

Abstract
Daily rhythms in physiology may affect the pharmacokinetics of a drug. The aim of this study was to evaluate 24-hour variation in the pharmacokinetics of the CYP3A substrate midazolam. Oral (2 mg) and intravenous (1 mg) midazolam was administered at six timepoints throughout the 24-hour period in 12 healthy volunteers. Oral bioavailability (population mean value [RSE%] of 0.28 (7.1%)) showed 24-hour variation that was best parameterized as a cosine function with an amplitude of 0.04 (17.3%) and a peak at 12:14 in the afternoon. The absorption rate constant was 1.41 (4.7%) times increased after drug administration at 14:00. Clearance (0.38 L/min (4.8%)) showed a minor 24-hour variation with an amplitude of 0.03 (14.8%) L/min and a peak at 18:50. Simulations show that dosing time minimally affects the concentration time profiles after intravenous administration, while concentrations are higher during the day compared to the night after oral dosing, reflecting considerable variation in intestinal processes.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC? ☑ The pharmacokinetics of the CYP3A4 substrate midazolam may be subject to 24-hour variation, but previous studies did not assess all pharmacokinetic parameters simultaneously and yielded conflicting results. • WHAT QUESTION DID THIS STUDY ADDRESS? ☑ How do the pharmacokinetics of oral and intravenous midazolam depend on time of administration? • WHAT THIS STUDY ADDS TO OUR KNOWLEDGE ☑ Oral bioavailability and absorption rate constant of midazolam show considerable 24-hour variation, while clearance shows minor fluctuations. Concentration–time profiles of midazolam are affected by dosing time after oral administration, but not after intravenous administration. • HOW THIS MIGHT CHANGE CLINICAL PHARMACOLOGY AND THERAPEUTICS ☑ Our design, with appropriate control for unperturbed circadian rhythmicity and semi-simultaneous oral and intravenous administration, combined with population pharmacokinetic modeling, can be applied to study 24-hour variation in the pharmacokinetics of other model compounds, yielding detailed information on the effect of time of administration on the concentration profile.
Many physiological processes, including gene expression, metabolism, and organ function, exhibit 24-hour variation.1 As a result of these rhythms, the pharmacokinetics of drugs may vary over the day.2 Although different chronopharmacological studies have shown that the pharmacokinetics of several drugs depend on the time of administration,3–5 this source of variability has not been evaluated systematically. A possible approach to methodically assess 24-hour variation in pharmacokinetic (PK) parameters is to study a model drug representing a group of drugs that are absorbed, distributed, metabolized, and/or eliminated in a similar way. Such an approach requires a strict standardized study protocol with external validators to ensure that the research is performed with minimal or no disturbance of the physiological rhythms.
Midazolam is extensively metabolized by both hepatic and intestinal cytochrome P450 3A (CYP3A) and is considered a probe of CYP3A enzyme activity.6–10 CYP3A is an important drug-metabolizing enzyme, metabolizing 30% of clinically used drugs.11 In vitro research shows that hepatic CYP3A activity fluctuates during the 24-hour period.12,13 Moreover, in vivo CYP3A activity in humans measured by urinary 6βhydroxy-cortisol to cortisol ratio showed diurnal variation by an average of 2.8-fold.14
Several chronopharmacokinetic studies on midazolam have been published.15–19 In most of these studies, however, midazolam was administered either orally18 or as an intravenous (i.v.) infusion,15,17,19 and therefore not all PK parameters (absorption rate constant, bioavailability, and clearance) could be assessed separately. To distinguish between bioavailability, systemic clearance, and volume of distribution, oral and i.v. administration should be combined in a single study. In the current study, we aimed to evaluate 24-hour variation in the PK parameters of midazolam after semi-simultaneous oral and i.v. administration in healthy volunteers.
METHODS
Study design and data
Healthy, nonsmoking Caucasian male subjects, aged between 18 and 50 and a body mass index (BMI) between 18 and 30 kg/m2, were recruited for this study, which took place at the Centre for Human Drug Research in Leiden, The Netherlands. Subjects were excluded from participation if any clinically significant abnormality was found in medical history, routine laboratory tests, or 12-lead ECG recordings or if they used any medication, could be characterized as an extreme morning or evening type as determined by the Horne-Ostberg Chronotype Questionnaire,20 made transmeridian flights, or did shift work from a month prior to the start of the study. The study was approved by the Medical Ethics Committee of the Leiden University Medical Center and was carried out according to the International Conference on Harmonization (ICH) guidelines for good clinical practice.21
From 1 week prior to each study visit, subjects were instructed to maintain a stable sleep–wake schedule (waking times between 07:00–08:00, bedtimes between 23:00–00:00). Subjects kept a sleep diary and wore an Actiwatch (CamNtech Actiwatch Light, UK) to monitor their daily activity profiles. Subjects refrained from heavy exercise for 24 hours prior to a scheduled study visit and were not allowed to use products that interfere with CYP3A metabolism (such as grapefruit, banpeiyu, pomegranate, star fruit, black berry, and wild grape) for 2 weeks prior to the study, and no caffeinated drinks, alcoholic drinks, honey, or cruciferous vegetables for 72 hours prior to the drug administration until 48 hours thereafter.
The study consisted of three study visits at which the subjects received a 2 mg oral midazolam solution and 1 mg i.v. midazolam (separated by 150 minutes) twice a day at a 12-hour interval. The clock times of midazolam administration differed for each study visit, so that data were collected at six different timepoints throughout the 24-hour period (oral administration at 10:00, 14:00, 18:00, 22:00, 02:00, and 06:00) in each of the 12 volunteers (Figure 1a), with a washout period of at least 2 weeks between the study visits. Throughout the study visits, subjects remained in a semirecumbent position. At night (23:30 until 07:30), lights were dimmed, and subjects wore an eye mask. From 2 hours prior to drug administration, subjects fasted. A light meal was served at t = 395 minutes and a snack at t = 540 minutes after oral administration. Water was allowed as required.
Figure 1.
