

Abstract
Aim:
Ca2+-release-activated Ca2+ (CRAC) channel, a subfamily of store-operated channels, is formed by calcium release-activated calcium modulator 1 (ORAI1), and gated by stromal interaction molecule 1 (STIM1). CRAC channel may be a novel target for the treatment of immune disorders and allergy. The aim of this study was to identify novel small molecule CRAC channel inhibitors.
Methods:
HEK293 cells stably co-expressing both ORAI1 and STIM1 were used for high-throughput screening. A hit, 1-phenyl-3-(1-phenylethyl)urea, was identified that inhibited CRAC channels by targeting ORAI1. Five series of its derivatives were designed and synthesized, and their primary structure-activity relationships (SARs) were analyzed. All derivatives were assessed for their effects on Ca2+ influx through CRAC channels on HEK293 cells, cytotoxicity in Jurkat cells, and IL-2 production in Jurkat cells expressing ORAI1-SS-eGFP.
Results:
A total of 19 hits were discovered in libraries containing 32 000 compounds using the high-throughput screening. 1-Phenyl-3-(1-phenylethyl)urea inhibited Ca2+ influx with IC50 of 3.25±0.17 μmol/L. SAR study on its derivatives showed that the alkyl substituent on the α-position of the left-side benzylic amine (R1) was essential for Ca2+ influx inhibition and that the S-configuration was better than the R-configuration. The derivatives in which the right-side R3 was substituted by an electron-donating group showed more potent inhibitory activity than those that were substituted by electron-withdrawing groups. Furthermore, the free N–H of urea was not necessary to maintain the high potency of Ca2+ influx inhibition. The N,N′-disubstituted or N′-substituted derivatives showed relatively low cytotoxicity but maintained the ability to inhibit IL-2 production. Among them, compound 5b showed an improved inhibition of IL-2 production and low cytotoxicity.
Conclusion:
1-Phenyl-3-(1-phenylethyl)urea is a novel CRAC channel inhibitor that specifically targets ORAI1. This study provides a new chemical scaffold for design and development of CRAC channel inhibitors with improved Ca2+ influx inhibition, immune inhibition and low cytotoxicity.
Supplementary information
The online version of this article (doi:10.1038/aps.2015.52) contains supplementary material, which is available to authorized users.
Keywords: CRAC channel, ORAI1, 1-phenyl-3-(1-phenylethyl)urea, YM58483, high-throughput screening, IL-2 production, structure modification, SAR
Introduction
Ca2+-release-activated Ca2+ (CRAC) channel belongs to a subfamily of store-operated channels that possess the characteristic features of an inwardly rectifying current-voltage relationship1 and rapid Ca2+-dependent inactivation2. The channel is formed by ORAI (CRACM, calcium release-activated calcium modulator), which is a 4-transmembrane protein and is gated by a single-pass transmembrane protein, STIM (stromal interaction molecule), which functions as the sensor of the endoplasmic reticulum (ER) luminal Ca2+ concentration3. Compared with other ion channels, CRAC channel currents have a substantially higher selectivity for Ca2+ over monovalent cations (PCa/PNa >1000)4, with low single-channel conductance (<1 pS)5 and a lack of significant voltage-dependent gating.
In recent years, several human diseases have been linked to abnormal CRAC channel activities, including severe combined immunodeficiency (SCID) disorders6, allergy7,8, inflammatory bowel disease (IBD)9, thrombosis10, breast cancer11,12,13, Kawasaki disease14 and organ transplant rejection. Specific CRAC channel inhibitors may be developed into new safe and potent immune suppressors, which would benefit patients with these diseases.
Thus far, a variety of small molecules blocking the CRAC channel have been identified, such as the imidazole derivative, SKF96365 (IC50 of approximately 4 μmol/L)15,16, 2-APB [at low concentrations of 1–5 μmol/L, 2-APB potentiates CRAC currents, whereas high concentrations (>10 μmol/L) cause a transient enhancement of CRAC currents followed by a complete block in S2 for mammalian cells]17,18, YM58483 (IC50 of approximately 150 nmol/L after 24 h of preincubation; IC50 of approximately 10 μmol/L immediately after addition)19,20, and Synta 66 (IC50 of approximately 3 μmol/L)21 (Figure 1); however, these compounds are not specific because they interfere with a variety of other transport processes. The only specific CRAC channel inhibitor tested in human is CM248922, the structure of which has not yet been disclosed. Recent studies have suggested that blocking CRAC channel activity by inhibiting STIM1 may inadvertently affect other channels23. Moreover, STIM1 mutations are associated with a syndrome of immunodeficiency and autoimmunity, whereas ORAI1 mutations cause only a major clinical syndrome of immunodeficiency24. Therefore, molecules that specifically block ORAI1 may have fewer side effects compared with molecules that target STIM1. This is the focus and priority of CRAC channel research (Figure 1).
Figure 1.
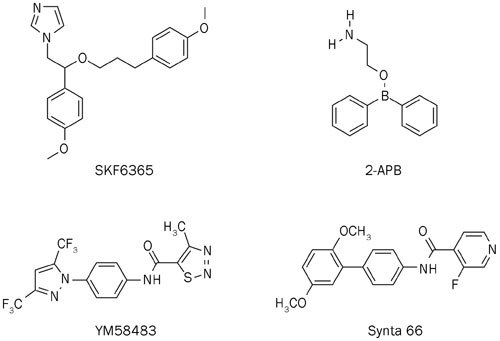
Structures of known CRAC channel inhibitors.
