, Popko B, Fukushima K, Fukushima J, Nagasawa T, Yoshida N, Wada Y, Taniguchi N. 2002. Paranodal junction formation and spermatogenesis require sulfoglycolipids. Proc Natl Acad Sci U S A 99:4227–4232. doi: 10.1073/pnas.032068299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martin-Kleiner I. 2012. BORIS in human cancers—a review. Eur J Cancer 48:929–935. doi: 10.1016/j.ejca.2011.09.009. [DOI] [PubMed] [Google Scholar]
- 20.Vatolin S, Abdullaev Z, Pack SD, Flanagan PT, Custer M, Loukinov DI, Pugacheva E, Hong JA, Morse H III, Schrump DS, Risinger JI, Barrett JC, Lobanenkov VV. 2005. Conditional expression of the CTCF-paralogous transcriptional factor BORIS in normal cells results in demethylation and derepression of MAGE-A1 and reactivation of other cancer-testis genes. Cancer Res 65:7751–7762. [DOI] [PubMed] [Google Scholar]
- 21.Hong JA, Kang Y, Abdullaev Z, Flanagan PT, Pack SD, Fischette MR, Adnani MT, Loukinov DI, Vatolin S, Risinger JI, Custer M, Chen GA, Zhao M, Nguyen DM, Barrett JC, Lobanenkov VV, Schrump DS. 2005. Reciprocal binding of CTCF and BORIS to the NY-ESO-1 promoter coincides with derepression of this cancer-testis gene in lung cancer cells. Cancer Res 65:7763–7774. [DOI] [PubMed] [Google Scholar]
- 22.Kang Y, Hong JA, Chen GA, Nguyen DM, Schrump DS. 2007. Dynamic transcriptional regulatory complexes including BORIS, CTCF and Sp1 modulate NY-ESO-1 expression in lung cancer cells. Oncogene 26:4394–4403. doi: 10.1038/sj.onc.1210218. [DOI] [PubMed] [Google Scholar]
- 23.Kholmanskikh O, Loriot A, Brasseur F, De Plaen E, De Smet C. 2008. Expression of BORIS in melanoma: lack of association with MAGE-A1 activation. Int J Cancer 122:777–784. doi: 10.1002/ijc.23140. [DOI] [PubMed] [Google Scholar]
- 24.Looijenga LH, Hersmus R, Gillis AJ, Pfundt R, Stoop HJ, van Gurp RJ, Veltman J, Beverloo HB, van Drunen E, van Kessel AG, Pera RR, Schneider DT, Summersgill B, Shipley J, McIntyre A, van der Spek P, Schoenmakers E, Oosterhuis JW. 2006. Genomic and expression profiling of human spermatocytic seminomas: primary spermatocyte as tumorigenic precursor and DMRT1 as candidate chromosome 9 gene. Cancer Res 66:290–302. doi: 10.1158/0008-5472.CAN-05-2936. [DOI] [PubMed] [Google Scholar]
- 25.Risinger JI, Chandramouli GV, Maxwell GL, Custer M, Pack S, Loukinov D, Aprelikova O, Litzi T, Schrump DS, Murphy SK, Berchuck A, Lobanenkov V, Barrett JC. 2007. Global expression analysis of cancer/testis genes in uterine cancers reveals a high incidence of BORIS expression. Clin Cancer Res 13:1713–1719. doi: 10.1158/1078-0432.CCR-05-2569. [DOI] [PubMed] [Google Scholar]
- 26.Woloszynska-Read A, James SR, Link PA, Yu J, Odunsi K, Karpf AR. 2007. DNA methylation-dependent regulation of BORIS/CTCFL expression in ovarian cancer. Cancer Immun 7:21. [PMC free article] [PubMed] [Google Scholar]
- 27.Link PA, Zhang W, Odunsi K, Karpf AR. 2013. BORIS/CTCFL mRNA isoform expression and epigenetic regulation in epithelial ovarian cancer. Cancer Immun 13:6. [PMC free article] [PubMed] [Google Scholar]
- 28.Hoivik EA, Kusonmano K, Halle MK, Berg A, Wik E, Werner HM, Petersen K, Oyan AM, Kalland KH, Krakstad C, Trovik J, Widschwendter M, Salvesen HB. 2014. Hypomethylation of the CTCFL/BORIS promoter and aberrant expression during endometrial cancer progression suggests a role as an Epi-driver gene. Oncotarget 5:1052–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cheema Z, Hari-Gupta Y, Kita GX, Farrar D, Seddon I, Corr J, Klenova E. 2014. Expression of the cancer-testis antigen BORIS correlates with prostate cancer. Prostate 74:164–176. doi: 10.1002/pros.22738. [DOI] [PubMed] [Google Scholar]