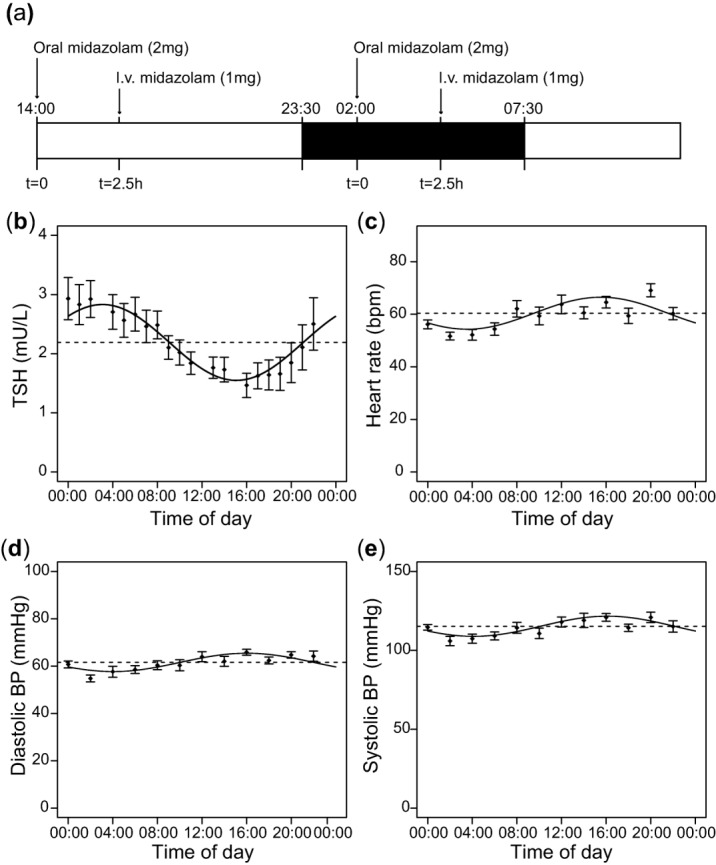
(a) Schematic representation of the drug administration protocol per study visit. Subjects completed two occasions, separated by 12 hours. At t = 0, subjects received 2 mg midazolam (MDZ) orally. At t = 2.5 hours, subjects received 1 mg midazolam intravenously. After 12 hours the procedure was repeated. In each of the three study visits drug administration took place at two different clock times (t = 0 at 14:00 and 02:00 in this example), so drug administration occurred at six different clock times throughout the 24-hour period. The order of time of drug administration was randomized. The dark box indicates the clock times during which the subjects were instructed to sleep. (b–e) Mean values of TSH levels (b), heart rate (c), diastolic (d), and systolic blood pressure (e) obtained during the study visits across the 24-hour period (n = 12 subjects). The solid lines show the cosine curve with a period of 24-hour that best fits the data, obtained through cosinor analysis.
Samples (2.7 mL) to determine midazolam concentrations in serum were collected at t = 0, 15, 30, 45, 58, 65, 70, 75, 80, 90, 120, 148, 155, 165, 180, 210, 240, 270, 330, and 390 minutes after oral administration, as well as at t = 715 minutes in case it involved the first 12 hours of a study visit. Midazolam concentrations were measured using a validated liquid chromatographic tandem mass spectrometric (LC-MS/MS) assay.22 Within-day and between-day inaccuracy and imprecision were less than 5% and the lower limit of quantitation (LLQ) was 0.3 μg/L.22
Samples to determine thyroid-stimulating hormone (TSH) concentrations in serum (1.2 mL) were collected hourly during the study visits. TSH concentrations (μIU/mL) were measured by an electrochemiluminescence immunoassay (ECLIA, Cobas, Roche Diagnostics, Mannheim, Germany) on an Elecsys immunoassay analyzer (Roche Diagnostics), calibrated against the World Health Organization Second Standard International Reference Preparation (80/558). The LLQ was 0.005 μIU/mL. Blood pressure and heart rate were measured every 2 hours during the study visits.
Single-component cosinor analysis was performed to evaluate the presence of a 24-hour rhythm in blood pressure, heart rate, and endogenous TSH levels using R software (v. 2.15; R Foundation for Statistical Computing, Vienna, Austria). Cosinor analysis is a statistical method to fit a cosine function to longitudinal data. If the period assumed to be known (in this case, 24 hours), a cosine function can be rewritten as a linear function and the data can be fitted via least squares regression.23 The mesor, amplitude, and acrophase can be calculated from the estimated intercept and coefficients.
Population PK modeling
The PK data were analyzed using nonlinear mixed effects modeling (NONMEM v. 7.2; ICON Development Solutions, Hanover, MD)24 and R (v. 2.15),25 Pirana (v. 2.7.1), Xpose (4.5.0), and PsN (3.6.2)26 were used to visualize the data. The first-order conditional estimation method with interaction was used throughout model development.
Structural and statistical model
PK models incorporating either two or three compartments with first-order, zero-order, or combined first- and zero-order oral absorption were investigated. Furthermore, the addition of one or more transit compartments or an oral absorption lag time was evaluated.27 Interindividual variability (IIV) in PK parameters was assumed to be log-normally distributed. Residual variability was investigated using proportional, additive, or combined proportional and additive error models.
Twenty-four hour variation
Twenty-four hour variation in the different structural PK parameters was first explored by incorporating interoccasion variability (IOV), representing the variability between the six different times of administration, on each of these parameters of interest using the following equation28:
| 1 |
where θij is the individual parameter estimate at the jth occasion, θmean is the population mean, ηi is a random variable for the ith individual (IIV) and kij is a random variable for the ith individual at the jth occasion (IOV). Both ηi and kij were assumed to be independently normally distributed with mean of zero and variances ω2 and π2, respectively. The k values used in IOV plots are empirical Bayes estimates (EBEs) of the interoccasional random effect (NONMEM ETA) of the parameter involved.
If a 24-hour rhythm was visually identified in IOV plots, a cosine function with a period of 24 hours (1,440 minutes) was implemented in the model as follows:
| 2 |
where P represents the studied PK parameter, θI the mesor (individual value of the PK parameter around which it oscillates), θAMP the amplitude, and θACROPHASE the acrophase (time of the peak of the cosine function). TIME represents the time in minutes starting at midnight of the first study visit and continuing until the end of the third study visit. It was assumed that the cosine function described the data accurately when no residual trend of diurnal variation was left in the IOV plots upon inclusion of the function and it resulted in a reduced IOV value. Twenty-four hour variation was also evaluated by estimation of different multiplication factors on the PK parameters for the six timepoints of administration (10:00, 14:00, 18:00, 22:00, 02:00, and 06:00).
