

Abstract
Background
The identification of oncogenic driver mutations has largely relied on the assumption that genes that exhibit more mutations than expected by chance are more likely to play an active role in tumorigenesis. Major cancer sequencing initiatives have therefore focused on recurrent mutations that are more likely to be drivers. However, in specific genetic contexts, low frequency mutations may also be capable of participating in oncogenic processes. Reliable strategies for identifying these rare or even patient-specific (private) mutations are needed in order to elucidate more personalized approaches to cancer diagnosis and treatment.
Methods
Here we performed whole-exome sequencing on three cases of childhood pre-B acute lymphoblastic leukemia (cALL), representing three cytogenetically-defined subgroups (high hyperdiploid, t(12;21) translocation, and cytogenetically normal). We applied a data reduction strategy to identify both common and rare/private somatic events with high functional potential. Top-ranked candidate mutations were subsequently validated at high sequencing depth on an independent platform and in vitro expression assays were performed to evaluate the impact of identified mutations on cell growth and survival.
Results
We identified 6 putatively damaging non-synonymous somatic mutations among the three cALL patients. Three of these mutations were well-characterized common cALL mutations involved in constitutive activation of the mitogen-activated protein kinase pathway (FLT3 p.D835Y, NRAS p.G13D, BRAF p.G466A). The remaining three patient-specific mutations (ACD p.G223V, DOT1L p.V114F, HCFC1 p.Y103H) were novel mutations previously undescribed in public cancer databases. Cytotoxicity assays demonstrated a protective effect of the ACD p.G223V mutation against apoptosis in leukemia cells. ACD plays a key role in protecting telomeres and recruiting telomerase. Using a telomere restriction fragment assay, we also showed that this novel mutation in ACD leads to increased telomere length in leukemia cells.
Conclusion
This study identified ACD as a novel gene involved in cALL and points to a functional role for ACD in enhancing leukemia cell survival. These results highlight the importance of rare/private somatic mutations in understanding cALL etiology, even within well-characterized molecular subgroups.
Electronic supplementary material
The online version of this article (doi:10.1186/s12885-015-1639-5) contains supplementary material, which is available to authorized users.
Background
Childhood acute lymphoblastic leukemia is a heterogeneous disease both biologically and clinically, and is the leading cause of cancer-related deaths among children. Despite significant advances in our understanding of the pathobiology of cALL leading to risk-based treatment regimens and increased survival rates, the etiological causes of this disease remain elusive. Approximately 75 % of pre-B cALL cases exhibit hyperdiploidy or a recurring gross chromosomal rearrangement, detection of which is central to disease diagnosis, risk stratification and management [1]. While these chromosomal alterations play an important role in driving the leukemic process by affecting molecular pathways that halt lymphoid progenitor cell differentiation and promote cell proliferation and survival, they are not sufficient for leukemic transformation and are often detected years before leukemia onset. This suggests a need for additional cooperating events in order to achieve overt leukemogenesis [2]. Thorough investigation of cALL genomes is crucial to better understand the underlying genomic complexity of this disease and thus better diagnose and treat it. Toward these goals, recent large-scale sequencing efforts revealed many somatic driver mutations/genes recurrently mutated in cALL [1]. The identification of these high frequency driver mutations is essential to better understand disease etiology and characterize prognostic subgroups. Yet accumulating evidence has also shown that low-frequency mutations within a cancer type can contribute to onset and progression of the disease [3], and play a role in intra-tumor and inter-patient heterogeneity. The identification of patient-specific mutations could provide crucial information regarding molecular pathways underlying cALL tumorigenesis and thus point to new therapeutic avenues. Here, we performed whole-exome sequencing of 3 pre-B cALL cases. Case 1 was diagnosed with high hyperdiploiy (>50 chromosome) cALL and Case 2 harboured the t(12;21) (ETV6-RUNX1) translocation. Together these two molecularly defined subgroups represent over 40 % of cALL cases. Case 3 was cytogenetically normal at diagnosis. Using a unique quartet design, we sequenced matched normal (blood following remission) and tumor (bone marrow at diagnosis) patient samples, and the parents of each case. We successfully identified known recurrent drivers, as well as novel patient-specific somatic mutations with high functional potential. Using in vitro assays, we showed that the private p.G223V mutation, adjacent to the TEL patch of the telomere protein ACD (also known as TPP1), leads to apoptosis resistance and may contribute to leukemia cell survival by promoting telomere maintenance and protection.
Methods
Patients
All study subjects were self-declared French-Canadians of European descent from the established Quebec cALL (QcALL) cohort [4]. The Sainte-Justine UHC Research Ethics Board approved the protocol. Informed consent was obtained from the parents of the patients to participate in this study and for publication of this report and any accompanying images. A copy of the written consent is available for review by the Editor of this journal.
Case 1, a 10 year old male, was classified as high risk based on his age. He presented with a platelet count of 15.0 × 109/L, a white blood cell count (WBC) of 9.0 × 109/L, and 63 % and 97 % lymphoblast cells in the blood and bone marrow samples respectively. The cytogenetic analysis revealed high hyperdiploidy (karyotype: 53,XY,+3,+4,+6,+10,+14,+17,+18). He was enrolled on the Dana Farber Cancer Institute (DFCI) ALL Consortium protocol 95–01. He achieved complete remission and has been out of treatment for over 60 months with leukemia free-survival (LFS).
