

Abstract
The automatic recognition of gene names and their associated database identifiers from biomedical text has been widely studied in recent years, as these tasks play an important role in many downstream text-mining applications. Despite significant previous research, only a small number of tools are publicly available and these tools are typically restricted to detecting only mention level gene names or only document level gene identifiers. In this work, we report GNormPlus: an end-to-end and open source system that handles both gene mention and identifier detection. We created a new corpus of 694 PubMed articles to support our development of GNormPlus, containing manual annotations for not only gene names and their identifiers, but also closely related concepts useful for gene name disambiguation, such as gene families and protein domains. GNormPlus integrates several advanced text-mining techniques, including SimConcept for resolving composite gene names. As a result, GNormPlus compares favorably to other state-of-the-art methods when evaluated on two widely used public benchmarking datasets, achieving 86.7% F1-score on the BioCreative II Gene Normalization task dataset and 50.1% F1-score on the BioCreative III Gene Normalization task dataset. The GNormPlus source code and its annotated corpus are freely available, and the results of applying GNormPlus to the entire PubMed are freely accessible through our web-based tool PubTator.
1. Introduction
With the rapid growth of biomedical literature, text-mining or biomedical natural language processing (BioNLP) becomes increasingly important for today's biomedical research [1–6]. BioNLP holds the promise to have computers to read the vast amount of the literature and extract key knowledge about specific topics, such as protein-protein/drug-drug interactions [7–11], protein functions and transport [12, 13], and genetic mutations [14–16]. To accomplish that, the first BioNLP task is often known as named entity recognition (NER): to automatically identify the names of biological entities (e.g., gene/protein) from unstructured texts [17]. Given the central role of gene/proteins in the biomedical research [18], the automatic recognition of gene (note that we use gene and protein interchangeably in this paper) names has received much more attention by the BioNLP researchers [19–26] than other entities such as diseases (e.g., DNorm [27]) and chemicals (e.g., tmChem [28]).
Despite many attempts in the past, the gene NER task remains challenging due to both language variation and ambiguity. First, the same gene is often described in multiple different ways by the authors including the orthographical variation (e.g., “ESR1” and “ESR-1”), morphological variation (e.g., “GHF-1 transcriptional factor” and “GHF-1 transcription factor”), variation with abbreviation (e.g., “estrogen receptor alpha (ERα)”), and composition mentions (e.g., “BRCA1/2” and “SMADs 1, 5, and 8”). With respect to ambiguity, the first challenge is multispecies (orthologous) gene ambiguity. That is, the same gene name can indicate different concept identifiers depending on its associated organism information (e.g., erbb2 can be either a human gene or mouse gene name). The second ambiguity arises because different genes can share the same name. For example, “AP-1” can refer to either “jun proto-oncogene” (Entrez Gene: 3725) or “FBJ murine osteosarcoma viral oncogene homolog” (Entrez Gene: 2353).
To advance the state of the art in NER, a number of community-wide shared tasks have been organized [29–31] (see Huang and Lu, 2015 [32], for a complete list). In particular, the Critical Assessment of Information Extraction Systems in Biology (BioCreative) has repeatedly organized both gene mention (GM) and gene normalization (GN) tasks where the former task involves finding the occurrence (i.e., string offsets) of gene names in text while the latter typically asks for returning gene concept identifiers per document. In BioCreative I [33] and BioCreative II [7], the GM tasks focused on four species (e.g., human, fly, mouse, and yeast) gene mentions. The best results obtained in the challenges are 83.2% of F-measure in BC I GM task [33] and 88.22% in BC II GM task [34]. In BioCreative II, the GN task was introduced which asked participants to return human gene/protein concept identifiers given target articles. The best performance in this task was 81.0% F-measure [7]. In BioCreative III, the GN task was reintroduced with the additional challenges of dealing with full text and multiple species. As a result, the best performance is lower (46.56% in F-measure [19]).