If no full 24-hour variation could be identified for a PK parameter, but only an increase at a certain time interval of the day, this was parameterized as half a cycle of a sine function:
| 3 |
where INC represents the increase in a parameter, θAMP the amplitude, θFR the frequency of the oscillations (minutes), TSIN the clock time in minutes after 12:00 (noon), and θON represents the onset of the increase in the parameter. The end of the increase in the PK parameter was calculated as follows:
| 4 |
Model selection and internal model evaluation
Model development and selection was guided by comparison of the objective function value (OFV, i.e., −2 log likelihood (–2LL)) between nested models, precision of parameter estimates, and visual improvement in goodness-of-fit plots split by the six times of administration (observed vs. individual-predicted concentrations, observed vs. population-predicted concentrations, conditional weighted residuals vs. time after dose, and conditional weighted residuals vs. population-predicted concentrations plots and individual plots). P < 0.05 (ΔOFV = −3.84 for one degree of freedom) was considered statistically significant.
For internal model evaluation, a bootstrap analysis was performed using 250 replicates and visual predictive checks (VPCs), stratified by the six times of administration, were created using 1,000 simulated datasets.
Simulations
The final population PK model was used to simulate the concentration-time curves of a subject dosed at six different administration times of a 7.5 mg oral dose or a 2 mg i.v. bolus dose.
RESULTS
Study participants
Twelve healthy Caucasian male volunteers participated in the study. Their demographics are summarized in Table 1. One subject withdrew consent during the study due to personal reasons and was replaced by another study subject who was dosed according the same randomization order.
Table 1.
Subject demographics
| N | Mean | SD | CV (%) | Median | Range | |
|---|---|---|---|---|---|---|
| Age (years) | 12 | 21.8 | 3.19 | 14.6 | 22 | 18–27 |
| Weight (kg) | 12 | 76.0 | 8.65 | 11.4 | 75.4 | 63.4–92.9 |
| Body mass index (kg/m2) | 12 | 22.3 | 2.37 | 10.6 | 21.9 | 18.8–25.8 |
CV, coefficient of variation; N, number of subjects; SD, standard deviation.
Physiological parameters
Several physiological variables, used to verify that the approach of our study is suited to assess diurnal rhythmicity in physiological processes, fluctuated over the 24-hour period (Figure 1b–e). TSH levels showed significant 24-hour variation with a relative amplitude of 29% and peak levels around 03:05 at night (r2 = 0.13, P < 0.0001). Heart rate and diastolic and systolic blood pressure also exhibited a significant 24-hour rhythm (r2 = 0.14, P < 0.0001 for all three parameters) with relative amplitudes of 10%, 6.3%, and 5.6%, respectively, and peaks around 16:00.
Population PK model and internal model evaluation
The mean concentration–time profiles of midazolam after oral and i.v. administration at the six timepoints is shown in Supplementary Figure 1. A three-compartment PK model with equalized peripheral volumes of distribution best described the data. The peripheral volumes were equalized, as these values were almost equal and the model resulted in a similar objective function (P > 0.05). Oral absorption of midazolam was best described by a one-transit compartment absorption model, where oral absorption rate constant and transit compartment rate constant were equalized. Residual variability was best described by using a proportional error model for both oral and i.v. data.
To explore 24-hour variation in the different PK parameters, IOV was sequentially incorporated on oral bioavailability, absorption rate constant, and systemic clearance (Supplementary Table 1). The presence of a 24-hour rhythm was most evident for oral bioavailability (Figure 2a, P < 0.001, ΔOFV −349). After implementation of IOV on absorption rate constant an increase in this parameter was identified after administration at 14:00 (Figure 2b, P < 0.001 ΔOFV −258). The magnitude of a possible 24-hour rhythm in clearance of midazolam seemed lower compared to oral bioavailability and absorption rate constant (Figure 2c, P < 0.001, ΔOFV −93). The η-shrinkage for the EBEs of the interoccasional random effect was higher than 30% for oral bioavailability and absorption rate constant (33% and 55%, respectively, Supplementary Table 1), resulting in potentially unreliable EBEs.29 Therefore, these observations necessitated further analysis by implementation of a cosine function on each of these parameters evaluated by objective function.
Figure 2.
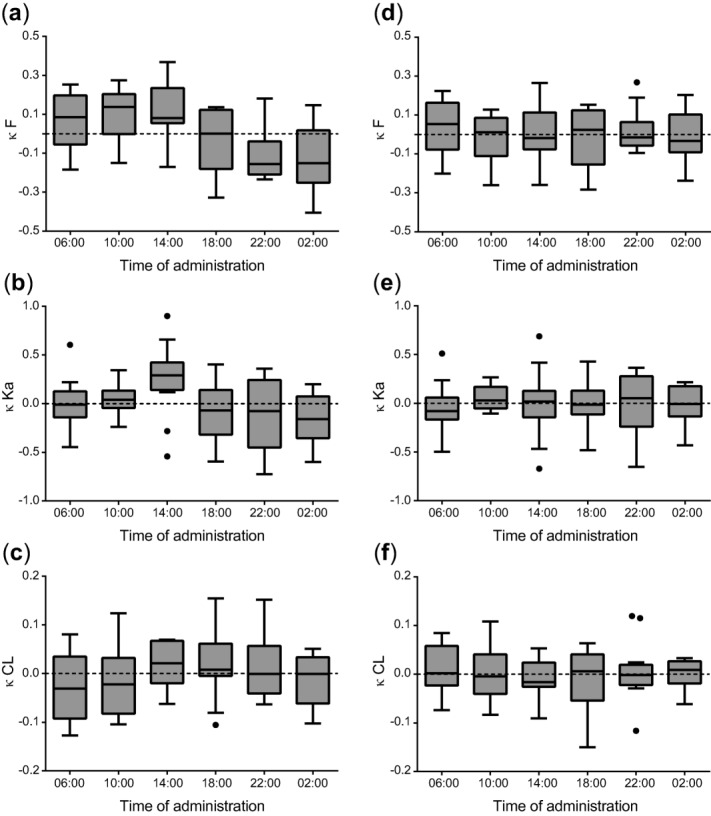
Interoccasion variability (κ, kappa) vs. time of administration of midazolam for oral bioavailability (F) (a,d), absorption rate constant (Ka) (b,e), and clearance (CL) (c,f). Left column represents IOV (κ) vs. time plots of the simple model in which no cosine function was incorporated (a–c) and right column represents IOV (κ) vs. time plots of the models after implementation of a cosine function for oral bioavailability (d), a multiplication factor at the 14:00 hour administration time for absorption rate constant (e) and a cosine function for clearance (f). The k values used in these IOV plots are empirical Bayes estimates (EBEs) of the interoccasional random effect (NONMEM ETA) in the parameter involved (oral bioavailability, absorption rate constant, or clearance).