Case 2 was a 4 year old female with a platelet count of 35.0 × 109/L, a WBC of 33.1 × 109/L, and 75 % and 96 % lymphoblast cells in the blood and bone marrow samples respectively. Cytogenetic analysis revealed a t(12;21) translocation. Despite her karyotype, usually associated with a good prognostic, she was classified as high risk based on the presence of leukemia cells in the cerebrospinal fluid. She was enrolled on DFCI/ALL Consortium protocol 2000–01. She also achieved complete remission and has been out of treatment for over 60 months with LFS.
Case 3 was a 6 year-old male with a WBC of 183.1 × 109/L, a platelet count of 29.0 × 109/L, 87 % and 95 % lymphoblast cells in the blood and bone marrow samples respectively, and presented with a normal karyotype according to the cytogenetic analysis. The patient was classified as high risk based on hyperleukocytosis and was treated on DFCI/ALL Consortium Protocol 2000–01. Case 3 experienced a first relapse 41 months post diagnosis, and following 2 subsequent relapses he died 73 months post diagnosis.
Whole exome sequence capture and sequencing
DNA was extracted from bone marrow samples (at diagnosis) and peripheral blood samples (obtained after remission) from the cases and their parents using standard protocols as described previously [5]. Whole exomes were captured in solution with Agilent’s SureSelect Human All Exon 50 Mb kits for SOLiD sequencing (Life Technologies) according to the manufacturer’s protocol, were sequenced on the Life Technologies SOLiD System (mean coverage =32X) and aligned to the Hg19 reference genome using LifeScope Genomic Analysis Software (see Fig. 1 for complete sequencing analysis workflow). Polymerase chain reaction (PCR) duplicates were removed using Picard [http://broadinstitute.github.io/picard/]. Base quality score recalibration was performed using the Genome Analysis ToolKit (GATK) [6] and reads that failed the quality control were removed. Cleaned BAM files were used to create pileup files using SAMtools [7]. Somatic single nucleotide variants (SNVs) were called from pileup files using SNooPer, an in-house variant caller that is based on a machine learning approach that integrates tumoral and normal data and that was specifically trained for optimal identification of somatic mutations in our low-depth SOLiD sequencing data (manuscript submitted and software available upon request). Furthermore, using the familial design, we were able to use parental sequence information to remove Mendelian inconsistencies, reduce false-positive sequencing and alignment errors and facilitate somatic variant identification. The robustness of this approach was further demonstrated using high-depth sequencing on an independent platform to confirm top-ranked somatic mutations (see below). Resulting somatic SNVs were queried against publically available datasets such as 1000 Genomes [8] and NHLBI GO Exome Sequencing Project (ESP) [http://evs.gs.washington.edu/EVS/] to filter out common polymorphism eventually remaining (minor allele frequency >0.01).
Fig. 1.
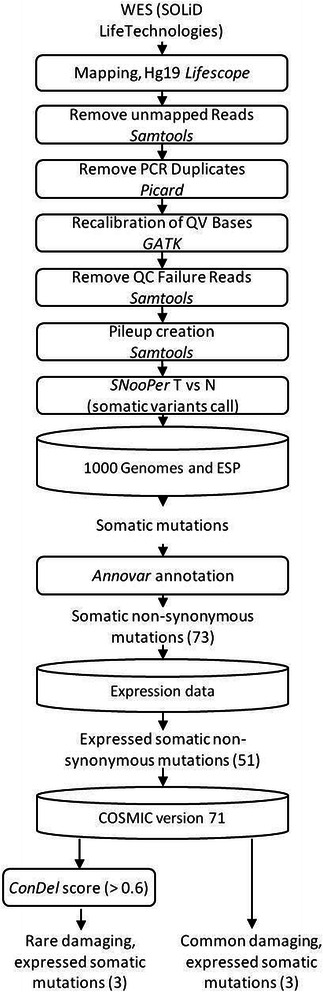
Whole-exome sequencing analysis workflow. Boxes represent the analysis/cleaning steps. Cylinders represent the SNV filtering steps used in the data reduction strategy to identify functional somatic mutations. The number of variations remaining after each step is shown in brackets. Note that only SNVs that passed a given filter were tested for in the subsequent step. Using public databases and variant annotation tools, we identified 6 top-ranked mutations among the pre-B cALL patients, including 3 SNVs referenced in COSMIC v71 and 3 candidate rare/private SNVs in ACD (p.G223V), DOT1L (p.V114F) and HCFC1 (p.Y103H) with a potential functional impact
Expression data filter
Publically available microarray expression data [9] were used to filter variants based on the assumption that expressed genes are more likely to carry functionally relevant mutations [10]. Mutated genes identified from each case were compared to the expression profiles of the corresponding cALL subgroup (hyperdiploidy, t(12;21), or “others”) and only expressed genes were subsequently retained in the somatic variant analysis.
Ultra-deep targeted re-sequencing
Top-ranked rare/private candidate mutations, as well as common somatic mutations identified in the pre-B cALL cases were validated on the Ion Torrent system. Selected variant regions (~125pb flanking the identified somatic mutation) were amplified and PCR products were sequenced on the Ion PGM Sequencer (Life Technologies) according to the manufacturer’s protocol with a mean coverage >1,800X. Primer sequences and PCR protocols are available upon request.