- 30.Chen K, Huang W, Huang B, Wei Y, Li B, Ge Y, Qin Y. 2013. BORIS, brother of the regulator of imprinted sites, is aberrantly expressed in hepatocellular carcinoma. Genet Test Mol Biomarkers 17:160–165. doi: 10.1089/gtmb.2012.0242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Okabayashi K, Fujita T, Miyazaki J, Okada T, Iwata T, Hirao N, Noji S, Tsukamoto N, Goshima N, Hasegawa H, Takeuchi H, Ueda M, Kitagawa Y, Kawakami Y. 2012. Cancer-testis antigen BORIS is a novel prognostic marker for patients with esophageal cancer. Cancer Sci 103:1617–1624. doi: 10.1111/j.1349-7006.2012.02355.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schick B, Wemmert S, Willnecker V, Dlugaiczyk J, Nicolai P, Siwiec H, Thiel CT, Rauch A, Wendler O. 2011. Genome-wide copy number profiling using a 100K SNP array reveals novel disease-related genes BORIS and TSHZ1 in juvenile angiofibroma. Int J Oncol 39:1143–1151. [DOI] [PubMed] [Google Scholar]
- 33.Biggers JD, McGinnis LK, Raffin M. 2000. Amino acids and preimplantation development of the mouse in protein-free potassium simplex optimized medium. Biol Reprod 63:281–293. doi: 10.1095/biolreprod63.1.281. [DOI] [PubMed] [Google Scholar]
- 34.Chen Z, Liu Z, Huang J, Amano T, Li C, Cao S, Wu C, Liu B, Zhou L, Carter MG, Keefe DL, Yang X, Liu L. 2009. Birth of parthenote mice directly from parthenogenetic embryonic stem cells. Stem Cells 27:2136–2145. doi: 10.1002/stem.158. [DOI] [PubMed] [Google Scholar]
- 35.Nagy A, Rossant J, Nagy R, Abramow-Newerly W, Roder JC. 1993. Derivation of completely cell culture-derived mice from early-passage embryonic stem cells. Proc Natl Acad Sci U S A 90:8424–8428. doi: 10.1073/pnas.90.18.8424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 37.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. 2013. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L. 2013. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol 31:46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang da W, Sherman BT, Lempicki RA. 2009. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang da W, Sherman BT, Lempicki RA. 2009. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4:44–57. [DOI] [PubMed] [Google Scholar]
- 41.Kawai J, Shinagawa A, Shibata K, Yoshino M, Itoh M, Ishii Y, Arakawa T, Hara A, Fukunishi Y, Konno H, Adachi J, Fukuda S, Aizawa K, Izawa M, Nishi K, Kiyosawa H, Kondo S, Yamanaka I, Saito T, Okazaki Y, Gojobori T, Bono H, Kasukawa T, Saito R, Kadota K, Matsuda H, Ashburner M, Batalov S, Casavant T, Fleischmann W, Gaasterland T, Gissi C, King B, Kochiwa H, Kuehl P, Lewis S, Matsuo Y, Nikaido I, Pesole G, Quackenbush J, Schriml LM, Staubli F, Suzuki R, Tomita M, Wagner L, Washio T, Sakai K, Okido T, Furuno M, Aono H, et al. . 2001. Functional annotation of a full-length mouse cDNA collection. Nature 409:685–690. doi: 10.1038/35055500. [DOI] [PubMed] [Google Scholar]
- 42.Findlater GS, McDougall RD, Kaufman MH. 1993. Eyelid development, fusion and subsequent reopening in the mouse. J Anat 183(Pt 1):121–129. [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang H, Hara M, Seki K, Fukuda K, Nishida T. 2005. Eyelid fusion and epithelial differentiation at the ocular surface during mouse embryonic development. Jpn J Ophthalmol 49:195–204. doi: 10.1007/s10384-004-0189-1. [DOI] [PubMed] [Google Scholar]
