

Abstract
Background
The pathological hallmarks of Parkinson’s disease are intracellular inclusions composed mainly of misfolded α-synuclein (αSYN). Under physiological conditions αSYN is mostly localized in synapses. In addition, a portion of αSYN is secreted to the extracellular space, where it may be sequestered by neighboring cells and could induce inflammatory responses. The mechanisms of αSYN internalization and signal transduction are not unequivocally clarified. In this work we investigated in primary mouse astrocytes the involvement of toll-like receptor 4 (TLR4) in the induction of inflammatory responses upon exposure to purified human αSYN produced in bacteria.
Results
The mRNA induction of pro-inflammatory cytokines, inducible nitric oxide synthase and cyclooxygenase-2 was significantly reduced in TLR4 knockout astrocytes. The αSYN-mediated activation of c-Jun N-terminal kinases and p38 mitogen-activated protein kinase tended to be diminished, and nuclear translocation of the p65 subunit of nuclear factor κB was abolished in TLR4 knockout astrocytes. In contrast, the uptake of exogenous αSYN was unaffected by TLR4 knockout.
Conclusions
Extracellular αSYN can activate pro-inflammatory TLR4 pathways in astrocytes, whereas αSYN uptake is independent of TLR4.
Electronic supplementary material
The online version of this article (doi:10.1186/s12868-015-0192-0) contains supplementary material, which is available to authorized users.
Keywords: Synuclein, Toll-like receptor TLR4, Neuroinflammation, Cytokines, Nitric oxide synthase, Cyclooxygenase, MAP kinases, NF-κB, Endocytosis, Astrocytes
Background
Inflammation in the central nervous system is characterized by increased activation of microglia and astrocytes, elevated production of cytokines and other pro-inflammatory mediators including nitric oxide (NO) and prostaglandins, enhanced blood–brain-barrier permeability and hence increased leukocyte invasion. Neuroinflammation is an acutely protective mechanism, however, long-lasting and persistent formation and accumulation of pro-inflammatory mediators can initiate neuronal damage, neuronal circuit impairments and neurodegeneration. Correspondingly prolonged activation of neuroinflammation is thought to play a destructive role in many neurodegenerative diseases [1].
Parkinson’s disease (PD) is the most common neurodegenerative movement disorder, which affects approximately 1 % of the population over 65 years of age. Postmortem studies have shown an increase in neuroinflammatory signals in the brains of PD patients. Activated microglia and astrogliosis have been found in the affected substantia nigra of PD patients [2, 3]. Moreover, a higher density of CD8+ and CD4+ T-cells was shown in PD brains than in healthy control brains [4]. Correspondingly, the neuroinflammatory pathology found in brains of PD patients has been reproduced in several PD animal models [5].
Most of the glial cells in the brain are astrocytes, which are present in all regions of the brain and are localized in strategic positions in close proximity to neurons. Astrocytes are important contributors to the inflammatory responses during brain injury and infection. Similar to microglia, astrocytes can express and secrete a variety of molecules that modulate inflammation, for example toll-like receptors (TLRs), proinflammatory cytokines and NO. TLRs are receptors that are expressed in cells of the innate immune system. They recognize pathogen-associated molecular patterns and certain endogenous molecular patterns. The induction of inflammatory responses by the bacterial endotoxin lipopolysaccharide (LPS) is mediated by TLR4 [6]. Interestingly, TLR4 has been shown to be upregulated in microglia in synucleinopathy brains [7].
The pathological hallmarks of PD are intraneuronal protein inclusions called Lewy bodies or Lewy neurites. These inclusions mainly consist of the protein α-synuclein (αSYN) [8]. In addition, although astrocytes express only very low levels of αSYN themselves [9], αSYN-containing inclusions have been found in astrocytes in postmortem brains from patients with Lewy body diseases [10–12]. Moreover, point mutations and genomic multiplications of the gene encoding αSYN (SNCA) are linked to autosomal-dominant PD [13–15].
αSYN contains 140 amino acids and can associate with lipids [16–19]. The non-amyloid component domain (amino acids 65–95) of αSYN is essential for its pathogenic oligomerization and fibril formation [20]. αSYN is highly abundant in the brain, but is also present in other tissues, for example in red blood cells [21]. It is mainly localized in pre-synaptic terminals [19], and a small portion of the protein is also secreted to the extracellular space [22]. Cell stress like proteasomal dysfunction or oxidative stress may increase the secretion of αSYN [23, 24]. Extracellularly applied αSYN is able to induce inflammatory responses in neurons and glial cells [25]. Primary astrocytes treated with media from αSYN overexpressing SH-SY5Y neuroblastoma cells show increased expression of cytokines, among others the interleukins IL-6 and IL-1β, as well as cyclooxygenase-2 (COX-2) [26]. However, the molecular mechanisms by which αSYN induces neuroinflammatory responses are not entirely resolved. TLR4 was reported to mediate αSYN induction of tumor necrosis factor-α (TNF-α) and IL-6 in both microglia and astroglia [27]. On the other hand, TLR2 was reported to mediate microglial activation (proliferation and cytokine production) in response to cell-secreted αSYN [28], and β1-integrin signaling mediated the morphological and motility responses [29]. Moreover, pre-conditioning microglia with αSYN alters TLR responses [30].
The aim of this study was to investigate the role of TLR4 in the activation of inflammatory responses in astrocytes in the presence of extracellular αSYN. Primary astrocyte-rich cultures from TLR4 knockout mice and littermate controls were treated with purified recombinant human αSYN produced in bacteria. Moreover the internalization of extracellular αSYN into primary astrocytes was investigated. Our results indicate that αSYN is able to activate TLR4 signal transduction and thereby induces the expression of pro-inflammatory cytokines, inducible NO synthase (Nos2) and Cox-2, to mediate nuclear translocation of nuclear factor κB (NF-κB) and to activate c-Jun N-terminal kinase (JNK) and p38 mitogen-activated protein kinase (MAPK) modules. In contrast, the uptake of extracellular αSYN into astrocytes appears to be mediated by TLR4-independent pathway(s).
Results
Treatment with recombinant αSYN induces TLR4-dependent gene expression in primary astrocyte cultures
To investigate if TLR4 plays a role in inflammatory reactions to exogenous αSYN, primary littermate Tlr4+/+ and Tlr4−/− astrocytes were treated with recombinant αSYN, and LPS as a positive control. The expression of Nos2, Il-6, Il1b, Cox-2, Tnfa and Ngf mRNA was investigated by semi-quantitative PCR. LPS dramatically induced the expression of Nos2, Il-6, Il1b, and Cox-2 in Tlr4+/+ astrocytes (Fig. 1). Tnfa expression was also enhanced by LPS treatment while Ngf expression was less consistently altered. Though significantly reduced (Fig. 1b), some induction by LPS could be detected also in Tlr4−/− astrocytes. This might reflect the ability of LPS to activate additional receptors, such as TLR2 [31]. Treatment with 0.7 µM αSYN induced the expression of Nos2, Il-6, Il1b, and Cox-2 in Tlr4+/+ but no significant induction was detected in Tlr4−/− astrocytes (Fig. 1b). Tnfa expression was elevated by αSYN in Tlr4+/+ astrocytes, but remained at basal levels in Tlr4−/− astrocytes.