The 24-hour variation in bioavailability was accurately described by a cosine function (Eq. 2), resulting in a significant improvement in OFV compared to the IOV on bioavailability model (P < 0.001, ΔOFV −28) and in a reduced IOV value (from 20 to 15.4%, Supplementary Table 1). Alternatively, 24-hour variation in bioavailability was estimated by implementing different multiplication factors on this parameter for each of the six timepoints of administration. This multiplication factor model showed a similar fluctuation over the 24-hour period compared to the cosine model (Supplementary Figure 2a) and had a similar OFV (2,431 for the cosine model with two additional parameters vs. 2,430 for the multiplication factor model with five additional parameters, P > 0.05 for 3 degrees of freedom). The cosine model was preferred over the multiplication factor model, because both the IOV model (Figure 2a) and multiplication factor model (Supplementary Figure 2a) revealed a cosine function in bioavailability and the cosine model required fewer parameters to be estimated, while having larger predictive value. After implementation of the cosine function for bioavailability, there was no remaining trend in IOV confirming the appropriateness of the cosine model for this parameter (Figure 2d).
After implementation of the cosine function for bioavailability, the variation in absorption rate constant was modeled, which was best described by the estimation of a multiplication factor at 14:00 (P < 0.01, ΔOFV −9, Supplementary Table 1). After implementation of this multiplication factor, IOV on absorption rate constant was removed from the model, because of the high η-shrinkage of the EBE of the interoccasional random effect (55%, Supplementary Table 1). Addition of multiplication factors on absorption rate constant at other timepoints of administration did not further improve the model (P > 0.05, Supplementary Figure 2b). Alternatively, a cosine function was tested, but this model did not result in adequate prediction of the increased absorption rate constant at 14:00. Furthermore, inclusion of half a cycle of a sine function to describe the peak in absorption rate constant (Eqs. 3 and 4) resulted in a peak at 14:59 and an amplitude of 0.056 min−1 (increase of 106%) and an onset and offset of the peak at 14:12 and 15:45, respectively. However, this model was very sensitive to initial parameter estimates and did not result in a significant improvement in OFV compared to the model with a multiplication factor at 14:00 (P > 0.05, ΔOFV −3.7, 2 degrees of freedom). Therefore, the model with a multiplication factor at 14:00 was selected. No rhythm remained in the IOV plot after implementation of this factor (Figure 2e). However, this plot should be viewed with caution because of the high ETA shrinkage and IOV on the absorption rate constant was therefore removed from the model, as described above. The multiplication factor estimated by this model was 1.46 (resulting in an absorption rate constant of 0.08 min−1), indicating a strong increase in absorption rate constant after administration at 14:00.
After implementation of a cosine function for bioavailability and a multiplication factor for absorption rate constant, 24-hour related changes in clearance were modeled. For this parameter, 24-hour variation was best described by a cosine function (Eq. 2), resulting in a significant decrease in OFV compared to the IOV model for clearance (P < 0.001, ΔOFV −26, Supplementary Table 1). Since the IOV value was substantially smaller than the IIV on clearance, IOV on clearance was removed from the model. Clearance could also be described by estimation of different multiplication factors for each of the six times of drug administration (Supplementary Figure 2c), resulting in similar variation over the 24-hour period as the cosine model. After implementation of the cosine function for clearance, there was no remaining trend in IOV on this parameter (Figure 2f) (η shrinkage of 20%), confirming the appropriateness of the cosine model for clearance.
Hence, the final model selected to describe 24-hour variation in midazolam concentration profiles included a cosine function for bioavailability and clearance and a multiplication factor to describe the increase in absorption rate constant at 14:00. The model parameter values are summarized in Table 2. Observed vs. individual predicted concentrations and observed vs. population predicted midazolam concentrations of the final PK model for all six timepoints of administration are shown in Supplementary Figure 3. The final model was evaluated using bootstrap analysis, confirming that the model parameters could be estimated with good precision (Table 2). Furthermore, VPCs stratified by time of administration indicated good predictive performance for both the oral and i.v. data with good agreement between observed data and model simulated confidence intervals for the median, 2.5th, and 97.5th percentiles (Figure 3). Figure 4 shows the 24-hour variation in bioavailability and in clearance of the final model. The cosine function on bioavailability has a relative amplitude of 14.7% with a peak at 12:14, while the cosine function on clearance has a relative amplitude of 7.2% and a peak at 18:50.
Table 2.
Population pharmacokinetic parameters of the final model for midazolam and results of the bootstrap analysis (250/250 resamples successful)
| Parameter | Model estimates (RSE%) | Bootstrap estimates (95% confidence interval) |
|---|---|---|
| CL= CLmesor+Amp x cos((2π/1440)*(Time-Acrophase)) | ||
| CLmesor (L/min) | 0.379 (4.8) | 0.380 (0.344–0.417) |
| Amp (L/min) | 0.027 (14.8) | 0.028 (0.017–0.039) |
| Acrophase (min) | 1,130 (2.9) | 1,130.2 (1,005.3–1,204.7) |
| Vcentral (L) | 18.2 (5.4) | 18.4 (15.3–20.9) |
| Vperipheral1 = Vperipheral2 (L) | 22.5 (2.5) | 22.4 (20.2–26.2) |
| Q (L/min) | 0.27 (6.8) | 0.269 (0.209–0.334) |
| Q2 (L/min) | 1.31 (8.5) | 1.29 (1.08–1.56) |
| Ka = Ktr (min−1) | 0.053 (5.8) | 0.053 (0.048–0.061) |
| Fraction Ka at 14:00 | 1.41 (4.7) | 1.41 (1.07–1.78) |
| F= Fmesor+Amp x cos((2π/1440)*(Time-Acrophase)) | ||
| F | 0.277 (7.1) | 0.275 (0.244–0.313) |
| Amp | 0.041 (17.3) | 0.041 (0.026–0.055) |
| Acrophase (min) | 734 (5.3) | 739.7 (667.0–821.0) |
| Interindividual variability | ||
| CL (%) | 16.2 (21) | 15.2 (9.7–19.6) |
| Ka (%) | 19.1 (21.9) | 18.7 (10.7–24.2) |
| F (%) | 23.3 (22.2) | 22.7 (15.8–28.8) |
| Interoccasion variability | ||
| F (%) | 14.8 (10.5) | 14.5 (11.5–17.9) |
| Residual proportional error | ||
| σ oral (%) | 18.0 (5.6) | 17.8 (15.8–19.8) |
| σ intravenous(%) | 15.4 (6.1) | 15.1 (13.2–17.3) |
| OFV (−2LL) | 2,299 | 2,242 (1,723–2,730) |
Acrophase, peak time of the cosine function in minutes after midnight; Amp, amplitude; CL, systemic clearance of midazolam; F, oral bioavailability; Ka, oral absorption rate constant; Ktr, transit compartment rate constant; OFV, objective function value; Q, intercompartmental clearance of midazolam between central and first peripheral compartment; Q2, intercompartmental clearance of midazolam between central and second peripheral compartment; RSE, relative standard error (%); V, volume of distribution.
