

Abstract
Hepatitis C virus (HCV) infection causes tremendous morbidity and mortality with over 170 million people infected worldwide. HCV gives rise to a sustained, chronic disease in the majority of infected individuals owing to a failure of the host immune system to clear the virus. In general, an adequate immune response is elicited by an efficient antigen presentation by dendritic cells (DCs), the cells that connect innate and adaptive immune system to generate a specific immune response against a pathogen. However, HCV seems to dysregulate the activity of DCs, making them less proficient antigen presenting cells for the optimal stimulation of virus-specific T cells, hence interfering with an optimal anti-viral immune response. There are discordant reports on the functional status of DCs in chronic HCV infection (CHC), from no phenotypic or functional defects to abnormal functions of DCs. Furthermore, the molecular mechanisms behind the impairment of DC function are even so not completely elucidated during CHC. Understanding the mechanisms of immune dysfunction would help in devising strategies for better management of the disease at the immunological level and help to predict the prognosis of the disease in the patients receiving antiviral therapy. In this review, we have discussed the outcomes of the interaction of DCs with HCV and the mechanisms of DC impairment during HCV infection with its adverse effects on the immune response in the infected host.
Keywords: Dendritic cells, Hepatitis C, Mechanism of functional impairment
Core tip: Infection with hepatitis C virus (HCV) is linked with serious outcome like chronic hepatitis in the majority of infected cases, leading to severe liver necrosis and an increased threat of cirrhosis and hepatocellular carcinoma. An aberrant signalling through an inefficient antigen presentation by dendritic cells (DCs) can lead to a subdued T cell immune response. There is a need to completely understand the mechanistic aspects of DC impairment during HCV infection so as to harness this critical arm of the immune system for successful resolution of disease.
INTRODUCTION
Hepatitis C virus (HCV) causes a persistent infection in the majority of infected humans, accounting for chronic liver diseases, cirrhosis, and hepatocellular carcinoma. Hepatitis may occur with limited or no symptoms, but often leads to jaundice, anorexia and malaise. The infection can be either acute which lasts for less than 6 mo or chronic hepatitis C (CHC), which lasts longer than 6 mo[1]. Till date, the reasons why some people are able to resolve the infection spontaneously, while others do not and go on to establish a chronic infection are not well defined. As the disease commences, the viral load (VL) increases rapidly, but the host immune response lags behind, with adaptive immune response appearing only after a month and humoral immune response after about 2 mo[2]. After few weeks of infection, the rate of increase in the VL slows down and in approximately 8-12 wk of infection, when serum alanine aminotransferase levels peak, the VL decline, HCV-specific antibodies may or may not become detectable at this stage. Most individuals develop a persistent, chronic infection with stable VL keeping 2-3 logs lower than the acute stage.
The mechanisms by which HCV establishes a chronic infection have yet not been completely delineated. Various theories have been set forth to explain the link between an inefficient cellular immune response and the establishment of a chronic infection, including rapid replication of HCV, which eviscerates the immune system[3]; the production of immunomodulatory proteins by HCV[4,5]; and inability of the body’s immune response to persuade opportune priming of naive-T-cells[6]. Moreover, HCV succeeds in disrupting the coordination between the components of the innate immune system, subsequently resulting in a deficient adaptive immune response[7]. Consequently, the host’s immune system is not able to clear the infection and fails to generate protective cellular immunity against the virus.
Dendritic cells (DCs) are the most potent antigen presenting cells (APCs), which connect innate and adaptive immune system to generate a specific immune response against a pathogen. HCV appears to disrupt the activity of DCs, making them less capable as an APC for the stimulation of virus-specific T cells and could thus delay the propagation of an effective immune response against the virus. There are discordant reports on the functional status of DCs in chronic HCV infection, with some studies reporting no phenotypic or functional defects in circulating DCs of chronically infected HCV patients[8-11], others indicate functionally and numerically impaired DCs[12-15]. Thus, it is important to reconcile these findings so as to reach a consensus on the status of DC functions to better explore its applicability in improving the overall immune response against the virus.
The molecular mechanisms behind the impairment of DC function are still not completely elucidated during chronic HCV infection. There are possibilities that the components of HCV through their interaction with DCs, may dysregulate their functional abilities or impair the maturation of DCs with a reduction in T helper 1 (Th1) cytokines or lead to apoptosis of these cells. These processes can also alter the expression of costimulatory/inhibitory receptors, thereby interfering with the allo-stimulatory abilities of DCs. The toll-like receptor (TLR)-nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signalling pathways are altered, leading to a downstream reduction of interleukin 12 (IL-12) secretion[16,17]. These pathways are also influenced by the suppressor of cytokine signalling (SOCS) family of proteins[18]. This review highlights the outcomes of the interaction of DCs with HCV and the mechanisms of DC impairment during HCV infection with its down-modulatory effects on the immune response of the infected host.
