

Abstract
The organ impairment and drug–drug interaction (OI-DDI) database is the first rigorously assembled database of pharmacokinetic drug exposure data from publicly available renal and hepatic impairment studies presented together with the maximum change in drug exposure from drug interaction inhibition studies. The database was used to conduct a systematic comparison of the effect of renal/hepatic impairment and pharmacologic inhibition on drug exposure. Additional applications are feasible with the public availability of this database.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC? ☑ The effect of DDI, RI, and HI are evaluated during drug development. However, there is a lack of a common database that summarizes results of these studies for the same drug that may help to determine if results from DDI or HI or RI studies can be used to inform the likely impact of other factors if such studies were not conducted. Data from publicly available articles and new drug applications usually present either OI data or drug interaction pharmacokinetic studies. • WHAT QUESTION DID THE STUDY ADDRESS? ☑ This database is the first dataset of quantitative data from organ impairment studies presented with the maximum change in drug exposure from a drug–drug inhibition study. • WHAT THIS STUDY ADDS TO OUR KNOWLEDGE ☑ For drugs that are highly cleared via CYP3A4, a well-conducted pharmacologic inhibition study may reflect the worst-case scenario for changes in AUC in patients with severe HI. For drugs that are renally eliminated, pharmacologic inhibition with transporter inhibitors does not reflect the worst-case scenario for changes in AUC in severe RI patients, but accurate estimation of the contribution of renal clearance can be useful in predicting a worst-case scenario. • HOW THIS MIGHT CHANGE CLINICAL PHARMACOLOGY AND THERAPEUTICS ☑ Public availability of this database will encourage additional analysis and assist development of regulatory recommendations for the evaluation of drug exposure in specific populations by leveraging information from DDI studies.
INTRODUCTION
Various intrinsic (e.g., organ impairment) and extrinsic factors (e.g., drug interactions) can affect a drug’s exposure and in some instances this may result in an altered response. In order to optimize efficacy and prevent toxicity in various subpopulations, particularly the growing populations affected by renal impairment (RI) or hepatic impairment (HI), the US Food and Drug Administration (FDA) recommends that drug sponsors evaluate new drug candidates in patients with organ impairment (OI) when a drug is likely to be used in these patients and OI may have an effect on systemic exposure.1,2 In addition to OI studies, drug–drug interaction (DDI) studies utilizing known inhibitors of involved metabolic or transport pathways are frequently conducted to assess the potential for drug interactions with coadministered medications to guide dosing and manage drug interactions.
Several clinically available strong and specific inhibitors have been identified for various cytochrome P450 (CYP) isoforms and some renal transporters. Due to the availability of these inhibitors, it may be possible to reach complete inhibition of the predominant elimination pathway with a pharmacologic agent, which may exceed the "worst-case scenario" that would be expected in a patient with severe OI, in which part of the enzyme (or transport) activity remains even at an advanced stage of the disease. Whether OI and DDIs result in concordant effects on drug pharmacokinetics is likely to be a function of a drug’s elimination pathways and the effects of OI on metabolism and transport. The concordance between OI and DDI studies on drug exposure pharmacokinetics has not been comprehensively assessed.
Herein, we describe the creation of an OI-DDI database that compiles changes in exposure and clearance values observed during inhibition and/or OI studies for drugs with available data, including new molecular entity (NME) drugs approved between 2007 and 2013. We also present examples of the use of the database to retrospectively analyze the association between renal or hepatic impairment and drug inhibition studies. This is the first time that key disposition parameters have been collected from OI and DDI studies and presented in tandem.