Fig. 1.
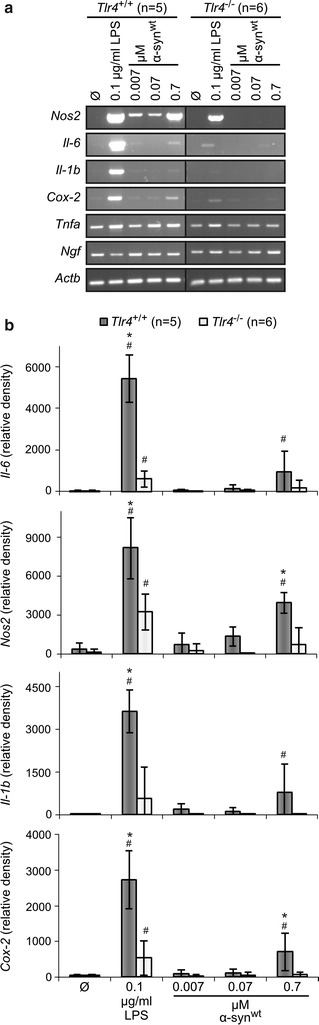
mRNA induction of pro-inflammatory mediators by extracellular αSYN is reduced in Tlr4 −/− primary astrocytes. a Primary astrocyte-rich cultures from littermate Tlr4 +/+ and Tlr4 −/− mice were treated for 72 h with indicated concentrations of LPS (as positive control) and αSYN, or left untreated (Ø). Total RNA was isolated from the cells and semi-quantitative PCR was performed with primers specific for the indicated gene products. Expression of β-actin mRNA (Actb) was measured as loading control. Images representative for 5–6 independent cultures are shown. b Ethidium bromide stained RT-PCR band signals were quantified with ImageJ software and normalized to Actb. Error bars indicate standard deviation, #p < 0.05 compared to untreated samples (ANOVA Fisher’s PLSD), *p < 0.05 compared to Tlr4 −/− (Student’s t test)
Time course experiments confirmed the inductions within few hours of Nos2 and Cox-2 as well as Il-6 and Il1b, which were abolished in Tlr4−/− astrocytes (Fig. 2). Tnfa was similarly induced by αSYN while again only basal expression remained in Tlr4−/− astrocytes. Washout of agonists showed the reversible nature of astrocyte stimulations (Fig. 2). We also measured NO release from stimulated astrocytes (Additional file 1: Figure S1). Similar to LPS, αSYN treatment clearly induced NO release, although overall the variance of this assay was high. Nevertheless, administration of viper peptide that inhibits TLR4 largely abolished NO release, in contrast to the control peptide (Additional file 1: Figure S1). In conclusion, the mRNA induction of pro-inflammatory mediators by extracellularly applied αSYN involves TLR4.
Fig. 2.
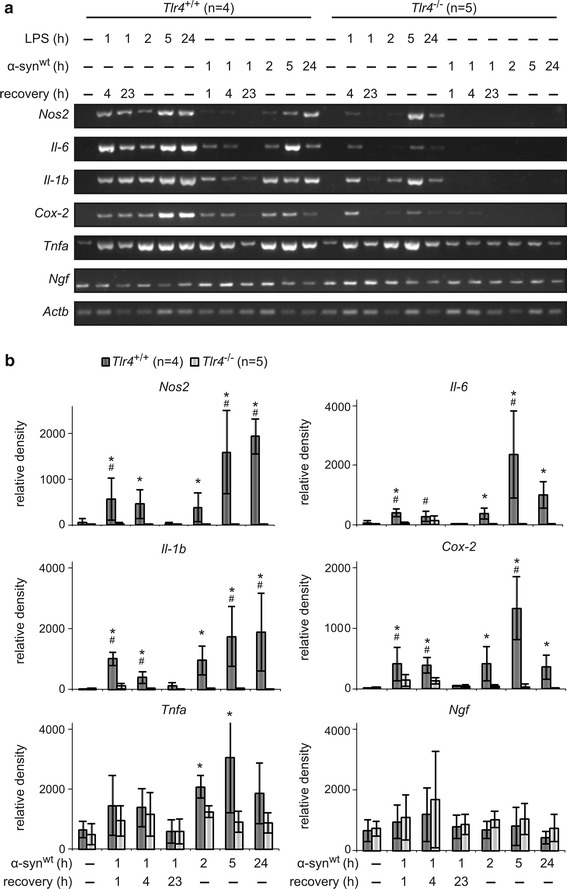
Time course and recovery of pro-inflammatory mediator expression. a Primary astrocyte-rich cultures from littermate Tlr4 +/+ and Tlr4 −/− mice were treated for 1 h with 1.0 µg/ml LPS (as positive control) or 0.7 µM αSYN, after which the cells were allowed to recover in growth media for 1, 4 or 23 h. Parallel cultures were continuously treated with 1.0 µg/ml LPS or 0.7 µM αSYN for 2, 5 or 24 h. Control cells were left untreated (-). Total RNA was isolated and semi-quantitative PCR was performed as in Fig. 1a. Images representative for 4–5 independent cultures are shown. b RT-PCR band quantifications were done as described for Fig. 1b. Error bars indicate standard deviation, #p < 0.05 compared to untreated samples (ANOVA Fisher’s PLSD), *p < 0.05 compared to Tlr4 −/− (Student’s t test)
Decreased p38 and JNK phosphorylation in Tlr4−/− astrocytes after αSYN treatment
Since mRNA inductions of inflammatory mediators by extracellular αSYN appeared to depend on the presence of TLR4 we wondered if other pathways that are activated by TLR4 were also affected. Therefore we treated primary astrocytes from littermate Tlr4+/+ and Tlr4−/− mice with LPS as positive control or recombinant human αSYN for 1 h. The cells were recovered in growth media for 0–24 h (Additional file 2: Figure S2A) and immunoblotting was performed using antibodies against phosphorylated (activated) p38 MAPK and JNK. In Tlr4+/+ cells treatment with αSYN led to phosphorylation of p38 MAPK and JNK whereas in Tlr4−/− less induction of the phosphorylation was seen. In another experiment the astrocytes were continuously treated with LPS and αSYN for 0–24 h (Additional file 2: Figure S2B). Also in this experiment we could detect increased phosphorylation of p38 MAPK and JNK after 2–6 h in Tlr4+/+ astrocytes. Again the induction of p38 MAPK and JNK phosphorylation tended to be reduced in Tlr4−/− astrocytes. In conclusion, treatment of astrocyte-rich primary cell cultures with recombinant αSYN induced phosphorylation of p38 MAPK and JNK, involving TLR4.