DCs as warriors against infection
The response of DCs to any infection in general and HCV in particular, at an early stage, is vital in shaping up the course and final outcome of the disease. DCs are the most efficient inducers of optimal immune responses and are capable of either inducing protective immunity against the non-self antigens or tolerance to self-antigens. DCs recognize microorganisms through pattern recognition receptors (PRRs) like TLRs, nucleotide-binding oligomerization domain-like receptors, retinoic acid inducible gene-I (RIG-I) like receptors and C-type lectin receptors[19]. The pathogen associated molecular pattern (PAMP) signature of HCV includes poly-uridine motifs and stem-loop double-stranded RNA (dsRNA) structures within its single-stranded RNA genome. The product of RIG-I which has been defined as a dsRNA PAMP receptor is critical for transducing HCV-induced signals in the host to activate immune responses towards HCV[20,21]. Following the interaction of PAMP with its cognate receptor there is downstream activation of genes like Interferon regulatory factor-3 and NF-κB, resulting in the expression of interferon beta (IFNβ) and its secretion from the infected cells. NF-κB activation and function is central to the chemokine and proinflammatory cytokine response to virus infection, which functions side by side with IFNβ to modulate the ensuing adaptive immune response. As soon as DCs encounter a pathogen, the expression of various molecules like the major histocompatibility complex (MHC) I and II, co-stimulatory molecules (CD80 and CD86) is increased on the DC surface along with increased secretion of Th1 cytokines like IL-12. As the DCs mature, they lose their ability to phagocytose the antigen and become good APCs. This process is guided by the changes in the expression of certain chemokine receptors that is required for their migration from the periphery to regional lymph nodes where they encounter naive-T cells. Endogenous antigens are processed and presented along with MHC I to CD8+ T cells, while the exogenous antigens are loaded onto MHC II for presentation to CD4+ T cells. DCs possess a unique ability whereby exogenous antigens can also be presented through the MHC I, known as cross-presentation of antigens. The processed peptides complexed with MHC molecules interact with T cell receptor accompanied by the binding of co-stimulatory molecules with CD28 on T cells providing appropriate signals for T cell activation. Eventually, the cytokines are produced from DCs, that determine the differentiation of effector cells into Th1, Th2 or cytotoxic T cells[22]. The interaction of CD40 on DCs with CD40L on T cells is also required by DCs for appropriate T cell activation[23]. These DCs also promote the survival and differentiation of cytotoxic T lymphocytes via the crosslinking of CD137L (4-1BBL), which is a co-stimulatory immune-checkpoint molecule, with CD137 on T cells[24].
Various subtypes of DCs are reported in the human body that perform different functions[25]. DCs are resident in tissues such as spleen and lymph nodes, those residing in the skin are known as Langerhans cells that migrate from non-lymphoid organs such as skin, intestines and lungs to lymph nodes to present tissue derived antigens to T cells. Plasmacytoid DCs (pDCs) and myeloid (mDCs) or monocyte-derived DCs (mo-DCs) may be present in various tissues, yet they mainly circulate in the blood. The pDCs are known as major producers of type I interferons in response to virus-associated molecules such as single-stranded RNA and unmethylated cytosine-phosphate-guanine-rich DNA that trigger TLR7 and TLR9, respectively[26]. Myeloid DCs on the other hand, represent the major fraction of APCs in the blood that responds to TLR ligation by producing IL-12[27].
Status of DCs during hepatitis C infection
DCs are known to get infected with the HCV as RNA of several genotypes have been previously detected in the blood of chronically infected subjects[28]. Moreover, the DCs express DC-specific intercellular adhesion molecule-3-grabbing nonintegrin receptor that is used for the uptake of HCV[29]. Patients with chronic HCV infection have been reported to have decreased frequencies of peripheral mDC and pDC[15,30]. The counts of DCs in circulation, however, do not necessarily reflect the total DC compartment because of the migration of DCs from the periphery to the site of infection.
Existing literature describes controversial reports regarding the interaction of DCs with HCV. Multiple defects like the reduced DC frequency, decreased expression of MHC molecules, deficient expression of co-stimulatory molecules, defects in the allo-stimulatory abilities, aberrant secretion of cytokines with a preponderance of immune-regulatory cytokines like IL-10 or transforming growth factor-beta that mainly induce the regulatory T cells have been observed in CHC patients[31,32]. The level of Th1 promoting cytokine like IL-12 is reportedly found at low levels, whereas the level of IL-10 is increased. This cytokine profile affects the allo-stimulatory abilities of DCs to induce lymphocyte proliferation as observed in cocultures of DCs with T cells. Patients with detectable HCV RNA had circulating DCs with significantly decreased capacity to stimulate allogeneic T lymphocytes and produce low IL-12 as compared to patients on anti-viral therapy with undetectable RNA, suggesting the important role of therapy in restoration of DC functions[33]. The effect of anti-viral therapy is augmented in patients with intact DC pathogen recognizing functions indicating a direct association of DC functional status with response to anti-viral treatment[34]. Our own study on CHC patients suggested that DCs of only those patients achieved sustained virological response, in whom the DCs exhibited mature and functional phenotype prior to therapy initiation, indicating functional modulation of defective DCs to be directly associated with successful response to therapy[35]. This also seems to predict the clinical efficacy of anti-viral drugs and is also influenced by the extent to which HCV inhibits DC functioning.