As a result of these challenge tasks, a number of annotated corpora were made available to the research community and have, in turn, enabled the development of a number of software tools. For instance, the BioCreative GM corpus was used to build several gene mention taggers, such as AIIA-GMT [35], BANNER [36], and BioTagger-GM [37]. However, existing gene corpora (e.g., BioCreative II GM/GN corpora [29, 30]) are annotated in either mention or document level as they were separately developed. The GM corpus (e.g., [34]) includes mention annotations but not gene identifiers of the target document; the GN corpus contains annotations for the gene identifiers but not their associated mentions. Training a supervised method on some GM data for the GN task is not ideal because different annotation criteria are often used (e.g., GM corpus may include mentions that cannot be mapped to gene identifiers). Thus, we propose developing a corpus that includes both gene mentions and concept identifiers for the same set of articles. To our best knowledge, the newly published IGN corpus [38] is the only other data set that includes both types of annotations. However, we differ from IGN in two main aspects. First, our newly developed corpus consists of more articles (694 versus 543). More importantly, we annotate gene-related concepts separately. That is, we distinguish gene, gene family, and protein domains and treat them as separate classes in our annotation (see Figure 1) as we believe such a distinction can help gene name disambiguation and improve performance. None of the current GM/GN corpora annotates these types separately. For instance, in the BioCreative II GM corpus, gene, protein family, protein domain, DNA, and RNA are all treated as gene mentions.
Figure 1.

A screenshot of gene, gene family, and protein domain annotation of PMID: 10828014 in PubTator.
Past GN systems are unable to distinguish between gene and gene families: they either completely ignored the problem or simply used a protein family name list as filters [24, 25, 39, 40]. However, the filtering strategy does not work once the family mention is not in this list. In this case, the family name becomes false positives in the results. Furthermore, detecting domain names can assist resolving ambiguous gene/protein names. As shown in Figure 2, the TEL1 and TEL2 proteins are both ETS-family transcription factors with the ETS finger domain and GGAA core motif. TEL1 also has the pointed (PNT) domain. When searching for the gene identifier in Entrez Gene, TEL1 can map to two different concepts: ATM serine/threonine kinase (gene ID: 472) and ETS translocation factor variant 6 (gene ID: 2120). But with extracted protein domain information, we can infer that in this case ETS translocation factor variant 6 is the correct answer because it is known to be associated with the PNT domain. Besides, the family name “ETS translocation factor” is also helpful to the disambiguation of TEL1/2 because it is included in the gene's official full name.
Figure 2.
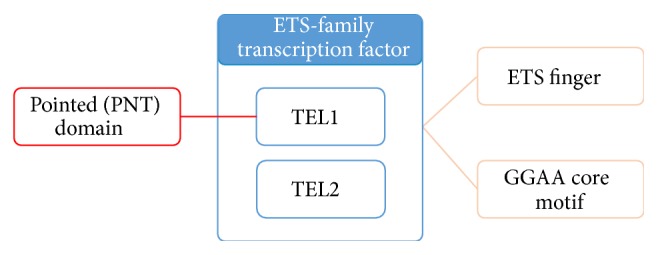
Relations between gene, gene family, and protein domains in PMID: 10828014.
Taken together, this research makes three major contributions. First, through reannotating two existing corpora, we are the first to build a new corpus that allows the development of new methods for distinguishing different gene-related entities: (gene, gene family, and protein domains). Second, we build a new end-to-end system that includes both GM and GN modules, together with several advanced BioNLP tools (e.g., GenNorm [19], SimConcept [41], SR4GN [42], and Ab3P [43]) for improved performance. Lastly, we show state-of-the-art performance on two separate benchmark data sets.
2. Materials and Methods
2.1. Corpus Development
We reannotated two existing gene corpora. The BioCreative II GN corpus is a widely used data set for benchmarking GN tools and includes document level annotations for a total of 543 articles (281 in its training set and 262 in test). The Citation GIA test collection was recently created for gene indexing at the NLM and includes 151 PubMed abstracts with both mention level and document level annotations. They are selected because both have a focus on human genes. For both corpora, we added annotations of gene families and protein domains. For the BioCreative GN corpus, we also added mention level gene annotations. As a result, in our new corpus, there are a total of 694 PubMed articles (see Table 1). PubTator [44, 45], a tool developed and evaluated through the BioCreative III Interactive Task [46], was used as our annotation software.