Extracellularly applied αSYN does not cause to nuclear translocation of NF-κB in Tlr4−/− astrocytes
Activation of TLR4 leads to phosphorylation and degradation of IκB and thus allows the transcription factor complex NF-κB to translocate to the nucleus. Primary Tlr4+/+ and Tlr4−/− astrocytes were treated with different concentrations of the positive control LPS or recombinant human αSYN for 6 h. The cells were immunostained for p65, a component of the class II NF-κB protein complex, and p65 positive nuclei were quantified. As expected, LPS treated cells showed an increased number of p65 positive nuclei in both Tlr4+/+ and Tlr4−/− cells, however to a lesser extent in the knock out astrocytes (Fig. 3). The recombinant αSYN induced NF-κB translocation in Tlr4+/+ cells in a concentration dependent manner, whereas in Tlr4−/− astrocytes very few p65 positive nuclei were detected. Thus indicating that extracellular αSYN can induce the nuclear translocation of NF-κB in a TLR4-dependent manner.
Fig. 3.
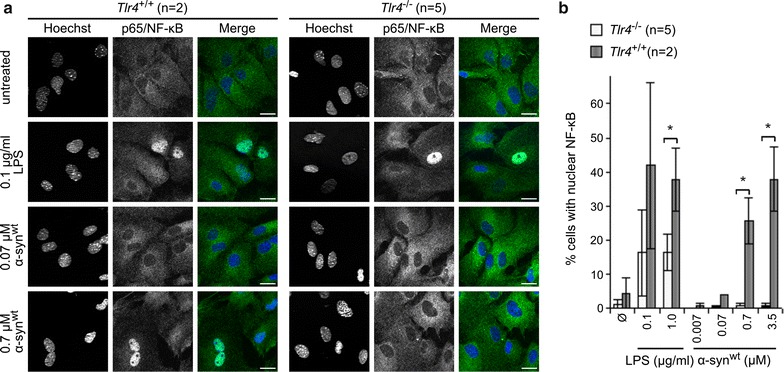
αSYN treatment induces TLR4 dependent nuclear translocation of NF-κB in primary astrocytes. a Primary astrocyte-rich cultures from littermate Tlr4 +/+ and Tlr4 −/− mice were treated with indicated concentrations of LPS (as positive control) and recombinant human αSYN for 6 h or left untreated. The cells were fixed and immunostained using an antibody specific for p65 (green). Nuclei were counterstained with Hoechst 33342 (blue). Images were obtained with 25× objective using Axioimager microscope equipped with ApoTome Imaging system (Carl Zeiss). Scale bars correspond to 20 µm. b p65/NF-κB positive versus total cells were quantified from images as shown in (a). 200–400 cells per sample were analyzed. Error bars show standard deviation, *p < 0.05 (Student’s t test), n is indicated in the figure
Primary astrocytes internalize αSYN from the medium in a TLR4-independent manner
Different studies have indicated that a variety of cell types, for example neuronal cells, microglia and astrocytes, are able to internalize extracellular αSYN [26, 32]. The mechanism of the uptake of the monomeric form has been suggested to differ from the uptake of oligomeric and fibrillar αSYN [33]. We investigated if primary Tlr4+/+ and Tlr4−/− astrocytes cultures were able to internalize the recombinant αSYN. Indeed, after 48 h of continuous incubation an immunoblot signal for human αSYN could be detected in samples that had been treated with 0.07 or 0.7 µM wild-type αSYNwt, mutant αSYNA30P or phosphorylation-deficient αSYNS129A (Fig. 4a). No considerable difference in the internalization of different αSYN variants or between the genotypes of astrocytes could be detected. Pathological αSYN is extensively phosphorylated at S129 and at this site phosphorylated αSYN is found in Lewy bodies [34]. To see if the internalized αSYN is phosphorylated in the astrocytes we treated primary astrocyte cultures with recombinant αSYN for 48 h. No signal could be detected with a phospho-S129 αSYN specific antibody (Fig. 4b). As a positive control for the antibody we used brain lysate from an aged SNCA transgenic mouse, which harbors excessive amounts of S129 phosphorylated αSYN [35]. Also the αSYNS129A mutant was internalized (Fig. 4b), suggesting that phosphorylation at S129 is not a predominant factor in astrocytic uptake of αSYN.
Fig. 4.
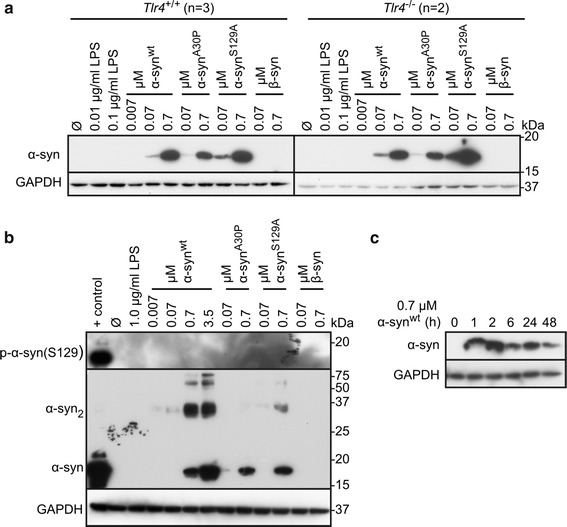
Extracellular αSYN is internalized by astrocytes in a TLR4 independent manner. Primary astrocytes from littermate Tlr4 +/+ and Tlr4 −/− mice (a) or Tlr4 +/+ mice (b) were continuously treated for 48 h with indicated concentrations of recombinant human synucleins or LPS or left untreated (Ø). Cells were washed twice with PBS prior to lysis. The protein lysates were immunoblotted and probed with human-specific anti-αSYN (a, b) and also antibody against αSYN phosphorylated at S129 (b). GAPDH serves as control for equal protein loading throughout. Positive control in (b) is brain lysate from a 18 month old Thy1[A30P]hSNCA transgenic mouse, which has high amounts of phosphorylated αSYN. N is indicated in the figure (a) or N = 8 (b, c). c Primary wild type astrocytes were treated continuously with 0.7 µM αSYN for indicated times. Protein lysates were examined by immunoblotting as above
Next we made a time course experiment where primary astrocyte cultures were treated continuously for 0–48 h with recombinant human αSYN. Already after 1 h the astrocytes had internalized detectable amounts of the recombinant αSYN. Interestingly, the intensity of the signal from internalized αSYN diminished with time (Fig. 4c). Thus, astrocytes seem to be able to purge extracellular αSYN.