CONSTRUCTION AND CONTENT
The OI-DDI database was created using data available from published articles curated into the University of Washington Metabolism and Transport Drug Interaction Database (DIDB, www.druginteractioninfo.org), publicly available new drug application (NDA) reviews, and FDA-approved labels from Drugs@FDA (http://www.accessdata.fda.gov/scripts/cder/drugsatfda/). The data collection protocol is available in the Supplementary Protocol, and an overview of the process is available in Figure 1. The starting point of the database was the availability of an OI study, either hepatic or renal, in the DIDB. The DIDB platform is updated daily and validated using established protocols to identify relevant citations; PubMed queries were performed several times per month using keywords including "hepatic impairment," "renal impairment," and "pharmacokinetics." Data extraction from the full text of the citation was performed by scientists with advanced degrees (e.g., MD, PharmD, PhD) with expertise in pharmaceutical sciences and the majority of entries were validated by a second expert reviewer. Only OI studies comparing impaired populations to healthy control subjects were included in the dataset, i.e., "impaired only" studies and "case reports" were not included. Pharmacokinetic parameters, changes in the area under the concentration–time curve (AUC), and/or clearance value (CL or CL/F) associated with OI along with degree of impairment were collected. In the event of multiple available studies, the maximum change in AUC or CL observed for each drug in an OI study was entered into the master dataset. Both renal and hepatic impairment entries captured the severity of impairment: hepatic impairment based on Child-Pugh classification, and renal impairment based on creatinine clearance (CLcr) or estimated glomerular filtration rate (eGFR). The percent changes of AUC or oral CL were calculated using mean AUC or CL values and are presented in the dataset. Due to the high degree of standardization in the conduct of OI studies (e.g., n = 6–8 subjects per impairment group as recommended by the FDA guidance) study size for individual studies was not noted in the database. The degree of OI presented in the dataset is based on the authors/sponsors of the studies and in most cases followed the criteria recommended by the FDA (2010 guidance)1,2 or European Medicines Agency (EMA) (2013 guidance).3,4 Occasionally, the classification by the study authors (mild, moderate, severe) was inconsistent with the FDA recommendations; in these instances, they were still reported as "mild, moderate, or severe groups." End Stage Renal Disease (ESRD) in the database was defined as subjects with a CLcr <15 mL/min or those requiring maintenance hemodialysis; note that the timing of drug administration (pre- or postdialysis) was not captured by the dataset due to inconsistent reporting in the review or literature. Compounds containing pooled subject groups (e.g., mild impairment and moderate impairment populations combined) were excluded from the analyses.
Figure 1.

Flowchart of the database construction and validation processes. AUC, area under the concentration-time curve; CL, clearance; CLiv, clearance after intravenous administration; CLr, renal clearance; DDI, drug–drug interaction; DIDB, Drug Interaction Database; F, absolute bioavailability; FDA, Food and Drug Administration; HI, hepatic impairment; OI, organ impairment; RI, renal impairment; UT, University of Tokyo; UW, University of Washington.
Drug characteristics (fraction metabolized [fm], fraction excreted unchanged into urine [fe], major enzymes or transporters) were extracted from the FDA website (Drugs@FDA), DIDB, Goodman and Gilman’s the Pharmacological Basis of Therapeutics,5 or separate PubMed sources (using keywords such as “pharmacokinetics,” “metabolism,” and “fraction metabolized” and “fraction excreted”); fe was defined as the fraction of the administered dose excreted unchanged into urine after an i.v. and/or oral dose (if available, fe values from both i.v. and oral were curated in the database). Extracted drug characteristic data were validated by a second reviewer to ensure data integrity. Definitions on the contributions of renal and nonrenal elimination pathways are described in the Supplementary Protocol.
Following the construction of the list of compounds with available OI data (Supplementary Table S1), the FDA labeling, reviews, and the DIDB were queried to obtain the drug interaction data and maximal AUC or CL changes and inhibitors used. The product labeling and NDA reviews from 2007–2013 (Drugs@FDA) were also systematically reviewed to obtain data from recently approved drugs that were not yet reported in the literature. Percent change in AUC was converted to the AUC ratio for compounds with available AUC change data; for compounds with only percent change in CL, these values were converted to the AUC ratio (AUCR) using the equations:
When AUC was not available, CL ratios were used:
The concordance between the results from the HI and pharmacologic inhibition studies was analyzed by categorizing drugs by the CYP fraction metabolized (fm,CYP), which is defined as the fraction of systemic clearance of the substrate mediated by the CYP enzyme. For CYP3A4, fm,CYP3A4 was back-calculated from the result of in vivo human ketoconazole DDI studies in which ketoconazole (a strong CYP3A inhibitor) was administered in a manner consistent with maximal inhibition (200–400 mg daily for more than 2 days) using the equation:
In the event of multiple available studies, the maximum change in AUC observed for each drug in a drug–drug inhibition study was entered into the master dataset. In cases when multiple routes of administration were available, the data from the same route of administration in the HI study were selected (Supplementary Table S2).