Table 1.
The statistic of our gene corpus.
| Data set | Articles | Gene mentions (gene/family/domains) | Gene identifiers |
|---|---|---|---|
| BioCreative II GN training set | 281 | 3,019/1,115/278 | 758 |
| BioCreative II GN test set | 262 | 3,233/1,252/361 | 928 |
| NLM Citation GIA test collection | 151 | 1,205/160/17 | 310 |
|
| |||
| Total | 694 | 7,457/2,527/656 | 1996 |
2.2. Method Overview
As shown in Figure 3, our proposed approach includes two main steps: mention recognition and concept normalization, respectively. In the mention recognition step, we developed a new module, together with our previous species recognition system (i.e., SR4GN) to recognize gene and species names and match them accordingly. In concept normalization step, we applied our previous system, GenNorm, combined with a composite mention simplification tool (i.e., SimConcept) and an abbreviation resolution tool (i.e., Ab3P) for optimized performance.
Figure 3.

The overview of our integration method (GNormPlus).
2.3. Mention Recognition Step
In this study, we propose a supervised approach to detect the mentions of gene, gene family, and protein domain from a target input (e.g., PubMed abstracts). We first translate this mention recognition problem as a sequence labeling task. Accordingly, we adapted a probability based sequence detection conditional random fields (CRF) model [47] provided by CRF++ (http://crfpp.googlecode.com/svn/trunk/doc/index.html) library by order 2 model. CRF++ applies L-BFGS [48] which is a Quasi-Newton algorithm for large scale numerical optimization problems. We chose BIEO (B: begin, I: inside, E: end, and O: outside) label set for this recognition model. We also used the tokenization module in our previous NER systems (i.e., tmChem [28] and tmVar [15]) here. More specifically, we applied tmVar's tokenization module which splits tokens not only at punctuation (e.g., “.,()+”) and spaces, but also at digits and transitions between uppercase and lowercase. For instance, “hTIF1” will be split into three individual tokens “h,” “TIF,” and “1.” We also reused the features in tmChem and tmVar as described below.
General Linguistic Features. We included the original tokens (e.g., genes), stemmed tokens (e.g., gene), and POS tagging result (e.g., “NN”). We also extracted the prefixes and suffixes as features (length: 1~5).
Character Features. Since many gene concepts include letters, digits, and special characters, we therefore detected the number of uppercases, lowercases, letters, digits, and special characters (“;:,.->+_”).
Semantic Features. We defined three types of features to recognize the difference between potential gene mentions and other concepts. We first use the gene vocabulary from ctdbase.org (http://ctdbase.org/downloads/#allgenes) to detect those strings which can match gene mentions. In general, literature usually uses abbreviation to describe bioconcepts. We therefore use Ab3P [43] to detect those abbreviation pairs. To help the CRF model to recognize the difference between bioconcepts (e.g., genes, disease, and chemical), we collected a list of semantic tokens for genes (e.g., strains), disease (e.g., “disorder”), chemical (e.g., “trivial ring”), domain (e.g., “region”), cell (e.g., “cell”), protein symbol (e.g., glutamine), and so forth.
Case Pattern Features. We applied the case pattern features from tmVar [15]. Each token is represented in four simplified forms. Uppercase alphabetic characters are replaced by “A” and lowercase characters are replaced by “a.” Likewise, digits (0–9) are replaced by “0.” Moreover, we also merged consecutive letters and numbers and generated additional single letter “a” and number “0” as features.
Contextual Features. In order to take advantage of contextual information, for a given token we included the dictionary and linguistic features of 3 neighboring tokens from each side.