Mechanism of DC impairment
The immunosuppressive strategies adopted by HCV to interfere with DC functioning and subsequent generation of adaptive effector cell responses indicate that the components of HCV including the HCV proteins interact with immune components of the host including DCs and possibly suppress the protective immunity against viral infection. In fact, the interaction of DCs with HCV core protein had a negative impact on the function of DCs as the exposure to this protein was able to inhibit TLR4-induced IL-12 secretion through its interaction with the gC1q receptor on the surface of mo-DCs by activating the phosphatidyl inositol 3-kinase (PI3K) pathway, leading to a hampered differentiation of Th1 cells[36,37]. Exposure of extracellular HCV core antigens to DCs also transduced signals leading to phosphorylation of signal transducer and activator of transcription (STAT)3, that dampened the T helper immune response through activation of PI3K/AKT signalling pathway[38]. STAT3 activation is also related to generation of myeloid suppressor cells, which through their immunosuppressive factors, restrains cell-mediated immune responses at the local inflammatory site[39].
HCV impairs the activation of DCs via select PRRs by reversibly interfering with Toll/IL-1 domain-containing adapter-inducing IFNγ (TRIF) and IFNβ promoter stimulator-1-dependent signal processing during chronic infection, which leads to the exhaustive functioning of HCV-specific CD8+ T cells (i.e., loss of IL-2 secretion and degranulation marker, CD107a)[40]. Thus, subjects in whom PRR signalling in DCs was intact exhibited enhanced polyfunctionality (i.e., increased secretion of IL-2 and expression of CD107a).
Another important parameter contributing to exhaustion of DCs during HCV infection is the expression profile of receptors with inhibitory function, such as programmed death ligand 1 (PD-L1)[41]. A balanced expression of costimulatory and inhibitory molecules on DCs governs the stimulatory signals delivered to T cells for their activation and regulates immunity vs tolerance[42]. Increased expression of co-stimulatory markers (such as CD80, CD86, and CD40) can promote T cell activation, while increased expression of co-inhibitory markers (PD-L1 or CTLA-4) is involved in T cell tolerance[43]. An increase in the expression of both costimulatory and coinhibitory markers was observed in CHC patients, however, it was only the expression of inhibitory molecule, PD-L1 that correlated with an altered ratio of PD-L1/CD86 expression that seem to be responsible for the DC dysfunction in these patients[44]. Thus, strategies that target the inhibitory molecules on DCs might represent tools to improve DC functions and more so for the better management of the disease. Clinical trials in many chronic diseases, including CHC, with an aim of investigating the efficiency of PD-1/PD-L1 modulation are underway.
An upregulation of tryptophan-catabolizing enzyme indolamine 2,3-dioxygenase (IDO), which is an inducer of immune tolerance was significantly upregulated in the myeloid DCs of CHC patients[45]. This enzyme seems to contribute to the attenuated functioning of DCs and has been reported to be associated with inhibition of T cell proliferation and function. Deprivation of tryptophan forms certain toxic metabolites that lead to cell-cycle arrest of both in vitro and in vivo activated human T cells making these cells susceptible to apoptosis[46]. Patients infected with human immunodeficiency virus, HCV and HBV have increased IDO activity and whether it has any role in facilitating long-term persistence of these viruses needs to be investigated further.
Further, in the presence of viral proteins, the DCs tend to upregulate many genes which might play a significant role in rendering them tolerogenic[47]. HCV core protein is shown to cause down regulation in host response by interfering with the downstream signalling pathway[48]. SOCS proteins, potent regulators of cytokine signalling also affect the DC differentiation, maturation and also act as a negative regulator of JAK/STAT signalling[49]. The SOCS proteins interfere with the binding of cytokines with their cognate receptors and downstream cell-signalling intracellular molecules. In a human hepatoma HepG2 cell line, over expression of SOCS 1 and SOCS 3 suppressed STAT activity and gene expression of various antiviral proteins[50]. The HCV core protein is shown to cause up-regulation of SOCS 3 which might be related to non-responsiveness to antiviral therapy[51,52]. Moreover, SOCS 3-tranduced DCs expressed low levels of MHC class II and CD86 molecules on their surface as was observed both in vitro and in vivo systems[53]. Besides, such DCs produced higher levels of IL-10 and lower levels of IL-12 and IFNα, suggesting a phenotype of tolerogenic DCs. The aforementioned mechanisms of DC impairment are summarized in Table 1.
Table 1.
Summary of mechanisms of dendritic cell impairment
| Mechanism | Effect on immune system | Ref. |
| STAT3 phosphorylation and activation of PI3K pathway | Reduced Th1 cell development | Tacke et al[38] |
| Interference with TRIF and IPS-1 signaling pathway | Exhaustive function of CD8+ T-cells | Rodrigue-Gervais et al[40] |
| Down modulation of costimulatory molecules | DC dysfunction | Bain et al[32], Rana et al[54] |
| Upregulation of inhibitory receptors | Impaired DC function and exhaustion of T cell functions | Freeman et al[42] |
| Upregulation of IDO | Immune tolerance | Schulz et al[45] |
| Increased expression of SOCS | Negative regulators of JAK/STAT signalling, | Kim et al[52] |
| differentiation of DCs to tolerogenic cell | Li et al[53] |
DC: Dendritic cell; Th1: T helper 1; SOCS: Suppressor of cytokine signalling; IDO: Indoleamine 2,3-dioxygenase; IPS: Interferon β promoter stimulator-1; TRIF: Toll/interleukin-1 domain-containing adapter-inducing interferon γ.