To ensure data integrity, all data in the OI-DDI dataset were curated by several evaluators. Data were subsequently validated by a second expert reviewer on the OI-DDI dataset team. Prior to publication, the entire dataset was again validated by verification of all values by expert reviewers from different study sites (University of Washington (UW) and FDA). Final validation of the database demonstrated that <7% of the drugs required modification (new data available since initial database construction or error correction) to one or more values in data cells ((HI: 6% (12 modified among 207 drugs), RI: 7% (14 modified among 206 drugs), DDI: 6% (15 modified among 271 drugs)). A total of 271 compounds were found to have dedicated pharmacokinetic (PK) studies in the HI population (n = 207) and/or RI population (n = 206). For HI studies, ∼24% (50/207) used a full study design (including mild, moderate, and severe impairment groups). For RI studies, 16% (33/206) used a full study design (including mild, moderate, severe impairment, and ESRD groups), and 41% (84/206) included patients with ESRD. The master database, including drugs that were initially listed and not included in the example applications, is publicly available at the DIDB, the FDA Drug Interaction website (http://www.fda.gov/Drugs/DevelopmentApprovalProcess/DevelopmentResources/DrugInteractionsLabeling/ucm080499.htm) and the Clinical Pharmacology and Therapeutics website. Examples of possible applications of the database are presented below.
UTILITY
As an example of the utility of this database, the OI-DDI database was used to assess whether data from DDI may help to estimate changes due to OI (either HI or RI) or vice versa by taking into consideration the major metabolism/transport pathways and comparing exposure changes between 1) hepatic impairment and pharmacologic inhibition studies, 2) renal impairment and pharmacologic inhibition studies. The underlying consideration is that if we have a good understanding of in vivo contribution of elimination pathways based on clinical DDIs, the information can be useful in quantitatively assessing the risk of pharmacokinetic change with OI, since the expected change with OI in each pathway is theoretically equal to or less than the pharmacologic strong inhibition of the particular pathway.
Case study 1: The AUC change comparison between hepatic impairment studies and the pharmacologic inhibition of CYP3A4 (Figure 2)
Fifty-seven drugs in the dataset were substrates of CYP3A4 and had AUCR data available from a hepatic impairment study as well as from a DDI study with the strong pharmacologic inhibitor ketoconazole, allowing estimation of the fraction metabolized by CYP3A4 (Supplementary Table S2). For most CYP3A4 substrates (76%), the percent change in the AUC values obtained in subjects with severe HI was less than those in subjects taking ketoconazole (Figure 2). The DDI effect was greatest for drugs with a fm,CYP3A4 >50%, wherein the AUCR of HI subjects was <30% of the AUCR observed in pharmacologically inhibited (ketoconazole) healthy subjects (Figure 2). The exception to this trend was cinacalcet, a calcimimetic that is used to treat secondary hyperparathyroidism in patients with kidney disease receiving dialysis. The impact of HI on the multiple enzymes involved and involvement of alternate clearance pathways for cinacalcet (fm,CYP3A4 = 0.51) may contribute to the observed unusual DDI-OI profile, where severe HI caused larger AUCR than DDI with ketoconazole (4.25 vs. 2.03).
Figure 2.
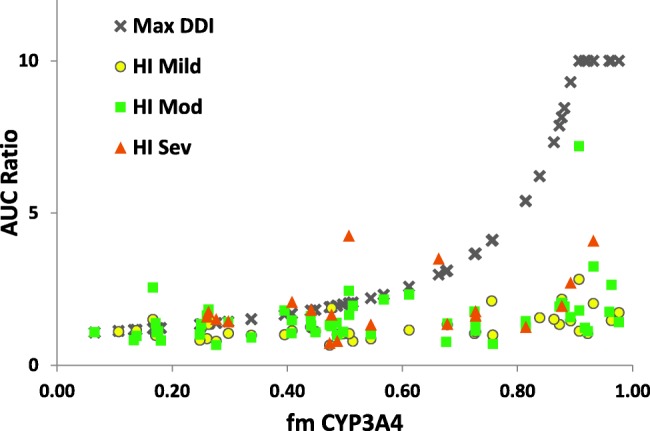
Relationship between fm,CYP3A4 and observed AUC ratios with various degrees of HI or maximum AUC ratios with DDIs. Each symbol represents AUC ratios for one compound in corresponding conditions. "Max DDI" represents the maximum AUC ratio in all the DDIs reported for each compound. All AUC ratio values >10 are shown on the graph as AUC = 10. AUC, area under the concentration–time curve; fm,CYP3A4, fraction of dose metabolized by CYP3A4; HI, hepatic impairment.