Site-directed mutagenesis and apoptosis assay
Identified mutations were introduced by site-directed mutagenesis into the complementary DNA (cDNA) sequence of each gene cloned into a pDONR221 vector. Using the pLenti-CMV-DEST Gateway Vector (w118-1), we subcloned the wild-type (WT) or mutant coding sequences and generated lentiviruses using a Third Generation Packaging System in 293 T cells. Lentiviral particles were then harvested and used to infect Nalm-6 (human leukemia pre-B cells) with 8ug/mL polybrene. Infected cells were selected with puromycin 1 μg/μL, seeded at 5 × 105 cells/mL in their culture medium and treated with 5 μM of camptothecin for 3 h. Apoptosis was measured using conventional Annexin V/Propidium iodide (PI) staining and quantified by flow cytometry (BD Biosciences FACS Fortessa). All experiments were done in triplicate.
Telomere restriction fragment (TRF) assay
Parental Nalm-6 cells and Nalm-6 cells overexpressing mutant ACD p.G223V (Nalm-6 ACD G223V), the wild-type ACD (Nalm-6 ACD WT) or Nalm-6 cells infected with the empty vector alone (Nalm-6 pLENTI) were used for TRF assays. For each cell line, extracted DNA at passage 16 (p16) (population doublings ≈ 50) and p27 (population doublings ≈ 80) after selection was digested with HinfI and RsaI restriction enzymes and fractionated on an electrophoresis gel apparatus. After drying, the gel was hybridized with a [γ -32P] adenosine triphosphate (ATP)end-labelled (T2AG3)3 probe and exposed on X-ray film. Mean TRF length was calculated as previously described [11]. The TRF assay was performed in duplicate.
Results/discussion
Assuming that functionally important genes can also be mutated more rarely and in specific tumor contexts, we performed whole-exome sequencing of three pre-B cALL patients and their parents and applied a data reduction strategy to identify both common and novel rare/private events with high functional potential (Fig. 1). SNVs were queried against public databases, annotated using ANNOVAR [12], and were binned according to frequency and function (Methods). Non-synonymous mutations that were expressed in cALL subgroups (based on microarray cALL expression data [9]) were then filtered based on a CONsensus DELeteriousness score >0.6 (measure of the degree of coherence of individual methods about the likelihood that a SNV is deleterious) [13]. This led to the identification of 6 expressed damaging non-synonymous somatic mutations among the 3 patients (FLT3 p.D835Y, NRAS p.G13D, BRAF p.G466A, ACD p.G223V, DOT1L p.V114F, HCFC1 p.Y103H) (Table 1) that were subsequently confirmed using targeted ultra-deep re-sequencing (mean coverage >1800X). Among these 6 somatic SNVs, FLT3 p.D835Y, NRAS p.G13D and BRAF p.G466A were referenced in the Catalogue Of Somatic Mutations In Cancer (COSMIC) database (v71) and previously shown to constitutively activate the mitogen-activated protein kinase (MAPK) pathway and to increase tumor proliferation [14–17]. The identification of previously reported driver mutations validates the robustness of our approach. The NRAS p.G13D gain of function mutation was identified in Case 1, which corroborates previous studies that report postnatal RAS activating mutations in ~30 % of pre-B hyperdiploid cALL patients [18]. The BRAF p.G466A mutation identified in Case 2 is associated with a mild increase of ERK activation [19]. While mutations in BRAF are identified in almost 70 % of patients with multiple myeloma [15], screening of childhood pre-B cohorts only identified a few cases harbouring these events [20]. Although alterations of the receptor tyrosine kinase FLT3 are frequent in hyperdiploid cALL and rare in other subtypes [21], FLT3 p.D835Y was identified in the cytogenetically normal Case 3. While Case 3 suffered a relapse and activated forms of FLT3 are usually associated with a poor outcome in acute myeloid leukemia patients [22], this association is not confirmed in cALL [23].
Table 1.
Candidate somatic mutations identified in each patient
| Patient | Subgroup | Gene | Genomic change | Protein change | Class | VAF | COSMIC v71 |
|---|---|---|---|---|---|---|---|
| Case 1 | HD | NRAS | g.chr1:115258744C > T | p.G13D | Missense | 0.48 | Haematopoietic and Lymphoid tissue |
| ACD/TPP1 | g.chr16:67693443C > A | p.G223V | Missense | 0.51 | - | ||
| DOT1L | g.chr19:2191086G > T | p.V114F | Missense | 0.46 | - | ||
| Case 2 | t(12;21) | BRAF | g.chr7:140481411C > G | p.G466A | Missense | 0.15 | Thyroid |
| HCFC1 | g.chrX:153230064A > G | p.Y103H | Missense | 0.51 | - | ||
| Case 3 | CN | FLT3 | g.chr13:28592642G > T | p.D835Y | Missense | 0.44 | Haematopoietic and Lymphoid tissue |
Variant allele frequencies (VAF) (number of supporting reads/coverage) were calculated based on ultra-deep targeted re-sequencing data (mean coverage >1800X). Only tissue types harbouring the highest occurrence of the mutation in the COSMIC database (v71) are presented. HD hyperdiploid, CN Cytogenetically normal
Our sequencing analysis pipeline also led to the identification of three candidate novel pathogenic mutations in Cases 1 and 2 (DOT1L p.V114F, HCFC1 p.Y103H, ACD p.G223V), that were neither previously reported nor referenced in public databases. Based on our strict filtering criteria, no private mutations were identified in Case 3. DOT1L is a histone writer and HCFC1 is a broad transcription regulator that plays a critical role in cell proliferation via its involvement in chromatin-modifying activities [24]. These results are consistent with recent cancer sequencing initiatives that highlighted the important role of chromatin remodeling genes in leukemogenesis [25]. Furthermore, DOT1L has recently been implicated in the development of MLL-rearranged leukemia and shown to be essential for leukemic transformation [26]. On the other hand, ACD is a core protein in the shelterin complex and mediates the access of telomerase to the telomere. It is essential for telomere homeostasis in hematopoietic stem and progenitor cell particularly [27, 28].