Figure 3.

Visual predictive checks of the final model stratified by time of midazolam administration (06:00, 10:00, 14:00, 18:00, 22:00, and 02:00). Observed concentrations are shown as open circles with solid and lower and upper dashed lines showing the median, 2.5th, and 97.5th percentiles of the observed data, respectively. The shaded areas represent 95% confidence intervals for the model predicted median, 2.5th, 97.5th percentiles constructed from 1,000 simulated datasets of individuals from the original dataset.
Figure 4.
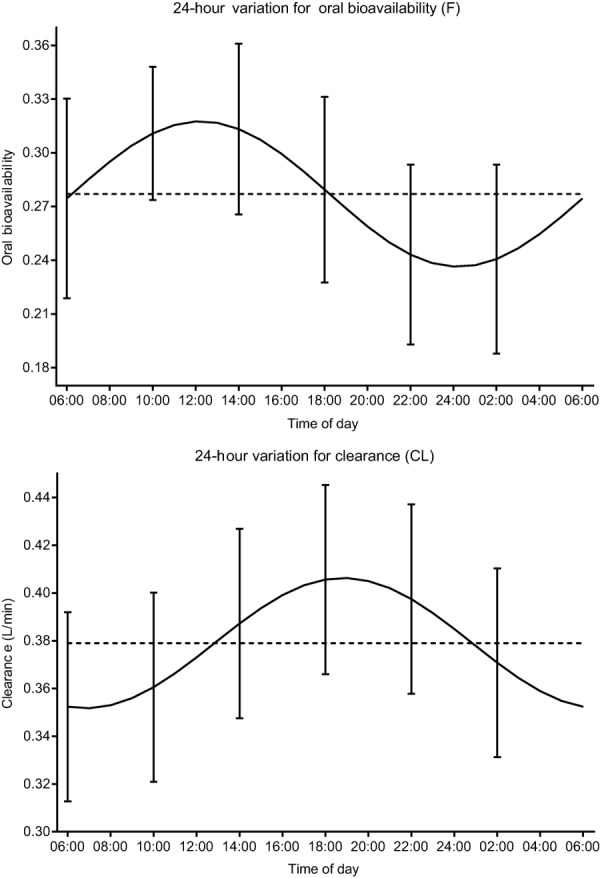
Twenty-four hour fluctuation for oral bioavailability (F) and clearance (CL) according to the final model with the 95% confidence interval of the empirical Bayes estimates (EBEs) for F (IIV+IOV) and CL (IIV) at each administration time. For oral bioavailability, the time of the peak was estimated at 12:14 with an estimated amplitude of 0.041 (14.7% increase) (upper panel). For clearance, the time of the peak was estimated at 18:50 with an estimated amplitude of 0.027 L/min (7.2% increase) (lower panel).
Simulations
Population predicted midazolam concentrations after a 7.5 mg oral dose and 2 mg i.v. bolus dose in a typical subject dosed at six different times during the day (10:00, 14:00, 18:00, 22:00, 02:00, and 06:00) were simulated using the final model (Figure 5). The oral midazolam dose simulations show that the concentrations after administration in the late morning and early afternoon (10:00 and 14:00) are higher compared to the concentrations after administration in the late evening and early night (22:00 and 02:00). In addition, the time to maximum concentration (Tmax) is shorter when midazolam is administered at 14:00. In contrast to the oral dose simulations, the i.v. dose simulations show almost no variation during the 24-hour period.
Figure 5.
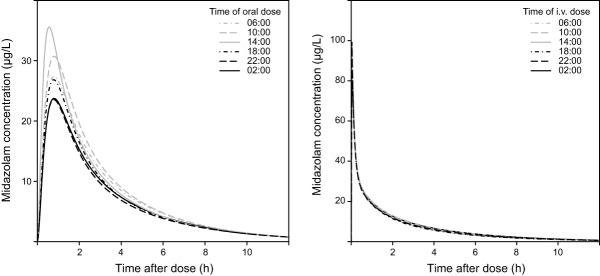
Population predicted midazolam concentrations over time after 7.5 mg oral administration (left panel) and a 2 mg intravenous bolus (right panel) at 06:00, 10:00, 14:00, 18:00, 22:00, and 02:00.
DISCUSSION
This study aimed to evaluate the 24-hour variation in the PK of the CYP3A substrate midazolam after semi-simultaneous oral and i.v. administration at six different timepoints during the day (06:00, 10:00, 14:00, 18:00, 22:00, and 02:00). It was found that oral bioavailability and clearance are subject to 24-hour variation that could both be described by a cosine function. The peak of oral bioavailability was found at 12:14, with a relative difference between peak and trough values of 29.4%. The effect for clearance was found to be small, with a peak at 18:50 and a relative difference between peak and trough levels of 14.4%. Furthermore, we found that absorption rate constant was increased 1.41 times after administration at 14:00.
Previous studies that investigated the diurnal variation of midazolam clearance in healthy volunteers did not yield consistent results.15–19 In agreement with our results, Klotz and Ziegler found a higher clearance value in the evening compared to the morning after i.v. administration.16 More recently, Tomalik-Scharte et al. reported a cosine function in midazolam clearance over the day with a 10% increase at 15:00.19 This is consistent with our results, as we found a 7.2% maximum increase in clearance at 18:50. The small difference in peak time may be explained by the nature of the study; where Tomalik-Scharte et al. evaluated midazolam concentrations during the day upon a continuous i.v. infusion, we studied an oral and i.v. bolus dose at six different times of administration. The fact that others found no influence of the time of administration on clearance may be explained by the low number of subjects in the study17 and the fact that intensive care patients were studied, showing a disrupted circadian rhythm.15 Hence, most chronopharmacokinetic studies about i.v. midazolam are in line with our findings of a relatively small 24-hour variation in midazolam clearance.
