

Abstract
There is a current and increasing demand for simple, robust, nonradioactive assays of protein tyrosine kinase activity with applications for clinical diagnosis and high-throughput screening of potential molecularly targeted therapeutic agents. One significant challenge is to detect and measure the activity of specific kinases with key roles in cell signaling as an approach to distinguish normal cells from cancer cells and as a means of evaluating targeted drug efficacy and resistance in cancer cells. Here, we describe a method in which kinase substrates fused to glutathione-S-transferase and immobilized on glutathione agarose beads are phosphorylated, eluted, and then assayed to detect kinase activity. The activity of recombinant, purified c-Abl kinase or Bcr-Abl kinase in whole cell extracts can be detected with equivalent specificity, sensitivity, and reproducibility. Similarly, inhibition of recombinant c-Abl or Bcr-Abl in cells or cell extracts by imatinib mesylate and other Bcr-Abl targeted kinase inhibitors is readily assayed. This simple kinase assay is sufficiently straightforward and robust for use in clinical laboratories and is potentially adaptable to high-throughput assay formats.
Keywords: Bcr-Abl, Protein tyrosine kinase, Kinase assay, Imatinib mesylate
Expression of the oncogenic protein tyrosine kinase (PTK)1 Bcr-Abl is commonly observed in chronic myelogenous leukemia (CML), acute lymphocytic leukemia (ALL), and other hematopoietic stem cell disorders [1]. Reciprocal translocation of the long arms of chromosomes 9 and 22 to form the Philadelphia chromosome replaces the N terminus of c-Abl with Bcr, resulting in constitutive Abl kinase activation [2,3]. Under normal conditions, c-Abl is tightly regulated, serving roles in diverse pathways that promote cell growth and survival. Deregulation of Abl kinase activity via fusion to Bcr induces cellular phenotypes associated with leukemia such as hyperproliferation and resistance to apoptosis [4].
Imatinib mesylate (IM, Gleevec, STI-571) is currently the frontline therapy for CML. IM has been shown to specifically inhibit Bcr-Abl activity in vivo, decreasing proliferation, and restoring apoptosis in CML cells [5]. In the clinic, IM induces hematological remission in nearly all chronic-phase CML patients treated [6–8]. Nonetheless, a subset of patients either fail to respond or rapidly develop drug resistance so that circulating CML cells persist despite treatment. Resistance is associated with amplification of the Bcr-Abl gene or, more commonly, point mutations in the Abl kinase catalytic domain that antagonize IM binding [9,10]. This limitation is being addressed by novel agents that can overcome most IM resistance mutations [5,11,12]. Regardless, no clinically useful assays for CML cell sensitivity to IM or other inhibitors that can guide clinical decision making are available. An in vitro or in vivo assay for Bcr-Abl kinase activity and inhibition in CML cells would directly address this challenge.
The large number of active kinases in most cells is often considered to obviate assays from unfractionated cell extract. Indeed, conventional assays typically require kinases to be immunopurified from cell extract or affinity-purified from recombinant expression systems and then combined with a peptide or recombinant protein substrate. Classically, kinase activity has been assayed via incorporation of radionuclides from ATP-γ-32P or ATP-γ-35S and then detected by filter binding or electrophoresis and autoradiography [13–15]. Recently, detection with phosphospecific antibodies has provided a viable alternative to radioactive nucleotides. Adapted for homogeneous scintillation proximity or fluorescence polarization detection, these assays have found application to high-throughput screening for small molecule inhibitors [16,17]. Nevertheless, assays based on purified kinases are poorly suited to tracking endogenous kinase activity in living cells such as in assessing the efficacy of small molecule inhibitors. Once purified, kinases may be released from inhibitors, lose regulatory phosphorylations, or be separated from regulatory partners, leading to altered activity and/or enzyme stability.
As an alternative, cell-based assays examine kinase activity by monitoring phosphorylation of endogenous kinase substrates, commonly detected by Western analysis or flow cytometry using phosphospecific antibodies [18–20]. The increasing range and quality of commercially available phosphospecific antibodies has popularized these methods. However, the drawbacks of such assays include the difficulty of assigning particular phosphorylations in vivo to a specific kinase and the uncertain correspondence between steady-state levels of protein phosphorylation and kinase activity, preventing useful quantitation. Few kinase assays reliably report a specific activity in unfractionated cell lysates. One important innovation has been to combine biotinylated peptide substrates with whole cell extracts and then to recover the peptides for assay [21]. As an alternative, Shults and coworkers [22] described a fluorescent chemosensor peptide for detecting protein kinase from crude cell extract in a homogeneous assay. However, these approaches have not been adapted to employ protein substrates, which are likely to provide better reporters of endogenous activity.
To address each of these challenges, we have developed a novel kinase assay that carries the advantages of in vitro assays with respect to direct measurement of kinase activity and of cell-based assays that do not require kinase purification. Our assay uses surface-immobilized protein substrates that are phosphorylated by active Bcr-Abl kinase present in a cell extract. Protein phosphorylation is determined by gelelectrophoresis and Western blotting with a commercially available antiphosphotyrosine antibody, generating semiquantitative kinase activity data with very high signal to noise. As an alternative, analysis of phosphorylation by proteolysis of the substrate and analysis by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI–TOF–MS) offers reproducible results and is similarly amenable to quantitation as is Western blotting, offering a potential advantage for use in high-throughput screening.
Materials and methods
Cloning
The optimal Abl peptide substrate EAIYAAPFAKKK [23], identical to Abltide (Upstate, Charlottesville, VA, USA), was cloned into BamHI–EcoRI digested pGEX2T vector using primers 5′-GATCCGCGGCGGAAGCGAT TTATGCGGCGCCGTTTGCGAAAAAAAAATAATA AG-3′ and 5′-AATTCTTATTATTTTTTTTTCGCAAAC GGCGCCGCATAAATCGCTTCCGCCGCG-3′. Abltide was also cloned into SalI–NotI digested pGEX2T vector with primers 5′-TCGACTGCGGCGGAAGCGAT TTATGCGGCGCCGTTTGCGAAAAAAAAATAATA AG-3′ and 5′-GCCTTATTATTTTTTTTTCGCAAA CGGCGCCGCATAAATCGCTTCCGCCGCAG-3′, creating a new plasmid, pGEX 4T-2-AP, in which various oligonucleotides could be inserted upstream of the Abltide sequence. The Abl SH3 ligand sequence was obtained by annealing primers 5′-GATCCGCGCCGACCTATAGC CCGCCGCCGCCGCCGGCGGCGGCGGCGCG-3′ and 5′ -AATTCGCGCCGCCGCCGCCGGCGGCGGC GGCGGGCTATAGGTCGGCGCG-3′ and inserted upstream of the Abltide sequence in BamHI-EcoRI digested pGEX 4T-2-AP. The CrkL SH3n domain was amplified with primers 5′-GCGTACGGATCCGCCGC CCTGGAATATGTACGGACTCTG-3′ and 5′-GCGTA CGAATTCGCTTTTCGACATAAGGGACAGG-3′, and inserted upstream of the Abltide sequence in BamHI-EcoRI digested pGEX 4T-2-AP.
Glutathione- S-transferase (GST)-CrkL constructs, including GST-CrkL full-length, GST-CrkL SH3-SH3, and GST-CrkL linker, were kindly provided by B. Druker (Oregon Health and Science University). GST-CrkL SH3n plus linker region was a gift from J. Groffen (University of Southern California).
