

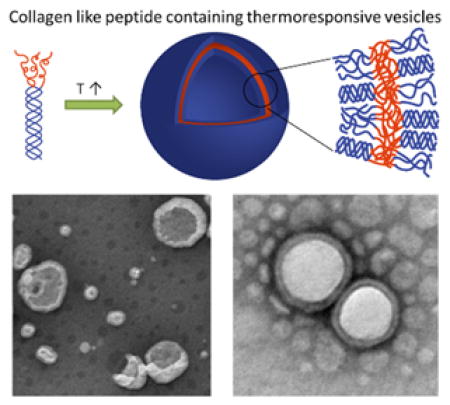
Abstract
Temperature-triggered formation of nanostructures with distinct biological activity offers opportunities in selective modification of matrices and in drug delivery. Toward these ends, diblock polymers comprising poly(diethylene glycol methyl ether methacrylate) (PDEGMEMA) conjugated to a triple helix-forming collagen-like peptide (CLP) is produced. The ability of the CLP domain to maintain its triple helix conformation after conjugation with the polymer is confirmed via circular dichroism (CD). Dynamic light scattering (DLS) measurements suggest the diblock conjugate undergo a reversible temperature-induced transition in aqueous solution to form nanoparticles with a diameter of approximately 100 nm, with a transition temperature of 37 °C. Transmission electron microscopy (TEM) suggests the formation of well-defined vesicles above the transition temperature, while no supramolecular assemblies are observed at room temperature. The self-assembly of PDEGMEMA-CLP diblock is triggered by the collapse of the thermoresponsive domain above its LCST. The incorporation of CLP domains in these nanostructures may offer opportunities for the selective targeting of collagen-containing matrices.
Keywords: Collagen-like peptide, thermoresponsive, self-assembly, vesicle, drug delivery
Graphical Abstract
1. Introduction
Collagen is the main component of the extracellular matrix (ECM) in mammals. It comprises 28 different types of proteins, which have widespread functions such as mediating cell adhesion, cell migration, and tissue scaffolding and repair.[1] Although the roles of these proteins vary widely, they all contain (at least in part) the same secondary structure, a collagen triple helix,[2] which comprises three polypropline-II helices. Each strand of the helix consists of a repetitive amino acid sequence, glycine-X-Y, where X and Y are frequently proline (Pro) and hydroxyproline (Hyp).[3] Due to their excellent gel-forming ability and biodegradability, collagens have been widely used as biomedical materials for the delivery of chemotherapeutic drugs such as paclitaxel,[4] doxorubicin,[5] and antibacterial drugs such as gentamicin,[6] as well as for tissue support and regeneration applications including skin replacement, bone substitutes, and artificial blood vessels.[7] However, some of the limitations of animal-derived collagens, such as their thermal instability, possible contamination with pathogenic substances, and relative difficulty in the introduction of specific sequence modifications, have limited their versatility. Accordingly, sequences modeled after the collagens, also known as collagen-like peptides (CLPs), collagen-mimetic peptides (CMPs), or collagen-related peptides (CRPs) have been explored.[8–10]
CLPs form a triple helical structure almost identical to that of natural collagen,[10] but because of their low molecular mass (<5 kDa), they exhibit reversible folding and unfolding behavior. Traditionally, CLPs were employed to elucidate the triple helix structure and the stabilization effect of different amino acid residues in collagens.[8,11–16] CLPs with novel sequences and interactions have also been developed to mimic collagen fibril formation[17–22] and to produce other higher-order supramolecular structures.[23–25] These include but are not limited to: (i) micrometer-scale fibrillar structure through noncovalent π-π end-to-end interactions;[20,21] (ii) fibrous structures with well-defined D-periodicity assembled from multi-domain CLPs such as (PRG)4(POG)4(EOG)4[22] and (PKG)4(POG)4(DOG)4;[17] (iii) metal cation-triggered assembled structures such as microflorettes, fiberlike meshes,[23,24] nanofibrils,[25] and microscale spheres;[25] (iv) hydrogel networks and scaffolds crosslinked by CLP triple helix association[26,27] or metal cation – CLP coordination[28].
More recently, Wang et al. have reported that unfolded single strand collagen-like peptides have a strong propensity to bind to natural collagen via a strand invasion process.[29] Short synthetic CLPs were successfully utilized as a staining reagent for natural collagens in human tissues including skin, cornea, bone[30] and liver.[31] In vivo studies[32] suggest that CLPs are able to permeate tumor vasculature and accumulate at tumor sites. A high level of accumulation of the CLP within the skeleton and joints has also been observed, especially in regions with high MMP and collagen remodeling activity, such as the articular cartilage of knee joints. These results suggest the great potential of CLPs as novel tool and material for tracking pathogenic collagens for diagnostic and drug delivery purposes.