- 44.Carroll JM, Romero MR, Watt FM. 1995. Suprabasal integrin expression in the epidermis of transgenic mice results in developmental defects and a phenotype resembling psoriasis. Cell 83:957–968. doi: 10.1016/0092-8674(95)90211-2. [DOI] [PubMed] [Google Scholar]
- 45.Vassalli A, Matzuk MM, Gardner HA, Lee KF, Jaenisch R. 1994. Activin/inhibin beta B subunit gene disruption leads to defects in eyelid development and female reproduction. Genes Dev 8:414–427. doi: 10.1101/gad.8.4.414. [DOI] [PubMed] [Google Scholar]
- 46.Matzuk MM, Kumar TR, Vassalli A, Bickenbach JR, Roop DR, Jaenisch R, Bradley A. 1995. Functional analysis of activins during mammalian development. Nature 374:354–356. doi: 10.1038/374354a0. [DOI] [PubMed] [Google Scholar]
- 47.Brown CW, Li L, Houston-Hawkins DE, Matzuk MM. 2003. Activins are critical modulators of growth and survival. Mol Endocrinol 17:2404–2417. doi: 10.1210/me.2003-0051. [DOI] [PubMed] [Google Scholar]
- 48.Liu Y, Kawai K, Khashabi S, Deng C, Liu YH, Yiu S. 2010. Inactivation of Smad4 leads to impaired ocular development and cataract formation. Biochem Biophys Res Commun 400:476–482. doi: 10.1016/j.bbrc.2010.08.065. [DOI] [PubMed] [Google Scholar]
- 49.Maddaluno L, Rudini N, Cuttano R, Bravi L, Giampietro C, Corada M, Ferrarini L, Orsenigo F, Papa E, Boulday G, Tournier-Lasserve E, Chapon F, Richichi C, Retta SF, Lampugnani MG, Dejana E. 2013. EndMT contributes to the onset and progression of cerebral cavernous malformations. Nature 498:492–496. doi: 10.1038/nature12207. [DOI] [PubMed] [Google Scholar]
- 50.Dünker N, Krieglstein K. 2003. Reduced programmed cell death in the retina and defects in lens and cornea of Tgfbeta2(−/−) Tgfbeta3(−/−) double-deficient mice. Cell Tissue Res 313:1–10. doi: 10.1007/s00441-003-0761-x. [DOI] [PubMed] [Google Scholar]
- 51.Renaud S, Pugacheva EM, Delgado MD, Braunschweig R, Abdullaev Z, Loukinov D, Benhattar J, Lobanenkov V. 2007. Expression of the CTCF-paralogous cancer-testis gene, brother of the regulator of imprinted sites (BORIS), is regulated by three alternative promoters modulated by CpG methylation and by CTCF and p53 transcription factors. Nucleic Acids Res 35:7372–7388. doi: 10.1093/nar/gkm896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pugacheva EM, Suzuki T, Pack SD, Kosaka-Suzuki N, Yoon J, Vostrov AA, Barsov E, Strunnikov AV, Morse HC III, Loukinov D, Lobanenkov V. 2010. The structural complexity of the human BORIS gene in gametogenesis and cancer. PLoS One 5:e13872. doi: 10.1371/journal.pone.0013872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yuan L, Shan J, De Risi D, Broome J, Lovecchio J, Gal D, Vinciguerra V, Xu HP. 1999. Isolation of a novel gene, TSP50, by a hypomethylated DNA fragment in human breast cancer. Cancer Res 59:3215–3221. [PubMed] [Google Scholar]
- 54.Shan J, Yuan L, Xiao Q, Chiorazzi N, Budman D, Teichberg S, Xu HP. 2002. TSP50, a possible protease in human testes, is activated in breast cancer epithelial cells. Cancer Res 62:290–294. [PubMed] [Google Scholar]
- 55.Kosaka-Suzuki N, Suzuki T, Pugacheva EM, Vostrov AA, Morse HC III, Loukinov D, Lobanenkov V. 2011. Transcription factor BORIS (brother of the regulator of imprinted sites) directly induces expression of a cancer-testis antigen, TSP50, through regulated binding of BORIS to the promoter. J Biol Chem 286:27378–27388. doi: 10.1074/jbc.M111.243576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yahata T, de Caestecker MP, Lechleider RJ, Andriole S, Roberts AB, Isselbacher KJ, Shioda T. 2000. The MSG1 non-DNA-binding transactivator binds to the p300/CBP coactivators, enhancing their functional link to the Smad transcription factors. J Biol Chem 275:8825–8834. doi: 10.1074/jbc.275.12.8825. [DOI] [PubMed] [Google Scholar]