GST fusion protein purification
GST fusion constructs were expressed in Escherichia coli strain BL21. Overnight cultures were diluted 10 times in Luria-Bertani (LB) media with 50 μg/ml ampicillin and were induced with 0.1 mM isopropylthio-β-d-galactosidase (IPTG) for 3h. Bacterial lysates were prepared in phosphate-buffered saline (PBS, 11.9mM phosphate, 137mM sodium chloride, 2.7 mM potassium chloride, pH 7.4) containing 0.5 mM dithiothreitol (DTT), 1 mM phenylmethylsulfonyl fluoride (PMSF), 1 mM orthovanadate, and 25× complete protease inhibitor (Roche Diagnostics, Mannheim, Germany). Lysates were sonicated for 20 s on ice and gently mixed with 10% Triton X-100 for 30min. The supernatant was collected after centrifugation for 10min at 14,000g. To purify the GST fusion proteins, bacterial lysates were incubated with SwellGel immobilized glutathione discs (Pierce, Rockford, IL, USA) for 3h at 4°C. GST fusions were eluted with 10mM reduced glutathione in 50 mM Tris-HCl (pH 8.0) for 20 min. Glutathione was removed from the purified protein by dialysis against ice-cold PBS. Purified protein concentrations were measured by Bradford analysis using Cytoskeleton protein assay reagent (Cytoskeleton, Denver, CO, USA).
Cells, cell culture, and drug treatment
The Ba/F3 cell line was obtained from N. Shah (University of California, Los Angeles). Ba/F3 cells were grown in suspension at 37 °C and 5% CO2 in RPMI 1640 medium containing 10% heat-inactivated fetal bovine serum, 1% penicillin/streptomycin, 0.3 mg/ml l-glutamine, and 5 ng/ml murine recombinant IL-3 (BD Biosciences, San Diego, CA, USA). K562 cells were cultured as above without IL-3 added to the medium. IM was provided by Novartis Pharmaceutical (East Hanover, NJ, USA). The compound was dissolved in dimethyl sulfoxide (DMSO) and then further diluted with H2O. Approximately 5 million K562 cells were plated in 10-cm culture dishes with fresh media 24h prior to treatment. Duplicate plates were treated for 2h with 0, 1, 10, or 100μM IM. Immediately following treatment, cells were washed with ice-cold PBS and prepared for protein isolation.
Cell extract preparation
Whole cell lysates were prepared in buffer containing 50 mM Hepes (pH 7.3), 150mM NaCl, 1.5 mM MgCl2, 1 mM ethylenediamine tetraacetic acid (EDTA, pH 7.4), 100mM sodium fluoride, 10mM sodium pyrophosphate, 200μM sodium orthovanadate, 1% Triton X-100, 10% glycerol, 2.5mM PMSF, and 1000× protease inhibitor cocktail (10.4mM AEBSF, 8μM aprotinin, 200μM leupeptin, 400μM bestatin, 150μM pepstatin A, and 140μM E-64, Sigma, St. Louis, MO, USA). Cell lysates were incubated on ice for 20 min, vortexed briefly, and centrifuged at 4°C for 10 min at 15,000g. The supernatant was collected, and protein yields were determined by Bradford analysis using Bio-Rad protein assay reagent (Bio-Rad Laboratories, Hercules, CA, USA).
Solution-phase kinase assay with recombinant c-Abl and Bcr-Abl
In vitro kinase assays were performed in 80-μl reaction volumes of kinase buffer (50 mM Tris-HCl [pH 7.5], 10mM MgCl2, 1 mM DTT), 10μ,M ATP, 1nmol of GST fusion substrates, and 1 U c-Abl (Upstate Cell Signaling Solutions, Lake Placid, NY, USA) or 50μg K562 cell extract. Reaction mixtures were incubated for 1 h at 30°C (for c-Abl) or at 37°C (for cell extract).
Solid-phase kinase assays with recombinant v-Abl, c-Abl, and Bcr-Abl
SwellGel discs (Pierce) were suspended in cold 50mM Tris-HCl (pH 7.5) so that 1 μl of bead suspension bound 1 μg of GST fusion protein. GST fusion protein (1 nmol) was incubated with the glutathione bead suspension for 1 h at 4 °C with constant mixing. The protein-bound beads were washed twice with ice-cold 50 mM Tris-HCl (pH 7.5) containing 10mM MgCl2. For the solid-phase kinase assays, substrate-bound beads were incubated with either recombinant v-Abl, c-Abl, or 50μg K562 cell extract, 10μM ATP, and kinase buffer in 80μl reactions for 1 h at 30°C (for Abl and c-Abl) or at 37°C (for cell extract). To observe inhibition of c-Abl or Bcr-Abl, solid-phase kinase assays were performed as above in the presence of the indicated inhibitors. PD 1666326 and PD 173955 were a kind gift from B. Clark-son (Sloan-Kettering Institute for Cancer Research, New York, NY, USA). The inhibitors IM (Novartis), PD 1666326, PD 173955, AG 957 (Calbiochem, San Diego, CA, USA), and Genestein (Calbiochem) were dissolved in DMSO. Following the reaction, the beads were washed twice with ice-cold 50 mM Tris-HCl (pH 7.5). GST fusion proteins were eluted with 10mM reduced glutathione in 50 mM Tris-HCl (pH 8.0) for 10 min. Concentrations of the eluted protein were measured by Bradford assay.
Western blotting
Kinase assay samples were separated on 12% SDS-PAGE gels and transferred to nitrocellulose membranes according to standard procedures. Uniform sample loading and transfer were confirmed using the Memcode reversible protein stain kit (Pierce). Membranes were blocked in 10% bovine serum albumin (BSA) for 1 h at 25 °C and then probed with 4G10 antiphosphotyrosine primary antibody (Upstate Cell Signaling Solutions) at 1:1000 in 5% BSA at 25 °C for 1 h and horseradish peroxidase-conjugated anti-mouse IgG secondary antibody (Amersham, Piscataway, NJ, USA) at 1:5000 in 5% BSA for 30 min. Blots were developed using Supersignal WestPico chemiluminescent substrate (Pierce) and were exposed to autoradiography film. Memcode-stained blots and developed films were scanned with a Microtek Scan-Maker 6800 at 600 ppi resolution. The integrated density of protein bands was determined with ImageJ software from the National Institutes of Health (http://rsb.info.nih.gov/ij).
Trypsin digestion and MALDI–TOF–MS analysis of phosphorylated proteins
Protein samples for MALDI–TOF–MS analysis were incubated with 1 mM DTT in 50 mM NH4CO3 (pH 8.9) for 10min at 22°C followed by 0.1% (v/v) Rapigest detergent (Waters, Milford, MA, USA) for 45min at 37°C. The samples were digested with sequencing-grade modified trypsin (Promega, Madison, WI, USA) for 90min at 37 °C and concentrated by vacuum centrifugation. Peptide fragments were reconstituted in 50 mM NH4CO3 buffer (pH 8.9) and purified by C18 Zip-Tip (Millipore, Billerica, MA, USA). Peptides were eluted in a saturated solution of α-cyano-4-hydroxycinnamic acid in acetonitrile:H2O: NH4OH (75:25:0.1), spotted in triplicate on a 196-well stainless-steel target (Applied Biosystems, Foster City, CA, USA), and analyzed in linear positive ion mode with an ABI 4700 MALDI [24]. Spectra shown represent the averaging of spectra from 3000 laser shots obtained by random uniform analysis of the entire sample spot surface to ensure that the results were representative of the actual sample composition. The spectra of tryptic digest fragments from phosphorylated and unphosphorylated samples were compared to identify phosphopeptides. For all quantification, the peaks representing the unphosphorylated and phosphorylated peptide were integrated and percentage phosphorylated was determined from the ratio of the peak area of phosphorylated peptide to the sum of peak areas of phosphorylated and unphosphorylated peptide. The Wnal percentage phosphorylation was determined by averaging this ratio for values derived from the three spots on the MALDI target plate.