To best distinguish the three gene-related mention types, gene versus gene family versus protein domains, we applied several postprocessing rules to the CRF results. (1) Set the type by suffix (e.g., “OSBP-related proteins” to family, “LIM1 domain” to domain). (2) If we find a mention (e.g., “TIF1”) which is also a prefix of another mention (e.g., “TIF1alpha”), then we set the type of the mention to be gene family. (3) When abbreviation pairs are found, use the mention type of the long form to the sort form (e.g., “TIF1” is tagged as protein family because of its long form “transcriptional intermediary factor 1 family”). (4) If a mention occurs multiple times in an article but is tagged with different types by the CRF module, we then apply the majority rule to determine its final type in the article. For example, if hif1 was tagged twice by the CRF as a gene but as gene family in three times, then all five occurrences of hif1 will be tagged as gene family names.
2.4. Concept Normalization Step
The second step of our system is to map gene mentions to specific concepts in Entrez Gene. To do that, we first applied our previous GN tool, GenNorm [19, 49], which is based on a statistical inference network model via two individual matching strategies (i.e., exact match and bag-of-words match). More specifically, the exact match strategy requires the input mention to be identical to the names in the controlled vocabulary. On the other hand, the bag-of-words approach matches tokens in both input text and target vocabulary. GenNorm achieved the best performance in the BioCreative III GN task [29].
For performance optimization, we also integrated an abbreviation resolution and composite mention simplification tool in this step. First, we applied Ab3P [43] to extract the long form and short form abbreviation pairs. When the short form and long form map to different gene candidates, we typically infer the candidate gene of long form to short form for improved performance. SimConcept [41] was used to identify and resolve composite named entities, where a single span refers to more than one concept (e.g., BRCA1/2). Most past NER studies have either ignored this issue, used simple ad hoc rules, or only handled coordination ellipsis, which is only one of the many types of composite mentions studied in this work. SimConcept was shown to successfully tag individual entities from composite mentions.
3. Evaluation and Results
The first evaluation is a species-specific experiment where only human genes are considered. In this evaluation, we trained our system using both BioCreative II GN training set and NLM Citation GIA test collection and tested it on the BioCreative II GN test set. As shown in Table 2, we compared GNormPlus with several previously reported systems, including our previous system, GenNorm [19]. The default setting of GenNorm uses AIIA-GMT [35] for gene mention recognition. AIIA-GMT is one of the high-performing gene mention recognition tools and provided web API service. Unfortunately, AIIA-GMT is no longer available since 2013.
Table 2.
The evaluation of human species gene normalization.
| Methods | Precision | Recall | F-measure | System availability |
|---|---|---|---|---|
| Our approach (GNormPlus) | 87.1% | 86.4% | 86.7% | Open source |
| GenNorm [19] + AIIA-GMT [35] | 78.9% | 81.4% | 80.1% | GenNorm is open source but AIIA-GMT is no longer available |
| GNAT [23] | 90.7% | 82.4% | 86.4% | Open source |
| GeNO [24] | 87.8% | 85.0% | 86.4% | N/A |
| Hu et al., 2012 [40] | 83.5% | 82.5% | 83.0% | N/A |
| Li et al., 2013 [39] | 88.1% | 92.3% | 90.1% | N/A |
In the second experiment (see Table 3), we evaluate GNormPlus in multispecies gene normalization using the BioCreative III GN task data set. In this evaluation, we used the whole set of 694 articles for system training. As can be seen, our proposed method significantly outperforms previously published results in both standard F-measure and the task-specific TAP-k measures. The new system also outperforms our previous GenNorm tool by a significant margin.
Table 3.