To identify small chemical molecules that block CRAC channel activity, we established an ORAI1 and STIM1 stably co-expressed single-cell clone human embryonic kidney 293 (HEK293) cell line and performed high-throughput screening of libraries containing 32 000 compounds using a fluorescence assay of Ca2+ influx. The average Z-factor of the screen was approximately 0.74, and the coefficient of variation was 4.7%. Hits in the screen were defined by a significantly reduced cytosolic Ca2+ level [inhibition rate ≥MEAN (signal means) +3×STDEV (standard deviations)] elevated by thapsigargin (TG). A total of 19 hits were identified in the screen. Of these, 8 compounds were lanthanide complexes, 3 compounds showed strong cytotoxicity, and 2 compounds exhibited weak inhibition using the patch clamp technique. We selected compound 1 from the remaining 6 compounds because this compound presents a novel chemical scaffold and good drug-like properties, which makes it different from the known CRAC channel inhibitors. In this study, we synthesized a series of structure closed analogs (2a-2h, 3a-3l, 4a-4j, and 5a-5j), with modifications of the left-side benzylamine subunit and right-side phenyl subunit of compound 1, and determined the primary structure-activity relationships (SARs). Several compounds showed improved potency and immune inhibitory activity. Compound 1 inhibits the CRAC channel by specifically targeting the ORAI1 protein. We propose this derivative as a potential scaffold for developing novel CRAC channel inhibitors (Figure 2).
Figure 2.
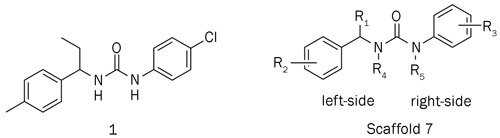
Structure of original hit compound 1.
Materials and methods
Chemistry
The experimental procedures and characterization of all compounds are provided in the Supplementary Information.
Biology experiment
[Ca2+]i measurement
The intracellular calcium level was measured in 96-well plates using an automated fluorometric imaging plate reader (FLIPR, Molecular Devices, Sunnyvave, CA, USA). Either HEK293 or Chinese hamster ovary (CHO) cells, which stably expressed ORAI1 and STIM1, were pre-incubated with 4 μmol/L Fluo-4/AM (Invitrogen) in standard extracellular Ringer's solution (145 mmol/L NaCl, 4.5 mmol/L KCl, 2 mmol/L CaCl2, 1 mmol/L MgCl2, 10 mmol/L D-glucose, and 5 mmol/L Hepes, pH adjusted to 7.4 with NaOH) supplemented with 2.5 mmol/L probenecid at 37 °C for 30–45 min. The cells were washed twice and then immersed in standard extracellular Ringer's solution. The change in Fluo-4 fluorescence was systematically assessed as follows: we first tested the basal calcium level for 30 s; we next added 1 μmol/L TG to open the CRAC channel and constantly measured the calcium level for 3 min; finally, we added a designated concentration (10 μmol/L) of compound and constantly measured the calcium level for an additional 6 min. To correct for variations in dye loading or cell numbers, we normalized the calcium level at 1 min prior to adding the inhibitors and selected the calcium level value at the final 1 min to calculate the inhibition rate of the compounds. A concentration of 10 μmol/L YM58483 was used as the positive control, and an equal amount of dimethylsulfoxide (DMSO) was the negative control.
Tetracycline was added to induce the expression of MSS (monomer ORAI1 covalently linked with two S336–485 domains, abbreviated as MSS)25 and the ORAI1 V102A mutant. The customized medium was used (no calcium) to decrease the toxicity effect of high calcium caused by the constitutively open MSS and V102A channel.
For the Ca2+ influx assay with the MSS and V102A mutants, after loading the cells with Fluo-4/AM, the cells were incubated in standard extracellular Ringer's solution with 2 mmol/L Ca2+ for approximately 10 min. When the calcium level was stable, the cells were treated with 10 μmol/L YM58483 as a positive control, an equal amount of DMSO as a negative control, and different doses of compound 1 to test its inhibitory effect. The inhibition rate of 10 μmol/L YM58483 was set to 100% and the inhibition rate of an equal amount of DMSO was 0 to normalize and calculate the inhibition rate of compound 1. In the Ca2+ influx assay with O1S1 and O2S1 cells, before treating the cells with compounds, 1 μmol/L TG was added to stimulate the opening of the CRAC channel.