Results
Solid-phase kinase assays with cell extract
The focus of these studies was to develop a simple and robust methodology for assay of a specific kinase compatible with using whole cell extracts as a source of enzyme activity. Cell extracts are desirable because they include components that promote enzyme stability and maintain proper kinase regulation and specificity that may be lost during kinase purification. On the other hand, extracts contain an undefined population of active kinases, phosphatases, and proteases as well as abundant phosphorylated proteins and unknown levels of endogenous substrates of the kinase to be assayed. These and other factors may negatively influence detection of a specific kinase activity via assays of substrate phosphorylation.
One proven approach to enhancing signal to noise in kinase assays is to employ substrate immobilization or capture, allowing soluble reaction components to be washed away prior to detection of phosphorylation [21,25]. We sought to exploit this approach by using the Schistosoma japonicum GST as a reversible tag for immobilization on glutathione (GT) agarose beads.
As an initial model system, we used a GST fusion to full-length CrkL, an SH2-SH3-SH3 adaptor protein that has been shown to be a highly phosphorylated in vivo substrate of Bcr-Abl [26]. GST-CrkL was expressed in E. coli, purified by GT-agarose chromatography, dialyzed, and concentrated. The protein was judged to be greater than 95% pure and full-length (∼60kDa) based on SDS-PAGE analysis. Aliquots of GST-CrkL were incubated in kinase buffer with ATP at 37 °C for 1 h in the presence of 5–400 μg of extract from Bcr-Abl expressing K562 erythroleukemia cells. The reactions were subjected to SDS–PAGE, transferred to nitrocellulose, and stained with Memcode, revealing the protein distribution expected for cell lysates. Western analysis of the reactions using 4G10 antiphosphotyrosine antibody and anti-mouse IgG horseradish peroxidase conjugate revealed increasing levels of phosphotyrosine immunoreactivity with increasing cell extract, but it was not possible to reliably distinguish the GST-CrkL substrate from background bands due to phosphoprotein components of the cell extract (Fig. 1 A, upper panel). As an alternative strategy, the GST-CrkL was rebound to GT-agarose before incubation with K562 cell extract under the same reaction conditions. Here the beads were washed free of soluble proteins before elution with 10mM GT, analysis by SDS–PAGE, and transfer to nitrocellulose. In contrast to the solution-phase assay, Memcode staining of the membrane showed the expected 60-kDa band corresponding to GST-CrkL and only low levels of coeluting proteins (Fig. 1A, lower panel). However, Western analysis with 4G10 antibody revealed several distinct bands greater than 40 kDa. The addition of GST-CrkL, extract, and ATP was required to observe 4G10 immunoreactivity in this assay. We inferred that the increase in immunoreactivity at 60 kDa with added extract represented tyrosine phosphorylation of GST-CrkL. The other bands may have resulted from association of one or more highly tyrosine phosphorylated proteins from the cell extract with the beads and/or GST-CrkL that persisted through the washing procedure. Alternatively, the (faster mobility) background bands may have resulted from partial degradation of the GST-CrkL substrate. We observed only slightly improved specificity using a phosphospecific antibody recognizing the physiological Bcr-Abl phosphorylation site on CrkL (anti-phospho-CrkL Tyr 207 antibody, Cell Signaling Technologies, not shown). significantly, immunoreactivity of either the 4G10 or anti-phospho-CrkL Tyr 207 antibodies to the 60-kDa band was lost when 100 μM IM was added to the reactions (not shown), suggesting that Bcr-Abl mediated the GST-CrkL phosphorylation. These data establish a simple protocol to detect specific phosphorylation of a substrate by kinases present in a whole cell extract.
Fig. 1.
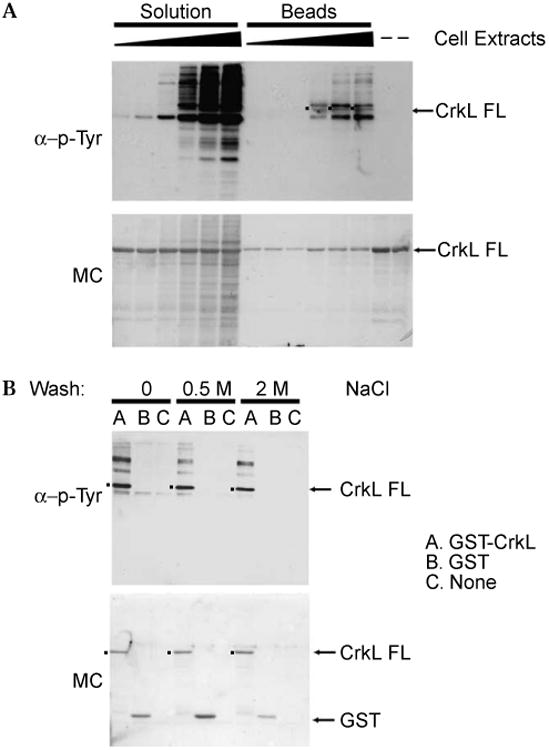
Bcr-Abl tyrosine kinase assays in solution and immobilized on beads. (A) GST-CrkL was incubated with 5, 10, 50, 100, 200, and 400 μg of K562 cell extracts in solution-phase and solid-phase kinase assays as described in the Materials and Methods. Specific quantities of the reaction mixture from solution reactions, the elution product from the solid-phase assays, and GST-CrkL alone were loaded onto a 12% SDS–PAGE gel. Following electrotransfer, the membrane was treated with Memcode reversible protein stain (MC, lower panel) to confirm equal loading and transfer, immunoblotted with 4G10 monoclonal antiphosphotyrosine antibody and horseradish peroxidase-linked secondary antibody, and then imaged with enhanced chemiluminescence (α-p-Tyr, upper panel). FL, full-length. (B) Comparison of Bcr-Abl tyrosine kinase assays with GST-CrkL or GST bound to glutathione beads or the beads alone suggests that background immunoreactivity results from phosphoproteins bound to the CrkL moiety. Following incubation with cell extract and washing as before, beads were washed with 0, 0.5, or 2 M NaCl in 50 mM Tris-HCl (pH 7.5), respectively, before elution and Western analysis. Lower panel: Memcode protein stain (MC). Upper panel: 4G10 antiphosphotyrosine antibody (α-p-Tyr). FL, full-length.
To further characterize the origin of the phosphorylated background bands that coeluted with GST-CrkL, the solid-phase kinase assays were performed with GST-CrkL, GST bound to beads, or beads alone. Bead eluates were subjected to Western analysis by antiphosphotyrosine antibody. The Western analysis (Fig. 1B, upper panel) demonstrated that multiple tyrosine phosphorylated proteins eluted along with GST-CrkL, whereas only a faint band around 55 kDa eluted from GST or beads only, indicating that much of the background derived from phosphorylated proteins in the cell extract might be associated with the immobilized CrkL. Note that the GST alone is not recognized by the 4G10 or secondary antibodies.
CrkL is reported to bind Bcr-Abl through its SH3 domain and to bind other signaling proteins, such as p120 Cas, p120 Cbl, and paxillin, through its SH2 domain [27,28]. Each of these is subject to tyrosine phosphorylation. To test for such noncovalent associations, we performed additional washes prior to elution using 0.5 or 2 M NaCl to disrupt nonspecific binding and protein–protein interactions. The NaCl wash removed the 55 kDa band from GST and beads and decreased the intensity of background bands coeluting with GST-CrkL.