The evaluation of cross species gene normalization.
| Methods | TAP-5 | TAP-10 | TAP-20 | F-measure | System availability |
|---|---|---|---|---|---|
| Our approach (GNormPlus) | 33.3% | 36.7% | 36.7% | 50.1% | Open source |
| GenNorm [42] + AIIA-GMT [23] | 32.8% | 35.5% | 35.5% | 46.9% | GenNorm is open source but AIIA-GMT is no longer available |
| GeneTuKit [22] | 29.7% | 31.4% | 32.5% | — | Open source |
| Kuo et al. [21] | 21.4% | 25.1% | 25.1% | 30.6% | N/A |
| Tsai et al. [20] | 19.0% | 22.9% | 23.9% | — | N/A |
4. Discussion and Conclusion
To assess the impact of using multiple gene-related mention types (i.e., gene versus family versus domain), we built a baseline model where all three types were treated as one. As shown in Table 4, the proposed multitype scheme significantly boosted the final GN performance as shown in this comparison.
Table 4.
The comparison of different mention recognition training corpus.
| Gene mention type scheme | Precision | Recall | F-measure |
|---|---|---|---|
| Gene/family/domain | 87.1% | 86.4% | 86.7% |
| Single gene type only | 78.4% | 79.2% | 78.8% |
Despite our best efforts, errors remain in our tagging results. Based on our results on the BioCreative II GN test set, we performed an error analysis including 127 false positive (FP) errors and 87 false negatives. In order to better understand the causes of different errors, we first separated the 214 errors by the GM step and GN step where the former accounts for 53% and the latter 47%. Among the errors in the GM step, many are due to gene/family/domain mention type confusion (e.g., assigning gene mentions to family/domain or assigning family/domain mentions to genes). Some gene mentions (e.g., TGF-beta) are particularly confusing when they refer to genes in some articles but to family/domain in other articles. In the GN step, failure in disambiguation is a frequent error (17.3%). A number of gene mentions can be associated with multiple identifiers. With only limited information in the abstract, sometimes it is very difficult to disambiguate and assign genes with correct identifiers. Another 8.9% of the errors are due to deficiencies of the gene name dictionary. Overall, as can be seen in Table 5, both the GM and GN results are important to the final performance.
Table 5.
The frequency of false negative and positive errors of GNormPlus.
| FN | FP | Total | Percentage | |
|---|---|---|---|---|
| Gene mention (GM) recognition | ||||
| Gene/family/domain mention type confusion | 38 | 18 | 56 | 27.1% |
| Wrong boundary or missed gene mention | 18 | 18 | 36 | 17.4% |
| Not a gene mention | 0 | 15 | 15 | 7.3% |
| Gene normalization (GN) | ||||
| Wrong gene identifier due to ambiguity | 19 | 18 | 37 | 17.9% |
| Insufficiency of the gene name dictionary | 19 | 0 | 19 | 9.2% |
| Not annotated in the gold standard | 0 | 17 | 17 | 8.2% |
| Nonhuman genes found | 0 | 11 | 11 | 5.3% |
| Others | 13 | 3 | 16 | 7.7% |
To conclude, we developed GNormPlus: an end-to-end gene recognition system which handles both GM and GN tasks. By integrating several advanced BioNLP tools (i.e., GenNorm, SR4GN, Ab3P, and SimConcept), GNormPlus achieved competitive results in our two benchmarking experiments when compared with the state of the art. Unlike our previous GenNorm system that relies on AIIA-GMT, GNormPlus is a stand-alone open source tool with no dependence on external tools (freely available at http://www.ncbi.nlm.nih.gov/CBBresearch/Lu/Demo/tmTools/#GNormPlus). GNormPlus is made interoperable with other BioC-compatible BioNLP tools. For convenience, we have also applied GNormPlus to PubMed and stored its results in PubTator (http://www.ncbi.nlm.nih.gov/CBBresearch/Lu/Demo/PubTator/) so that users can readily access gene data via PubTator. In the future, we plan to explore its applications in real-world uses such as biocuration [50] and also investigate the automatic recognition of other gene-related biological entities such as microRNAs [51].