Electrophysiology
Electrophysiology and patch clamp experiments were performed at room temperature using the standard whole cell recording configuration25. Cells were plated on poly-L-lysine-coated coverslips 12–24 h prior to the experiments. The extracellular solution contained 120 mmol/L NaCl, 10 mmol/L CaCl2, 2 mmol/L MgCl2, 10 mmol/L tetraethylammonium chloride and 10 mmol/L Hepes (pH 7.4 adjusted with NaOH). For recording the constitutive ICRAC, the pipette was filled with a solution containing 110 mmol/L cesium glutamate, 6 mmol/L MgCl2, 10 mmol/L BAPTA, and 10 mmol/L Hepes (pH 7.2 adjusted with CsOH). The GFP fluorescence of each cell was measured before patch clamp recording. After establishing the whole-cell configuration, voltage stimuli consisting of a 10-ms step to −100 mV followed by a 50-ms voltage ramp from −100 to +100 mV were delivered from the holding potential of 0 mV every 2 s. The currents were digitized at a rate of 20 kHz and filtered offline at 2 kHz. All current traces were leak-subtracted. After seal formation but before break-in, the I–V currents were collected by delivering the voltage ramps, and those with a current magnitude at +50 mV equal to the sustained outward current after break-in were assigned as the leak current. Capacitive currents were determined and corrected before each voltage ramp. The current amplitude at -80 mV of the individual ramp was extracted to monitor the development of ICRAC, and current amplitudes at -80 mV were used for statistical analysis. Data were analyzed using the IGOR Pro 5.01 (Wavemetrics). The averaged results are presented as the mean value±SEM, with the number of experiments indicated.
Cytotoxicity assay
A Cell Counting Kit-8 (CCK-8) assay (Dojindo Laboratories, Kumamoto, Japan) was used to measure the cytotoxicity of compounds on Jurkat cells. Jurkat cells (2×104 cells/well) were seeded into 96-round-bottom-well plates and treated with 10 or 30 μmol/L compound or the corresponding amount of DMSO as a negative control for 48 h. After the cells were washed with PBS once, 10% (v/v) CCK-8 solution was added to each well, followed by incubation for 4 h at 37 °C. The absorbance at 450 nm was determined using an ELISA (enzyme-linked immunosorbent assay) reader (Wallac 1420 Victor2 Microplate Reader, Perkin Elmer).
Immune inhibition assay
Jurkat cells were nucleo-transfected with ORAI1-SS-eGFP plasmids25 and cultured overnight with customized calcium free RPMI-1640 media. Then, the cells were washed, resuspended with complete RPMI-1640 media, and seeded (2×104 cells/well) into 96-round-bottom-well plates. A concentration of 10 μmol/L compound, 10 μmol/L YM58483 (positive control), or the corresponding amount of DMSO (negative control) was added to the wells. The Jurkat cells that express ORAI1-SS-eGFP produce IL-2 because of the constitutively opened CRAC channels. After 24 h, the amount of IL-2 in the cell supernatant was determined using the human IL-2 ELISA kit (human IL-2 duoset, R&D).
Results
The mechanistic studies of compound 1
Compound 1 was evaluated to determine the inhibitory mechanism on the CRAC channel (Figure 3). As shown in Figure 3A and 3B, 10 μmol/L of compound 1 effectively decreased the high intracellular calcium level induced by the TG-opened CRAC channels in the ORAI1 and STIM1 stably co-expressed HEK293 cells. To analyze the target protein and inhibitory effect of compound 1, we tested it in constitutively opened CRAC channels that were formed by ORAI1-SS (monomer ORAI1 covalently linked with two S336–485 domains, MSS)25 and the ORAI1 mutant, V102A26. In MSS cells, the Ca2+ influx data showed that 30 μmol/L of compound 1 maximally inhibited 78% of the calcium level mediated by the MSS construct and that the calculated IC50 was approximately 0.2 μmol/L. Patch clamp data confirmed that 10 μmol/L of compound 1 inhibited 54% of the total MSS current. Although the inhibitory effect of compound 1 on the MSS channels was partial, this result indicated that the possible target site of compound 1 was located on ORAI1, the S (336–485) domain, or both, instead of other regions, except 336-485, on STIM1. The ORAI1 mutant, V102A, produces no Ca2+ selective, constitutively opened CRAC channels, even in the absence of STIM126. The inhibitory effect of compound 1 on these STIM1-free V102A mutant channels is shown in Figure 3C and indicates that 10 μmol/L of compound 1 completely, inhibits the calcium level and the current mediated by the opened V102A channel, which further demonstrates that the target protein of compound 1 is ORAI1.
Figure 3.