Substrate optimization for Abl and Bcr-Abl detection
The above experiment used full-length CrkL, which contains multiple tyrosine residues and thus may be a specific or nonspecific substrate for several kinases. In turn, the sites on CrkL that determine Bcr-Abl phosphorylation might be distinct from those that participate in background binding. Taken together, these considerations suggested that a simpler substrate might be developed. To enhance specificity and sensitivity of the assay and to identify determinants of phosphorylation within CrkL, GST fusions to full-length and deletion constructs (Fig. 2A) were expressed, purified, and immobilized on GT-agarose as above. As a reference Abl substrate, we cloned and expressed the “optimal” peptide substrate EAIYAAPFAKKK (Abltide [23]) fused to the carboxyl terminus of GST. We hypothesized that the CrkL “linker” sequence between the two SH3 domains that encodes the in vivo phosphorylation site Tyr 207 [29] would be both necessary and sufficient for the phosphorylation observed in our GST-CrkL experiments and would have high specificity and sensitivity compared with the shorter Abltide substrate. Indeed, previously published radiometric kinase assays using recombinant Abl catalytic domain in vitro [29] showed that GST fusions to full-length CrkL, the SH3-SH3 domains, the SH3n domain plus linker region, and the linker alone all are efficiently and similarly phosphorylated.
Fig. 2.
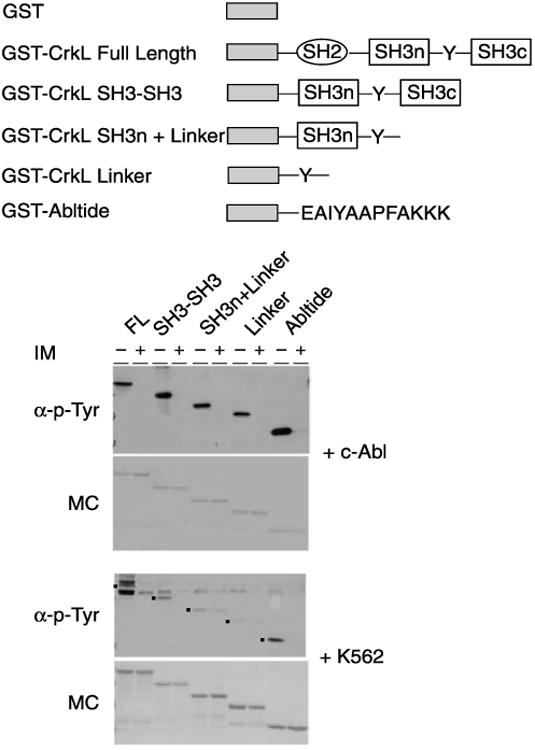
Performance of the bead-immobilized kinase assay for c-Abl and Bcr-Abl activity with different GST-substrate fusion constructs. (A) Schematic of GST kinase substrate fusion protein constructs. (B) Antiphosphotyrosine Western blot of GST fusion substrates incubated with c-Abl alone or inhibited with IM (α-p-Tyr) and Memcode stain of the same blot (MC) (upper) and antiphosphotyrosine Western blot of GST fusion substrates incubated with K562 cell extract alone or inhibited with IM (α-p-Tyr) and Memcode stain of the same blot (MC) (lower). FL, GST-CrkL full-length; SH3-SH3, GST-CrkL SH3-SH3; SH3n+Linker, GST-CrkL SH3n plus linker; Linker, GST-CrkL linker; Abltide, GST-Abltide.
Initially, we used a recombinant c-Abl kinase (Calbio-chem) lacking the amino-terminal myristoylated regulatory domain and carboxyl terminal domains of the native c-Abl-1A or c-Abl-1B but retaining the SH2 and SH3 domains and the Abl catalytic domain. Using this enzyme, we recapitulated the published data, demonstrating efficient phosphorylation of all GST fusion substrates containing the linker region, and of GST-Abltide, and observed effective inhibition by 100 μM IM (Fig. 2B). However, parallel assays performed with cell extracts yielded comparatively weak phosphorylation of the GST-CrkL SH3n linker and the GST-CrkL linker constructs relative to GST-CrkL full-length, GST-CrkL SH3-SH3, and the GST-Abltide control (Fig. 2B). Like c-Abl, the K562 extract could be efficiently inhibited by 100μM IM. We were particularly struck that GST-CrkL, GST-CrkL SH3-SH3, and GST-Abltide were comparably efficient substrates of Bcr-Abl in cell extract under these conditions. Furthermore, GST-Abltide showed reduced background of coeluted phosphoproteins. These considerations led us to focus on optimization of Abltide for further studies.
Developing a novel substrate for Bcr-Abl kinase assays in cell extract
Although previous work using solution-based assays of peptide phosphorylation [23] would suggest that Abltide has limited affinity for Abl, we reasoned that a modular approach based on adding domains that promote association with Abl kinase outside the substrate binding site should enhance sensitivity. In studies of Src family tyrosine kinases, linking a substrate peptide to ligands that bind the Src SH2 or SH3 domains increased the apparent affinity of the kinase for the substrate and enhances phosphorylation [30,31]. Based on this model, two novel GST fusions were designed as artificial substrates. For one substrate (APTYSPPPPP), a sequence encoding an artificial Abl SH3 ligand [32], and for the second substrate, the CrkL SH3n (SH3n) domain, were cloned in frame between the GST coding sequence and the Abltide sequence in the pGEX 4T-2-AP vector. This formed GST-Abl SH3L-Abltide and GST-SH3n-Abltide proteins, respectively (Fig. 3A). The Abl SH3 ligand has been previously shown to have high affinity for the Abl SH3 domain [32], whereas the CrkL SH3n domain interacts with a proline-rich region in Bcr-Abl [33].
Fig. 3.
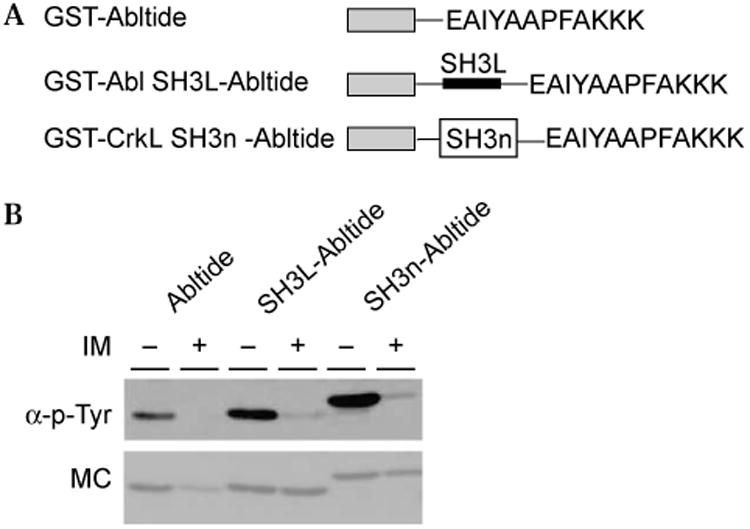
Comparison of the phosphorylation of GST-Abltide, GST-Abl SH3L-Abltide, and GST-CrkL SH3n-Abltide by Bcr-Abl. (A) The structure of GST-Abltide, GST-Abl SH3L-Abltide, bearing an artificial pep-tide ligand for the Abl SH3 domain, and GST-CrkL SH3n-Abltide, bearing the CrkL amino-terminal SH3 domain. (B) Antiphosphotyrosine Western blot of substrate phosphorylation (upper) compared with Mem-code stain (MC, lower) reveals enhanced phosphorylation with the addition of a protein–protein association domain.
Using the solid-phase assay and K562 cell extract, we compared the phosphorylation and inhibition by IM of GST-Abltide, GST-Abl SH3L-Abltide, and GST-SH3n-Abltide (Fig. 3B). The integrated density of each band in the Western blot was normalized to that of the corresponding band in the Memcode stain (Fig. 3B). Based on normalized densities of the Western bands, phosphorylation of GST-Abl SH3L-Abltide was approximately twice that of GST-Abltide, whereas the phosphorylation of GST-SH3n-Abltide was approximately three times that of GST-Abltide. Based on these and similar data, we selected GST-SH3n-Abltide for further analysis as a potential substrate for measuring Bcr-Abl activity in cell extract.