While these CLPs and other (poly)peptide-based biomaterials exhibit well defined hierarchical structures and variety of biological functions such as cell recognition, adhesion and proliferation,[33–35] their use in therapeutics can be limited due to elevated immune responses, rapid degradation by enzymes, and short circulation times in vivo.[36,37] Chemical conjugation of (poly)peptides with synthetic polymers thus has become a widely used method to overcome these limitations while maintaining desirable bioactivities. For instance, the attachment of polyethylene glycol (PEG) – or PEGylation – to protein-based drugs, enzymes, and antibodies greatly improves the solubility of the biomolecules, reducing their immunogenicity and increasing their blood circulation time.[36] In addition, a variety of (poly)peptides including α-helical coiled-coil peptide domains,[38–41] β-sheet peptide motifs,[42–44] and elastin-mimetic peptides[45–48] have been successfully employed to produce polymer-peptide hybrid materials, which show potential in biomedical applications such as drug and gene delivery. However, compared with the recent surge in research on these peptide-polymer conjugates, collagen-like peptide hybrid materials remain relatively less explored. Yu et al.[49] attached one or two C12 head groups to a collagen-like peptide and found that it improved the thermal stability of the collagen triple helix, and self-assembly of the amphiphiles into various structures, dependent on the length of the amphiphile, was also observed.[50] Luo et al.[51] reported that after a C16 lipophilic tail was attached to a CLP with the sequence (GPO)3GFOGER(GPO)3 (F: phenylalanine; O: hydroxyproline), the amphiphile self-assembled into micrometer-long nanofibers with a diameter of approximately 16 nm. More recently, our groups have reported a polymer-CLP-polymer triblock,[52] which self-assembled into micron-scale hollow spheres at 37 °C and transformed into nanofibers at higher temperature.
In this study, we have conjugated a thermoresponsive polymer, poly (diethylene glycol methyl ether methacrylate) (PDEGMEMA), to the N-terminus of a triple-helix forming collagen-like peptide ((GPO)7GG), to synthesize a polymer-CLP diblock (Scheme 1). Due to the thermoresponsiveness of the polymer domain, we anticipated temperature-triggered assembly of the diblock into nanoparticles in aqueous conditions above the lower critical solution temperature (LCST) of the polymer, in a two-step process (Scheme 1). The first level of assembly involved the formation of collagen triple helix from diblock monomers, with a second level of assembly triggered by increasing the temperature of the solution, leading to the collapse of the polymer domain and assembly into higher-order nanoparticles. A poly (ethylene glycol) - CLP diblock copolymer was studied as a non-thermoresponsive control. The thermally responsive diblock polymers provide a novel approach to the formation of nanoparticles that may be uniquely responsive to biological environments.
Scheme 1.
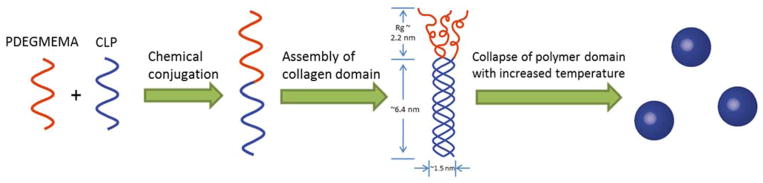
Proposed two-step assembly mechanism for PDEGMEMA-CLP conjugates.
2. Experimental Section
2.1 Materials
Fmoc-protected amino acids, Rink amide MBHA resin, N,N,N′,N′-tetramethyl-O-(1H-benzotriazol-1-yl) uronium hexafluorophosphate (HBTU), and piperidine for solid-phase peptide synthesis were purchased from AAPPTEC Inc. (Louisville, KY). HPLC-grade acetonitrile and dimethylformamide (DMF) were purchased from Fisher Scientific (Fairlawn, NJ). N-Methyl-2-pyrrolidone (NMP), trifluoroacetic acid (TFA), triisopropylsilane (TIS), triethylamine (TEA), and diisopropylethylamine (DIEA) were purchased from Sigma-Aldrich (St. Louis, MO).
2.2 Peptide Synthesis
A collagen-like peptide with the sequence (GPO)7GG was synthesized via traditional solid-phase peptide synthesis methods (SPPS) using a Focus XC automatic peptide synthesizer (AAPPTec Inc., Louisville, KY). Rink amide MBHA resin with a loading capacity of 0.52 mmol/g was used for the synthesis. The amino acids were activated for coupling with HBTU in the presence of 2 M diisopropylethylamine (DIEA) in NMP. Deprotection of the Fmoc group was conducted using 20% piperidine in DMF. One-hour coupling cycles were used for all the residues. Cleavage of the peptide from the resin was conducted in 95:2.5:2.5 (v:v:v) trifluoroacetic acid (TFA) /triisopropylsilane (TIS) /water for 3 hours. The TFA was mostly evaporated and the cleaved peptide was precipitated in cold ether. The peptide was then redissolved in water and lyophilized.
Crude peptide was purified via reverse-phase HPLC (Waters Inc., Milford, MA) on a Waters Symmetry 300, C-18 column. The mobile-phase comprised gradients of degassed deionized water with 0.1% TFA and acetonitrile with 0.1% TFA, at a flow rate of 5 ml/min. Peptide was detected by UV absorbance at 214 nm; fractions with product were collected and lyophilized. The molecular weight of the peptide was confirmed via electrospray ionization mass spectrometry (ESI-MS, AutospecQ, VG Analytical, Manchester, UK) and the purity of the peptide was confirmed via analytical scale, reverse-phase HPLC (Waters 2996; Symmetry C18, 3.5 μm, 4.6 × 75 mm).