Our results about absorption processes of midazolam (24-hour variation in oral bioavailability and increase in absorption rate constant at 14:00) are not consistent with earlier chronopharmacokinetic studies on oral midazolam, finding no influence on Cmax, Tmax, or oral bioavailability.16,18 These discrepancies may be due to methodological differences. Klotz and Ziegler administered midazolam only at two different timepoints during the day,16 and therefore the peak and trough may easily be missed. In the study of Koopmans et al., subjects were not allowed to lie down or sleep from 1 hour before to 8 hours after dosage,18 which could have disrupted the circadian rhythms in physiological processes of the subjects.30 However, our finding of 24-hour variation in oral bioavailability of midazolam is supported by chronopharmacokinetic studies of other CYP3A substrates, such as nifedipine, tacrolimus, and cyclosporin.31,32 Lemmer et al. showed an increased Cmax and 35% increase in oral bioavailability after a morning dose of immediate release nifedipine compared to an evening dose.31 Furthermore, studies with oral tacrolimus and cyclosporin showed in general an increased Cmax and area under the curve (AUC) after morning dose compared to evening dosing.32–36 Therefore, it seems that our findings on 24-hour variation in absorption processes are strengthened by the advanced study design that we used in comparison to previous oral midazolam studies that did not report these changes, and are supported by chronopharmacological studies of other CYP3A substrates.
Twenty-four hour variation in clearance and oral bioavailability as well as the increase in absorption rate constant can be explained by several physiological factors. Since midazolam is a typical probe for CYP3A activity,6,7,9 the rhythm in systemic clearance of midazolam may be explained by minor 24-hour variation in CYP3A activity. Multiple lines of evidence show that hepatic CYP3A activity fluctuates during the 24-hour period.12–14,19,37 Like systemic clearance, 24-hour variation in oral bioavailability of midazolam may also be explained by variation in intestinal CYP3A activity, since CYP3A is present both in the gut wall and liver.8 Another explanation for the variation in oral bioavailability may be the variation in splanchnic blood flow during the 24-hour period, which is supported by the findings of Lemmer and Nold, who demonstrated a 24-hour rhythm in hepatic blood flow (as a proxy for splanchnic blood flow) with a peak at 08:00.38 This supports our finding that oral bioavailability is increased from the early morning until the end of the afternoon (Figure 4). An increased splanchnic blood flow will decrease the intestinal first-pass effect, as it will carry the drug away from the enterocyte and the CYP3A enzyme.39,40 In contrast to oral bioavailability, the clearance of midazolam is not expected to be influenced by hepatic blood flow to such an extent, because midazolam is a low to intermediate extraction drug (extraction rate of 35%), making it relatively independent of hepatic blood flow.9 The increase in absorption rate constant after oral administration at 14:00 may be explained by 24-hour variation in gastric emptying, gastrointestinal mobility, and splanchnic blood flow,2,38,41,42 even though we could not identify a cosine function for absorption rate constant.
In this study we utilized a semi-simultaneous design in which midazolam was administered as an oral and i.v. dose separated by 150 minutes.6 An advantage of this crossover approach is that intraindividual variability is limited, since the oral and i.v. dose are administered to the same individual at a relatively short time frame.43 By using six different timepoints of oral and i.v. midazolam administration, 24-hour variation in absorption parameters as well as clearance could be accurately identified. Moreover, we ensured that subjects had stable rest/activity patterns between the study days and controlled for the influence of eating and physical activity, both of which are known to have an impact on physiological rhythms.44 Another strength of our study design is that several endogenous markers, with known diurnal variation (heart rate, systolic/diastolic blood pressure, and serum TSH levels) were used as external validators to verify that our approach, including the low dose of midazolam, did not interfere with normal circadian physiology of the subjects. We found that these endogenous markers show clear diurnal variation, with peak and trough times that are comparable to values reported in the literature.45,46 These findings indicate that the study population and design were well suited to study diurnal variation of midazolam exposure.
As the PK of midazolam have been shown to be linear over a wide dose range,47,48 we performed simulations on the basis of the final PK model using therapeutic doses. These simulations illustrate the findings of the current study by showing a substantial effect of time of administration on midazolam concentration–time profiles after oral administration, whereas this effect is minimal after i.v. administration. Midazolam concentrations after oral administration are higher in the morning and afternoon compared to concentrations after administration in the evening and night. In addition, the time to maximum concentration (Tmax) is shorter after oral administration at 14:00. In the clinic, midazolam is mainly given as an i.v. dose, for example, as premedication or for induction of anesthesia, upon which the time of administration will have no clinical impact. However, midazolam is also prescribed as a hypnotic to patients with insomnia. For these patients, who take an oral dose in the evening, lower serum concentrations should be anticipated.
In conclusion, this study shows that oral bioavailability of midazolam is subject to 24-hour variation and that the absorption rate constant is increased at 14:00 in the afternoon. The clearance of midazolam is also subject to 24-hour variation, although its magnitude is small and without clinical significance. As a result, the 24-hour variation in oral bioavailability results in higher serum concentrations during the day compared to the night upon oral midazolam dosing, while the concentration–time profiles are hardly affected by time of administration after i.v. dosing. Future research should elucidate the specific processes that contribute to the 24-hour variation in the PK of midazolam, and of other drugs with similar physicochemical properties, for example, by using markers for intestinal motility or blood flow.
Acknowledgments
This research was supported by the Dutch Technology Foundation (STW), which is the applied science division of NWO, and the Technology Programme of the Ministry of Economic Affairs and by a grant from the Leiden University Medical Center. We thank LAP&P Consultants for their technical support with NONMEM, Rick Admiraal for help with the VPC analysis and simulations, and Marieke de Kam for input on earlier drafts of the article. We thank Catherijne Knibbe for supervising the development of the population pharmacokinetic model performed by A.V.R and M.J.E.B. Current affiliation F van Oosterhout: Center for Sleep and Wake Disorders, Slotervaart Medical Center, Amsterdam, the Netherlands.