Background and nonspecific phosphorylation of the novel Bcr-Abl substrate
We have shown that GST-SH3n-Abltide is highly phosphorylated in cell extract in an IM-dependent manner. To rule out the possibility that this reflects significant nonspecific background phosphorylation of the substrate, immobilized GST-SH3n-Abltide was incubated with K562 and Ba/F3 cell extracts (Fig. 4). Ba/F3 cells do not express Bcr-Abl. Any substrate phosphorylation in this extract might be due to nonspecific activity of other tyrosine kinases and/or background phosphorylation by endogenous c-Abl and/or c-Arg. GST-SH3n-Abltide phosphorylation in Ba/F3 extract was approximately one-tenth that in K562 cell extract. However, this phosphorylation was inhibited by IM (Fig. 4), indicating that any background phosphorylation of GST-SH3n-Abltide, if it occurs, is likely to be due to Abl-related kinases.
Fig. 4.
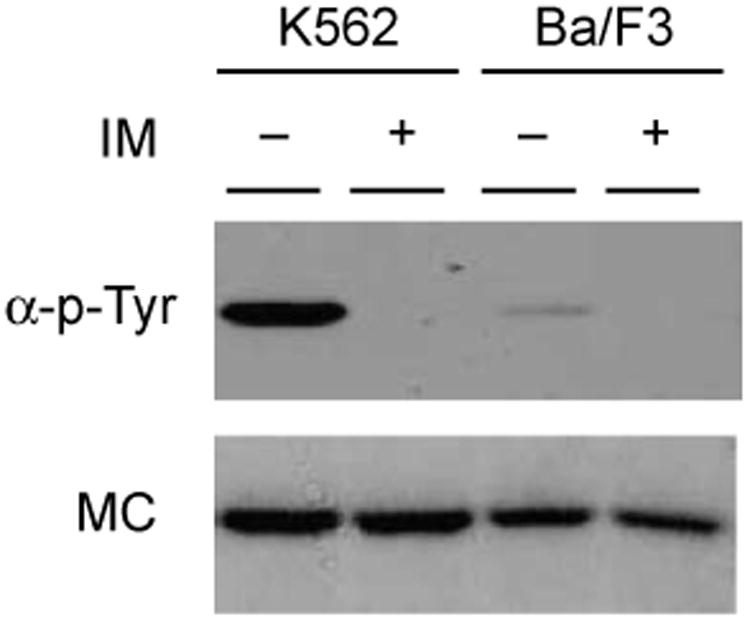
Low background of GST-SH3n-Abltide phosphorylation by Ba/F3 cell extract: Antiphosphotyrosine Western blot of phosphorylation of GST-SH3n-Abltide by K562 and Ba/F3 cell extracts alone or inhibited with IM (upper) and Memcode stain of the same blot (MC, lower). Note IM inhibition of “background” phosphorylation by Ba/F3 extract.
Quantification of kinase activity inhibition in extracts of cells treated with imatinib mesylate
To this point, our motivation was to develop a PTK assay that is both highly sensitive and specific for Bcr-Abl so as to efficiently determine kinase activity within cells. One potential application of this assay is to detect changes of kinase activity in cells that have been treated with PTK inhibitors as a means of predicting drug efficacy. Here extracts prepared from K562 cells treated with IM prior to cell lysis would be examined for decreased phosphorylation of bead-immobilized GST-SH3n-Abltide. Lysates were prepared from K562 cells treated with 1, 10, and 100μM IM for 2h in culture media. First, Western analysis was performed with anti-phospho-CrkL antibody and anti-eIF4E as loading control (Fig. 5A). The integrated density of phosphorylated CrkL bands was normalized to the eIF4E bands. Compared with untreated K562 cell extract, the phosphorylation of endogenous CrkL was unchanged when the cells were treated with 1μM IM. Incubation with 10μM IM reduced phosphorylation of CrkL by approximately 10%, whereas 100μM IM yielded nearly an 85% reduction. In the bead-based assay, the amount of phosphorylation of GST-SH3n-Abltide was similar to that of control extracts when the extracts of the K562 cells treated with 1 or 10μM IM were assayed. When GST-SH3n-Abltide was incubated with extracts of the K562 cells treated with 100μM IM, substrate phosphorylation was reduced by approximately 90% (Fig. 5B). These data suggest that measuring the phosphorylation of surface-immobilized kinase-specific substrates by cell extract may be a reasonable reporter of kinase activity and inhibition as they occur in the cell.
Fig. 5.

Western analysis of K562 cell extract pretreated with IM and kinase activity of Bcr-Abl from pretreated K562 cell extract. (A) Western analysis of whole cell lysate using a mixed polyclonal antibody recognizing phospho-Tyr207 in CrkL and total eIF4E. Treatment of K562 cells with IM at the indicated concentrations decreases phosphorylation of endogenous CrkL. The relative steady-state phosphorylation of CrkL is estimated by the ratio of immunoreactivity of anti-phospho-CrkL to anti-eIF4E. (B) Upper: Antiphosphotyrosine Western blot of GST-SH3n-Abl-tide phosphorylation by K562 cell extracts in panel A. Pretreating cells with 100 μM IM yields similar kinase inhibition to adding 100 μM IM- to the kinase reaction. Lower: Memcode stain of the same membrane (MC).
Using the solid-phase assay to detect the efficacy of diverse PTK inhibitors
To test the versatility of the solid-phase kinase assay with the GST-SH3n-Abltide substrate, we evaluated the inhibition of Bcr-Abl with a variety of protein tyrosine kinase inhibitors. AG 957, Genestein, PD 1666326, and PD 173955 were compared with IM. AG 957 and Genestein are considered to be nonspecific PTK inhibitors, whereas PD 1666326 and PD 173955 are Src/Abl dual-specificity inhibitors that have been evaluated as alternatives to IM [34,35]. As shown in Fig. 6, AG 957 and Genestein did not inhibit Bcr-Abl activity in K562 cell extract even at 1 mM concentrations. Substrate phosphorylation was significantly inhibited, however, by 100μM IM, 0.01 μM PD 1666326, and 0.1 μM PD 173955. These results correspond to published data showing that PD 1666326 and PD 173955 are more potent inhibitors than IM, whereas IM is more potent than AG 957 and Genestein [12,36,37]. The solid-phase kinase assay offers adequate dynamic range to compare candidate Bcr-Abl kinase inhibitors in a simple format that approximates in vivo conditions.
Fig. 6.
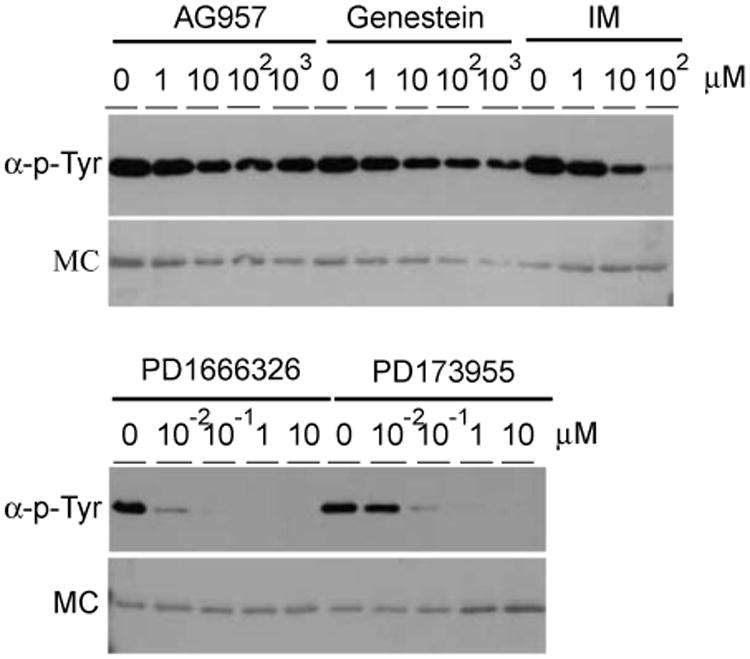
Analysis of candidate Bcr-Abl inhibitor activity by the in vitro assay. Upper panels: Antiphosphotyrosine Western blot of GST-SH3n-Abltide phosphorylation by K562 cell extract treated with inhibitors as indicated. Lower panels: Memcode stain of the same membrane (MC).