Discussion
The molecular mechanisms that trigger degeneration of the dopaminergic neurons in PD are still under investigation. It is currently heavily debated if αSYN can act as an agent that spreads neurodegeneration in PD brains and thus cause the progression of the disease. The role of the portion of αSYN that is secreted from neurons is largely unknown. Previous studies have shown that treatment with recombinant αSYN produced in bacteria as well as cell culture medium from cells that overexpress αSYN induces neuroinflammatory responses in microglia and astroglia [26, 27, 36–38]. In agreement with this we could see an induction of cytokines, Cox-2 and Nos2 mRNA, phosphorylation of p38 MAPK and JNK, and nuclear translocation of NF-κB in primary astrocyte rich cultures that had been treated with recombinant human αSYN purified from bacteria. Previous studies have suggested that TLR4 may be involved in synucleinopathies. Upregulation of TLR4 has been detected in multiple system atrophy, both in the brains of patients with the disease and in brains from a transgenic mouse model of the disease [7, 39]. Specifically, Fellner et al. [27] recently showed that αSYN preparations greatly enhanced rapid secretion of TNF-α and C-X-C motif chemokine ligand 1, and a more delayed secretion of IL-6 in Tlr4+/+ astrocytes, which was strongly suppressed in Tlr4−/− astrocytes. Our present study confirms these findings at the mRNA level for the delayed Il-6 response, and adds Il1b to the list of TLR4-dependent αSYN responses. Moreover, we identify Nos2 and Cox-2 as potential signaling targets, and show the activation of MAPK and NF-κB pathways by which TLR4 appears to mediate pro-inflammatory responses to extracellular αSYN in astrocytes.
Astrocytes are in close proximity to neurons and neuronal synapses. These cells have been shown to be important scavengers of different potentially neurotoxic molecules that are secreted from neurons like glutamate, potassium and calcium. In accordance with this, we could detect a fast astrocytic internalization of the applied extracellular αSYN followed by degradation. These findings are supported by a previous study where cell secreted αSYN was readily endocytosed by primary rat astrocytes [26]. This implicates that astrocytes are decreasing the amount of potentially proinflammatory extracellular αSYN. The exact mechanism of the uptake of αSYN is still being investigated. In addition, the mechanism of uptake of monomeric and protofibrillar αSYN has been suggested to differ [33]. Monomeric αSYN has been suggested to be able to passively diffuse through the plasma membrane [40]. Thus αSYN would be localized in the cytosol and be directly available for proteasomes. However, also lipid-raft mediated endocytosis has been suggested for the uptake of monomeric αSYN [41]. The reproducible TLR4-independent αSYN uptake route in astroglial cells observed here and by Fellner et al. [27] remains to be further characterized. It is presently unknown if other receptor(s) mediate αSYN endocytosis in astrocytes, or if it is a receptor-independent form of pinocytosis, or a non-classical internalization or membrane penetration mechanism.
The formation and biological activities of oligomeric and fibrillar αSYN species are a vast topic in the field. Specifically, pre-formed fibrils are suggested to propagate cell-to-cell spread of αSYN [42, 43], and specific types of αSYN oligomers were reported to activate TLR2 on microglia [28]. On the other hand, “soluble” αSYN was a powerful stimulator of TLR4 both on microglia and astrocytes [27]. Cell type and receptor specificities, experimental conditions or conceptual issues could account for such differences. The present study did not focus on the nature of αSYN species involved in TLR4 responses and uptake in astrocytes. No procedures were undertaken to pre-aggregate our αSYN preparations, but we cannot rule out the presence of some αSYN oligomeric or aggregated species. Moreover, it is hard to predict what happens on the membrane surface, and if αSYN oligomers assemble near or on TLR4, or if αSYN oligomerizes during or after uptake. More work is necessary to unravel the different roles of the various αSYN assemblies in the brain.
The αSYN level has been measured to be 0.01 or 0.035 nM in the interstitial fluid of control or transgenic SNCA mice, respectively [44]. The inflammatory responses were seen when we used 0.7 µM of αSYN, which is a more than 20,000 times higher concentration. However, it is possible that the local concentration within synapses, which are touched by astrocytic endfeet, can be at least temporarily increased. It would be very interesting to be capable of measuring the αSYN concentration within synapses and to investigate the effect of physiologically relevant extracellular αSYN concentrations.
Conclusions
Extracellular αSYN can induce inflammatory responses in astrocytes in a TLR4-dependent manner and this may worsen the stress conditions in synucleinopathy brains. On the other hand, astrocytes may also play a protective role in the disease progression by efficiently sequestering and degrading the proinflammatory extracellular αSYN.
Methods
Antibodies
Primary antibodies used were: rat monoclonal against human αSYN (15G7 [45]; provided by Dr. Elisabeth Kremmer, Helmholtz Center, Munich, Germany), mouse monoclonal anti-βSYN (Syn207; Millipore), mouse monoclonal against glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (6C5; Meridian Life Sciences), rabbit monoclonal 3D7 against phospho-p38 MAPK (T180/Y182), rabbit polyclonal anti-p38 MAPK, rabbit polyclonal against phospho-JNK (T183/Y185), rabbit monoclonal 56G8 anti-JNK (all from Cell Signaling Technology), F-6 mouse monoclonal against NF-κB p65 (Santa Cruz Biotechnology) and rabbit monoclonal EP1536Y against αSYN phosphorylated at S129 (Abcam). Secondary peroxidase conjugated antibodies were purchased from Jackson ImmunoResearch Laboratories and secondary Alexa-Fluor conjugated antibodies were purchased from Invitrogen GmbH.
Synuclein expression constructs
pET30a SNCB was a kind gift from Hilal Lashuel at the Swiss federal institute of technology in Lausanne, Switzerland. To create bacterial expression vectors SNCA wt, A30P and S129A were cloned from pCDNA3.1-SNCA [46] into NheI/HindIII site of pET21a vector (Novagen). The nucleic acid sequences of the constructs were confirmed by sequencing using BigDye Terminator v3.1 according to the manufacturer’s instructions. Reaction products were analyzed using an ABI 3100 Genetic Analyzer.
Preparation of recombinant αSYN
DYT medium (1.6 % (w/v) Tryptone, 1 % (w/v) Yeast extract, 0.5 % (w/v) NaCl) supplied with 100 µg/ml ampicillin was inoculated with Escherichia Coli BL21 Rosetta2 (Stratagene) transformed with pET21a-SNCA wt or mutants or pET30a-SCNB. The bacteria were grown at 37 °C at 200 rpm to an optical density between 0.5 and 0.8 at 590 nm. Protein expression was induced by 0.5 mM isopropyl-β-d-thiogalactopyranoside for 4–6 h at 37 °C at 200 rpm. The bacteria were centrifuged at 3500×g for 10 min at 4 °C and the cell pellet was stored at −20 °C.