The high variant allele frequency (VAF) of these rare/private mutations (DOT1L, VAF =0.46; HCFC1, VAF =0.51; ACD, VAF =0.51), calculated based on our ultra-deep targeted re-sequencing data (Table 1), was indicative of early clonal selection supporting a possible functional role in leukemia development. Functional validation of the ACD p.G223V mutation (Fig. 2a) was further pursued using in vitro expression assays, however our in vitro lentiviral expression system did not permit functional characterization of neither DOT1L nor HCFC1, due to the size of the open reading frames (ORFs) (4.6 kb and 6.1 kb, respectively) that were beyond the viral packaging capacity of the capside [29]. Further investigation of these two novel mutations using alternative functional screening methods is ongoing. In vitro expression assays of ACD using the topoisomerase I inhibitor camptothecin, showed that leukemic cells overexpressing mutant p.G223V ACD (Nalm-6 ACD G223V) exhibit lower levels of apoptosis compared with cells overexpressing wild-type ACD (Nalm-6 ACD WT) (P =0.014, Mann–Whitney U test) and the empty vector (Nalm-6 pLenti) (P =0.014, Mann–Whitney U test) (Fig. 2b). The p.G223V mutation is adjacent to the TEL patch in the oligonucleotide/oligosaccharide-binding (OB) fold domain of ACD that interacts directly with the catalytic subunit of telomerase. Within the shelterin complex, ACD was specifically shown to interact with POT1 to protect telomeres and recruit telomerase at chromosome ends [30–32]. Interestingly, recurrent somatic mutations in the OB domains of POT1 have been shown to cause telomere dysfunction in chronic lymphocytic leukemia suggesting that alteration of shelterin-mediated protein-telomere binding could lead to genomic instability and cancer [33]. Furthermore, very recently, germline mutations in the POT1 binding domain and the TEL patch of ACD were respectively associated with familial melanomas and bone marrow failure disorders [34–36] (Fig. 2a). Although further studies are needed to decipher the underlying molecular mechanisms implicating ACD in apoptosis inhibition, taken together, these data strongly support a role for ACD p.G223V in promoting leukemia cell maintenance.
Fig. 2.
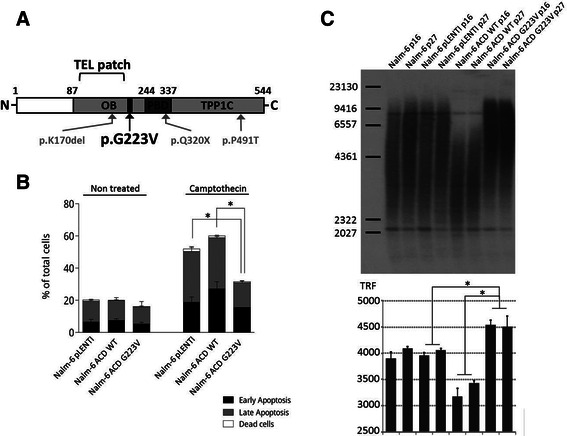
ACD p.G223V protects from camptothecin-induced apoptosis and increases telomere length. a. Schematic representation of the ACD protein. The p.G223V mutation, depicted in black, is adjacent to the TEL patch of the OB-fold domain involved in telomerase recruitment and composed of seven critical amino acids located in a region defined by the curly bracket (E168, E169, E171, R180, L183, L212 and E215) [30]. p.Q320X, p.P491T and p.K170del, depicted in grey, are three germline mutations recently identified and associated with familial melanomas and bone marrow failure disorders [34–36]. TPP1C corresponds to the TIN2-binding domain. Together, the OB (oligonucleotide/oligosaccharide-binding) and PBD (POT1 binding domain) domains form the ACDN domain necessary for POT1 binding to telomeric DNA and the stimulation of telomerase processivity. b. In vitro apoptosis assays show overall reduced levels of apoptosis associated with ACD p.G223V. The c.659 g > t mutation was introduced into the ACD transgene by site-directed mutagenesis and expressed in Nalm-6 cells. The graph shows annexin V/PI staining for 3 h on Nalm-6 pLenti (empty vector), Nalm-6 ACD WT and Nalm-6 ACD G223V cells. c. The telomere restriction fragment assay (TRF) showed a quantitative increase in telomere size for Nalm-6 ACD G223V cells at passage 16 (p16) and p27 after selection. Mean TRF length = ∑ (ODi)/∑ (ODi/Li) where ODi is the radioactive signal, Li is the TRF fragment length at position i. The bar chart of Fig. 2c (bottom) represents the mean TRF length for each condition directly quantified from each corresponding lane of the TRF gel presented at the top of Fig. 2c Significance (in b and c) was determined by a Mann–Whitney U test; p-values <0.05 are represented by an asterisk
To further investigate the effect of p.G223V mutant ACD on telomere structure, we performed a TRF assay (Fig. 2c) and showed that decreased apoptosis correlates with increased telomere length in Nalm-6 ACD G223V at both tested passages compared to Nalm-6 ACD WT and Nalm-6 pLenti (P =0.029, Mann–Whitney U test), confirming stable alteration of telomere homeostasis. ACD mutant induced telomere elongation in leukemia cells is consistent with reports demonstrating disrupted shelterin complex function and telomere lengthening due to mutations in POT1 in chronic lymphocytic leukemia [33, 37]. Furthermore, as observed for POT1 at the same population doubling point [33], overexpression of WT ACD did not increase telomere length (Fig. 2c), confirming that the observed telomere lengthening is due to the p.G223V mutation and not caused by global overexpression of ACD. Concomitant lengthening of the telomeres and decreased apoptosis levels in NALM-6 ACD G223V following camptothecin treatment corroborates previous findings that altered telomerase activity can lead to hypersensitivity of tumor cells to topoisomerase inhibitors [38, 39]. Further investigation is required to characterize the influence of the p.G223V mutation on the recruitment and processivity of telomerase and on telomere-length regulation. Missense or frameshift mutations in ACD, many of which are located in the OB fold, adjacent to the TEL patch, and in