To deduce the mechanism of DC impairment during CHC infection, we exposed the monocytes of healthy individuals to the HCV-3 specific core and NS5 antigens during differentiation to immature DCs in vitro, and by further inducing maturation in the presence of lipopolysaccharide (LPS)[54]. We observed that both core and NS5 antigens induced maturation and activation defects in healthy mo-DCs as they failed to upregulate surface expression of HLA-DR, CD83, CD80 and CD86 upon LPS stimulation. Further, we found that in the presence of NS5 and core antigens, the expression of PD-L1 and IDO got upregulated while only NS5, and not core, caused the increase in expression of SOCS 3 in the mo-DCs that were differentiated in the presence of these viral proteins right from day one. Based on our findings, we proposed a model of DC dysfunction during CHC infection, indicating the upregulation and the role of various negative regulatory factors in rendering the defective phenotype of mDCs during CHC as summarized in Figure 1.
Figure 1.
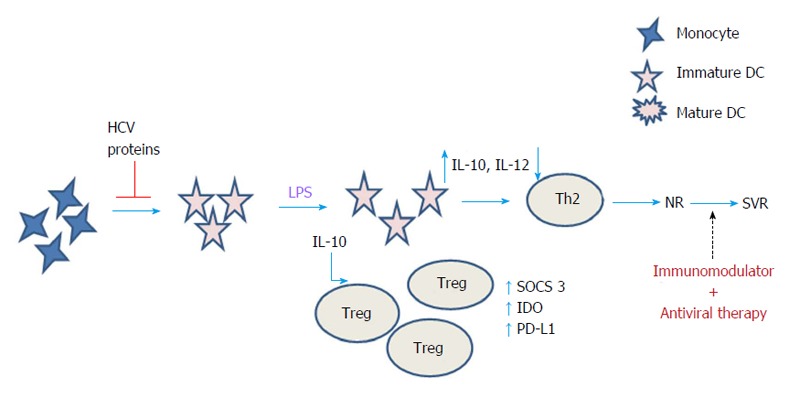
Proposed mechanism of dendritic cell impairment during chronic hepatitis infection and its relationship with antiviral therapy. Exposure of mo-DCs to HCV proteins ex vivo upregulated the expression of SOCS 3, IDO and PD-L1 that may be responsible for the observed maturation and activation defects in DCs including decreased secretion of IL-12 and IFNγ on LPS stimulation leading to the differentiation of Th2 cells. Such DCs also produced increased levels of IL-10 that promote the differentiation of regulatory T cells. Antiviral therapy along with some immunomodulation targeting these inhibitory molecules would help in the reconstitution of DC function in the antiviral therapy non-responders and would help to achieve SVR. SVR: Sustained virological response; NR: Non-responders to therapy; IL: Interleukin; IFN: Interferon; Th: T helper; SOCS: Suppressor of cytokine signalling; IDO: Indoleamine 2,3-dioxygenase; PD-L1: Programmed death ligand 1; LPS: Lipopolysaccharide; CHC: Chronic hepatitis C; DC: Dendritic cells; HCV: Hepatitis C virus; mo-DC: Monocyte-derived DCs; Treg: Regulatory T cells.
Strategies to improve DC functions: Future aspect
The host immune response takes on a cardinal role in virus control, recovery from the disease and provides protective immunity. Evidence from literature indicates that HCV, like many other viruses that cause chronic disease in humans, targets the DCs and interfere not only with its functioning but also use them for their dissemination within host tissues. An effective HCV vaccine would limit the number of new infections and in that way cut the burden on healthcare organizations. Nonetheless, on that path there are many hurdles and challenges since an effective vaccine is confronted with many factors associated with the HCV. This includes the subsistence of an array of HCV genotypes, limited availability of animal models and the gaps in the existing knowledge regarding the immunological mechanisms to HCV. Combinatorial approaches that would simultaneously enhance immunogenicity of vaccines and negate immunoregulatory pathways may significantly impact the nature of immune response. In this context, DC-based vaccines could be combined with immuno-modulatory molecules to be useful as both prophylactic and therapeutic vaccination against HCV[55]. These include the use of adjuvants with DC-based vaccines such as synthetic TLR agonists (Glucopyranosyl lipid A, polyinosinic:polycytidylic acid or synthetic oligodeoxynucleotides) that have been used to improve the function of DCs against many other infections[56-58]. The functions of cytotoxic CD8+ T cells have been rescued effectively by blocking the expression of PD-L1 and CTLA-4 in vitro using blocking antibodies with profound improvement in DC functions as well[59]. Therefore, reinvigorating the immune response through blocking/down-modulating inhibitory molecules on DCs are currently proposed to be innovative strategies during chronic HCV infection. Furthermore, silencing SOCS proteins with either siRNA based approaches or via the use of antagonists may improve TLR-mediated STAT-1 activation and IL-12 production in monocytes/macrophages. Besides, blocking SOCS proteins along with blocking PD-L1 in DCs would abrogate HCV-induced inhibition as has been reported for T cell function reconstitution[60]. In future, such synergistic strategies could be employed with the aim of eliminating the pathogenic effects of HCV, although a deeper insight into all these mechanistic aspects of DC dysfunction would be essential.