The bacterial pellet was gently diluted in a 10 mM phosphate buffer (pH 7.4) containing 25 mM NaCl and EDTA-free Cømplete protease inhibitor (Roche Diagnostics). The cells were homogenized in a French press (EmulsiFlex-C5; AVESTIN) and sonicated three times for 30 s at 50 % input (SONOPULS HD 2070; BANDELIN). The solutions were incubated for 15 min at 95 °C. The denatured proteins were removed by two time centrifugation at 17,000×g at 4 °C for 30 min. The supernatant was applied on a Q-Sepharose column (50 ml, diameter 23 mm) and eluted with a two column volumes long 25 mM to 1000 mM NaCl gradient in 10 mM phosphate buffer (pH 7.4). Fractions with strong synuclein signals on dot blots were desalted on a Sephadex S-200 column (column volume 180 ml, diameter 16 mm) using a 10 mM phosphate buffer containing 150 mM NaCl, (pH 7.4). The synuclein content in the eluted fractions was monitored on dot blots. Absence of contaminating proteins was examined in 2–5 µl samples by a sensitive variant of Coomassie staining (10 % (v/v) ethanol, 30 mM HCl and 0.3 mM Coomassie Brilliant Blue G250). Fractions that were positive for an immunoblot synuclein signal and had only one visible protein band in the Coomassie gel were pooled, aliquoted and stored at −20 °C. The absorbance at 280 nm was measured and the synuclein concentrations were calculated using the extinction factors ε(280, 0.1 %) 0.354 for αSYN and 0.417 for βSYN (calculated with the ProtParam tool of ExPASy, Swiss Institute of Bioinformatics). Characterization of recombinant synucleins is shown in Additional file 3: Figure S3.
Endotoxin levels of the recombinant proteins were estimated using PYROGENT Plus Single Tests (sensitivity 0.06 EU/ml) following the manufacturer’s instructions (LONZA). Briefly, Limulus Amebocyte Lysate was dissolved in 250 μl of diluted protein samples (1:1, 1:10 and 1:25), or endotoxin-free water, then incubated for 60 min at 37 °C.
Mouse maintenance and preparation of primary astrocytes
Mice were kept under standard conditions with free access to food and water in a cycle of 12 h of light and 12 h of dark. The local animal welfare committee, Referat 35, Regierungspräsidium Tübingen approved all experiments and procedures. Number of animals used and their suffering was kept to a minimum.
Heterozygous Tlr4+/− mice [47] were intercrossed. Astrocyte-rich primary cultures were prepared as described previously [48]. Newborn pups were decapitated, and whole brains were removed. The brain from each pup was handled separately. Cells were separated in preparation buffer (137 mM NaCl, 5.4 mM KCl, 0.2 mM KH2PO4, 0.2 mM Na2HPO4, 1 g/l glucose, 20 g/l sucrose, 50 µg/ml gentamycin) by mechanically forcing the brains through nets with a mesh width of first 250 µM and second 135 µm. The cells were resuspended in Dulbecco’s modified Eagle’s medium (Biochrom) supplemented with 10 % fetal calf serum, 10 U/ml penicillin G and 10 µg/ml streptomycin sulfate. Cells were cultured in 75 cm2 cell culture bottles at 37 °C in 5 % CO2 in a humidified atmosphere. Each individual culture was genotyped by PCR of genomic DNA (primers Tlr4+/+rev 5′-CGTGTAAACCAGCCAGGTTTTGAAGGC-3′, Tlr4+/+for 5′-TGTTGCCCTTCAGTCACAGAGACTCTG-3′, Tlr4−/−rev 5′-TGTTGGGTCGTTTGTTCGGATCCGTCG-3′). After 1 week in culture the flasks containing the astrocytes were shaken at 37 °C at 180 rpm over night. Then the cells were washed once with phosphate buffered saline (PBS, pH 7.4; 2.2 mM KH2PO4, 7.8 mM Na2HPO4, 150 mM NaCl) and fresh growth medium was supplied. After one additional week the cells were plated in appropriate cell culture dishes. In order to avoid stress the astrocytes were allowed to normalize for 4–7 days and they were supplied with fresh media 1 or 2 days before use in experiments. The cells were incubated in growth media unless stated otherwise supplemented with indicated concentrations of LPS or recombinant synuclein for indicated times.
Semi-quantitative PCR
Cells were lysed in RLT-RNA lysis buffer at −20 °C. RNA was isolated using the RNeasy Mini kit (Qiagen). cDNA was produced with Transcriptor High Fidelity cDNA Synthesis kit (Roche Diagnostics) and anchored-oligo(dT)18 primer. The cDNA was used as template for PCR reactions with given primers (Actb: 5′-CTAAGGCCAACCGTGAA-3′ and 5′-CCGGAGTCCATCACAAT-3′, Il1b: 5′-CAGGCAGGCAGTATCACTCA-3 and 5′-AGGCCACAGGTATTTTGTCG-3′, Il-6: 5′-GTTCTCTGGGAAATCGTGGA-3 and 5′-GGAAATTGGGGTAGGAAGGA-3′, Cox-2: 5′-TCCTCCTGGAACATGGACTC-3′ and 5′-CCCCAAAGATAGCATCTGGA-3′, Tnfa: 5′-AGCCCCCAGTCTGTATCCTT-3′ and 5′-AGCAAAAGAGGAGGCAACAA-3′, Ngf: 5′-GCAGTGAGGTGCATAGCGTA-3′ and 5′-CACTGAGAACTCCCCCATGT-3′, Nos2: 5′-GTGGTGACAAGCACATTTGG-3′ and 5′-GGCTGGACTTTTCACTCTGC-3′). The DNA fragments were separated in 1.5 % (w/v) agarose gels containing ethidium bromide. DNA bands were detected with a Vilber Lourmat. Signals were quantified by densitometry using ImageJ software.
Western immunoblot analysis
Cells were lysed for 30 min on ice in lysis buffer (1 % Triton X-100, 50 mM Tris–HCl (pH 7.6), 150 mM sodium chloride, 10 mM sodium pyrophosphate, 2 mM EDTA and Cømplete protease inhibitor cocktail from Roche Diagnostics). Cell debris was removed by centrifugation (15 min at 14,000×g and 4 °C) and protein concentrations of lysates were determined using a bicinchoninic acid protein assay kit (Pierce). Samples were diluted in Laemmli buffer, boiled at 95 °C for 5 min, and centrifuged for 20 s at 16,100×g before loading. A total of 10–25 µg proteins were then subjected to denaturing 10 or 15 % polyacrylamide gel electrophoresis and transferred onto Immobilon-P polyvinylidene difluoride membranes (EMD Millipore). Membranes were pre-incubated for 1 h at 20 °C in 5 % skim milk in TBS-T [50 mM Tris–HCl (pH 7.4), 150 mM NaCl and 0.1 % Tween-20]. Primary antibodies diluted in Western Blocking Reagent (Roche Diagnostics) were incubated over night at 4 °C. Then membranes were washed 3–5 times in TBS-T. Horseradish peroxidase-conjugated secondary antibodies in TBS-T supplemented with 5 % milk were incubated for 1 h at 20 °C and membranes washed again 3–5 times with TBS-T. Detection of proteins was performed with the Immobilon Western Chemiluminescent HRP Substrate (EMD Millipore) on Amersham Hyperfilm™ for enhanced chemiluminescence (GE Healthcare).