This analysis has several limitations—first, not all drugs had complete datasets—notably, only 19 of 57 drugs had data from severely impaired subjects, which limited the ability to draw firm conclusions for this population. For this analysis, we used the Child-Pugh scoring system to group the HI subjects, but recognize that this system may not be the ideal method to classify HI subjects. However, this categorization is standardized and approved by global regulatory agencies and is widely available in published HI studies. The utility of the Child-Pugh scoring system to predict hepatic drug metabolism has been previously discussed by Johnson et al.,6 who showed no significant difference between observed and predicted (using Child-Pugh scores) clearance ratios, with the exception of two studies of i.v. omeprazole. Although there is certainly some decreased expression/activity of CYPs in cirrhosis, the mechanism of decreased hepatic drug metabolic clearance in vivo is more complex and may have differing effects on different enzymes. In addition, the greater changes in exposure observed in our dataset after inhibition by ketoconazole as compared to an HI study may in some instances be partly explained by additional inhibition of intestinal CYP3A in the DDI studies. Due to insufficient data, one cannot generalize from this example (drugs with a high fm,CYP3A4 evaluated with strong CYP3A4 inhibitors) to other CYP isoforms (e.g., CYP2D6, which only had eight compounds available for analysis).
These analyses showed that, for drugs that are highly cleared via CYP3A4 (i.e., fm > 50%), a well-conducted pharmacologic inhibition study with a strong and specific inhibitor for CYP3A like ketoconazole may reflect the "worst-case scenario" for changes in AUC when enzyme activities are impaired by either DDI or OI. When data are not available from HI patients, the DDI results may help to project the AUC changes that would be observed in patients with severe HI. Additional confirmatory studies will be required to validate this finding. Note that in 2013 the FDA limited the use of ketoconazole due to potentially fatal liver injury and risk of drug interactions and adrenal gland problems.7 Recently, Ke et al.8 suggested itraconazole and clarithromycin to be acceptable alternatives to ketoconazole as strong CYP3A inhibitors in drug interaction studies.
Case study 2: The AUC change comparison between renal impairment and pharmacologic inhibition studies (Figure 3)
Forty-seven compounds were identified to have fe,iv or fe,po/F values ≥30% with at least one DDI report (Figure 3, Supplementary Table S3). For this group of compounds, the mean AUC ratio for DDI was 2.2. The effect of RI on exposure correlates with the severity of the impairment, i.e., the more severe the dysfunction, the greater the effect on drug exposure (Figure 3). The mean AUC ratios for the mild, moderate, severe, and ESRD groups were: 1.5, 2.3, 3.5, and 4.8, respectively. Severity of the effect on AUC also correlates with fe,iv or fe,po/F values; the mean of maximum AUC ratio in RI subjects for 31 compounds with fe,iv or fe,po/F ≥50% was 5.0, while that of 16 compounds with fe,iv or fe,po/F values between 30 and 50% was 2.3. When comparing observed AUC ratios for RI patients with theoretical maximum AUC ratios by assuming complete block of renal elimination (represented as dotted line in Figure 3), most of the observed data showed lower or similar values to the theoretical maximum. Accurate estimation of the contribution of renal clearance is of great importance for predicting the effect of renal impairment. It is also important to distinguish RI effects among drugs that undergo passive filtration, active reabsorption, and active tubular secretion. Although we did not examine this issue, Sayama et al.9 recently demonstrated that the changes in CLR were similar among these three types of renally eliminated drugs.
Figure 3.