the POT1 interaction domain, have been described in multiple cancer types (Additional file 1 Table S1) with the highest prevalence found in melanoma (2.7 %), suggesting that ACD mutations may participate in a common underlying cancer-promoting pathway that involves telomere dysfunction. Case 1 who carries the ACD p.G223V mutation had high hyperdiploid cALL and although telomere dysfunction can cause chromosome destabilization and aneuploidy [40, 41], it is unlikely that this mutation precluded the mitotic event producing the hyperdiploid phenotype. However, our results do support a role for ACD p.G223V in the earlier stages of clonal expansion contributing to telomere maintenance and apoptosis resistance, at least in vitro. Further investigation is required in order to characterize the mechanistic role of ACD p.G223V within this patient’s specific tumor context.
Conclusions
The prenatal origins of cALLs are well established, along with the need for additional postnatal mutations in order to drive overt leukemogenesis [42, 43]. The extent to which rare/private genetic events contribute to the onset and progression of cALL however is largely unknown. Through whole-exome sequencing of 3 cALL cases, we successfully identified not only common drivers (NRAS p.G13D, FLT3 p.D835Y, and BRAF p.G466A), but also rare/private somatic mutations (DOT1L p.V114F, HCFC1 p.Y103H, ACD p.G223V), in well-characterized cALL molecular subgroups. The identification of patient-specific events with a functional potential is not surprising and further confirms the underlying genetic complexity of this disease. We went on to demonstrate the functional impact of ACD p.G223V on apoptosis resistance and telomere-length regulation in pre-B ALL cells. The high VAF of this somatic mutation suggests that it is likely present in the major subclone; while this does not necessarily imply functionality, it does support an early role for ACD p.G223V in driving the leukemic process. Though further investigation is needed to fully characterize the influence of the identified mutation on telomere homeostasis, this study is the first to describe the functional implications of a somatic mutation in ACD on leukemic cell behaviour, supporting a role for ACD and telomere regulation in leukemia cell resistance to apoptosis. In conclusion, these results support the need of thorough investigation of rare/private mutations to reveal the underlying complexity of cALL landscapes, including within well-characterized subgroups, and the inter-patient variability that may influence diagnosis and prognosis.
Consent
The Sainte-Justine UHC Research Ethics Board approved the protocol. Informed consent was obtained from the parents of the patients to participate in this study and for publication of this report and any accompanying images. A copy of the written consent is available for review by the Editor of this journal.
Acknowledgments
The authors are indebted to the patients and their parents for participating in this study. This study was supported by research funds provided by the Terry Fox Research Institute and the Canadian Institutes for Health Research. JFS is the recipient of a Réseau de médecine génétique appliquée (RMGA) Fellowship. NG is the recipient of postdoctoral Cole Foundation Fellowship. DS holds the François-Karl-Viau Research Chair in Pediatric Oncogenomics. Whole exome sequencing was performed at the Child Health Genomics Platform of the Sainte-Justine UHC Research Center; Targeted re-sequencing was performed at the McGill University and Genome Quebec Innovation Centre; Computations were made on the supercomputer Briarée from Université de Montréal, managed by Calcul Québec and Compute Canada. The operation of this supercomputer is funded by the Canada Foundation for Innovation (CFI), NanoQuébec, RMGA and the Fonds de recherche du Québec - Nature et technologies (FRQ-NT).
Abbreviations
- cALL
childhood acute lymphoblastic leukemia
- WBC
White blood cell count
- DFCI
Dana Farber Cancer Institute
- LFS
Leukemia free-survival
- GATK
Genome Analysis ToolKit
- SNVs
Somatic single nucleotide variants
- ESP
NHLBI GO Exome Sequencing Project
- TRF
Telomere restriction fragment
- COSMIC
Catalogue of Somatic Mutations In Cancer
- VAF
Variant allele frequency
- ConDel score
CONsensus DELeteriousness score
- PCR
Polymerase chain reaction
- WT
Wild-type
- PI
Propidium iodide
- OB
Oligonucleotide/oligosaccharide-binding fold domain
- PBD
POT1 binding domain
- TPP1C
TIN2-binding domain
- HD
hyperdiploid
- CN
Cytogenetically normal
Additional file
Description of data: Non-synonymous and frame-shift mutations in ACD referenced in the public cancer database COSMIC version 71. (PDF 120 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
DS is the principle investigator and takes primary responsibility for the paper. JFS, JH, and DS contributed to the conception and design of the study. JFS, PC, MO and CR were involved in sample and library preparation. VS analyzed raw sequence data and provided bioinformatics support. JFS performed whole-exome and targeted sequencing analysis. PC, PR, NG, and CD performed the functional validations: PC, NG and CD performed the apoptosis resistance assays, PR performed the telomere restriction fragment assays. JFS and JH wrote the paper and PC, NG, CA and DS contributed to the interpretation of the data and were involved in critical manuscript revision. All authors approved the final version.