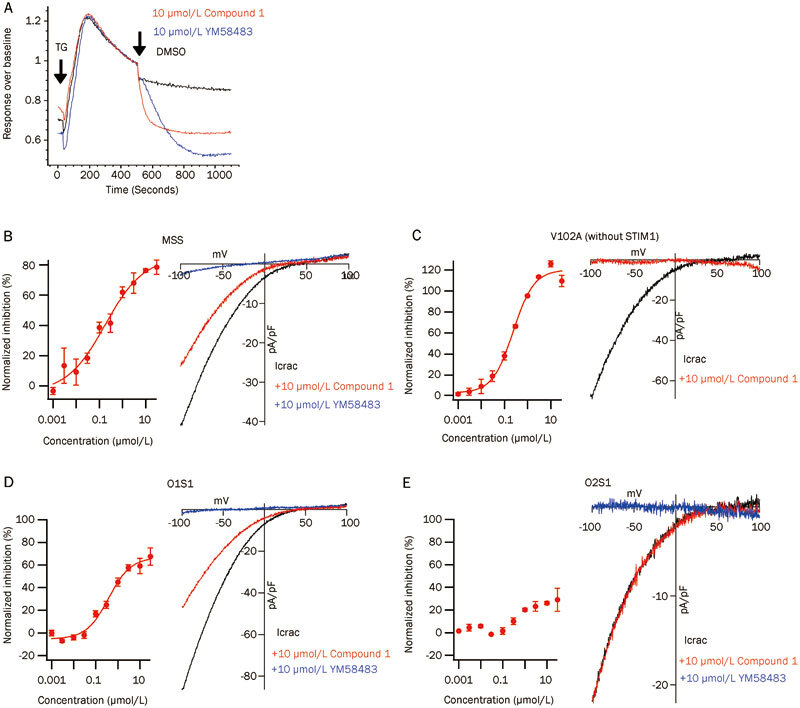
Compound 1 inhibits the CRAC channel by specifically targeting the ORAI1 protein. (A) Ca2+ influx data indicate that Compound 1 decreases the calcium level in the O1S1 opened CRAC channel. (B) Compound 1 partially inhibits the constitutively opened MSS CRAC channel. (C) Compound 1 completely inhibits the constitutively opened V102A CRAC channel. (D) Compound 1 partially inhibits the O1S1 CRAC channel. (E) Compound 1 does not inhibit the O2S1 CRAC channel. CRAC current was recorded, and the data were processed into the leak-subtracted current-voltage relationship at −100 mV. The black, red and blue lines represent the current trace in untreated cells (MSS, n=10; V102A, n=6; O1S1, n=7; O2S1, n=7), the current trace in cells treated with 10 μmol/L of compound 1 (MSS, n=10; V102A, n=6; O1S1, n=9; O2S1, n=7), and the current trace in cells treated with 10 μmol/L of YM58483 (MSS, n=7; O1S1, n=7; O2S1, n=7), respectively.
Human ORAI protein has three homologs, ORAI1, ORAI2, and ORAI3. When these proteins are co-expressed together with STIM1 in HEK293 cells, ORAI1 or ORAI2 generates substantial CRAC current, but ORAI3 fails to produce any detectable Ca2+ selective currents27. To determine whether compound 1 specifically targets ORAI1 but not other ORAI proteins, we used an ORAI1 and STIM1 stably expressed cell line (O1S1) and an ORAI2 and STIM1 stably expressed cell line (O2S1) to test the inhibitory effect of compound 1. As shown in Figure 3D and 3E, compound 1 partially inhibits O1S1 channels. The Ca2+ influx data indicate that 30 μmol/L of compound 1 reaches the maximum inhibition rate of 66.98% for the O1S1 channel and that the calculated IC50 is 0.38 μmol/L. Patch clamp data confirm that 10 μmol/L of compound 1 inhibits 47.00% of the O1S1 current. However, both the Ca2+ influx assay and electrophysiological experiments showed that compound 1 did not inhibit the O2S1 channel. Therefore, these results indicate that compound 1 inhibits the CRAC channel by specifically targeting the ORAI1 protein (Figure 3).
Structure and activity relationship of compound 1 derivatives
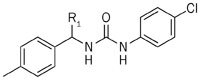
The specificity of compound 1 led us to further explore this class of compounds. We first examined the effect of substitutions at the α-position in the benzyl group of compound 1 on the CRAC channel inhibitory activity, cytotoxicity and IL-2 production (Table 1). Un-substituted compound 2a and methyl substituted compound 2b did not exhibit CRAC channel inhibition at a 10 μmol/L concentration, whereas longer side-chain compounds, such as n-propyl 2c and n-butyl 2d, showed better inhibition of 74.09% and 48.86%, respectively; however, the cytotoxicity of these two compounds was increased significantly compared with compound 1. These results indicate that the length of the side-chain has a large effect on the inhibitory activity and cytotoxicity and that the ?-ethyl phenyl subunit may be the best choice to balance potency and cytotoxicity.
Table 1.
Inhibitory activity for CRAC channel of compounds 2a–2h.
 | |||||
|---|---|---|---|---|---|
| Compound | R1 | Inhibition of Ca2+ influx | Cytotoxicity (%) | IL-2 production | |
| (10 μmol/L) | (10 μmol/L) | (30 μmol/L) | (10 μmol/L) | ||
| 1 | Et |
IC50=3.25±0.17a Imax=82.50%c |
9.49 | 51.17 | 85.82% |
| 2a | H | 0.92% | NTb | NTb | NTb |
| 2b | Me | 7.05% | NTb | NTb | NTb |
| 2c | nPr | 74.09% | 47.65 | 94.82 | 96.35% |
| 2d | nBu | 48.86% | 48.41 | 59.17 | 96.39% |
| 2e | (S)-Et |
IC50=0.75±0.02a Imax=77.71%c |
19.83 | 46.99 | 87.57% |
| 2f | (R)-Et |
IC50=7.60±0.24a Imax=67.99%c |
22.41 | 54.22 | 82.23% |
| 2g | (S)-Ethylene | 37.09% | NTb | NTb | NTb |
| 2h | (S)-Ethynyl | 16.33% | NTb | NTb | NTb |
a IC50 value for Ca2+ influx (μmol/L). IC50 values were estimated by inhibition at eight concentrations. Assays were performed in triplicate, and data represent similar results.
b Not tested.
c Imax value stands for maximum inhibition of Ca2+ influx.