Footnotes
P- Reviewer: Ji FP, Lo SY, Shier MK S- Editor: Qiu S L- Editor: A E- Editor: Liu SQ
Conflict-of-interest statement: The authors declare no conflict of interest.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: June 26, 2015
First decision: August 3, 2015
Article in press: August 31, 2015
References
- 1.Alter MJ, Margolis HS, Krawczynski K, Judson FN, Mares A, Alexander WJ, Hu PY, Miller JK, Gerber MA, Sampliner RE. The natural history of community-acquired hepatitis C in the United States. The Sentinel Counties Chronic non-A, non-B Hepatitis Study Team. N Engl J Med. 1992;327:1899–1905. doi: 10.1056/NEJM199212313272702. [DOI] [PubMed] [Google Scholar]
- 2.Major ME, Dahari H, Mihalik K, Puig M, Rice CM, Neumann AU, Feinstone SM. Hepatitis C virus kinetics and host responses associated with disease and outcome of infection in chimpanzees. Hepatology. 2004;39:1709–1720. doi: 10.1002/hep.20239. [DOI] [PubMed] [Google Scholar]
- 3.Kantzanou M, Lucas M, Barnes E, Komatsu H, Dusheiko G, Ward S, Harcourt G, Klenerman P. Viral escape and T cell exhaustion in hepatitis C virus infection analysed using Class I peptide tetramers. Immunol Lett. 2003;85:165–171. doi: 10.1016/s0165-2478(02)00224-9. [DOI] [PubMed] [Google Scholar]
- 4.Large MK, Kittlesen DJ, Hahn YS. Suppression of host immune response by the core protein of hepatitis C virus: possible implications for hepatitis C virus persistence. J Immunol. 1999;162:931–938. [PubMed] [Google Scholar]
- 5.Kittlesen DJ, Chianese-Bullock KA, Yao ZQ, Braciale TJ, Hahn YS. Interaction between complement receptor gC1qR and hepatitis C virus core protein inhibits T-lymphocyte proliferation. J Clin Invest. 2000;106:1239–1249. doi: 10.1172/JCI10323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bertoletti A, Ferrari C. Kinetics of the immune response during HBV and HCV infection. Hepatology. 2003;38:4–13. doi: 10.1053/jhep.2003.50310. [DOI] [PubMed] [Google Scholar]
- 7.Saha B, Szabo G. Innate immune cell networking in hepatitis C virus infection. J Leukoc Biol. 2014;96:757–766. doi: 10.1189/jlb.4MR0314-141R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Longman RS, Talal AH, Jacobson IM, Albert ML, Rice CM. Presence of functional dendritic cells in patients chronically infected with hepatitis C virus. Blood. 2004;103:1026–1029. doi: 10.1182/blood-2003-04-1339. [DOI] [PubMed] [Google Scholar]
- 9.Longman RS, Talal AH, Jacobson IM, Rice CM, Albert ML. Normal functional capacity in circulating myeloid and plasmacytoid dendritic cells in patients with chronic hepatitis C. J Infect Dis. 2005;192:497–503. doi: 10.1086/431523. [DOI] [PubMed] [Google Scholar]
- 10.Piccioli D, Tavarini S, Nuti S, Colombatto P, Brunetto M, Bonino F, Ciccorossi P, Zorat F, Pozzato G, Comar C, et al. Comparable functions of plasmacytoid and monocyte-derived dendritic cells in chronic hepatitis C patients and healthy donors. J Hepatol. 2005;42:61–67. doi: 10.1016/j.jhep.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 11.Barnes E, Salio M, Cerundolo V, Francesco L, Pardoll D, Klenerman P, Cox A. Monocyte derived dendritic cells retain their functional capacity in patients following infection with hepatitis C virus. J Viral Hepat. 2008;15:219–228. doi: 10.1111/j.1365-2893.2007.00934.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Szabo G, Dolganiuc A. Subversion of plasmacytoid and myeloid dendritic cell functions in chronic HCV infection. Immunobiology. 2005;210:237–247. doi: 10.1016/j.imbio.2005.05.018. [DOI] [PubMed] [Google Scholar]
- 13.Kanto T, Hayashi N, Takehara T, Tatsumi T, Kuzushita N, Ito A, Sasaki Y, Kasahara A, Hori M. Impaired allostimulatory capacity of peripheral blood dendritic cells recovered from hepatitis C virus-infected individuals. J Immunol. 1999;162:5584–5591. [PubMed] [Google Scholar]
- 14.Tsubouchi E, Akbar SM, Horiike N, Onji M. Infection and dysfunction of circulating blood dendritic cells and their subsets in chronic hepatitis C virus infection. J Gastroenterol. 2004;39:754–762. doi: 10.1007/s00535-003-1385-3. [DOI] [PubMed] [Google Scholar]