NF-κB nuclear translocation
Primary astrocytes were cultured on poly-d-lysine and collagen coated coverslips. Cells were treated with 0.007–3.5 µM recombinant αSYN or 0.1–1.0 µg/ml LPS in growth media for 6 h. After which the cells were fixed in 4 % (w/v) paraformaldehyde in PBS and permeabilized with 1 % (v/v) Triton X-100 for 5 min and blocked in 10 % (v/v) normal goat serum in PBS solution for 60 min at room temperature. Anti-p65 NF-κB (1:100) and anti-mouse-Alexa488 (1:1000) were diluted in 1 % (w/v) BSA in PBS and incubated for 1 h at room temperature or over night at 4 °C in a humidified chamber. Nuclei were counterstained with 2 µg/ml Hoechst 33342 in PBS for 10 min at room temperature. Coverslips were mounted in Mowiol/DABCO solution onto glass slides. Images were acquired with 25× objective with AxioImager microscope equipped with ApoTome Imaging system and processed with AxioVision software. A blinded observer manually counted the number of nuclei positive for NF-κB as well as total nuclei in 200–400 cells per condition.
Uptake of exogenous αSYN
Primary astrocytes were plated in six-well cell culture plates. The cells were incubated with 0.007–3.5 µM recombinant synuclein, which was added to the growth media. The cells were treated for 1–48 h. In some experiments a 1 h treatment was followed by a recovery time of 0.5–48 h, where the medium was replaced with fresh medium. The cells were washed twice in cold PBS, lysed and processed for Western immunoblot analysis as described above.
Statistical analyses
Each experiment was performed independently at least three times unless stated otherwise. The statistical analyses were done with JMP 9 or StatView software (both from SAS Institute Inc). Comparisons were analyzed with either unpaired Student’s t test (when comparing differences between genotypes) or two-way ANOVA followed by Fisher’s unprotected least significant difference (PLSD) post hoc test (when assessing inductions after treatments), as stated in the figure legends. P values less than 0.05 were considered as statistically significant.
Authors’ contributions
EHR designed and performed the study and analyses, and drafted the manuscript. SSW provided technical assistance and experimental help. PJK conceived the study, participated in its design and coordination, and finalized the manuscript. All authors read and approved the final manuscript.
Acknowledgements
15G7 anti-αSYN was obtained Elisabeth Kremmer (Helmholtz Center, Munich, Germany). The βSYN expressing construct was provided by Hilal Lashuel (EPFLausanne, Switzerland). Tlr4+/− mouse breeder pairs were provided by Tilo Biedermann (Department of Dermatology, University Clinics Tübingen, Germany) with permission from Shizuo Akira (Osaka University, Japan). We thank Cindy Boden for animal assistance and Naoto Sugeno for critically reading the manuscript. The study was funded by the German National Genome Research Network (Parkinson’s Disease), the Hertie Foundation, and supported by the Deutsche Forschungsgemeinschaft and Open Access Publishing Fund of the University of Tübingen.
Compliance with ethical guidelines
Competing interests The authors declare that they have no competing interests.
Abbreviations
- COX-2
cyclooxygenase-2
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- IL
interleukin
- JNK
c-Jun N-terminal kinase
- LPS
lipopolysaccharide
- MAPK
mitogen-activated protein kinase
- NF-κB
nuclear factor-κB
- NO
nitric oxide
- NOS
NO synthase
- PBS
phosphate-buffered saline
- PD
Parkinson’s disease
- SYN
synuclein
- TLR
toll-like receptor
- TNF-α
tumor necrosis factor-α
Additional files
{kind=link}
Figure S1. NO production in αSYN and TLR4-inhibitor treated astrocytes. Primary astrocytes were treated in OptiMEM (supplemented with penicillin G and streptomycin) with 5 µM of control peptide (ctrl pep; CP7: RNTISGNIYSARRRRRRRRR) or TLR4 inhibitor peptide (viper: KYSFKLILAEYRRRRRRRRR) (both from Imgenex/Biomol) two hours prior to the addition of recombinant αSYN (0.7 µM or 3.5 µM, as indicated) or LPS (0.1 µg/ml) as positive control or left untreated (Ø). 48 h later NO released to the media was measured using Griess reagent (Fluka/Sigma-Aldrich). The amount of NO was normalized to total protein amount in the sample well. The samples were incubated protected from light for 15 min, after which the optic absorption at 550 nm was measured (Model 680 Microplate Reader, Bio-Rad). A standard curve made from 0 to 50 µM sodium nitrite was used as reference. Measurements were normalized against the total protein content of the well. Error bars are standard deviation; n = 16.
{kind=link}
Figure S2. Extracellular αSYN induces less p38 MAPK and JNK phosphorylation in Tlr4 −/− than in Tlr4 +/+ astrocytes. Primary astrocyte-rich cultures from littermate Tlr4 −/− and Tlr4 +/+ mice were left untreated (-) or treated with LPS (as positive control) and recombinant human αSYN for one hour, after which medium was replaced and cells let to recover for indicated times (A), or continuously for indicated times (B). Immediately after lysis the cell lysates were immunoblotted for phosphorylated p38 MAPK and JNK. Total p38 MAPK, total JNK and GAPDH serve as controls for equal protein loading. Arrows indicate the heights of JNK2 α2 and β2 (48 kDa, black arrow), JNK1, JNK2α1 and JNK2β1 (44 kDa, grey arrow), and a potentially cross-reacting band possibly from phosphorylated ERK2 (41 kDa, open arrow). Several representative experiments shown for a total number of (A) N = 2 (Tlr4 +/+) and N = 9 (Tlr4 −/−) and (B) N = 4 (Tlr4 +/+) AND N = 5 (Tlr4 −/−) mice, respectively.
{kind=link}
Figure S3. Low levels of contaminating endotoxins and proteins in the recombinant synuclein preparations. (A) Endotoxin levels in the purified protein samples were measured with the 0.06 EU/ml cut off using three dilutions of the samples (1:1, 1:10 and 1:25). Protein concentration was measured with nanodrop using the extinction factors ε(280, 0.1 %) 0.354 for αSYN and 0.417 for βSYN. The table indicates the concentrations of synuclein and endotoxin levels in the protein stocks used in this work. (B) Samples of purified recombinant αSYN and βSYN were tested for purity by a sensitive variant of Coomassie stained polyacrylamide gels. (C) Samples of purified synuclein proteins were immunoblotted using antibodies specific for αSYN and βSYN, respectively. (D) Samples that were cut out from Coomassie stained polyacrylamide gels (indicated by red dashed boxes in the upper right panel) were trypsinized and the peptides were analyzed by mass spectroscopy.