Notably, compound 2f, the R-enantiomer of compound 1 (IC50 of 1 is 3.25 μmol/L), showed weaker inhibition (IC50 of 2f is 7.60 μmol/L) compared with the S-enantiomer compound, 2e (IC50 of 2e is 0.75 μmol/L). In addition, the (S)-ethylene compound, 2g (37.09% inhibition at 10 μmol/L), and the (S)-ethynyl compound, 2h (16.33% inhibition at 10 μmol/L), reduced the inhibitory effect on the CRAC channel compared with the alkyl compound 1. From these results, we propose that the ability to inhibit the CRAC channel is sensitive to the size of the α-position substitutions on the left-side benzyl group. Small alkyl groups at the α-position on the left-side benzyl group were favorable for inhibiting the CRAC channel, and bulky groups may interfere with interactions between the target and molecules (Table 1).
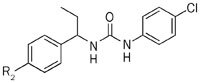
Then, we examined the effects of substitutions at the 4-position in the left-side phenyl group of compound 1 on the CRAC channel inhibitory activity, cytotoxicity and IL-2 production (Table 2). We found that hydrogen (3a) reduced the inhibitory effects on the CRAC channel. Furthermore, the compounds substituted with cyano (3e), methoxycarbonyl (3f), trifluoro (3g), chloro (3h) and fluoro (3k) groups did not change the potency of inhibiting the Ca2+ influx, whereas the cytotoxicity was increased compared with compound 1. The compounds substituted with hydroxyl (3b), methoxyl (3c) and benzyloxyl (3d) groups increased the inhibitory effects of Ca2+ efflux but showed much higher cytotoxicity.
Table 2.
Inhibitory activity for CRAC channel of compounds 3a–3l.
 | |||||
|---|---|---|---|---|---|
| Compound | R2 | Inhibition of Ca2+ influx | Cytotoxicity (%) | IL-2 production | |
| (10 μmol/L) | (10 μmol/L) | (30 μmol/L) | (10 μmol/L) | ||
| 1 | 4-Me |
IC50=3.25±0.17a Imax=82.50%c |
9.49 | 51.17 | 85.82% |
| 3a | 4-H | 19.83% | NTb | NTb | NTb |
| 3b | 4-OH | 70.05% | 40.76 | 37.48 | 85.09% |
| 3c | 4-OMe |
IC50=4.52±0.07a Imax=95.24%c |
32.40 | 56.22 | 87.18% |
| 3d | 4-OBn | 63.08% | 65.50 | 58.16 | 73.46% |
| 3e | 4-CN | 34.50% | 55.07 | 76.54 | 68.06% |
| 3f | 4-COOMe | 38.22% | 75.14 | 79.63 | 72.72% |
| 3g | 4-CF3 | 29.85% | 60.92 | 100.00 | 76.73% |
| 3h | 4-Cl | 45.59% | 41.41 | 90.89 | 87.92% |
| 3i | 2,4-Di-F | 37.42% | 39.91 | 80.75 | 76.45% |
| 3je | 44.07% | NTb | NTb | NTb | |
| 3ke | NAd | NTb | NTb | NTb | |
| 3le | NAd | NTb | NTb | NTb | |
aIC50 value for Ca2+ influx (μmol/L). IC50 values were estimated by inhibition at eight concentrations. Assays were performed in triplicate, and data represent similar results.
bNot tested.
cImax value stands for maximum inhibition of Ca2+ influx.
dNot available.
e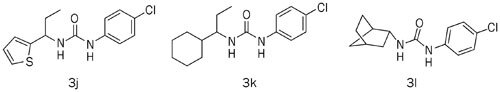
We also investigated the effects of various replacements of the left-side phenyl group of compound 1 by substituting phenyl, hetero aromatic and aliphatic groups and found that compounds with hetero aromatic groups, such as 2-thiophenyl (3j), showed comparable inhibitory effects on the CRAC channel, whereas compounds with aliphatic replacements (3k and 3l) completely lost the inhibitory activity. These results demonstrate that left-side aromatic groups are essential features of CRAC channel inhibitors (Table 2).
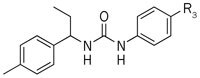
Because the majority of the compounds in Table 2 have high cytotoxicity, the focus was changed to the right-side phenyl ring subunit of compound 1 to obtain improved potency with low cytotoxicity. Compounds (4a−4j) were synthesized and tested (Table 3). The activities of these compounds varied greatly. In the series of compounds possessing chloro groups (4a, 4b, and 4c), the 3-chloro group (4c) reduced the inhibitory activity to 11.14% at 10 μmol/L. The 2-chloro group (4b) reduced the inhibitory activity to 26.90% at 10 μmol/L, and the 4-chloro derivative 1 showed 47.13% inhibition at 10 μmol/L. The inhibitory effects on the CRAC channel of the compound with a 4-position chloride (1) is greater than fluoride (4c) and bromide (4d). Compounds with electron-withdrawing groups (4e and 4f) exhibited better inhibitory activity, whereas compounds with electron-donating groups (4g, 4h, and 4j) showed decreased inhibitory activity. By contrast, removal of the chloro group (4i) significantly decreased the inhibitory activity (Table 3).