Contributor Information
Jean-François Spinella, Email: jf.spinella@gmail.com.
Pauline Cassart, Email: cassart.pauline@gmail.com.
Nicolas Garnier, Email: nicolasgarnier3000@gmail.com.
Philippe Rousseau, Email: pmp.rousseau@gmail.com.
Claire Drullion, Email: claire.drullion@gmail.com.
Chantal Richer, Email: cricher.hsj@gmail.com.
Manon Ouimet, Email: manon.ouimet@recherche-ste-justine.qc.ca.
Virginie Saillour, Email: vr.saillour@gmail.com.
Jasmine Healy, Email: jasmine.healy@umontreal.ca.
Chantal Autexier, Email: chantal.autexier@mcgill.ca.
Daniel Sinnett, Phone: (514) 345-4931(2990), Email: daniel.sinnett@umontreal.ca.
References
- 1.Inaba H, Greaves M, Mullighan CG. Acute lymphoblastic leukaemia. Lancet. 2013;381(9881):1943–55. doi: 10.1016/S0140-6736(12)62187-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Greaves MF, Wiemels J. Origins of chromosome translocations in childhood leukaemia. Nat Rev Cancer. 2003;3(9):639–49. doi: 10.1038/nrc1164. [DOI] [PubMed] [Google Scholar]
- 3.Cancer Genome Atlas Research Network Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474(7353):609–15. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Healy J, Bélanger H, Beaulieu P, Larivière M, Labuda D, Sinnett D. Promoter SNPs in G1/S checkpoint regulators and their impact on the susceptibility to childhood leukemia. Blood. 2007;109(2):683–92. doi: 10.1182/blood-2006-02-003236. [DOI] [PubMed] [Google Scholar]
- 5.Baccichet A, Qualman SK, Sinnett D. Allelic loss in childhood acute lymphoblastic leukemia. Leuk Res. 1997;21(9):817–23. doi: 10.1016/S0145-2126(97)00075-1. [DOI] [PubMed] [Google Scholar]
- 6.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. 1000 Genome Project Data Processing Subgroup. The Sequence alignment/map (SAM) format and SAMtools. Bioinformatics. 2009;25:2078–9. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.1000 Genomes Project Consortium. Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, Gibbs RA, Hurles ME, McVean GA. A map of human genome variation from population-scale sequencing. Nature. 2010;467(7319):1061–73. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ross ME, Zhou X, Song G, Shurtleff SA, Girtman K, Williams WK, Liu HC, Mahfouz R, Raimondi SC, Lenny N, Patel A, Downing JR. Classification of pediatric acute lymphoblastic leukemia by gene expression profiling. Blood. 2003;102(8):2951–9. doi: 10.1182/blood-2003-01-0338. [DOI] [PubMed] [Google Scholar]
- 10.D’Antonio M, Ciccarelli FD. Integrated analysis of recurrent properties of cancer genes to identify novel drivers. Genome Biol. 2013;14(5):R52. doi: 10.1186/gb-2013-14-5-r52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.D’Souza Y, Lauzon C, Chu TW, Autexier C. Regulation of telomere length and homeostasis by telomerase enzyme processivity. J Cell Sci. 2013;126:676–87. doi: 10.1242/jcs.119297. [DOI] [PubMed] [Google Scholar]
- 12.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16) doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gonzalez-Perez A, Lopez-Bigas N. Improving the assessment of the outcome of nonsynonymous SNVs with a consensus deleteriousness score. Condel Am J Hum Genet. 2011;88(4):440–9. doi: 10.1016/j.ajhg.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yamamoto Y, Kiyoi H, Nakano Y, Suzuki R, Kodera Y, Miyawaki S, Asou N, Kuriyama K, Yagasaki F, Shimazaki C, Akiyama H, Saito K, Nishimura M, Motoji T, Shinagawa K, Takeshita A, Saito H, Ueda R, Ohno R, Naoe T. Activating mutation of D835 within the activation loop of FLT3 in human hematologic malignancies. Blood. 2001;97(8):2434–9. doi: 10.1182/blood.V97.8.2434. [DOI] [PubMed] [Google Scholar]
- 15.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, Davis N, Dicks E, Ewing R, Floyd Y, Gray K, Hall S, Hawes R, Hughes J, Kosmidou V, Menzies A, Mould C, Parker A, Stevens C, Watt S, Hooper S, Wilson R, Jayatilake H, Gusterson BA, Cooper C, Shipley J, Hargrave D, Pritchard-Jones K, Maitland N, Chenevix-Trench G, Riggins GJ, Bigner DD, Palmieri G, Cossu A, Flanagan A, Nicholson A, Ho JW, Leung SY, Yuen ST, Weber BL, Seigler HF, Darrow TL, Paterson H, Marais R, Marshall CJ, Wooster R, Stratton MR, Futreal PA. Mutations of the BRAF gene in human cancer. Nature. 2002;417(6892):949–54. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 16.Boguski MS, McCormick F. Proteins regulating Ras and its relatives. Nature. 1993;366(6456):643–54. doi: 10.1038/366643a0. [DOI] [PubMed] [Google Scholar]