2.3 PDEGMEMA-CLP Conjugate Synthesis
The synthesis of the conjugate was performed via the reaction of N-terminal amine of the collagen-like peptide with activated-ester end-functionalized PDEGMEMA, following a recently developed protocol.[53] The activated ester end-functionalized PDEGMEMA was synthesized via RAFT polymerization described earlier.[54] The PDEGMEMA-CLP diblock copolymer was synthesized by mixing 3 equiv of PDEGMEMA per primary amine group of the peptide. The reaction was conducted in 2 mL 50:50 (v:v) NMP/DMSO, with the addition of 6 μL triethylamine (TEA), for 3 days at 35 °C (Scheme 2). The resulting hybrid polymer was precipitated into a 5-fold volume of cold diethyl ether and redissolved in 50:50 (v:v) NMP/DMSO (repeated 5 times) to removed excessive PDEGMEMA starting material, followed by dissolution in water and lyophilization.
Scheme 2.
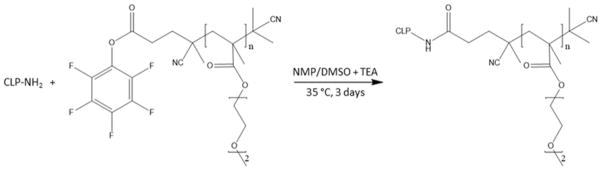
Chemical synthesis of PDEGMEMA-CLP diblock via the reaction of the activated esterterminated PDEGMEMA with the N-terminal amine of a collagen-like peptide. The reaction was carried out in a mixture of dimethyl sulfoxide (DMSO) and N-methyl-2-pyrrolidone (NMP) (v/v=1:1) in the presence of triethylamine at 35 °C for 3 days.
2.4 Nuclear Magnetic Resonance (NMR)
1H NMR spectra were recorded under standard quantitative conditions on a Bruker AVIII spectrometer operating at 600 MHz, using at least 64 scans. All samples were dissolved in deuterated dimethyl sulfoxide (δ (d6-DMSO) = 2.50 ppm) at a concentration of 2 mg/mL. The resulting spectra were analyzed using Mnova software (Mestrelab Research, Santiago de Compostela, Spain).
2.5 Gel Permeation Chromatography (GPC)
GPC measurement of the PDEGMEMA-CLP conjugate was performed in DMF with 0.01 M lithium chloride at 30 °C. The system was operated at 1 mL/min with a Sonntek HPLC pump (K-501), one 50 mm × 7.5 mm PL gel mixed guard column, one 300 mm × 7.5 mm PL gel 5 μm mixed C column, one 300 mm × 7.5 mm PL gel 5 μm mixed D column, a Knauer refractive index detector (K-2301), and an Alltech solvent recycler 3000. Poly (methyl methacrylate) (PMMA) standards with Mw 875 Da, 3070 Da, 10570 Da and 30620 Da were used for molecular weight calibration. All samples were dissolved in DMF at 1 mg/mL and 100 μL of solution was injected.
GPC measurement of the PEG-CLP conjugate was performed in 10 mM PBS buffer (pH 7.4, 137 mM NaCl and 2.7 mM KCl) using a combination of two Waters ultrahydro linear column (WAT011545 and WAT011525) with a nominal flow rate of 1 mL/min. A refractive index detector (Waters 2414), a UV absorbance detector (Waters 2996) were used. PEG standards with Mw 400 Da, 1970 Da, 6430 Da and 21030 Da were used to calculate molecular weights of the products. CLP and PEG starting material samples were dissolved in PBS at a concentration of 2 mg/mL, PEG-CLP diblock conjugate sample was dissolved in PBS at a concentration of 1 mg/mL. 100 μL solution was injected for all samples.
2.6 Circular Dichroic Spectroscopy (CD)
Characterization of the secondary structure of the CLP domain was conducted via circular dichroic spectroscopy (Jasco 810 circular dichroism spectropolarimeter, Jasco Inc., Easton, MD, USA). Either CLP or PDEGMEMA-CLP conjugate were dissolved at a concentration of 100 μM in PBS (10 mM, pH 7.4, 137 mM NaCl and 2.7 mM KCl ) and incubated overnight to allow triple helix formation. The CD spectra were recorded using quartz cells with a 0.2 cm optical path length.
Full wavelength scans were collected to study the conformation of the peptide domain at selected temperatures. The sample was incubated at each temperature for 10 min before measurement. The scanning rate was 50 nm/min, with a response time of 4 s. The wavelength scans were obtained from 200 nm to 250 nm and were recorded every 1 nm. In order to precisely measure the melting temperature of the CLP domain, variable temperature experiments were conducted at a constant wavelength of 225 nm with a 0.25 °C/min heating rate. Refolding kinetics were studied via temperature-jump experiments. The sample solution was incubated at 80 °C for 30 min followed by quenching to 5 °C in less than 2 minutes. The ellipticity at 225 nm as a function of time was monitored at 5 °C beginning right after the temperature jump.
2.7 Dynamic Light Scattering (DLS)
Analysis of particles sizes in solution was conducted via dynamic light scattering (DLS) on a ZetaSizer Nano Series (Nano ZS, Malvern Instruments, U.K.) at a scattering angle of 173°, and data fitting using the cumulant method. PEG-CLP and PDEGMEMA-CLP samples were prepared at 1 mg/mL in PBS (10 mM, pH 7.4, 137 mM NaCl and 2.7 mM KCl). PDEGMEMA-CLP samples were also prepared in water at a concentration of 1 mg/mL and 5 mg/mL. A PDEGMEMA sample was prepared in water as the polymer alone was not soluble in PBS. All samples were incubated at 4 °C overnight before measurement. The lower critical solution temperature (LCST) of the polymer and conjugate was assessed by measurement of the average size of particles at temperatures from 5 °C to 80 °C. Samples were incubated at each temperature for 2 minutes before measurements. The LCST was assigned as the temperature at which the intensity of scattered light began to increase. For particle size distribution studies at 5 °C, 25 °C, 37 °C, 50 °C, and 70 °C, each sample was incubated for 10 min at the desired temperature before measurements. The reported data represent an average of at least three measurements.