Acknowledgments
The authors would like to thank Robert Leaman for his proofreading of the paper. This research was supported by the NIH Intramural Research Program, National Library of Medicine.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Lu Z. PubMed and beyond: a survey of web tools for searching biomedical literature. Database. 2011;2011 doi: 10.1093/database/baq036.baq036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zweigenbaum P., Demner-fushman D., Yu H., Cohen K. B. Frontiers of biomedical text mining: current progress. Briefings in Bioinformatics. 2007;8(5):358–375. doi: 10.1093/bib/bbm045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rzhetsky A., Seringhaus M., Gerstein M. Seeking a new biology through text mining. Cell. 2008;134(1):9–13. doi: 10.1016/j.cell.2008.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shatkay H., Feldman R. Mining the biomedical literature in the genomic era: an overview. Journal of Computational Biology. 2003;10(6):821–855. doi: 10.1089/106652703322756104. [DOI] [PubMed] [Google Scholar]
- 5.Rebholz-Schuhmann D., Kirsch H., Couto F. Facts from text—is text mining ready to deliver? PLoS Biology. 2005;3(2, article e65) doi: 10.1371/journal.pbio.0030065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ananiadou S., Kell D. B., Tsujii J.-I. Text mining and its potential applications in systems biology. Trends in Biotechnology. 2006;24(12):571–579. doi: 10.1016/j.tibtech.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 7.Krallinger M., Leitner F., Rodriguez-Penagos C., Valencia A. Overview of the protein-protein interaction annotation extraction task of BioCreative II. Genome Biology. 2008;9(2, article S4) doi: 10.1186/gb-2008-9-s2-s4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Krallinger M., Vazquez M., Leitner F., et al. The Protein-Protein Interaction tasks of BioCreative III: classification/ranking of articles and linking bio-ontology concepts to full text. BMC Bioinformatics. 2011;12(supplement 8, article S3) doi: 10.1186/1471-2105-12-s8-s3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baumgartner W. A., Jr., Lu Z., Johnson H. L., et al. Concept recognition for extracting protein interaction relations from biomedical text. Genome Biology. 2008;9(supplement 2, article S9) doi: 10.1186/gb-2008-9-s2-s9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Segura-Bedmar I., Martínez P., Herrero-Zazo M. Lessons learnt from the DDIExtraction-2013 shared task. Journal of Biomedical Informatics. 2014;51:152–164. doi: 10.1016/j.jbi.2014.05.007. [DOI] [PubMed] [Google Scholar]
- 11.Segura-Bedmar I., Martínez P., de Pablo-Sánchez C. Using a shallow linguistic kernel for drug-drug interaction extraction. Journal of Biomedical Informatics. 2011;44(5):789–804. doi: 10.1016/j.jbi.2011.04.005. [DOI] [PubMed] [Google Scholar]
- 12.Gobeill J., Pasche E., Vishnyakova D., Ruch P. Closing the loop: from paper to protein annotation using supervised Gene Ontology classification. Database. 2014;2014 doi: 10.1093/database/bau088.bau088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mao Y., Van Auken K., Li D., et al. Overview of the gene ontology task at BioCreative IV. Database. 2014;2014 doi: 10.1093/database/bau086.bau086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yepes A. J., Verspoor K. Mutation extraction tools can be combined for robust recognition of genetic variants in the literature. F1000Research. 2014;3, article 18 doi: 10.12688/f1000research.3-18.v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wei C.-H., Harris B. R., Kao H.-Y., Lu Z. TmVar: a text mining approach for extracting sequence variants in biomedical literature. Bioinformatics. 2013;29(11):1433–1439. doi: 10.1093/bioinformatics/btt156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Doughty E., Kertesz-Farkas A., Bodenreider O., et al. Toward an automatic method for extracting cancer- and other disease-related point mutations from the biomedical literature. Bioinformatics. 2011;27(3):408–415. doi: 10.1093/bioinformatics/btq667.btq667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baumgartner W. A., Jr., Lu Z., Johnson H. L., et al. An integrated approach to concept recognition in biomedical text. Proceedings of the 2nd BioCreative Challenge Evaluation Workshop; 2007; Madrid, Spain. Centro Nacional de Investigaciones Oncologicas (CNIO); pp. 257–271. [Google Scholar]