- 17.Bos JL, Toksoz D, Marshall CJ, Verlaan-de Vries M, Veeneman GH, van der Eb AJ, van Boom JH, Janssen JW, Steenvoorden AC. Amino-acid substitutions at codon 13 of the N-ras oncogene in human acute myeloid leukaemia. Nature. 1985;315(6022):726–30. doi: 10.1038/315726a0. [DOI] [PubMed] [Google Scholar]
- 18.Wiemels JL, Kang M, Chang JS, Zheng L, Kouyoumji C, Zhang L, Smith MT, Scelo G, Metayer C, Buffler P, Wiencke JK. Backtracking RAS mutations in high hyperdiploid childhood acute lymphoblastic leukemia. Blood Cells Mol Dis. 2010;45(3):186–91. doi: 10.1016/j.bcmd.2010.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wan PT, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, Jones CM, Marshall CJ, Springer CJ, Barford D, Marais R. Cancer Genome Project. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004;116(6):855–67. doi: 10.1016/S0092-8674(04)00215-6. [DOI] [PubMed] [Google Scholar]
- 20.Knight T, Irving JA. Ras/Raf/MEK/ERK pathway activation in childhood acute lymphoblastic leukemia and its therapeutic targeting. Front Oncol. 2014;4:160. doi: 10.3389/fonc.2014.00160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Armstrong SA, Mabon ME, Silverman LB, Li A, Gribben JG, Fox EA, Sallan SE, Korsmeyer SJ. FLT3 mutations in childhood acute lymphoblastic leukemia. Blood. 2004;103(9):3544–6. doi: 10.1182/blood-2003-07-2441. [DOI] [PubMed] [Google Scholar]
- 22.Ravandi F, Kantarjian H, Faderl S, Garcia-Manero G, O’Brien S, Koller C, Pierce S, Brandt M, Kennedy D, Cortes J, Beran M. Outcome of patients with FLT3-mutated acute myeloid leukemia in first relapse. Leuk Res. 2010;34(6):752–6. doi: 10.1016/j.leukres.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Taketani T, Taki T, Sugita K, Furuichi Y, Ishii E, Hanada R, Tsuchida M, Sugita K, Ida K, Hayashi Y. FLT3 mutations in the activation loop of tyrosine kinase domain are frequently found in infant ALL with MLL rearrangements and pediatric ALL with hyperdiploidy. Blood. 2004;103(3):1085–8. doi: 10.1182/blood-2003-02-0418. [DOI] [PubMed] [Google Scholar]
- 24.Wysocka J, Myers MP, Laherty CD, Eisenman RN, Herr W. Human Sin3 deacetylase and trithorax-related Set1/Ash2 histone H3-K4 methyltransferase are tethered together selectively by the cell-proliferation factor HCF-1. Genes Dev. 2003;17(7):896–911. doi: 10.1101/gad.252103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Holmfeldt L, Wei L, Diaz-Flores E, Walsh M, Zhang J, Ding L, Payne-Turner D, Churchman M, Andersson A, Chen SC, McCastlain K, Becksfort J, Ma J, Wu G, Patel SN, Heatley SL, Phillips LA, Song G, Easton J, Parker M, Chen X, Rusch M, Boggs K, Vadodaria B, Hedlund E, Drenberg C, Baker S, Pei D, Cheng C, Huether R, Lu C, Fulton RS, Fulton LL, Tabib Y, Dooling DJ, Ochoa K, Minden M, Lewis ID, To LB, Marlton P, Roberts AW, Raca G, Stock W, Neale G, Drexler HG, Dickins RA, Ellison DW, Shurtleff SA, Pui CH, Ribeiro RC, Devidas M, Carroll AJ, Heerema NA, Wood B, Borowitz MJ, Gastier-Foster JM, Raimondi SC, Mardis ER, Wilson RK, Downing JR, Hunger SP, Loh ML, Mullighan CG. The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat Genet. 2013;45(3):242–52. doi: 10.1038/ng.2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McLean CM, Karemaker ID, van Leeuwen F. The emerging roles of DOT1L in leukemia and normal development. Leukemia. 2014;28(11):2131–8. doi: 10.1038/leu.2014.169. [DOI] [PubMed] [Google Scholar]
- 27.Jones M, Osawa G, Regal JA, Weinberg DN, Taggart J, Kocak H, Friedman A, Ferguson DO, Keegan CE, Maillard I. Hematopoietic stem cells are acutely sensitive to Acd shelterin gene inactivation. J Clin Invest. 2014;124(1):353–66. doi: 10.1172/JCI67871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nakashima M, Nandakumar J, Sullivan KD, Espinosa JM, Cech TR. Inhibition of telomerase recruitment and cancer cell death. J Biol Chem. 2013;288(46):33171–80. doi: 10.1074/jbc.M113.518175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Campeau E, Ruhl VE, Rodier F, Smith CL, Rahmberg BL, Fuss JO, Campisi J, Yaswen P, Cooper PK, Kaufman PD. A versatile viral system for expression and depletion of proteins in mammalian cells. PLoS One. 2009;4(8) doi: 10.1371/journal.pone.0006529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nandakumar J, Bell CF, Weidenfeld I, Zaug AJ, Leinwand LA, Cech TR. The TEL patch of telomere protein TPP1 mediates telomerase recruitment and processivity. Nature. 