- 15.Wertheimer AM, Bakke A, Rosen HR. Direct enumeration and functional assessment of circulating dendritic cells in patients with liver disease. Hepatology. 2004;40:335–345. doi: 10.1002/hep.20306. [DOI] [PubMed] [Google Scholar]
- 16.Abe T, Kaname Y, Hamamoto I, Tsuda Y, Wen X, Taguwa S, Moriishi K, Takeuchi O, Kawai T, Kanto T, et al. Hepatitis C virus nonstructural protein 5A modulates the toll-like receptor-MyD88-dependent signaling pathway in macrophage cell lines. J Virol. 2007;81:8953–8966. doi: 10.1128/JVI.00649-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dolganiuc A, Kodys K, Kopasz A, Marshall C, Do T, Romics L, Mandrekar P, Zapp M, Szabo G. Hepatitis C virus core and nonstructural protein 3 proteins induce pro- and anti-inflammatory cytokines and inhibit dendritic cell differentiation. J Immunol. 2003;170:5615–5624. doi: 10.4049/jimmunol.170.11.5615. [DOI] [PubMed] [Google Scholar]
- 18.Baetz A, Frey M, Heeg K, Dalpke AH. Suppressor of cytokine signaling (SOCS) proteins indirectly regulate toll-like receptor signaling in innate immune cells. J Biol Chem. 2004;279:54708–54715. doi: 10.1074/jbc.M410992200. [DOI] [PubMed] [Google Scholar]
- 19.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 20.Hemmi H, Akira S. TLR signalling and the function of dendritic cells. Chem Immunol Allergy. 2005;86:120–135. doi: 10.1159/000086657. [DOI] [PubMed] [Google Scholar]
- 21.Simmonds P, Tuplin A, Evans DJ. Detection of genome-scale ordered RNA structure (GORS) in genomes of positive-stranded RNA viruses: Implications for virus evolution and host persistence. RNA. 2004;10:1337–1351. doi: 10.1261/rna.7640104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reis e Sousa C. Dendritic cells in a mature age. Nat Rev Immunol. 2006;6:476–483. doi: 10.1038/nri1845. [DOI] [PubMed] [Google Scholar]
- 23.Ma DY, Clark EA. The role of CD40 and CD154/CD40L in dendritic cells. Semin Immunol. 2009;21:265–272. doi: 10.1016/j.smim.2009.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lippert U, Zachmann K, Ferrari DM, Schwarz H, Brunner E, Mahbub-Ul Latif AH, Neumann C, Soruri A. CD137 ligand reverse signaling has multiple functions in human dendritic cells during an adaptive immune response. Eur J Immunol. 2008;38:1024–1032. doi: 10.1002/eji.200737800. [DOI] [PubMed] [Google Scholar]
- 25.Idoyaga J, Steinman RM. SnapShot: Dendritic Cells. Cell. 2011;146:660–660.e2. doi: 10.1016/j.cell.2011.08.010. [DOI] [PubMed] [Google Scholar]
- 26.Zhang Z, Wang FS. Plasmacytoid dendritic cells act as the most competent cell type in linking antiviral innate and adaptive immune responses. Cell Mol Immunol. 2005;2:411–417. [PubMed] [Google Scholar]
- 27.Kadowaki N, Ho S, Antonenko S, Malefyt RW, Kastelein RA, Bazan F, Liu YJ. Subsets of human dendritic cell precursors express different toll-like receptors and respond to different microbial antigens. J Exp Med. 2001;194:863–869. doi: 10.1084/jem.194.6.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Navas MC, Fuchs A, Schvoerer E, Bohbot A, Aubertin AM, Stoll-Keller F. Dendritic cell susceptibility to hepatitis C virus genotype 1 infection. J Med Virol. 2002;67:152–161. doi: 10.1002/jmv.2204. [DOI] [PubMed] [Google Scholar]
- 29.Pöhlmann S, Zhang J, Baribaud F, Chen Z, Leslie GJ, Lin G, Granelli-Piperno A, Doms RW, Rice CM, McKeating JA. Hepatitis C virus glycoproteins interact with DC-SIGN and DC-SIGNR. J Virol. 2003;77:4070–4080. doi: 10.1128/JVI.77.7.4070-4080.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kanto T, Inoue M, Miyatake H, Sato A, Sakakibara M, Yakushijin T, Oki C, Itose I, Hiramatsu N, Takehara T, et al. Reduced numbers and impaired ability of myeloid and plasmacytoid dendritic cells to polarize T helper cells in chronic hepatitis C virus infection. J Infect Dis. 2004;190:1919–1926. doi: 10.1086/425425. [DOI] [PubMed] [Google Scholar]
- 31.Ryan EJ, Stevenson NJ, Hegarty JE, O’Farrelly C. Chronic hepatitis C infection blocks the ability of dendritic cells to secrete IFN-α and stimulate T-cell proliferation. J Viral Hepat. 2011;18:840–851. doi: 10.1111/j.1365-2893.2010.01384.x. [DOI] [PubMed] [Google Scholar]