Author Contributions
L.K., A.V.R., M.J.E.B., J.H.M., J.B., and F.V.O. wrote the manuscript; H.V.M., H.J.G., J.H.M., J.B., and F.V.O. designed the research; H.V.M., J.D.H., and F.V.O. performed the research; L.K., A.V.R., M.J.E.B., and J.B. analyzed the data; J.D.H. contributed new reagents/analytical tools. A.V.R. and L.K. contributed equally to this work.
Conflict of Interest
The authors declare no conflicts of interest.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
References
- Meijer JH, Colwell CS, Rohling JHT, Houben T. Michel S. Dynamic neuronal network organization of the circadian clock and possible deterioration in disease. Prog. Brain Res. 2012;199:143–162. doi: 10.1016/B978-0-444-59427-3.00009-5. [DOI] [PubMed] [Google Scholar]
- Dallmann R, Brown SA. Gachon F. Chronopharmacology: new insights and therapeutic implications. Annu. Rev. Pharmacol. Toxicol. 2014;54:339–361. doi: 10.1146/annurev-pharmtox-011613-135923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baraldo M. The influence of circadian rhythms on the kinetics of drugs in humans. Expert Opin. Drug Metab. Toxicol. 2008;4:175–192. doi: 10.1517/17425255.4.2.175. [DOI] [PubMed] [Google Scholar]
- Kaur G, Phillips C, Wong K. Saini B. Timing is important in medication administration: a timely review of chronotherapy research. Int. J. Clin. Pharm. 2013;35:344–358. doi: 10.1007/s11096-013-9749-0. [DOI] [PubMed] [Google Scholar]
- Bruguerolle B, Boulamery A. Simon N. Biological rhythms: a neglected factor of variability in pharmacokinetic studies. J. Pharm. Sci. 2008;97:1099–1108. doi: 10.1002/jps.21044. [DOI] [PubMed] [Google Scholar]
- Lee J-I, et al. Application of semisimultaneous midazolam administration for hepatic and intestinal cytochrome P450 3A phenotyping. Clin. Pharmacol. Ther. 2002;72:718–728. doi: 10.1067/mcp.2002.129068. [DOI] [PubMed] [Google Scholar]
- Gorski JC, et al. The contribution of intestinal and hepatic CYP3A to the interaction between midazolam and clarithromycin. Clin. Pharmacol. Ther. 1998;64:133–143. doi: 10.1016/S0009-9236(98)90146-1. [DOI] [PubMed] [Google Scholar]
- Thummel KE, et al. Oral first-pass elimination of midazolam involves both gastrointestinal and hepatic CYP3A-mediated metabolism. Clin. Pharmacol. Ther. 1996;59:491–502. doi: 10.1016/S0009-9236(96)90177-0. [DOI] [PubMed] [Google Scholar]
- Tsunoda SM, Velez RL, Moltke, von LL. Greenblatt DJ. Differentiation of intestinal and hepatic cytochrome P450 3A activity with use of midazolam as an in vivo probe: effect of ketoconazole. Clin. Pharmacol. Ther. 1999;66:461–471. doi: 10.1016/S0009-9236(99)70009-3. [DOI] [PubMed] [Google Scholar]
- Fuhr U, Jetter A. Kirchheiner J. Appropriate phenotyping procedures for drug metabolizing enzymes and transporters in humans and their simultaneous use in the “cocktail” approach. Clin. Pharmacol. Ther. 2007;81:270–283. doi: 10.1038/sj.clpt.6100050. [DOI] [PubMed] [Google Scholar]
- Zanger UM. Schwab M. Cytochrome P450 enzymes in drug metabolism: regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 2013;138:103–141. doi: 10.1016/j.pharmthera.2012.12.007. [DOI] [PubMed] [Google Scholar]
- Froy O. Cytochrome P450 and the biological clock in mammals. Curr. Drug Metab. 2009;10:104–115. doi: 10.2174/138920009787522179. [DOI] [PubMed] [Google Scholar]
- Takiguchi T, et al. Molecular basis for rhythmic expression of CYP3A4 in serum-shocked HepG2 cells. Pharmacogenet. Genomics. 2007;17:1047–1056. doi: 10.1097/FPC.0b013e3282f12a61. [DOI] [PubMed] [Google Scholar]
- Ohno M, et al. Circadian variation of the urinary 6β-hydroxycortisol to cortisol ratio that would reflect hepatic CYP3A activity. Eur. J. Clin. Pharmacol. 2000;55:861–865. doi: 10.1007/s002280050708. [DOI] [PubMed] [Google Scholar]
- Bienert A, et al. Assessing circadian rhythms during prolonged midazolam infusion in the pediatric intensive care unit (PICU) children. Pharmacol. Rep. 2013;65:107–121. doi: 10.1016/s1734-1140(13)70969-1. [DOI] [PubMed] [Google Scholar]
- Klotz U. Ziegler G. Physiologic and temporal variation in hepatic elimination of midazolam. Clin. Pharmacol. Ther. 1982;32:107–112. doi: 10.1038/clpt.1982.133. [DOI] [PubMed] [Google Scholar]
- Klotz U. Reimann IW. Chronopharmacokinetic study with prolonged infusion of midazolam. Clin. Pharmacokinet. 1984;9:469–474. doi: 10.2165/00003088-198409050-00006. [DOI] [PubMed] [Google Scholar]
- Koopmans R, Dingemanse J, Danhof M, Horsten GP. van Boxtel CJ. The influence of dosage time of midazolam on its pharmacokinetics and effects in humans. Clin. Pharmacol. Ther. 1991;50:16–24. doi: 10.1038/clpt.1991.99. [DOI] [PubMed] [Google Scholar]
- Tomalik-Scharte D, et al. Population pharmacokinetic analysis of circadian rhythms in hepatic CYP3A activity using midazolam. J. Clin. Pharmacol. 2014;54:1162–1169. doi: 10.1002/jcph.318. [DOI] [PubMed] [Google Scholar]
- Horne JA. Ostberg O. A self-assessment questionnaire to determine morningness-eveningness in human circadian rhythms. Int. J. Chronobiol. 1976;4:97–110. [PubMed] [Google Scholar]
- ICH. The International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use< http://www.ich.org >. [DOI] [PMC free article] [PubMed]
- Erp NP, van, et al. Mitotane has a strong and a durable inducing effect on CYP3A4 activity. Eur. J. Endocrinol. 2011;164:621–626. doi: 10.1530/EJE-10-0956. [DOI] [PubMed] [Google Scholar]
- Cornelissen G. Cosinor-based rhythmometry. Theor. Biol. Med. Model. 2014;11:16. doi: 10.1186/1742-4682-11-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beal S, Sheiner LB, Boeckmann A. Bauer RJ. NONMEM User’s Guides (1989-2009) Ellicott City, MD: Icon Development Solutions; 2009. [Google Scholar]
- R Development Core Team. 2008. R: a language and environment for statistical computing. R Foundation for Statistical Computing(Vienna, Austria, ). < http://www.r-project.org >.