2.8 Transmission Electron Microscopy (TEM)
Samples for TEM were prepared on carbon-coated copper grids (CF300-Cu, Electron Microscopy Sciences Inc.). The grids, pipette tips, and samples were incubated in an isothermal oven (VWR Signature™ Forced Air Safety Ovens, VWR Inc.) at desired temperature (25 °C and 50 °C) for at least 30 min before sample preparation, which was also conducted in the oven. PDEGMEMA-CLP and PEG-CLP diblock samples were dissolved in water at concentrations of 1 mg/mL and 5 mg/mL. 5 μL of the sample solution was drop cast on the grid and blotted after 60 seconds. For staining, 1% phosphotungstic acid (PTA) (pH adjusted to 7.0 using 1 M NaOH) as a negative stain was used. 5 μL of the PTA solution was drop cast on the grid and blotted after 10 seconds. The sample was allowed to dry in the oven at the desired temperature for 30 minutes and then was air-dried for 2 hours. TEM images were taken on a Tecnai G2 12 TEM (FEI Company, Hillsboro, OR) at an acceleration voltage of 120 keV.
3. Results and Discussion
3.1 Synthesis of PDEGMEMA-CLP Diblock Polymer
The collagen-like peptide with the sequence (GPO)7GG was synthesized via traditional Fmoc-based, solid-phase peptide synthesis methods (SPPS). Two glycines at the C-terminus were introduced to avoid the formation of diketopiperazine.[55] After purification with reverse-phase HPLC, peptide with purity greater than 99% was obtained (Figure S1). The molecular weight of the peptide was verified via ESI-MS (Figure S2).
The thermoresponsive polymer, PDEGMEMA, was synthesized via reversible addition–fragmentation chain-transfer polymerization (RAFT) using a standard procedure described earlier.[54] The polymer was conjugated to the CLP via the reaction of the activated ester end group of PDEGMEMA with the N-terminal amine of the peptide. The molecular weight distribution of purified products was studied via gel permeation chromatography (GPC) using DMF as the mobile phase (Figure 1). DMF was used instead of water because both the polymer and the diblock showed aggregation behavior in aqueous solution (see below). The traces shown were obtained using refractive index detection and were normalized to give a better comparison between the diblock and starting material. The chromatogram for the collagen-like peptide was not collected because the CLP was insoluble in DMF. As shown in Figure 1, the elution time of the diblock was clearly shifted to the higher molecular weight region compared with the PDEGMEMA starting material. The results suggest successful conjugation of the two building blocks and the single product peak without any shoulders indicates complete removal of excess PDEGMEMA starting material. A rough estimation of molecular weight from a calibration employing linear poly (methyl methacrylate) (PMMA) standards yielded a Mw of 7.1 kDa of the PDEGMEMA, with a polydispersity index (PDI) of 1.16. Interestingly, the results also suggested a PDI of 1.13 for the diblock and a Mw of 22.1 kDa, which was larger than anticipated, likely due to the potential physicochemical differences between the CLP domain of the diblock and PMMA standards, which would affect their mobility and/or hydrodynamic volume in the mobile phase.
Figure 1.

GPC trace of PDEGMEMA-CLP diblock and PDEGMEMA starting material.
1H NMR spectroscopy was utilized to confirm the presence of both the peptide and polymer domain in the diblock product. Figure 2a provides the 1H NMR spectrum of the collagen-like peptide with typical regions of interest: protons from peptide backbone amide bonds from 7.0 ppm to 8.2 ppm (10H, CONH-CHR and CO-NH2, labeled He,e′,e″ in the figure); α-protons from amino acid residues located from 4.2 ppm to 5.3 ppm (30H, NH-CHR-CO and NH-CH2-CO, Hc,c′,c″,c‴); and methylene protons from the pyrrolidine ring from 1.5 ppm to 2.3 ppm (42H, CONR-CH2CH2CH2-CH and CH(OH)-CH2-CH, Hd,d′,d″). The spectrum of PDEGMEMA given in Figure 2b shows strong peaks for side-chain ethylene protons at 4.02 ppm (2H, COO-CH2-CH2, Hc) as well as 3.62 ppm, 3.54 ppm and 3.47 ppm (6H, COO-CH2CH2O-CH2CH2O-CH3, Hd,d′,d″), side-chain methyl group protons at 3.28 pm (3H, OCH3, He), backbone methylene protons from 1.7 ppm to 1.9 ppm (2H, -CRCH3-CH2-, Hb), and methyl group protons at 0.81 ppm and 0.97 ppm (3H, -CRCH3-CH2-, Ha). A solvent peak located at 5.76 ppm arises from dichloromethane (DCM), which was introduced to the sample when concentrating the polymer. The spectrum of the PDEGMEMA-CLP diblock, given in Figure 2c, showed all corresponding characteristic signals from the polymer as well as the peptide building block. The peaks from DCM observed in the polymer (Figure 2b) were completely removed, indicating complete separation by precipitating into cold ether. These results, together with the higher MW suggested by GPC, indicate successful synthesis and purification of the diblock. Comparison between the integrals from peptide α-protons (Hc,c′,c″,c‴ in Figure 2a, 30 protons per molecule) with protons from the polymer side-chain methylene group (Hc in Figure 2b, 79 protons per molecule) suggested the molecular weight of the polymer domain in the diblock to be 7.4 kDa, which was consistent with the molecular weight determined via GPC.