Contributor Information
Emmy H. Rannikko, Email: emmy.rannikko@ki.se
Stephanie S. Weber, stephanie.sara.weber@googlemail.com
Philipp J. Kahle, Email: philipp.kahle@uni-tuebingen.de
References
- 1.Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010;140(6):918–934. doi: 10.1016/j.cell.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McGeer PL, Itagaki S, Boyes BE, McGeer EG. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology. 1988;38(8):1285–1291. doi: 10.1212/WNL.38.8.1285. [DOI] [PubMed] [Google Scholar]
- 3.Damier P, Hirsch EC, Zhang P, Agid Y, Javoy-Agid F. Glutathione peroxidase, glial cells and Parkinson’s disease. Neuroscience. 1993;52(1):1–6. doi: 10.1016/0306-4522(93)90175-F. [DOI] [PubMed] [Google Scholar]
- 4.Brochard V, Combadiere B, Prigent A, Laouar Y, Perrin A, Beray-Berthat V, Bonduelle O, Alvarez-Fischer D, Callebert J, Launay JM, et al. Infiltration of CD4 + lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J Clin Invest. 2009;119(1):182–192. doi: 10.1172/JCI36470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hirsch EC, Hunot S. Neuroinflammation in Parkinson’s disease: a target for neuroprotection? Lancet Neurol. 2009;8(4):382–397. doi: 10.1016/S1474-4422(09)70062-6. [DOI] [PubMed] [Google Scholar]
- 6.Poltorak A, He X, Smirnova I, Liu M-Y, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282(5396):2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 7.Stefanova N, Reindl M, Neumann M, Kahle PJ, Poewe W, Wenning GK. Microglial activation mediates neurodegeneration related to oligodendroglial α-synucleinopathy: implications for multiple system atrophy. Mov Disord. 2007;22(15):2196–2203. doi: 10.1002/mds.21671. [DOI] [PubMed] [Google Scholar]
- 8.Spillantini MG, Schmidt ML, Lee VM-Y, Trojanowski JQ, Jakes R, Goedert M. α-Synuclein in Lewy bodies. Nature. 1997;388(6645):839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 9.Solano SM, Miller DW, Augood SJ, Young AB, Penney JB., Jr Expression of α-synuclein, parkin, and ubiquitin carboxy-terminal hydrolase L1 mRNA in human brain: genes associated with familial Parkinson’s disease. Ann Neurol. 2000;47(2):201–210. doi: 10.1002/1531-8249(200002)47:2<201::AID-ANA10>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 10.Braak H, Sastre M, Del Tredici K. Development of α-synuclein immunoreactive astrocytes in the forebrain parallels stages of intraneuronal pathology in sporadic Parkinson’s disease. Acta Neuropathol. 2007;114(3):231–241. doi: 10.1007/s00401-007-0244-3. [DOI] [PubMed] [Google Scholar]
- 11.Terada S, Ishizu H, Yokota O, Tsuchiya K, Nakashima H, Ishihara T, Fujita D, Ueda K, Ikeda K, Kuroda S. Glial involvement in diffuse Lewy body disease. Acta Neuropathol. 2003;105(2):163–169. doi: 10.1007/s00401-002-0622-9. [DOI] [PubMed] [Google Scholar]
- 12.Wakabayashi K, Hayashi S, Yoshimoto M, Kudo H, Takahashi H. NACP/α-synuclein-positive filamentous inclusions in astrocytes and oligodendrocytes of Parkinson’s disease brains. Acta Neuropathol. 2000;99(1):14–20. doi: 10.1007/PL00007400. [DOI] [PubMed] [Google Scholar]
- 13.Krüger R, Kuhn W, Müller T, Woitalla D, Graeber M, Kösel S, Przuntek H, Epplen JT, Schöls L, Riess O. Ala30Pro mutation in the gene encoding α-synuclein in Parkinson’s disease. Nat Genet. 1998;18(2):106–108. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- 14.Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, et al. Mutation in the α-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276(5321):2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 15.Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, et al. α-Synuclein locus triplication causes Parkinson’s disease. Science. 2003;302(5646):841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- 16.Davidson WS, Jonas A, Clayton DF, George JM. Stabilization of α-synuclein secondary structure upon binding to synthetic membranes. J Biol Chem. 1998;273(16):9443–9449. doi: 10.1074/jbc.273.16.9443. [DOI] [PubMed] [Google Scholar]
- 17.Fortin DL, Troyer MD, Nakamura K, Kubo S, Anthony MD, Edwards RH. Lipid rafts mediate the synaptic localization of α-synuclein. J Neurosci. 2004;24(30):6715–6723. doi: 10.1523/JNEUROSCI.1594-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jo E, McLaurin J, Yip CM, St George-Hyslop P, Fraser PE. α-Synuclein membrane interactions and lipid specificity. J Biol Chem. 2000;275(44):34328–34334. doi: 10.1074/jbc.M004345200. [DOI] [PubMed] [Google Scholar]
- 19.Maroteaux L, Campanelli JT, Scheller RH. Synuclein: a neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J Neurosci. 1988;8(8):2804–2815. doi: 10.1523/JNEUROSCI.08-08-02804.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Giasson BI, Murray IVJ, Trojanowski JQ, Lee VM-Y. A hydrophobic stretch of 12 amino acid residues in the middle of α-synuclein is essential for filament assembly. J Biol Chem. 2001;276(4):2380–2386. doi: 10.1074/jbc.M008919200. [DOI] [PubMed] [Google Scholar]
- 21.Barbour R, Kling K, Anderson JP, Banducci K, Cole T, Diep L, Fox M, Goldstein JM, Soriano F, Seubert P, et al. Red blood cells are the major source of alpha-synuclein in blood. Neurodegener Dis. 2008;5(2):55–59. doi: 10.1159/000112832. [DOI] [PubMed] [Google Scholar]
- 22.Stefanis L. α-Synuclein in Parkinson’s disease. Cold Spring Harb Perspect Med. 2012;2(2):a009399. doi: 10.1101/cshperspect.a009399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jang A, Lee H-J, Suk J-E, Jung J-W, Kim K-P, Lee S-J. Non-classical exocytosis of α-synuclein is sensitive to folding states and promoted under stress conditions. J Neurochem. 2010;113(5):1263–1274. doi: 10.1111/j.1471-4159.2010.06695.x. [DOI] [PubMed] [Google Scholar]