MALDI analysis of phosphorylated substrates from the PTK assay
The solid-phase assay developed here provides high signal to noise data that report Bcr-Abl activity and inhibition in cells. These data are generated by Western blot analysis, a standardized but time-consuming assay that is difficult to exploit as a quantitative method. We have evaluated analyzing the solid-phase kinase assay with MALDI–TOF–MS, a technique that has the potential to generate quantitative phosphoproteomic data in a high-throughput manner. To compare Western analysis to MS, we performed a basic kinetic experiment on Bcr-Abl in cell extract. The immobilized substrate was incubated with K562 cell extract in the presence of increasing concentrations of ATP (1 μM, 10μM, 100 μM, and 1 mM). Eluted substrate was subjected to Western analysis and, in parallel, digested with trypsin for MALDI–TOF analysis.
Complete trypsin digestion of unphosphorylated GST-SH3n-Abltide would generate 42 fragments varying in mass from 147 to 4060 Da. The predicted fragment containing the tyrosine phosphorylation site has a mass of 1676 Da when unphosphorylated and 1756 Da when phosphorylated, corresponding to the 80 Da addition of phosphate. To test this prediction, GST-SH3n-Abltide was reacted with excess c-Abl and ATP for 3 h. Then unphosphorylated and “fully” phosphorylated GST-SH3n-Abltide were digested and analyzed by MALDI–TOF. As shown in Fig. 7A, unphosphorylated GST-SH3n-Abltide displays only the 1676-m/z peak, whereas in the c-Abl phosphorylated protein this peak disappears and is replaced by a 1756-m/z peak. The 1516-m/z peak comes from the digestion of GST, serving as an internal control.
Fig. 7.
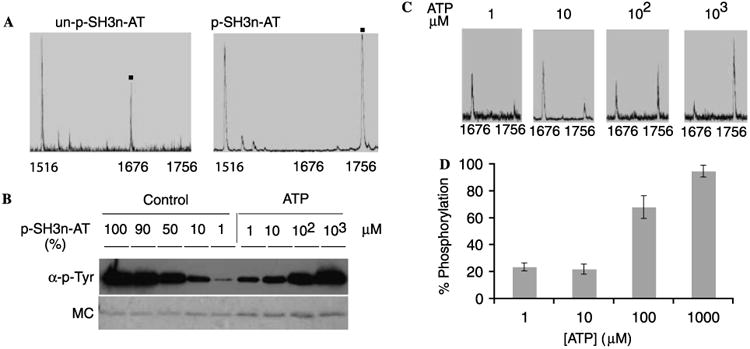
MALDI spectra of trypsin digested GST-SH3n-Abltide and Western blot of the same samples. (A) MALDI spectra of trypsin digested unphosphorylated (un-p) GST-SH3n-Abltide and fully phosphorylated (p) GST-SH3n-Abltide. The fragment at 1516 m/z is from the digestion of GST. (B) Upper: Western blot of the phosphorylated (p) GST-SH3n-Abltide with the positive controls. In the positive control, the phosphorylations of the substrate are approximately 99, 90, 50, 10, and 1%. (C) GST-SH3n-Abltide was phosphorylated by K562 cell extract with 1 μM, 10 μM, 100 μM, and 1 mM ATP and was subject to trypsin digestion and MALDI analysis. The fragment containing unphosphorylated peptide is located at 1676 m/z, and the fragment containing phosphorylated peptide is located at 1756 m/z. (D) Corresponding to the MALDI–TOF spectra in panel C, apparent phosphorylation was calculated from the ratio of the area of phosphopeptide peak to the sum of the areas of the phosphopeptide and unphosphorylated peptide peaks from three independent spots. The histogram shows the mean apparent phosphorylation and standard deviation at each ATP concentration, suggesting a Kapp[ATP] of less than 100 μM.
To provide a calibration for the phospho-GST-SH3n-Abltide bands on the Western blot, fully phosphorylated GST-SH3n-Abltide was mixed with unphosphorylated protein in 100:1, 10:1, 1:1, 1:10, and 1:100 ratios to yield 99, 90, 50, 10, and 1% phosphorylated positive control lanes, respectively. Compared with these positive controls, the phosphorylation of GST-SH3n-Abltide by K562 cell extract was proportional to added ATP, so that 1 μM, 10 μM, 100μM, and 1 mM yielded less than 10%, approximately 10%, approximately 50%, and approximately 100% phosphorylation in 1 h, respectively (Fig. 7B).
Digestion and MALDI analysis of these samples showed that the intensity of the peak at 1756 amu also increased in an ATP-dependent manner (Fig. 7C). Using the ratio of the area of phosphopeptide peak to the sum of the areas of the phosphopeptide and unphosphorylated peptide peaks, the apparent phosphorylation of peptide was calculated among the triplicate spots and the ratios were 24, 22, 68, and 95% for the 1 μM, 10 μM, 100 μM, and 1 mM ATP reactions, respectively (Figs. 7C and D), demonstrating a strong correspondence between the results of Western blot and MALDI analyses. Standard deviations between triplicate MALDI analyses of the samples were 3, 4, 8, and 5% (Fig. 7D). These data suggest that analyzing the solid-phase kinase assay with MALDI–TOF–MS may offer a straight-forward approach to obtaining relative kinase activity data with comparatively high-throughput. However, it should be noted that the ratios of peak areas in MALDI–MS between unphosphorylated and phosphorylated peptides might not be representative of the actual ratios of these peptides in the sample, reflecting known differences in ionization efficiency caused by the negatively charged phosphate modification [38,39]. External calibration of the relationship between degree of phosphorylation and MALDI–TOF peak ratios for each informative peptide (e.g., using HPLC or another independent method) might be necessary for quantifying degree of phosphorylation for each site on a protein substrate.
Discussion
We have developed a solid-phase kinase assay that allows the detection of endogenous Bcr-Abl activity from cell extract using highly specific immobilized substrates. High levels of kinase activity are observed using novel protein substrates that combine the artificial specificity of a peptide substrate with the in vivo sensitivity of a physiological protein substrate. The result is a substrate that is specifically phosphorylated by Bcr-Abl in cell extract. This method can report Bcr-Abl kinase activity and inhibition as it occurs within cells as well as predict the efficacy of various Bcr-Abl inhibitors in vitro. These results imply that the assay may be applicable to the general screening of Bcr-Abl inhibitors using cells and cell extracts and can be used to gauge their effects in cells from patients with CML and Philadelphia chromosome-positive ALL.
This solid-phase assay using whole cell extract has performed well in a semiquantitative Western format, precluding tedious kinase purification protocols, the use of radioactive tracers, and antibodies specific for phosphorylated Bcr-Abl substrates. Although not tested here, this solid-phase kinase assay in cell extract appears to be compatible with other assay geometries exploiting antibody binding such as enzyme-linked immunosorbent assay (ELISA), “on-bead” detection via flow cytometry, and protein microarray analysis. Moreover, we demonstrated that the assay is amenable to “probe-free” methods of phosphorylation detection such as MALDI–TOF–MS. MS has the potential for ready development of multiplexed assays that would provide internal controls or quantitation of other kinases in parallel.