Table 3.
Inhibitory activity for CRAC channel of compounds 4a–4j.
 | |||||
|---|---|---|---|---|---|
| Compound | R3 | Inhibition of Ca2+ influx | Cytotoxicity (%) | IL-2 production | |
| (10 μmol/L) | (10 μmol/L) | (30 μmol/L) | (10 μmol/L) | ||
| 1 | 4-Cl |
IC50=3.25±0.17a Imax=82.50%c |
9.49 | 51.17 | 85.82% |
| 4a | 2-Cl | 26.90% | 3.25 | 34.04 | 67.66% |
| 4b | 3-Cl | 11.14% | 31.18 | 73.80 | 98.26% |
| 4c | 4-F |
IC50=1.62±0.11a Imax=71.43%c |
15.60 | 15.01 | 75.54% |
| 4d | 4-Br | 21.10% | NTb | NTb | NTb |
| 4e | 4-NO2 |
IC50=6.95±0.27a Imax=58.97%c |
13.47 | 68.30 | 75.85% |
| 4f | 4-COOEt | 42.86% | 10.91 | 9.45 | 84.18% |
| 4g | 4-OMe | 19.80% | 21.46 | 15.36 | 24.16% |
| 4h | 4-Me | 31.69% | 26.11 | 31.61 | 80.79% |
| 4i | H | 0.26% | 27.82 | 24.28 | 29.66% |
| 4j | 4-cyclopropyl | 11.18% | NTb | NTb | NTb |
aIC50 value for Ca2+ influx (μmol/L). IC50 values were estimated by inhibition at eight concentrations. Assays were performed in triplicate, and data represent similar results.
bNot tested.
cImax value stands for maximum inhibition of Ca2+ influx.
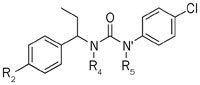
Because modification on the phenyl ring makes it difficult to balance activity and cytotoxicity, we focused on the effect of N–H on the inhibitory activity. We found that N' single-methylated compounds 5a (36.30%), 5c (32.52%) and N,N'-dimethylated compounds 5b (44.30%), 5d (48.35%) exhibited considerable activity compared with compound 1 (47.13%). However, because the introduction of larger moieties (5e, 5f) or a cyclized nitrogen with an ortho-substitution (5g, 5h, 5i, and 5j) can lead to reduced inhibitory activity on the CRAC channel, it is possible that cyclization with nitrogen and a left-side phenyl ortho-substitution influences the torsional angle between the left-side phenyl ethylamine and the right-side phenyl ring. This finding indicates that free N–H is not necessary to maintain the high potency of the inhibitory activity and thus provides a suitable point for further modification to improve the activity (Table 4).
Table 4.
Inhibitory activity for CRAC channel of compounds 5a–5j.
 | |||||||
|---|---|---|---|---|---|---|---|
| Compound | R2 | R4 | R5 | Inhibition of Ca2+ influx | Cytotoxicity (%) | IL-2 production | |
| (10 μmol/L) | (10 μmol/L) | (30 μmol/L) | (10 μmol/L) | ||||
| 1 | Me | H | H |
IC50=3.25±0.17a Imax=82.50%c |
9.49 | 51.17 | 85.82% |
| 5a | Me | H | Me |
IC50=2.13±0.00a Imax=65.20%c |
2.83 | 10.85 | 61.67% |
| 5b | Me | Me | Me |
IC50=11.95±0.83a Imax=95.23%c |
6.25 | 28.02 | 86.76% |
| 5c | OMe | H | Me | 1.46% | −2.13 | 8.86 | 52.86% |
| 5d | OMe | Me | Me |
IC50=6.50±0.31a Imax=91.74%c |
5.88 | 29.13 | 87.38% |
| 5e | OMe | H | Allyl | 4.26% | −1.93 | −6.54 | 47.25% |
| 5f | OMe | (CH2)4 | 13.87 | NTb | NTb | NTb | |
| 5g |
|
H | 15.08% | 3.40 | 35.88 | 75.77% | |
| 5h | Me | 7.48% | −0.51 | 21.92 | 31.57% | ||
| 5i |
|
H | 6.23% | 1.37 | 12.87 | 28.64% | |
| 5j | Me | 22.07% | 17.34 | 19.49 | 77.19% | ||
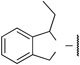
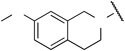
aIC50 value for Ca2+ influx (μmol/L). IC50 values were estimated by inhibition at eight concentrations. Assays were performed in triplicate, and data represent similar results.
bNot tested.
cImax value stands for maximum inhibition of Ca2+ influx.
Interestingly, we discovered that the majority of N-substituted compounds showed relatively low cytotoxicity while retaining their potency to inhibit IL-2 production. For example, the cytotoxicity of compound 5b is less than 30% at 30 μmol/L, but it inhibits IL-2 production by 86% at 10 μmol/L. This compound has improved Ca2+ influx inhibition, immune inhibition and low cytotoxicity and may be a lead in the further development of small molecule inhibitors of the CRAC channel.