Figure 2.

1H NMR spectra (600 MHz) of: a) collagen-like peptide; b) PDEGMEMA; c) PDEGMEMA-CLP diblock in d6-DMSO.
3.2 Triple Helix Formation
Although native collagens have a variety of biological functions, at the molecular level, they all share the characteristic triple helical secondary structure. The ability of the collagen-like peptide, as well as the CLP domain in the diblock, to form triple helix is fundamental for specific binding with native collagen via triple helix hybridization. Circular dichroic spectroscopy (CD) was utilized to characterize the conformation of the peptide products; representative data are shown in Figure 3. The CD spectra of the collagen-like peptide and PDEGMEMA-CLP diblock at variable temperatures ranging from 5 °C to 80 °C are plotted in Figure 3a and 3d, respectively. Both the peptide and diblock exhibited a triple helix characteristic peak at 225 nm and a crossover at 213 nm,[17,19,56] confirming triple helix formation of the CLP, as well as the ability of the CLP domain to maintain the structure after conjugation to the polymer. The intensity of the 225 nm peak was reduced with an increase in temperature (black arrows in Figure 3a and 3d), consistent with the triple helix domain gradually unfolding upon heating.
Figure 3.

a) CD spectra showing the wavelength scans for the collagen like peptide; b) Blue curve with dots (Y axis on the left side): thermal unfolding profile for CLP plotted as [θ]MRE225nm versus temperature. The red curve (Y axis on the right side) is the first derivative of the unfolding curve with respect to temperature; c) Refolding profile of CLP after quenching to 5 °C; d) CD spectra showing the wavelength scans for PDEGMEMA-CLP diblock; e) Blue curve with dots (Y axis on the left side): thermal unfolding profile for PDEGMEMA-CLP diblock plotted as [θ]MRE225nm versus temperature. The red curve (Y axis on the right side) is the first derivative of the unfolding curve with respect to temperature; f) Refolding profile of the PDEGMEMA-CLP diblock after quenching to 5 °C, with an inserted image from 0 – 7 minutes.
The thermal stability of the triple helices was investigated via monitoring the mean residue ellipticity at λ = 225 nm as a function of temperature while the sample was heated at a slow rate (0.25 °C/min). The thermal unfolding profile (red) and the first derivative of the melting curve (blue) for the CLP and diblock are plotted in Figure 3b and 3e respectively. The data in Figure 3b yield a melting temperature (Tm) of 39.2 °C for the peptide and those in Figure 3e yield a melting temperature of 38.8 °C for the diblock. The melting temperature of the CLP is consistent with previously reported results for (Gly-Pro-Hyp)n collagen-like peptides.[31,57,58] By comparing the thermal unfolding curves of the CLP and the diblock (blue curves in Figure 3b and 3e), it is apparent that the triple helix domain in the diblock sample started to melt at a much higher temperature (32 °C) than the peptide alone (20 °C), indicating the greater energy required for the unfolding of the triple helix in the conjugate. Additionally, while the peak at 225 nm for the peptide vanished upon heating (Figure 3a), the unfolding process for the triple helix domain in the conjugate was not complete even at 80 °C (Figure 3d and 3e). These results indicate that the conjugation of PDEGMEMA to the peptide constricts the unfolding of the triple helix. The constriction is likely attributed to the anchoring effect of the polymer domain when it collapsed above its LCST (32 °C in PBS, Figure 4a, described below). Such triple helix stability enhancements via hydrophobic conjugation were also reported in our previous studies,[52,53] as well as by the Fields and the Tong research groups.[49,51]
Figure 4.
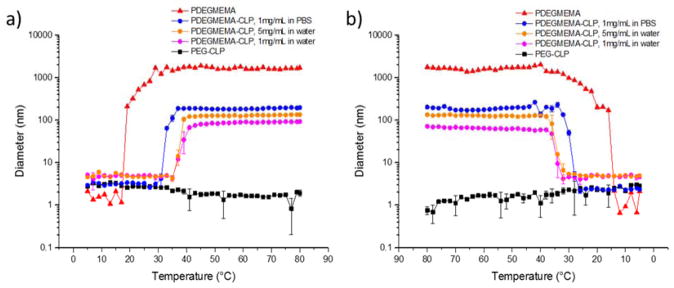
Study of transition temperature via dynamic light scattering. a) Hydrodynamic diameter of assemblies as a function of temperature upon heating; b) Hydrodynamic diameter of assemblies as a function of temperature upon cooling. Samples were heated at each temperature for 2 minutes before measurement. Red curve with triangles: PDEGMEMA sample dissolved in water at 1 mg/mL; blue curve with circles: PDEGMEMA-CLP diblock dissolved in PBS buffer (pH = 7.4) at 1 mg/mL; orange curve with circles: PDEGMEMA-CLP diblock dissolved in water at 5 mg/mL; pink curve with circles: PDEGMEMA-CLP diblock dissolved in water at 1 mg/mL; black curve with squares: PEG-CLP diblock dissolved in water at 1 mg/mL.