- 18.Dogan R. I., Murray G. C., Névéol A., Lu Z. Understanding PubMed user search behavior through log analysis. Database. 2009;2009 doi: 10.1093/database/bap018.bap018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wei C.-H., Kao H.-Y. Cross-species gene normalization by species inference. BMC Bioinformatics. 2011;12(supplement 8, article S5) doi: 10.1186/1471-2105-12-s8-s5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tsai R. T., Lai P.-T. Multi-stage gene normalization for full-text articles with context-based species filtering for dynamic dictionary entry selection. BMC Bioinformatics. 2011;12(supplement 8, article S7) doi: 10.1186/1471-2105-12-s8-s7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kuo C.-J., Ling M. H. T., Hsu C.-N. Soft tagging of overlapping high confidence gene mention variants for cross-species full-text gene normalization. BMC Bioinformatics. 2011;12(8, article S6) doi: 10.1186/1471-2105-12-s8-s6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang M., Liu J., Zhu X. GeneTUKit: a software for document-level gene normalization. Bioinformatics. 2011;27(7):1032–1033. doi: 10.1093/bioinformatics/btr042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hakenberg J., Gerner M., Haeussler M., et al. The GNAT library for local and remote gene mention normalization. Bioinformatics. 2011;27(19):2769–2771. doi: 10.1093/bioinformatics/btr455.btr455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wermter J., Tomanek K., Hahn U. High-performance gene name normalization with GeNo. Bioinformatics. 2009;25(6):815–821. doi: 10.1093/bioinformatics/btp071. [DOI] [PubMed] [Google Scholar]
- 25.Hakenberg J., Plake C., Leaman R., Schroeder M., Gonzalez G. Inter-species normalization of gene mentions with GNAT. Bioinformatics. 2008;24(16):i126–i132. doi: 10.1093/bioinformatics/btn299. [DOI] [PubMed] [Google Scholar]
- 26.van Landeghem S., Björne J., Wei C.-H., et al. Large-scale event extraction from literature with multi-level gene normalization. PLoS ONE. 2013;8(4) doi: 10.1371/journal.pone.0055814.e55814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leaman R., Doğan R. I., Lu Z. DNorm: disease name normalization with pairwise learning to rank. Bioinformatics. 2013;29(22):2909–2917. doi: 10.1093/bioinformatics/btt474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leaman R., Wei C.-H., Lu Z. tmChem: a high performance approach for chemical named entity recognition and normalization. Journal of Cheminformatics. 2015;7(supplement 1, article S3) doi: 10.1186/1758-2946-7-S1-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lu Z., Kao H.-Y., Wei C.-H., et al. The gene normalization task in BioCreative III. BMC Bioinformatics. 2011;12(8, article S2) doi: 10.1186/1471-2105-12-s8-s2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Morgan A. A., Lu Z., Wang X., et al. Overview of BioCreative II gene normalization. Genome Biology. 2008;9(supplement 2, article S3) doi: 10.1186/gb-2008-9-s2-s3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hirschman L., Colosimo M., Morgan A., Yeh A. Overview of BioCreAtIvE task 1B: normalized gene lists. BMC Bioinformatics. 2005;6(supplement 1, article S11) doi: 10.1186/1471-2105-6-s1-s11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang C.-C., Lu Z. Community challenges in biomedical text mining over 10 years: success, failure and the future. Briefings in Bioinformatics. 2015 doi: 10.1093/bib/bbv024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yeh A., Morgan A., Colosimo M., Hirschman L. BioCreAtIvE task 1A: gene mention finding evaluation. BMC Bioinformatics. 2005;6(supplement 1, article S2) doi: 10.1186/1471-2105-6-s1-s2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smith L., Tanabe L. K., Ando R., et al. Overview of BioCreative II gene mention recognition. Genome Biology. 2008;9(2, article S2) doi: 10.1186/gb-2008-9-s2-s2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hsu C.-N., Chang Y.-M., Kuo C.-J., Lin Y.-S., Huang H.-S., Chung I.-F. Integrating high dimensional bi-directional parsing models for gene mention tagging. Bioinformatics. 2008;24(13):i286–i294. doi: 10.1093/bioinformatics/btn183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Leaman R., Gonzalez G. BANNER: an executable survey of advances in biomedical named entity recognition. Proceedings of the Pacific Symposium on Biocomputing; January 2008; Kohala Coast, Hawaii, USA. pp. 652–663. [PubMed] [Google Scholar]