- 24.Lee H-J, Patel S, Lee S-J. Intravesicular localization and exocytosis of α-synuclein and its aggregates. J Neurosci. 2005;25(25):6016–6024. doi: 10.1523/JNEUROSCI.0692-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marques O, Outeiro TF. Alpha-synuclein: from secretion to dysfunction and death. Cell Death Dis. 2012;3:e350. doi: 10.1038/cddis.2012.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee H-J, Suk J-E, Patrick C, Bae E-J, Cho J-H, Rho S, Hwang D, Masliah E, Lee S-J. Direct transfer of α-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. J Biol Chem. 2010;285(12):9262–9272. doi: 10.1074/jbc.M109.081125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fellner L, Irschick R, Schanda K, Reindl M, Klimaschewski L, Poewe W, Wenning GK, Stefanova N. Toll-like receptor 4 is required for α-synuclein dependent activation of microglia and astroglia. Glia. 2013;61(3):349–360. doi: 10.1002/glia.22437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim C, Ho D-H, Suk J-E, You S, Michael S, Kang J, Joong Lee S, Masliah E, Hwang D, Lee H-J, et al. Neuron-released oligomeric α-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat Commun. 2013;4:1562. doi: 10.1038/ncomms2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim C, Cho E-D, Kim H-K, You S, Lee H-J, Hwang D, Lee S-J. β1-integrin-dependent migration of microglia in response to neuron-released α-synuclein. Exp Mol Med. 2014;46:e91. doi: 10.1038/emm.2014.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roodveldt C, Labrador-Garrido A, Gonzalez-Rey E, Lachaud CC, Guilliams T, Fernandez-Montesinos R, Benitez-Rondan A, Robledo G, Hmadcha A, Delgado M, et al. Preconditioning of microglia by α-synuclein strongly affects the response induced by toll-like receptor (TLR) stimulation. PLoS One. 2013;8(11):e79160. doi: 10.1371/journal.pone.0079160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Netea MG, van Deuren M, Kullberg BJ, Cavaillon JM, Van der Meer JW. Does the shape of lipid A determine the interaction of LPS with Toll-like receptors? Trends Immunol. 2002;23(3):135–139. doi: 10.1016/S1471-4906(01)02169-X. [DOI] [PubMed] [Google Scholar]
- 32.Lee H-J, Suk J-E, Bae E-J, Lee S-J. Clearance and deposition of extracellular α-synuclein aggregates in microglia. Biochem Biophys Res Commun. 2008;372(3):423–428. doi: 10.1016/j.bbrc.2008.05.045. [DOI] [PubMed] [Google Scholar]
- 33.Lee H-J, Suk J-E, Bae E-J, Lee J-H, Paik SR, Lee SJ. Assembly-dependent endocytosis and clearance of extracellular α-synuclein. Int J Biochem Cell Biol. 2008;40(9):1835–1849. doi: 10.1016/j.biocel.2008.01.017. [DOI] [PubMed] [Google Scholar]
- 34.Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, Shen J, Takio K, Iwatsubo T. α-Synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol. 2002;4(2):160–164. doi: 10.1038/ncb748. [DOI] [PubMed] [Google Scholar]
- 35.Neumann M, Kahle PJ, Giasson BI, Ozmen L, Borroni E, Spooren W, Müller V, Odoy S, Fujiwara H, Hasegawa M, et al. Misfolded proteinase K-resistant hyperphosphorylated α-synuclein in aged transgenic mice with locomotor deterioration and in human α-synucleinopathies. J Clin Invest. 2002;110(10):1429–1439. doi: 10.1172/JCI200215777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Klegeris A, Pelech S, Giasson BI, Maguire J, Zhang H, McGeer EG, McGeer PL. α-Synuclein activates stress signaling protein kinases in THP-1 cells and microglia. Neurobiol Aging. 2008;29(5):739–752. doi: 10.1016/j.neurobiolaging.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 37.Su X, Maguire-Zeiss KA, Giuliano R, Prifti L, Venkatesh K, Federoff HJ. Synuclein activates microglia in a model of Parkinson’s disease. Neurobiol Aging. 2008;29(11):1690–1701. doi: 10.1016/j.neurobiolaging.2007.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang W, Wang T, Pei Z, Miller DS, Wu X, Block ML, Wilson B, Zhou Y, Hong J-S, Zhang J. Aggregated α-synuclein activates microglia: a process leading to disease progression in Parkinson’s disease. FASEB J. 2005;19(6):533–542. doi: 10.1096/fj.04-2751com. [DOI] [PubMed] [Google Scholar]
- 39.Brudek T, Winge K, Agander TK, Pakkenberg B. Screening of Toll-like receptors expression in multiple system atrophy brains. Neurochem Res. 2013;38(6):1252–1259. doi: 10.1007/s11064-013-1020-5. [DOI] [PubMed] [Google Scholar]
- 40.Ahn KJ, Paik SR, Chung KC, Kim J. Amino acid sequence motifs and mechanistic features of the membrane translocation of α-synuclein. J Neurochem. 2006;97(1):265–279. doi: 10.1111/j.1471-4159.2006.03731.x. [DOI] [PubMed] [Google Scholar]
- 41.Park J-Y, Kim KS, Lee S-B, Ryu J-S, Chung KC, Choo Y-K, Jou I, Kim J, Park SM. On the mechanism of internalization of α-synuclein into microglia: roles of ganglioside GM1 and lipid raft. J Neurochem. 2009;110(1):400–411. doi: 10.1111/j.1471-4159.2009.06150.x. [DOI] [PubMed] [Google Scholar]
- 42.Luk KC, Kehm VM, Zhang B, O’Brien P, Trojanowski JQ, Lee VM-Y. Intracerebral inoculation of pathological α-synuclein initiates a rapidly progressive neurodegenerative α-synucleinopathy in mice. J Exp Med. 2012;209(5):975–986. doi: 10.1084/jem.20112457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Luk KC, Song C, O’Brien P, Stieber A, Branch JR, Brunden KR, Trojanowski JQ, Lee VM-Y. Exogenous α-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc Natl Acad Sci USA. 2009;106(47):20051–20056. doi: 10.1073/pnas.0908005106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Emmanouilidou E, Elenis D, Papasilekas T, Stranjalis G, Gerozissis K, Ioannou PC, Vekrellis K. Assessment of α-synuclein secretion in mouse and human brain parenchyma. PLoS One. 2011;6(7):e22225. doi: 10.1371/journal.pone.0022225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kahle PJ, Neumann M, Ozmen L, Müller V, Jacobsen H, Schindzielorz A, Okochi M, Leimer U, van Der Putten H, Probst A, et al. Subcellular localization of wild-type and Parkinson’s disease-associated mutant α-synuclein in human and transgenic mouse brain. J Neurosci. 2000;20(17):6365–6373. doi: 10.1523/JNEUROSCI.20-17-06365.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schell H, Hasegawa T, Neumann M, Kahle PJ. Nuclear and neuritic distribution of serine-129 phosphorylated α-synuclein in transgenic mice. Neuroscience. 2009;160(4):796–804. doi: 10.1016/j.neuroscience.2009.03.002. [DOI] [PubMed] [Google Scholar]
- 47.Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, Takeda Y, Takeda K, Akira S. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J Immunol. 1999;162(7):3749–3752. [PubMed] [Google Scholar]
- 48.Hamprecht B, Löffler F. Primary glial cultures as a model for studying hormone action. Methods Enzymol. 1985;109:341–345. doi: 10.1016/0076-6879(85)09097-8. [DOI] [PubMed] [Google Scholar]