Temperature jump experiments were conducted to analyze the refolding kinetics of the peptide domain. In these experiments, the samples were incubated at 80 °C for 30 min, followed by quenching to 5 °C to allow triple helix refolding. The ratio of mean residue ellipticity recovered compared to the original value at 5 °C was plotted as a function of time. Figure 3c shows the refolding profile for the CLP. After quenching to 5 °C, the CLP gradually refolded to nearly 90% of its original value, following third order reaction kinetics, consistent with previous reports.[59] A rate constant of k = 3.43 × 107 M−2·s−1 was derived from the curve using a third-order reaction model. Interestingly, the PDEGMEMA-CLP diblock reached 80% of its original CD intensity within only 3 minutes (Figure 3f), indicating the refolding of the peptide domain within the diblock was much faster than that for the peptide alone. The refolding profile also exhibited a constant refolding rate in the first three minutes, suggesting a zero-order process with a rate constant derived as 1.5 × 10−7 M· s−1. This difference in the order of the folding process in the free peptide versus the diblock likely originates from the collapsed polymer domain acting as an anchoring point, which significantly increased the local concentration of the peptide strands and refolding rate of the triple helix. Additionally, once the CLPs were anchored at close proximity, long range molecular collision of three CLP single strands driven by Brownian motion was not the main prerequisite for the refolding process, which changed the order of the reaction. Similar stabilization of secondary structures from such anchoring effects has also been observed in poly(N-isopropylacrylamide)-containing copolymers. For example, the stability of an α-helical peptide domain was increased in a diblock copolymer comprising poly(N-isopropylacrylamide)-block-poly(N5-(4-hydroxybutyl)-L-glutamine) (PNIPAAm-b-PHBG) when compared with that of the PHBG homopolymer.[60]
3.3 Temperature Triggered Self-Assembly into Nanoparticles
Diblock copolymers consisting of one thermoresponsive block and one hydrophilic block were expected to exhibit reversible self-assembly behaviors as temperature changes. One typical example is poly(ethylene glycol)-b-poly(N-isopropylacrylamide) (PEG-PNIPAAM), which self assembles into micelles in water above the lower critical solution temperature (LCST) of PNIPAAM.[61–63] The diblock in this work consists of a hydrophilic collagen-like peptide block and a thermoresponsive PDEGMEMA block, thus is anticipated to undergo self-assembly in aqueous conditions when the polymer domain collapses above its LCST. Dynamic light scattering studies were conducted to determine the LCST of the PDEGMEMA-CLP diblock as well as that of the polymer itself by observing the temperature-induced increase in hydrodynamic size. All samples were prepared at concentrations similar to those in the CD experiments, pre-incubated at 4 °C to allow triple helix formation, and filtered to remove dust before the measurements.
The transition temperature of the conjugate and PDEGMEMA were tested via monitoring of the hydrodynamic diameter as a function of increasing temperature (Figure 4a). Diblock sample dissolved in PBS (10 mM, pH 7.4, 137 mM NaCl and 2.7 mM KCl) at a concentration of 1 mg/mL, as well as samples dissolved in water at concentrations of 1 mg/mL and 5 mg/mL were measured. The results show an increase in intensity upon heating and suggested a transition temperature of 18 °C for the polymer and 32 °C for the diblock. As expected, the PEG-CLP control does not exhibit any phase transition behavior in the entire temperature range. The increase in the transition temperature for the PDEGMEMA-CLP diblock compared with the polymer is consistent with the conjugation of the hydrophilic peptide block to the polymer. Changes in LCST due to hydrophobicity differences have also been reported in PNIPAAM containing copolymers modified with short hydrophilic DNA segments[64] or poly(lysine) segments,[65] as well as elastin-like peptides (ELP) with amino acid valine (V) substituted by more hydrophobic residues such as leucine, isoleucine and phenylalanine.[66,67]
Due to practical difficulties in studies of assembly morphology with transmission electron microscopy using samples in PBS buffer, samples dissolved in water (at either 1 mg/mL or 5 mg/mL) were also prepared. Both samples exhibited a transition temperature of 37 °C, which was higher than that for samples dissolved in PBS. The lower LCST observed for diblock sample in PBS buffer is likely attributed to the salting out effect of kosmotropic ions added to the solution.[68,69] Cooling experiments were conducted to test the reversibility of this transition; the data are shown in Figure 4b. As illustrated in the figure, each sample shows only a small hysteresis in the transition, with LCSTs determined as 15 °C for PDEGMEMA, 29 °C for the diblock in PBS, and 34 °C for the diblock in H2O. The consistency of these values with those of the heating experiments confirms the reversibility of these transitions under the experimental conditions tested.