2012;492(7428):285–9. doi: 10.1038/nature11648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhong FL, Batista LF, Freund A, Pech MF, Venteicher AS, Artandi SE. TPP1 OB-Fold domain controls telomere maintenance by recruiting telomerase to chromosome ends. Cell. 2012;150(3):481–94. doi: 10.1016/j.cell.2012.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sexton AN, Youmans DT, Collins K. Specificity requirements for human telomere protein interaction with telomerase holoenzyme. J Biol Chem. 2012;287(41):34455–64. doi: 10.1074/jbc.M112.394767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ramsay AJ, Quesada V, Foronda M, Conde L, Martínez-Trillos A, Villamor N, Rodríguez D, Kwarciak A, Garabaya C, Gallardo M, López-Guerra M, López-Guillermo A, Puente XS, Blasco MA, Campo E, López-Otín C. POT1 mutations cause telomere dysfunction in chronic lymphocytic leukemia. Nat Genet. 2013;45(5):526–30. doi: 10.1038/ng.2584. [DOI] [PubMed] [Google Scholar]
- 34.Aoude LG, Pritchard AL, Robles-Espinoza CD, Wadt K, Harland M, Choi J, Gartside M, Quesada V, Johansson P, Palmer JM, Ramsay AJ, Zhang X, Jones K, Symmons J, Holland EA, Schmid H, Bonazzi V, Woods S, Dutton-Regester K, Stark MS, Snowden H, van Doorn R, Montgomery GW, Martin NG, Keane TM, López-Otín C, Gerdes AM, Olsson H, Ingvar C, Borg A, Gruis NA, Trent JM, Jönsson G, Bishop DT, Mann GJ, Newton-Bishop JA, Brown KM, Adams DJ, Hayward NK. Nonsense Mutations in the Shelterin Complex Genes ACD and TERF2IP in Familial Melanoma. J Natl Cancer Inst. 2014;107 (2). pii: dju408. [DOI] [PMC free article] [PubMed]
- 35.Guo Y, Kartawinata M, Li J, Pickett HA, Teo J, Kilo T, Barbaro PM, Keating B, Chen Y, Tian L, Al-Odaib A, Reddel RR, Christodoulou J, Xu X, Hakonarson H, Bryan TM. Inherited bone marrow failure associated with germline mutation of ACD, the gene encoding telomere protein TPP1. Blood. 2014;124(18):2767–74. doi: 10.1182/blood-2014-08-596445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kocak H, Ballew BJ, Bisht K, Eggebeen R, Hicks BD, Suman S, O’Neil A, Giri N, NCI DCEG Cancer Genomics Research Laboratory; NCI DCEG Cancer Sequencing Working Group. Maillard I, Alter BP, Keegan CE, Nandakumar J, Savage SA. Hoyeraal-Hreidarsson syndrome caused by a germline mutation in the TEL patch of the telomere protein TPP1. Genes Dev. 2014;28(19):2090–102. doi: 10.1101/gad.248567.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hockemeyer D, Sfeir AJ, Shay JW, Wright WE, de Lange T. POT1 protects telomeres from a transient DNA damage response and determines how human chromosomes end. EMBO J. 2005;24(14):2667–78. doi: 10.1038/sj.emboj.7600733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cerone MA, Londoño-Vallejo JA, Autexier C. Mutated telomeres sensitize tumor cells to anticancer drugs independently of telomere shortening and mechanisms of telomere maintenance. Oncogene. 2006;25(56):7411–20. doi: 10.1038/sj.onc.1209727. [DOI] [PubMed] [Google Scholar]
- 39.Ludwig A, Saretzki G, Holm PS, Tiemann F, Lorenz M, Emrich T, Harley CB, von Zglinicki T. Ribozyme cleavage of telomerase mRNA sensitizes breast epithelial cells to inhibitors of topoisomerase. Cancer Res. 2001;61(7):3053–61. [PubMed] [Google Scholar]
- 40.Pathak S, Multani AS, Furlong CL, Sohn SH. Telomere dynamics, aneuploidy, stem cells, and cancer (review) Int J Oncol. 2002;20(3):637–41. [PubMed] [Google Scholar]
- 41.Martinez P, Blasco MA. Telomeric and extra-telomeric roles for telomerase and the telomere-binding proteins. Nat Rev. 2011;11(3):161–76. doi: 10.1038/nrc3025. [DOI] [PubMed] [Google Scholar]
- 42.Maia AT, van der Velden VH, Harrison CJ, Szczepanski T, Williams MD, Griffiths MJ, van Dongen JJ, Greaves MF. Prenatal origin of hyperdiploid acute lymphoblastic leukemia in identical twins. Leukemia. 2003;17:2202–6. doi: 10.1038/sj.leu.2403101. [DOI] [PubMed] [Google Scholar]
- 43.Mori H, Colman SM, Xiao Z, Ford AM, Healy LE, Donaldson C, Hows JM, Navarrete C, Greaves M. Chromosome translocations and covert leukemic clones are generated during normal fetal development. Proc Natl Acad Sci U S A. 2002;99(12):8242–7. doi: 10.1073/pnas.112218799. [DOI] [PMC free article] [PubMed] [Google Scholar]