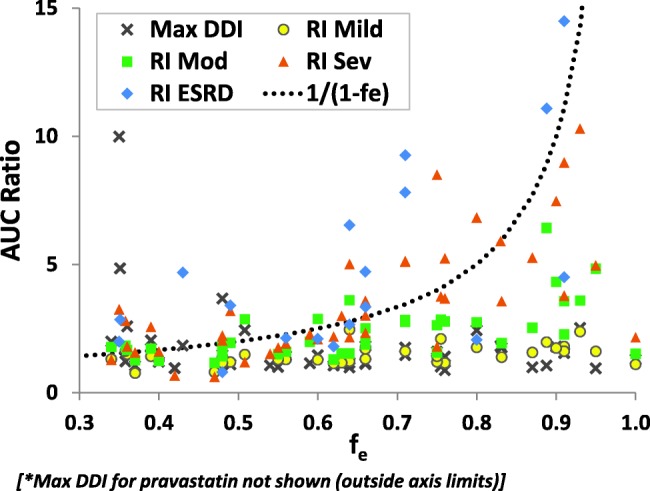
Relationships between fe (fe,iv or fe,po/F) and observed AUCR with various degrees of RI or maximum AUC ratios with DDIs for compounds with fe ≥ 0.3. Each symbol represents AUCR for one compound in corresponding conditions. "Max DDI" represents the maximum AUCR in all the DDIs reported for each compound. The dotted lines represent theoretical maximum AUC ratios by assuming complete block of renal elimination, calculated by 1/(1-fe). AUC, area under the concentration–time curve; F, absolute bioavailability; fe, fraction of dose excreted unchanged into urine; RI, renal impairment.
When comparing AUC ratios with DDI data, the maximum AUC ratio for RI was greater than that for DDI in all but seven drugs (Supplementary Table S3). The mechanisms of DDI for these seven drugs can be attributed to the inhibition of hepatic or intestinal metabolic enzymes or transporters. On the other hand, for 11 compounds with coadministration of well-known inhibitors of renal transporters (probenecid, cimetidine, and gemfibrozil), the mean maximum AUC ratio of renally impaired/healthy control subjects was 454% greater than the AUC ratio from DDI studies. There are two major factors that might contribute to greater AUC increase for RI than for DDI with pharmacological inhibitors of renal transporters for drugs with fe ≥30%. The first factor to consider is that RI can affect not only active secretion, but also the glomerular filtration of drugs, as both processes appeared to be affected similarly in RI.9 The second factor is the lack of potent and specific inhibitors for renal transporters, and therefore the effect of complete transporter inhibition cannot be ascertained with pharmacologic inhibition studies. Feng et al.10 demonstrated that clinical inhibitors of renal transporters do not completely inhibit tubular secretion of their substrates; the clinical effect of renal transporter inhibitors can be approximately predicted, assuming 75% inhibition for active transport of organic anions by probenecid or 50% inhibition for active transport of organic cations by cimetidine. It is possible that RI results in greater effect on active section of the transporter substrates than pharmacologic transporter inhibitors.
Our analyses show that, for drugs that are mainly renally eliminated (fe ≥30%), accurate estimation of the contribution of renal clearance from mass-balance studies may still be the most reliable indicator of the effect of the changes in AUC with RI, while pharmacologic inhibition studies with currently available transporter inhibitors do not reflect the worst-case scenario. Further research is needed to provide a mechanistic explanation of greater AUC ratios observed in RI studies than in DDI studies.
In summary, the analyses presented in this article provide examples of the utility of a complex dataset that incorporates both OI and DDI data and were not intended as comprehensive meta-analyses. The public availability of this database will facilitate additional analyses, including the examination of elimination-pathway dependencies in HI. Although we only performed analyses with CYP3A4, additional studies using this database could relate the magnitude of effects of different degrees of HI and DDI data to highlight differences in the effect of HI for other CYP enzymes. This database can also be used to examine the effects of RI on drugs that are nonrenally cleared.
DISCUSSION
Here an OI and DDI database is presented. It utilizes data available from published articles curated into the University of Washington Metabolism and Transport Drug Interaction Database (DIDB, www.druginteractioninfo.org) and publicly available NDA reviews and labeling from the FDA (http://www.accessdata.fda.gov/scripts/cder/drugsatfda/) (new molecular entity NDAs approved 2007–2013). This database includes 271 drugs with corresponding OI data as well as the maximum observed change in AUC or CL from a pharmacologic inhibition study (when available). This database will be useful to investigators seeking a comprehensive resource for renal and hepatic impairment pharmacokinetic study data, as well as those seeking to correlate RI, HI, and DDI data. The database can be found at the DIDB website, the FDA Drug Interaction website, and the Clinical Pharmacology and Therapeutics website (.xls format).