This article demonstrates the development of an artificial kinase substrate and its use in a solid-phase kinase assay that is sufficiently robust and reproducible to detect Bcr-Abl activity and inhibition in cell extracts. We imagine that this approach could be generalized to specifically detect the activity of other protein tyrosine kinases. Using the modular substrate approach presented in this article, kinase-specific optimized peptide substrates derived from combinatorial (e.g., one bead, one compound, and SPOT arrays) [40–42] or phage display [43] libraries might be linked to GST via protein–protein interaction tags and/or domains of endogenous kinase substrates. This method would generate “super” substrates that are specifically phosphorylated in cell extract. These novel substrates may enable detection of endogenous kinase activity for purposes ranging from biological analysis to therapeutic decisions and drug discovery.
Acknowledgments
We thank B. Druker and J. Groffen for providing GST-CrkL constructs, N. Shah for providing Ba/F3 cells, B. Clarkson for providing PD 1666326 and PD 173955 inhibitors, and Novartis Pharmaceuticals for providing imatinib mesylate. Funding for this work was provided by NIH NCI IMAT R33 Grant CA10323 and the University of Chicago MRSEC. Laurie Parker is supported by the University of Chicago NIH T32 Cardiovascular Pathophysiology and Biochemistry training grant. Stephen Kron is a Fletcher scholar of the Cancer Research Foundation and a Leukemia and Lymphoma Society scholar.
Footnotes
Abbreviations used: PTK, protein tyrosine kinase; CML, chronic myelogenous leukemia; ALL, acute lymphocytic leukemia; IM, imatinib mesylate; MALDI–TOF–MS, matrix-assisted laser desorption/ionization time-of-flight mass spectrometry; GST, glutathione-S-transferase; LB, Luria–Bertani; IPTG, isopropylthio-β-d-galactosidase; PBS, phosphate-buffered saline; DTT, dithiothreitol; PMSF, phenylmethylsulfonyl fluoride; DMSO, dimethyl sulfoxide; EDTA, ethylenediamine tetraacetic acid; BSA, bovine serum albumin; GT, glutathione; ELISA, enzyme-linked immunosorbent assay; MS, mass spectrometry.
References
- 1.Deininger MW, Goldman JM, Melo JV. The molecular biology of chronic myeloid leukemia. Blood. 2000;96:3343–3356. [PubMed] [Google Scholar]
- 2.Nagar B, Hantschel O, Young MA, Scheffzek K, Veach D, Bornmann W, Clarkson B, Superti-Furga G, Kuriyan J. Structural basis for the autoinhibition of c-Abl tyrosine kinase. Cell. 2003;112:859–871. doi: 10.1016/s0092-8674(03)00194-6. [DOI] [PubMed] [Google Scholar]
- 3.Rowley JD. Letter: a new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature. 1973;243:290–293. doi: 10.1038/243290a0. [DOI] [PubMed] [Google Scholar]
- 4.Saglio G, Cilloni D. Abl: the prototype of oncogenic fusion proteins. Cell Mol Life Sci. 2004;61:2897–2911. doi: 10.1007/s00018-004-4271-0. [DOI] [PubMed] [Google Scholar]
- 5.Druker BJ, Tamura S, Buchdunger E, Ohno S, Segal GM, Fanning S, Zimmermann J, Lydon NB. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med. 1996;2:561–566. doi: 10.1038/nm0596-561. [DOI] [PubMed] [Google Scholar]
- 6.Deininger MW. Basic science going clinical: molecularly targeted therapy of chronic myelogenous leukemia. J Cancer Res Clin Oncol. 2004;130:59–72. doi: 10.1007/s00432-003-0502-2. [DOI] [PubMed] [Google Scholar]
- 7.Deininger M, Buchdunger E, Druker BJ. The development of imatinib as a therapeutic agent for chronic myeloid leukemia. Blood. 2005;105:2640–2653. doi: 10.1182/blood-2004-08-3097. [DOI] [PubMed] [Google Scholar]
- 8.Tibes R, Trent J, Kurzrock R. Tyrosine kinase inhibitors and the dawn of molecular cancer therapeutics. Annu Rev Pharmacol Toxicol. 2005;45:357–384. doi: 10.1146/annurev.pharmtox.45.120403.100124. [DOI] [PubMed] [Google Scholar]
- 9.Gorre ME, Mohammed M, Ellwood K, Hsu N, Paquette R, Rao PN, Sawyers CL. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001;293:876–880. doi: 10.1126/science.1062538. [DOI] [PubMed] [Google Scholar]
- 10.Nardi V, Azam M, Daley GQ. Mechanisms and implications of imatinib resistance mutations in BCR-ABL. Curr Opin Hematol. 2004;11:35–43. doi: 10.1097/00062752-200401000-00006. [DOI] [PubMed] [Google Scholar]
- 11.Shah NP, Tran C, Lee FY, Chen P, Norris D, Sawyers CL. Overriding imatinib resistance with a novel ABL kinase inhibitor. Science. 2004;305:399–401. doi: 10.1126/science.1099480. [DOI] [PubMed] [Google Scholar]
- 12.Huron DR, Gorre ME, Kraker AJ, Sawyers CL, Rosen N, Moasser MM. A novel pyridopyrimidine inhibitor of abl kinase is a picomolar inhibitor of Bcr-abl-driven K562 cells and is effective against STI571-resistant Bcr-abl mutants. Clin Cancer Res. 2003;9:1267–1273. [PubMed] [Google Scholar]
- 13.Engstrom L, Ekman P, Humble E, Ragnarsson U, Zetterqvist O. Detection and identification of substrates for protein kinases: use of proteins and synthetic peptides. Methods Enzymol. 1984;107:130–154. doi: 10.1016/0076-6879(84)07008-7. [DOI] [PubMed] [Google Scholar]
- 14.Casnellie JE. Assay of protein kinases using peptides with basic residues for phosphocellulose binding. Methods Enzymol. 1991;200:115–120. doi: 10.1016/0076-6879(91)00133-h. [DOI] [PubMed] [Google Scholar]
- 15.Sawyers CL. Research on resistance to cancer drug Gleevec. Science. 2001;294:1834. doi: 10.1126/science.294.5548.1834b. [DOI] [PubMed] [Google Scholar]
- 16.Sundberg SA. High-throughput and ultra-high-throughput screening: solution- and cell-based approaches. Curr Opin Biotechnol. 2000;11:47–53. doi: 10.1016/s0958-1669(99)00051-8. [DOI] [PubMed] [Google Scholar]
- 17.al-Obeidi FA, Wu JJ, Lam KS. Protein tyrosine kinases: structure, substrate specificity, and drug discovery. Biopolymers. 1998;47:197–223. doi: 10.1002/(SICI)1097-0282(1998)47:3<197::AID-BIP2>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 18.Desplat V, Lagarde V, Belloc F, Chollet C, Leguay T, Pasquet JM, Praloran V, Mahon FX. Rapid detection of phosphotyrosine proteins by flow cytometric analysis in Bcr-Abl-positive cells. Cytometry A. 2004;62:35–45. doi: 10.1002/cyto.a.20030. [DOI] [PubMed] [Google Scholar]
- 19.Krutzik PO, Irish JM, Nolan GP, Perez OD. Analysis of protein phosphorylation and cellular signaling events by flow cytometry: techniques and clinical applications. Clin Immunol. 2004;110:206–221. doi: 10.1016/j.clim.2003.11.009. [DOI] [PubMed] [Google Scholar]