Discussion
Recently, several human diseases have been linked to abnormal CRAC channel activities; therefore, interest in this area is growing rapidly, but most of the present chemotypes of small-molecule CRAC channel inhibitors are similar. Thus, it is necessary to diversify the chemical classes that inhibit CRAC channels.
Moreover, previous data indicate that a STIM1 mutation is associated with a syndrome of immunodeficiency and autoimmunity but that a ORAI1 mutation only causes a major clinical syndrome of immunodeficiency. Therefore, molecules that specifically block ORAI1 may have fewer side effects than do molecules that target STIM1; however, the majority of the small molecules used previously were unselective. For example, they blocked the transient receptor potential channel (TRP) C and TRP M channels with a similar potency as CRAC channels. The only specific CRAC channel inhibitor tested in human is CM2489, the structure of which has not yet been disclosed. Therefore, the issue of selectivity has been a central theme in CRAC channel inhibition, and the discovery of molecules that specifically block ORAI1 will be the focus and priority for future CRAC channel research.
In this study, we designed and synthesized a series of novel 1-phenyl-3-(1-phenylethyl)urea derivatives and evaluated their inhibition of Ca2+ influx through CRAC channels. Of these CRAC channel inhibitors, several showed an improved inhibition of IL-2 production in the Jurkat cell line with low cytotoxicity. Mechanistic studies of inhibition indicated that compound 1 inhibits CRAC channels by specifically targeting the ORAI1 protein, which makes these compounds advantageous for further development compared with other non-specific compounds that target both the ORAI1 and STIM1 proteins of the CRAC channel.
Additional mechanistic studies and biological evaluations are currently ongoing. The discovery of this novel, potent and ORAI1-specific chemotype will provide a new strategy for the further design and development of CRAC channel inhibitors.
Author contribution
Fa-jun NAN and Tao XU designed the research; Hai-zhen ZHANG, Xiao-lan XU, Hua-yan CHEN, Sher ALI, Dan WANG and Jun-wei YU performed the research; all authors analyzed data; Hai-zhen ZHANG and Xiao-lan XU wrote the manuscript.
Supplementary information
Synthesis and data of all the compounds (DOC 306 kb)
Acknowledgements
This work was supported by grants from the National Science and Technology Major Projects for Major New Drugs Innovation and Development (2012ZX09304011 and 2013ZX09507002), Chinese Academy of Science for Technological Innovation and Cross-Team Collaboration, the State Key Laboratory of Drug Research, National Key laboratory of Biomacromolecules and the National Natural Science Foundation of China (81102456 and 81373422).
PowerPoint slides
Footnotes
Supplementary Information Synthesis and datafiles of all compounds is available at Acta Pharmacologica Sinica's website.
Hai-zhen Zhang and Xiao-lan Xu: These authors contributed equally to this article.
Contributor Information
Tao Xu, Email: xutao@ibp.ac.cn.
Fa-jun Nan, Email: fjnan@simm.ac.cn.
References
- 1.Hoth M, Penner R. Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature. 1992;355:353–6. doi: 10.1038/355353a0. [DOI] [PubMed] [Google Scholar]
- 2.Zweifach A, Lewis RS. Rapid inactivation of depletion-activated calcium current (ICRAC) due to local calcium feedback. J Gen Physiol. 1995;105:209–26. doi: 10.1085/jgp.105.2.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hogan PG, Lewis RS, Rao A. Molecular basis of calcium signaling in lymphocytes: STIM and ORAI. Annu Rev Immunol. 2010;28:491–533. doi: 10.1146/annurev.immunol.021908.132550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hoth M. Calcium and barium permeation through calcium release-activated calcium (CRAC) channels. Pflugers Arch. 1995;430:315–22. doi: 10.1007/BF00373905. [DOI] [PubMed] [Google Scholar]
- 5.Zweifach A, Lewis RS. Mitogen-regulated Ca2+ current of T lymphocytes is activated by depletion of intracellular Ca2+ stores. Proc Natl Acad Sci U S A. 1993;90:6295–9. doi: 10.1073/pnas.90.13.6295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Feske S. Immunodeficiency due to defects in store-operated calcium entry. Ann N Y Acad Sci. 2011;1238:74–90. doi: 10.1111/j.1749-6632.2011.06240.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Di Capite JL, Bates GJ, Parekh AB. Mast cell CRAC channel as a novel therapeutic target in allergy. Curr Opin Allergy Clin Immunol. 2011;11:33–8. doi: 10.1097/ACI.0b013e32834232b0. [DOI] [PubMed] [Google Scholar]
- 8.Chang WC. Store-operated calcium channels and pro-inflammatory signals. Acta Pharmacol Sin. 2006;27:813–20. doi: 10.1111/j.1745-7254.2006.00395.x. [DOI] [PubMed] [Google Scholar]