The OI-DDI database has several limitations. The primary limitation is the availability of the published data. Many published NDA reviews or labeling lack sufficient granularity regarding nonspecific classification of impairment groups and presenting only pooled mean changes in Cmax, AUC, or CL for all impaired study subjects without presenting data stratified by degree of impairment. We encourage subsequent publication of these OI studies after the public release of the NDA, similar to the case of DDI studies conducted as part of an NDA, that are frequently submitted for peer-reviewed publication. Also, publication/presentation bias may influence the quantity and quality of available data: studies with no change in exposure may not be presented in as much depth as a study with a significant change in exposure.
The creation of a detailed protocol that was used by all investigators that identified potential sources of data and classification criteria was critical for the successful development of this database. In this document, we specified working definitions of ADME criteria (e.g., defining fe as fraction excreted unchanged from either oral or i.v. administration, but listing the data separately by route of administration in the master data file) and described the procedure for including data that did not meet established definitions. However, inclusion of these data were still useful (e.g., inclusion of total radioactivity excreted when data regarding excretion of unchanged parent compound was not available).
This database is the first dataset composed of compiled quantitative data from available DDI and OI studies. This database has been used successfully to analyze the relationship between RI or HI studies and pharmacologic inhibition studies. We believe that the public availability of this database will encourage additional analysis and assist in the development of regulatory recommendations for the evaluation of drug exposure in specific populations by leveraging information from DDI studies.
Acknowledgments
The article reflects the views of the authors and should not be construed to represent the FDA’s views or policies. The authors thank Drs Darrell Abernethy and Issam Zineh for critical review of the article. This research was supported by the US Food and Drug Administration’s (FDA’s) Medical Countermeasures initiative. K.Y., L.L. and P.C. were supported in part by an appointment to the Research Participation Program at the Center for Drug Evaluation and Research, administered by the Oak Ridge Institute for Science and Education through an interagency agreement between the US Department of Energy and the FDA.
Conflict of Interest
The authors have no conflicts of interest to report.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
References
- Guidance for Industry: Pharmacokinetics in Patients with Impaired Hepatic Function: Study Design, Data Analysis, and Impact on Dosing and Labeling, U.S. Food and Drug Administration. < http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM072123.pdf > (2003). Accessed January 21, 2015.
- Draft Guidance for Industry Pharmacokinetics in Patients with Impaired Renal Function — Study Design, Data Analysis, and Impact on Dosing and Labeling, U.S. Food and Drug Adminstration. < http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM204959.pdf > (2010). Accessed January 21, 2015.
- Guideline on the evaluation of the pharmacokinetics of medicinal products in patients with decreased renal function, European Medicines Agency. < http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2014/02/WC500162133.pdf > (2014). Accessed January 21, 2015.
- Guideline on the Evaluation of the Pharmacokinetics of Medicinal Products in Patients with Impaired Hepatic Function, European Medicine Agency. < http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003122.pdf > (2005). Accessed January 21, 2015.
- Goodman and Gilman’s the Pharmacological Basis of Therapeutics. 11th ed. New York, NY: McGraw-Hill; 2006. [Google Scholar]
- Johnson TN, et al. A semi-mechanistic model to predict the effects of liver cirrhosis on drug clearance. Clin. Pharmacokinet. 2010;49:189–206. doi: 10.2165/11318160-000000000-00000. < http://www.fda.gov/Drugs/DrugSafety/ucm362415.htm > ( ). Accessed 21 January 2015. [DOI] [PubMed] [Google Scholar]
- FDA Drug Safety Communication: FDA limits usage of Nizoral (ketoconazole) oral tablets due to potentially fatal liver injury and risk of drug interactions and adrenal gland problems. < http://www.fda.gov/Drugs/DrugSafety/ucm362415.htm >. Accessed January 21, 2015.
- Ke AB, Zamek-Gliszczynski MJ, Higgins JW. Hall, Itraconzaole SD, clarithromycin as ketoconazole alternatives for clinical CYP3A inhibition studies. Clin. Pharmacol. Ther. 2014;95:473–476. doi: 10.1038/clpt.2014.41. [DOI] [PubMed] [Google Scholar]
- Sayama H, et al. Application of a physiologically based pharmacokinetic model informed by a top-down approach for the prediction of pharmacokinetics in chronic kidney disease patients. AAPS J. 2014;16:1018–1028. doi: 10.1208/s12248-014-9626-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng B, et al. Quantitative prediction of renal transporter-mediated clinical drug-drug interactions. Mol. Pharm. 2013;10:4207–4215. doi: 10.1021/mp400295c. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information