- 20.Nisitani S, Kato RM, Rawlings DJ, Witte ON, Wahl MI. In situ detection of activated Bruton's tyrosine kinase in the Ig signaling complex by phosphopeptide-specific monoclonal antibodies. Proc Natl Acad Sci USA. 1999;96:2221–2226. doi: 10.1073/pnas.96.5.2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schaefer EM, Guimond S. Detection of protein tyrosine kinase activity using a high-capacity streptavidin-coated membrane and optimized biotinylated peptide substrates. Anal Biochem. 1998;261:100–112. doi: 10.1006/abio.1998.2716. [DOI] [PubMed] [Google Scholar]
- 22.Shults MD, Janes KA, Lauffenburger DA, Imperiali B. A multiplexed homogeneous fluorescence-based assay for protein kinase activity in cell lysates. Nat Methods. 2005;2:277–283. doi: 10.1038/nmeth747. [DOI] [PubMed] [Google Scholar]
- 23.Songyang Z, Carraway KL, III, Eck MJ, Harrison SC, Feldman RA, Mohammadi M, Schlessinger J, Hubbard SR, Smith DP, Eng C, Lorenzo MJ, Ponder BAJ, Mayer BJ, Cantley L. Catalytic specificity of protein-tyrosine kinases is critical for selective signalling. Nature. 1995;373:536–539. doi: 10.1038/373536a0. [DOI] [PubMed] [Google Scholar]
- 24.Matsumoto H, Kahn ES, Komori N. Nonradioactive phosphopeptide assay by matrix-assisted laser desorption ionization time-of-flight mass spectrometry: application to calcium/calmodulin-dependent protein kinase II. Anal Biochem. 1998;260:188–194. doi: 10.1006/abio.1998.2691. [DOI] [PubMed] [Google Scholar]
- 25.Mosier J, Olesen CE, Voyta JC, Bronstein I. Immunoassay protocol for quantitation of protein kinase activities. Methods Enzymol. 2000;305:410–416. doi: 10.1016/s0076-6879(00)05503-8. [DOI] [PubMed] [Google Scholar]
- 26.ten Hoeve J, Arlinghaus RB, Guo JQ, Heisterkamp N, Groffen J. Tyrosine phosphorylation of CRKL in Philadelphia+ leukemia. Blood. 1994;84:1731–1736. [PubMed] [Google Scholar]
- 27.Salgia R, Pisick E, Sattler M, Li JL, Uemura N, Wong WK, Burky SA, Hirai H, Chen LB, Griffin JD. p130CAS forms a signaling complex with the adapter protein CRKL in hematopoietic cells transformed by the BCR/ABL oncogene. J Biol Chem. 1996;271:25198–25203. doi: 10.1074/jbc.271.41.25198. [DOI] [PubMed] [Google Scholar]
- 28.Feller SM. Crk family adaptors–signalling complex formation and biological roles. Oncogene. 2001;20:6348–6371. doi: 10.1038/sj.onc.1204779. [DOI] [PubMed] [Google Scholar]
- 29.de Jong R, ten Hoeve J, Heisterkamp N, Groffen J. Tyrosine 207 in CRKL is the BCR/ABL phosphorylation site. Oncogene. 1997;14:507–513. doi: 10.1038/sj.onc.1200885. [DOI] [PubMed] [Google Scholar]
- 30.Pellicena P, Stowell KR, Miller WT. Enhanced phosphorylation of Src family kinase substrates containing SH2 domain binding sites. J Biol Chem. 1998;273:15325–15328. doi: 10.1074/jbc.273.25.15325. [DOI] [PubMed] [Google Scholar]
- 31.Scott MP, Miller WT. A peptide model system for processive phosphorylation by Src family kinases. Biochemistry. 2000;39:14531–14537. doi: 10.1021/bi001850u. [DOI] [PubMed] [Google Scholar]
- 32.Pisabarro MT, Serrano L. Rational design of specific high-affinity peptide ligands for the Abl-SH3 domain. Biochemistry. 1996;35:10634–10640. doi: 10.1021/bi960203t. [DOI] [PubMed] [Google Scholar]
- 33.Heaney C, Kolibaba K, Bhat A, Oda T, Ohno S, Fanning S, Druker BJ. Direct binding of CRKL to BCR-ABL is not required for BCR-ABL transformation. Blood. 1997;89:297–306. [PubMed] [Google Scholar]
- 34.Kraker AJ, Hartl BG, Amar AM, Barvian MR, Showalter HD, Moore CW. Biochemical and cellular effects of c-Src kinase-selective pyrido[2,3-d] pyrimidine tyrosine kinase inhibitors. Biochem Pharmacol. 2000;60:885–898. doi: 10.1016/s0006-2952(00)00405-6. [DOI] [PubMed] [Google Scholar]
- 35.Tipping AJ, Baluch S, Barnes DJ, Veach DR, Clarkson BM, Bornmann WG, Mahon FX, Goldman JM, Melo JV. Efficacy of dual-specific Bcr-Abl and Src-family kinase inhibitors in cells sensitive and resistant to imatinib mesylate. Leukemia. 2004;18:1352–1356. doi: 10.1038/sj.leu.2403416. [DOI] [PubMed] [Google Scholar]
- 36.Okabe M, Kawamura K, Miyagishima T, Itaya T, Goodwyn D, Shoji M, Vogler WR, Sakurada K, Uehara M, Miyazaki T. Effect of herbimycin A, an inhibitor of tyrosine kinase, on protein tyrosine kinase activity and phosphotyrosyl proteins of Ph1-positive leukemia cells. Leuk Res. 1994;18:213–220. doi: 10.1016/0145-2126(94)90117-1. [DOI] [PubMed] [Google Scholar]
- 37.Sun X, Layton JE, Elefanty A, Lieschke GJ. Comparison of effects of the tyrosine kinase inhibitors AG957, AG490, and STI571 on BCR-ABL: expressing cells, demonstrating synergy between AG490 and STI571. Blood. 2001;97:2008–2015. doi: 10.1182/blood.v97.7.2008. [DOI] [PubMed] [Google Scholar]
- 38.Craig AG, Engstrom A, Bennich H, Hoffmann-Posorske E, Meyer HE. Plasma desorption mass spectrometry of phosphopeptides: an investigation to determine the feasibility of quantifying the degree of phosphorylation. Biol Mass Spectrom. 1991;20:565–574. doi: 10.1002/bms.1200200910. [DOI] [PubMed] [Google Scholar]
- 39.Janek K, Wenschuh H, Bienert M, Krause E. Phosphopeptide analysis by positive and negative ion matrix-assisted laser desorption/ionization mass spectrometry. Rapid Commun Mass Spectrom. 2001;15:1593–1599. doi: 10.1002/rcm.417. [DOI] [PubMed] [Google Scholar]
- 40.Frank R. The SPOT-synthesis technique: synthetic peptide arrays on membrane supports—Principles and applications. J Immunol Methods. 2002;267:13–26. doi: 10.1016/s0022-1759(02)00137-0. [DOI] [PubMed] [Google Scholar]
- 41.Lam KS, Lehman AL, Song A, Doan N, Enstrom AM, Maxwell J, Liu R. Synthesis and screening of “one-bead one-compound” combinatorial peptide libraries. Methods Enzymol. 2003;369:298–322. doi: 10.1016/S0076-6879(03)69017-8. [DOI] [PubMed] [Google Scholar]
- 42.Lam KS, Liu R, Miyamoto S, Lehman AL, Tuscano JM. Applications of one-bead one-compound combinatorial libraries and chemical microarrays in signal transduction research. Acc Chem Res. 2003;36:370–377. doi: 10.1021/ar0201299. [DOI] [PubMed] [Google Scholar]
- 43.Schmitz R, Baumann G, Gram H. Catalytic specificity of phosphotyrosine kinases Blk, Lyn, c-Src, and Syk as assessed by phage display. J Mol Biol. 1996;260:664–677. doi: 10.1006/jmbi.1996.0429. [DOI] [PubMed] [Google Scholar]