- 9.Di Sabatino A, Rovedatti L, Kaur R, Spencer JP, Brown JT, Morisset VD. Targeting gut T cell Ca2+ release-activated Ca2+ channels inhibits T cell cytokine production and T-box transcription factor T-bet in inflammatory bowel disease. J Immunol. 2009;183:3454–62. doi: 10.4049/jimmunol.0802887. [DOI] [PubMed] [Google Scholar]
- 10.Varga-Szabo D, Braun A, Kleinschnitz C, Bender M, Pleines I, Pham M. The calcium sensor STIM1 is an essential mediator of arterial thrombosis and ischemic brain infarction. J Exp Med. 2008;205:1583–91. doi: 10.1084/jem.20080302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Prevarskaya N, Skryma R, Shuba Y. Calcium in tumour metastasis: new roles for known actors. Nat Rev Cancer. 2011;11:609–18. doi: 10.1038/nrc3105. [DOI] [PubMed] [Google Scholar]
- 12.Ritchie MF, Yue C, Zhou Y, Houghton PJ, Soboloff J. Wilms tumor suppressor 1 (WT1) and early growth response 1 (EGR1) are regulators of STIM1 expression. J Biol Chem. 2010;285:10591–6. doi: 10.1074/jbc.M109.083493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang S, Zhang JJ, Huang XY. Orai1 and STIM1 are critical for breast tumor cell migration and metastasis. Cancer Cell. 2009;15:124–34. doi: 10.1016/j.ccr.2008.12.019. [DOI] [PubMed] [Google Scholar]
- 14.Kuo HC, Chang WC. Genetic polymorphisms in Kawasaki disease. Acta Pharmacol Sin. 2011;32:1193–8. doi: 10.1038/aps.2011.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chung SC, McDonald TV, Gardner P. Inhibition by SK&F 96365 of Ca2+ current, IL-2 production and activation in T lymphocytes. Br J Pharmacol. 1994;113:861–8. doi: 10.1111/j.1476-5381.1994.tb17072.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Franzius D, Hoth M, Penner R. Non-specific effects of calcium entry antagonists in mast cells. Pflugers Arch. 1994;428:433–8. doi: 10.1007/BF00374562. [DOI] [PubMed] [Google Scholar]
- 17.Maruyama T, Kanaji T, Nakade S, Kanno T, Mikoshiba K. 2APB, 2-aminoethoxydiphenyl borate, a membrane-penetrable modulator of Ins(1,4,5)P3-induced Ca2+ release. J Biochem. 1997;122:498–505. doi: 10.1093/oxfordjournals.jbchem.a021780. [DOI] [PubMed] [Google Scholar]
- 18.Prakriya M, Lewis RS. Potentiation and inhibition of Ca2+ release-activated Ca2+ channels by 2-aminoethyldiphenyl borate (2-APB) occurs independently of IP3 receptors. J Physiol. 2001;536:3–19. doi: 10.1111/j.1469-7793.2001.t01-1-00003.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ishikawa J, Ohga K, Yoshino T, Takezawa R, Ichikawa A, Kubota H. A pyrazole derivative, YM-58483, potently inhibits store-operated sustained Ca2+ influx and IL-2 production in T lymphocytes. J Immunol. 2003;170:4441–9. doi: 10.4049/jimmunol.170.9.4441. [DOI] [PubMed] [Google Scholar]
- 20.Zitt C, Strauss B, Schwarz EC, Spaeth N, Rast G, Hatzelmann A. Potent inhibition of Ca2+ release-activated Ca2+ channels and T-lymphocyte activation by the pyrazole derivative BTP2. J Biol Chem. 2004;279:12427–37. doi: 10.1074/jbc.M309297200. [DOI] [PubMed] [Google Scholar]
- 21.Ng SW, di Capite J, Singaravelu K, Parekh AB. Sustained activation of the tyrosine kinase Syk by antigen in mast cells requires local Ca2+ influx through Ca2+ release-activated Ca2+ channels. J Biol Chem. 2008;283:31348–55. doi: 10.1074/jbc.M804942200. [DOI] [PubMed] [Google Scholar]
- 22.Pevarello P, Cainarca S, Liberati C, Tarroni P, Piscitelli F, Severi E. Ca2+-release-activated Ca2+ channel inhibitors. Pharm Pat Anal. 2014;3:171–82. doi: 10.4155/ppa.14.7. [DOI] [PubMed] [Google Scholar]
- 23.Wang Y, Deng X, Mancarella S, Hendron E, Eguchi S, Soboloff J. The calcium store sensor, STIM1, reciprocally controls Orai and CaV1.2 channels. Science. 2010;330:105–9. doi: 10.1126/science.1191086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Picard C, McCarl CA, Papolos A, Khalil S, Lüthy K, Hivroz C. STIM1 mutation associated with a syndrome of immunodeficiency and autoimmunity. N Engl J Med. 2009;360:1971–80. doi: 10.1056/NEJMoa0900082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li Z, Liu L, Deng Y, Ji W, Du W, Xu P. Graded activation of CRAC channel by binding of different numbers of STIM1 to Orai1 subunits. Cell Res. 2011;21:305–15. doi: 10.1038/cr.2010.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McNally BA, Somasundaram A, Yamashita M, Prakriya M. Gated regulation of CRAC channel ion selectivity by STIM1. Nature. 2012;482:241–5. doi: 10.1038/nature10752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.DeHaven WI, Smyth JT, Boyles RR, Putney JW., Jr Calcium inhibition and calcium potentiation of Orai1, Orai2, and Orai3 calcium release-activated calcium channels. J Biol Chem. 2007;282:17548–56. doi: 10.1074/jbc.M611374200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Synthesis and data of all the compounds (DOC 306 kb)