- 57.Casanova ML, Bravo A, Ramirez A, Morreale de Escobar G, Were F, Merlino G, Vidal M, Jorcano JL. 1999. Exocrine pancreatic disorders in transgenic mice expressing human keratin 8. J Clin Invest 103:1587–1595. doi: 10.1172/JCI5343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Böttinger EP, Jakubczak JL, Roberts IS, Mumy M, Hemmati P, Bagnall K, Merlino G, Wakefield LM. 1997. Expression of a dominant-negative mutant TGF-beta type II receptor in transgenic mice reveals essential roles for TGF-beta in regulation of growth and differentiation in the exocrine pancreas. EMBO J 16:2621–2633. doi: 10.1093/emboj/16.10.2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lasorella A, Noseda M, Beyna M, Yokota Y, Iavarone A. 2000. Id2 is a retinoblastoma protein target and mediates signalling by Myc oncoproteins. Nature 407:592–598. doi: 10.1038/35036504. [DOI] [PubMed] [Google Scholar]
- 60.Xu H, Beasley MD, Warren WD, van der Horst GT, McKay MJ. 2005. Absence of mouse REC8 cohesin promotes synapsis of sister chromatids in meiosis. Dev Cell 8:949–961. doi: 10.1016/j.devcel.2005.03.018. [DOI] [PubMed] [Google Scholar]
- 61.Tachibana-Konwalski K, Godwin J, van der Weyden L, Champion L, Kudo NR, Adams DJ, Nasmyth K. 2010. Rec8-containing cohesin maintains bivalents without turnover during the growing phase of mouse oocytes. Genes Dev 24:2505–2516. doi: 10.1101/gad.605910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kudo NR, Anger M, Peters AH, Stemmann O, Theussl HC, Helmhart W, Kudo H, Heyting C, Nasmyth K. 2009. Role of cleavage by separase of the Rec8 kleisin subunit of cohesin during mammalian meiosis I. J Cell Sci 122:2686–2698. doi: 10.1242/jcs.035287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Miles DC, Wakeling SI, Stringer JM, van den Bergen JA, Wilhelm D, Sinclair AH, Western PS. 2013. Signaling through the TGF beta-activin receptors ALK4/5/7 regulates testis formation and male germ cell development. PLoS One 8:e54606. doi: 10.1371/journal.pone.0054606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Scanlan MJ, Simpson AJ, Old LJ. 2004. The cancer/testis genes: review, standardization, and commentary. Cancer Immun 4:1. [PubMed] [Google Scholar]
- 65.Saylam G, Yucel OT, Sungur A, Onerci M. 2006. Proliferation, angiogenesis and hormonal markers in juvenile nasopharyngeal angiofibroma. Int J Pediatr Otorhinolaryngol 70:227–234. doi: 10.1016/j.ijporl.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 66.Ritter MR, Dorrell MI, Edmonds J, Friedlander SF, Friedlander M. 2002. Insulin-like growth factor 2 and potential regulators of hemangioma growth and involution identified by large-scale expression analysis. Proc Natl Acad Sci U S A 99:7455–7460. doi: 10.1073/pnas.102185799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hark AT, Schoenherr CJ, Katz DJ, Ingram RS, Levorse JM, Tilghman SM. 2000. CTCF mediates methylation-sensitive enhancer-blocking activity at the H19/Igf2 locus. Nature 405:486–489. doi: 10.1038/35013106. [DOI] [PubMed] [Google Scholar]
- 68.Yu Y, Wylie-Sears J, Boscolo E, Mulliken JB, Bischoff J. 2004. Genomic imprinting of IGF2 is maintained in infantile hemangioma despite its high level of expression. Mol Med 10:117–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.