- 32.Bain C, Fatmi A, Zoulim F, Zarski JP, Trépo C, Inchauspé G. Impaired allostimulatory function of dendritic cells in chronic hepatitis C infection. Gastroenterology. 2001;120:512–524. doi: 10.1053/gast.2001.21212. [DOI] [PubMed] [Google Scholar]
- 33.Tsubouchi E, Akbar SM, Murakami H, Horiike N, Onji M. Isolation and functional analysis of circulating dendritic cells from hepatitis C virus (HCV) RNA-positive and HCV RNA-negative patients with chronic hepatitis C: role of antiviral therapy. Clin Exp Immunol. 2004;137:417–423. doi: 10.1111/j.1365-2249.2004.02544.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rodrigue-Gervais IG, Rigsby H, Jouan L, Willems B, Lamarre D. Intact dendritic cell pathogen-recognition receptor functions associate with chronic hepatitis C treatment-induced viral clearance. PLoS One. 2014;9:e102605. doi: 10.1371/journal.pone.0102605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rana D, Chawla YK, Duseja A, Dhiman R, Arora SK. Functional reconstitution of defective myeloid dendritic cells in chronic hepatitis C infection on successful antiviral treatment. Liver Int. 2012;32:1128–1137. doi: 10.1111/j.1478-3231.2011.02754.x. [DOI] [PubMed] [Google Scholar]
- 36.Waggoner SN, Cruise MW, Kassel R, Hahn YS. gC1q receptor ligation selectively down-regulates human IL-12 production through activation of the phosphoinositide 3-kinase pathway. J Immunol. 2005;175:4706–4714. doi: 10.4049/jimmunol.175.7.4706. [DOI] [PubMed] [Google Scholar]
- 37.Waggoner SN, Hall CH, Hahn YS. HCV core protein interaction with gC1q receptor inhibits Th1 differentiation of CD4+ T cells via suppression of dendritic cell IL-12 production. J Leukoc Biol. 2007;82:1407–1419. doi: 10.1189/jlb.0507268. [DOI] [PubMed] [Google Scholar]
- 38.Tacke RS, Tosello-Trampont A, Nguyen V, Mullins DW, Hahn YS. Extracellular hepatitis C virus core protein activates STAT3 in human monocytes/macrophages/dendritic cells via an IL-6 autocrine pathway. J Biol Chem. 2011;286:10847–10855. doi: 10.1074/jbc.M110.217653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cheng P, Corzo CA, Luetteke N, Yu B, Nagaraj S, Bui MM, Ortiz M, Nacken W, Sorg C, Vogl T, et al. Inhibition of dendritic cell differentiation and accumulation of myeloid-derived suppressor cells in cancer is regulated by S100A9 protein. J Exp Med. 2008;205:2235–2249. doi: 10.1084/jem.20080132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rodrigue-Gervais IG, Rigsby H, Jouan L, Sauvé D, Sékaly RP, Willems B, Lamarre D. Dendritic cell inhibition is connected to exhaustion of CD8+ T cell polyfunctionality during chronic hepatitis C virus infection. J Immunol. 2010;184:3134–3144. doi: 10.4049/jimmunol.0902522. [DOI] [PubMed] [Google Scholar]
- 41.Urbani S, Amadei B, Tola D, Pedrazzi G, Sacchelli L, Cavallo MC, Orlandini A, Missale G, Ferrari C. Restoration of HCV-specific T cell functions by PD-1/PD-L1 blockade in HCV infection: effect of viremia levels and antiviral treatment. J Hepatol. 2008;48:548–558. doi: 10.1016/j.jhep.2007.12.014. [DOI] [PubMed] [Google Scholar]
- 42.Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, Fitz LJ, Malenkovich N, Okazaki T, Byrne MC, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192:1027–1034. doi: 10.1084/jem.192.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Steinman RM. Some interfaces of dendritic cell biology. APMIS. 2003;111:675–697. doi: 10.1034/j.1600-0463.2003.11107802.x. [DOI] [PubMed] [Google Scholar]
- 44.Shen T, Chen X, Chen Y, Xu Q, Lu F, Liu S. Increased PD-L1 expression and PD-L1/CD86 ratio on dendritic cells were associated with impaired dendritic cells function in HCV infection. J Med Virol. 2010;82:1152–1159. doi: 10.1002/jmv.21809. [DOI] [PubMed] [Google Scholar]