- 37.Torii M., Hu Z., Wu C. H., Liu H. BioTagger-GM: a gene/protein name recognition system. Journal of the American Medical Informatics Association. 2009;16(2):247–255. doi: 10.1197/jamia.m2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dai H.-J., Wu J. C.-Y., Tsai R. T.-H. Collective instance-level gene normalization on the IGN corpus. PLoS ONE. 2013;8(11) doi: 10.1371/journal.pone.0079517.e79517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li L., Liu S., Fan W., Huang D., Zhou H. A multistage gene normalization system integrating multiple effective methods. PLoS ONE. 2013;8(12) doi: 10.1371/journal.pone.0081956.e81956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hu Y., Li Y., Lin H., Yang Z., Cheng L. Integrating various resources for gene name normalization. PLoS ONE. 2012;7(9) doi: 10.1371/journal.pone.0043558.e43558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wei C.-H., leaman R., Lu Z. SimConcept: a hybrid approach for simplifying composite named entities in biomedicine. Proceedings of the ACM Conference on Bioinformatics Computational Biology and Health Informatics; 2014; Newport Beach, Calif, USA. ACM; pp. 138–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wei C.-H., Kao H.-Y., Lu Z. SR4GN: a species recognition software tool for gene normalization. PLoS ONE. 2012;7(6) doi: 10.1371/journal.pone.0038460.e38460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sohn S., Comeau D. C., Kim W., Wilbur J. W. Abbreviation definition identification based on automatic precision estimates. BMC Bioinformatics. 2008;9(1, article 402) doi: 10.1186/1471-2105-9-402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wei C.-H., Kao H.-Y., Lu Z. PubTator: a web-based text mining tool for assisting biocuration. Nucleic Acids Research. 2013;41:W518–W522. doi: 10.1093/nar/gkt441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wei C.-H., Harris B. R., Li D., et al. Accelerating literature curation with text-mining tools: a case study of using PubTator to curate genes in PubMed abstracts. Database. 2012;2012 doi: 10.1093/database/bas041.bas041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Arighi C. N., Carterette B., Cohen K. B., et al. An overview of the BioCreative 2012 Workshop Track III: interactive text mining task. Database. 2013;2013 doi: 10.1093/database/bas056.bas056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lafferty J., McCallum A., Pereira F. Conditional random fields: probabilistic models for segmenting and labeling sequence data. Proceedings of the 18th International Conference on Machine Learning (ICML '01); June-July 2001; Williamstown, Mass, USA. ACM; pp. 282–289. [Google Scholar]
- 48.Liu D. C., Nocedal J. On the limited memory BFGS method for large scale optimization. Mathematical Programming B. 1989;45(3):503–528. doi: 10.1007/bf01589116. [DOI] [Google Scholar]
- 49.Wei C.-H., Huang I.-C., Hsu Y.-Y., Kao H.-Y. Normalizing biomedical name entities by similarity-based inference network and de-ambiguity mining. Proceedings of the 9th IEEE International Conference on Bioinformatics and Bioengineering; June 2009; Taichung, Taiwan. pp. 461–466. [DOI] [Google Scholar]
- 50.Lu Z., Hirschman L. Biocuration workflows and text mining: overview of the BioCreative 2012 Workshop Track II. Database. 2012;2012 doi: 10.1093/database/bas043.bas043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xie B., Ding Q., Han H., Wu D. miRCancer: a microRNA-cancer association database constructed by text mining on literature. Bioinformatics. 2013;29(5):638–644. doi: 10.1093/bioinformatics/btt014. [DOI] [PubMed] [Google Scholar]