DLS was also utilized to study the size distribution of the nanostructures formed by the conjugate. Figure 5a and 5b show the size distributions for PDEGMEMA-CLP diblock samples dissolved in water, at 1 mg/mL and 5 mg/mL respectively. In both cases, no higher order assembly was observed at 25 °C. The small objects with diameters of approximately 5 nm were attributed to the soluble trimers of the diblock, consistent with previous reports.[18,21] For the 1 mg/mL sample, nanostructures with average diameter of approximately 45 nm were formed at higher temperatures, while nanostructures with diameters around 110 nm were observed in the 5 mg/mL sample. Larger structures formed at 5 mg/mL may be attributed to a fusion process of smaller structures at higher concentrations (see below). Although CLP peptides alone have been reported to aggregate at high concentrations,[19,58] a PEG-CLP diblock control at the lower concentrations here did not show any aggregation at any temperatures measured (Figure S6a), while on the other hand, heating a PDEGMEMA solution above its LCST (18 °C) yielded large aggregates with diameters of approximately 1.7 μm (Figure S6b). These observations confirm both the role of the PDEGMEMA in the thermally induced aggregation and the role of the CLP in stabilization of the observed nanostructures.
Figure 5.
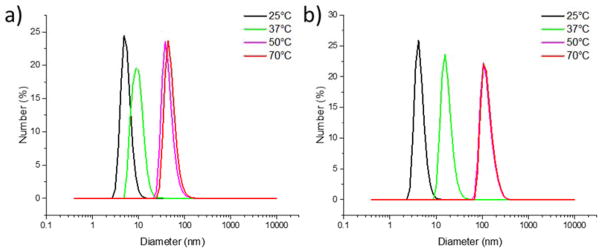
Size distribution of assemblies at different temperatures. a) PDEGMEMA-CLP diblock, 1 mg/mL in water; b) PDEGMEMA-CLP diblock, 5 mg/mL in water. Correlation functions and cumulant fits are provided in the Supporting Information (Figure S5).
To obtain insight into the morphologies of the nanostructures observed via DLS, transmission electron microscopy (TEM) measurements were also conducted. To better visualize the nanostructures, a 1% phosphotungstic acid (PTA, pH adjusted to 7.0 using NaOH) aqueous solution was used as negative stain. At 25 °C, which was below the LCST of PDEGMEMA domain in the diblock, no nanostructures were detected under any solution conditions (data not shown). When the diblock solutions were heated above the transition temperature, the polymer domain became hydrophobic and collapsed in aqueous solution, causing the solution to immediately become cloudy. Representative TEM images of the nanostructures formed at 50 °C by the 5 mg/mL sample are shown in Figure 6a and 6b. Nanostructures with an average diameter of approximately 70 – 200 nm were observed, which is consistent with the DLS measurements discussed above (Figure 5c). As observed in the images, both the exterior and interior surface of the nanoparticles are clearly observed, suggesting that the particles may have a hollow vesicular structure. It is possible that the solvent evaporation process employed during sample preparation destabilized the vesicles, and exposed the interior of the vesicles to the staining agent. Similar observations were also reported for bilayer vesicles formed from synthetic lipids 1,2-di-n-hexadecyloxypropyl-4-(beta-nitrostyryl) phosphate (DHPBNS) under aqueous conditions.[70] Some large particles with d ≈300 – 400 nm were also observed, which were likely generated via fusion of smaller particles during solvent evaporation (indicated by arrows in Figure 6a). For samples prepared at 1 mg/mL, smaller nanoparticles with an average diameter of approximately 50 nm are observed (Figure 6c and 6d), which is also consistent with DLS results (Figure 5b). The vesicular morphology of these particles was resolved more clearly when the sample grid was washed with deionized water after staining (Figure 6e and 6f). TEM analysis of PDEGMEMA solutions was found to be difficult practically because PDEGMEMA with Mw = 7 kDa is a liquid. As in the DLS experiments, no vesicular structures were observed for the hydrophilic-hydrophilic PEG-CLP diblock conjugate investigated under the same conditions, indicating that the thermally responsive collapse of the PDEGMEMA is crucial for the assembly of the vesicles.
Figure 6.

TEM images of nanoparticles formed from PDEGMEMA-CLP diblock at 50 °C, stained with phosphotungstic acid (1% PTA in water, pH adjusted to 7.0). a,b) 5 mg/mL; c,d) 1 mg/mL; e,f) 1 mg/mL, washed with deionized water after staining. Scale bars: 200 nm.
The nanoscale vesicular structures observed in this work are similar to those previously reported from other thermoresponsive polymer-based systems. For example, due to its high biocompatibility and its LCST of approximately 32 °C,[71] poly(N-isopropylacrylamide) (PNIPAAM) has become one of the most widely used building blocks for producing copolymers with thermoresponsive behavior. Otsuka et al.[72] observed vesicular morphologies with a diameter of 300 nm from the assembly of maltoheptose-block-poly(N-isopropylacrylamide) (Mal7-b-PNIPAAMn) hybrid diblock copolymers at 90 °C. More recently, Zhao et al.[73] synthesized polyethylene-graft-poly(N-isopropylacrylamide) copolymers (PE-g-PNIPAM) using coordination copolymerization and RAFT copolymerization. The amphiphilic graft copolymer formed vesicles with average diameters of 170–190 nm in aqueous solution when incubated at temperatures between 20–32 °C.