- Keizer RJ, Karlsson MO. Hooker A. Modeling and simulation workbench for NONMEM: tutorial on Pirana, PsN, and Xpose. CPT Pharmacometrics Syst. Pharmacol. 2013;2:e50. doi: 10.1038/psp.2013.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savic RM, Jonker DM, Kerbusch T. Karlsson MO. Implementation of a transit compartment model for describing drug absorption in pharmacokinetic studies. J. Pharmacokinet. Pharmacodyn. 2007;34:711–726. doi: 10.1007/s10928-007-9066-0. [DOI] [PubMed] [Google Scholar]
- Karlsson MO. Sheiner LB. The importance of modeling interoccasion variability in population pharmacokinetic analyses. J. Pharmacokinet. Biopharm. 1993;21:735–750. doi: 10.1007/BF01113502. [DOI] [PubMed] [Google Scholar]
- Karlsson MO. Savic RM. Diagnosing model diagnostics. Clin. Pharmacol. Ther. 2007;82:17–20. doi: 10.1038/sj.clpt.6100241. [DOI] [PubMed] [Google Scholar]
- Mullington JM, Haack M, Toth M, Serrador JM. Meier-Ewert HK. Cardiovascular, inflammatory, and metabolic consequences of sleep deprivation. Prog. Cardiovasc. Dis. 2009;51:294–302. doi: 10.1016/j.pcad.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmer B, Nold G, Behne S. Kaiser R. Chronopharmacokinetics and cardiovascular effects of nifedipine. Chronobiol. Int. 1991;8:485–494. doi: 10.3109/07420529109059184. [DOI] [PubMed] [Google Scholar]
- Baraldo M. Furlanut M. Chronopharmacokinetics of ciclosporin and tacrolimus. Clin. Pharmacokinet. 2006;45:775–788. doi: 10.2165/00003088-200645080-00002. [DOI] [PubMed] [Google Scholar]
- Iwahori T, et al. Pharmacokinetic differences between morning and evening administration of cyclosporine and tacrolimus therapy. Transplant. Proc. 2005;37:1739–1740. doi: 10.1016/j.transproceed.2005.02.104. [DOI] [PubMed] [Google Scholar]
- Min DI, et al. Circadian variation of tacrolimus disposition in liver allograft recipients. Transplantation. 1996;62:1190–1192. doi: 10.1097/00007890-199610270-00031. [DOI] [PubMed] [Google Scholar]
- Min DI, Chen HY, Lee MK, Ashton K. Martin MF. Time-dependent disposition of tacrolimus and its effect on endothelin-1 in liver allograft recipients. Pharmacotherapy. 1997;17:457–463. [PubMed] [Google Scholar]
- Tada H, et al. Chronopharmacokinetics of tacrolimus in kidney transplant recipients: occurrence of acute rejection. J. Clin. Pharmacol. 2003;43:859–865. doi: 10.1177/0091270003254797. [DOI] [PubMed] [Google Scholar]
- Lu Y-F, et al. Sex differences in the circadian variation of cytochrome p450 genes and corresponding nuclear receptors in mouse liver. Chronobiol. Int. 2013;30:1135–1143. doi: 10.3109/07420528.2013.805762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmer B. Nold G. Circadian changes in estimated hepatic blood flow in healthy subjects. Br. J. Clin. Pharmacol. 1991;32:627–629. doi: 10.1111/j.1365-2125.1991.tb03964.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Jamei M, Yeo KR, Tucker GT. Rostami-Hodjegan A. Prediction of intestinal first-pass drug metabolism. Curr. Drug Metab. 2007;8:676–684. doi: 10.2174/138920007782109733. [DOI] [PubMed] [Google Scholar]
- Patel N, Polak S, Jamei M. Rostami Hodjegan A. 2013. 5th WCDATD/OrBiTo Meet& Quantitative prediction of circadian variation in pharmacokinetics based on in vitro data: dependence on route of administration in the case of BCS/BDDCS Class II and CYP3A4 substrate drug nifedipine. In )
- Kumar D, Wingate D. Ruckebusch Y. Circadian variation in the propagation velocity of the migrating motor complex. Gastroenterology. 1986;91:926–930. doi: 10.1016/0016-5085(86)90696-7. [DOI] [PubMed] [Google Scholar]
- Hoogerwerf WA. Role of clock genes in gastrointestinal motility. Am. J. Physiol. Gastrointest. Liver Physiol. 2010;299:G549–555. doi: 10.1152/ajpgi.00147.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson MO. Bredberg U. Estimation of bioavailability on a single occasion after semisimultaneous drug administration. Pharm. Res. 1989;6:817–821. doi: 10.1023/a:1015939917646. [DOI] [PubMed] [Google Scholar]
- Froy O. Metabolism and circadian rhythms—implications for obesity. Endocr. Rev. 2010;31:1–24. doi: 10.1210/er.2009-0014. [DOI] [PubMed] [Google Scholar]
- Guo Y-F. Stein PK. Circadian rhythm in the cardiovascular system: chronocardiology. Am. Heart J. 2003;145:779–786. doi: 10.1016/S0002-8703(02)94797-6. [DOI] [PubMed] [Google Scholar]
- Andersen S, Bruun NH, Pedersen KM. Laurberg P. Biologic variation is important for interpretation of thyroid function tests. Thyroid. 2003;13:1069–1078. doi: 10.1089/105072503770867237. [DOI] [PubMed] [Google Scholar]
- Misaka S, et al. Pharmacokinetics and pharmacodynamics of low doses of midazolam administered intravenously and orally to healthy volunteers. Clin. Exp. Pharmacol. Physiol. 2010;37:290–295. doi: 10.1111/j.1440-1681.2009.05285.x. [DOI] [PubMed] [Google Scholar]
- Halama B, et al. A nanogram dose of the CYP3A probe substrate midazolam to evaluate drug interactions. Clin. Pharmacol. Ther. 2013;93:564–571. doi: 10.1038/clpt.2013.27. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information