- 45.Schulz S, Landi A, Garg R, Wilson JA, van Drunen Littel-van den Hurk S. Indolamine 2,3-dioxygenase expression by monocytes and dendritic cell populations in hepatitis C patients. Clin Exp Immunol. 2015;180:484–498. doi: 10.1111/cei.12586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fallarino F, Grohmann U, Vacca C, Bianchi R, Orabona C, Spreca A, Fioretti MC, Puccetti P. T cell apoptosis by tryptophan catabolism. Cell Death Differ. 2002;9:1069–1077. doi: 10.1038/sj.cdd.4401073. [DOI] [PubMed] [Google Scholar]
- 47.Krishnadas DK, Ahn JS, Han J, Kumar R, Agrawal B. Immunomodulation by hepatitis C virus-derived proteins: targeting human dendritic cells by multiple mechanisms. Int Immunol. 2010;22:491–502. doi: 10.1093/intimm/dxq033. [DOI] [PubMed] [Google Scholar]
- 48.Melén K, Fagerlund R, Nyqvist M, Keskinen P, Julkunen I. Expression of hepatitis C virus core protein inhibits interferon-induced nuclear import of STATs. J Med Virol. 2004;73:536–547. doi: 10.1002/jmv.20123. [DOI] [PubMed] [Google Scholar]
- 49.Krebs DL, Hilton DJ. SOCS proteins: negative regulators of cytokine signaling. Stem Cells. 2001;19:378–387. doi: 10.1634/stemcells.19-5-378. [DOI] [PubMed] [Google Scholar]
- 50.Vlotides G, Sörensen AS, Kopp F, Zitzmann K, Cengic N, Brand S, Zachoval R, Auernhammer CJ. SOCS-1 and SOCS-3 inhibit IFN-alpha-induced expression of the antiviral proteins 2,5-OAS and MxA. Biochem Biophys Res Commun. 2004;320:1007–1014. doi: 10.1016/j.bbrc.2004.06.051. [DOI] [PubMed] [Google Scholar]
- 51.Persico M, Capasso M, Persico E, Svelto M, Russo R, Spano D, Crocè L, La Mura V, Moschella F, Masutti F, et al. Suppressor of cytokine signaling 3 (SOCS3) expression and hepatitis C virus-related chronic hepatitis: Insulin resistance and response to antiviral therapy. Hepatology. 2007;46:1009–1015. doi: 10.1002/hep.21782. [DOI] [PubMed] [Google Scholar]
- 52.Kim KA, Lin W, Tai AW, Shao RX, Weinberg E, De Sa Borges CB, Bhan AK, Zheng H, Kamegaya Y, Chung RT. Hepatic SOCS3 expression is strongly associated with non-response to therapy and race in HCV and HCV/HIV infection. J Hepatol. 2009;50:705–711. doi: 10.1016/j.jhep.2008.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li Y, Chu N, Rostami A, Zhang GX. Dendritic cells transduced with SOCS-3 exhibit a tolerogenic/DC2 phenotype that directs type 2 Th cell differentiation in vitro and in vivo. J Immunol. 2006;177:1679–1688. doi: 10.4049/jimmunol.177.3.1679. [DOI] [PubMed] [Google Scholar]
- 54.Rana D, Chawla YK, Duseja A, Dhiman RK, Arora SK. Viral Proteins Mediate Upregulation Of Negative Regulatory Factors Causing Down-Modulated Dendritic Cell Functions In Chronic Hepatitis C Virus Infection. Hepatology. 2013;1:68–76. [Google Scholar]
- 55.Zhou Y, Zhang Y, Yao Z, Moorman JP, Jia Z. Dendritic cell-based immunity and vaccination against hepatitis C virus infection. Immunology. 2012;136:385–396. doi: 10.1111/j.1365-2567.2012.03590.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pantel A, Cheong C, Dandamudi D, Shrestha E, Mehandru S, Brane L, Ruane D, Teixeira A, Bozzacco L, Steinman RM, et al. A new synthetic TLR4 agonist, GLA, allows dendritic cells targeted with antigen to elicit Th1 T-cell immunity in vivo. Eur J Immunol. 2012;42:101–109. doi: 10.1002/eji.201141855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Spranger S, Javorovic M, Bürdek M, Wilde S, Mosetter B, Tippmer S, Bigalke I, Geiger C, Schendel DJ, Frankenberger B. Generation of Th1-polarizing dendritic cells using the TLR7/8 agonist CL075. J Immunol. 2010;185:738–747. doi: 10.4049/jimmunol.1000060. [DOI] [PubMed] [Google Scholar]
- 58.Bode C, Zhao G, Steinhagen F, Kinjo T, Klinman DM. CpG DNA as a vaccine adjuvant. Expert Rev Vaccines. 2011;10:499–511. doi: 10.1586/erv.10.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lee J, Suh WI, Shin EC. T-cell dysfunction and inhibitory receptors in hepatitis C virus infection. Immune Netw. 2010;10:120–125. doi: 10.4110/in.2010.10.4.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Frazier AD, Zhang CL, Ni L, Ma CJ, Zhang Y, Wu XY, Atia AN, Yao ZQ, Moorman JP. Programmed death-1 affects suppressor of cytokine signaling-1 expression in T cells during hepatitis C infection. Viral Immunol. 2010;23:487–495. doi: 10.1089/vim.2010.0010. [DOI] [PMC free article] [PubMed] [Google Scholar]