Nanoscale vesicles have also been observed from the assembly of other polymer-peptide conjugates, although to our knowledge not from samples in which the polymer block shows thermally responsive behavior. For example, Marsden et al.[74] reported vesicles with a diameter of 100 – 400 nm from the self-assembly, in aqueous conditions, of a conjugate diblock comprising poly(γ-benzyl L-glutamate) (PBLG) and a coiled-coil forming peptide with the sequence G(EIAALEK)3. Similar vesicular structures with diameter of 30 – 100 nm were also reported by Koga et al.,[75] using a β-sheet peptide-inserted amphiphilic block copolymer, polystyrene-block-tetra(leucine)-block-poly(ethylene glycol) (PS-b-L4-b-PEG). More recently, Bacinello et al.[76] reported the self-assembly of amphiphilic peptide-polymer hybrids comprising poly(trimethylene carbonate) (PTMC) and a matrix metalloproteinase-2 (MMP-2)-degradable peptide PVGLIG. Various morphologies including core-shell micelles and nanoscale vesicles with diameters of approximately 40 – 70 nm were reported. In one study involving CLP-based conjugates, Luo et al.[51] reported the self-assembly of a CLP-containing amphiphile with a C16 lipophilic tail attached to the C-terminus. Micrometer-long nanofibers with a diameter of approximately 16 nm were observed under aqueous conditions.
Compared with the surge in research on thermoresponsive polymers and other peptide-polymer conjugates, there are few reports focused on the assembly of collagen-like peptide containing copolymers, and none to our knowledge in which nanovesicles are produced. Scheme 3 illustrates the potential molecular organization of the vesicles observed at 50 °C in deionized water. Although other assembly mechanisms are possible, the proposed bilayer vesicular assembly pathway is likely for the nanostructures that we observed; the parallel orientation of the CLP domains are expected on the basis of previous reports.[24,27,30,77,78] At 25 °C, both building blocks of the diblock are hydrophilic, thus the diblock is fully soluble in deionized water, in the form of the triple helix-containing trimer. As the solution is heated to above the LCST of PDEGMEMA (37 °C), the polymer domain collapses and triggers coacervation. The radius of gyration (Rg) of 7 kDa PDEGMEMA in aqueous solution is calculated to be 2.2 nm below its LCST (good solvent) and 0.83 nm above LCST (bad solvent).[79] The collagen triple helix of (GPO)7GG is a semi-flexible rod with the length of 6.4 nm and diameter of 1.5 nm.[80] When the solution is heated above 37 °C, the diblock trimer thus can be treated as a rod-coil structure with dimensions of roughly 6.4 nm (rod)-1.2 nm (coil) (Rg of three anchored polymer chains). For a conventional coil-coil amphiphilic diblock, assemblies adopt rounded interfaces in order to minimize the interfacial contact between the solvent and the less soluble block. However, for rod-coil copolymers, the packing of the rods at a highly curved surface creates liquid crystalline defects, which increases the free energy of the assemblies.[81] It is likely that the tradeoffs between the liquid crystalline defect energy and surface energy give rise to the formation of vesicles, which have a lower interface curvature compared with micelles. Further heating of the solution to a higher temperature unfolds the collagen triple helix, but due to the stabilization of the collapsed PDEGMEMA as an anchoring point, this unfolding process is not completed until above 80 °C (Figure 3b and 3e). Thus at 50 °C, the CLP domain remains partially unfolded (Scheme 3). It is noteworthy that once the particles have been formed, their size does not change upon heating (Figure 4a), suggesting that unfolding of the CLP domain on the vesicle exterior and interior surfaces does not change the morphology of the vesicle. Although the size of the CLP may change during unfolding, the changes must be small compared to the size of the nanostructures. According to the TEM results (Figure 6f), the thickness of the vesicle surfaces are of approximately 10.5 – 19 nm, which is consistent with the calculated thickness of the bilayer proposed in Scheme 3 (approximately 15nm based on the length of the GPO7 triple helix (6.4 nm) and the Rg of three collapsed PDEGMEMA chains (~1.2 nm)).
Scheme 3.
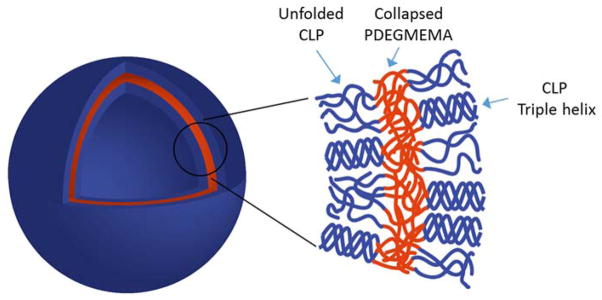
Schematic illustration of the potential structure of vesicles formed at 50 °C in aqueous condition.
Instead of using a conventional thermoresponsive polymer like PNIPAAM, the use of PDEGMEMA offers the improvement of a much lower LCST (18 °C in this case), so that after conjugation with the hydrophilic peptide, the polymer remains soluble at room temperature, with higher order assemblies being triggered at physiological temperature. Although understanding of the detailed role of the peptide trimerization and unfolding on the formation of observed nanostructures will require further investigation, the introduction of CLP domains at the exterior surface of the vesicles may serve as a specific means to target native collagens via triple helix hybridization. These peptide-based, dually thermally responsive conjugates thus offer intriguing opportunities for potential applications in targeted delivery of therapeutics for a range of pathologic conditions associated with collagen denaturation and reconstitution, such as tumor and rheumatoid arthritis.[82]
4. Conclusions
The conformational properties and assembly of a new class of thermoresponsive diblock conjugates, containing collagen-like peptides and PDEGMEMA, were introduced in this work. Circular dichroism (CD) experiments confirmed the ability for the peptide domain to adopt a triple helix conformation after conjugation with the polymer. The engineered LCST of these conjugates has enabled temperature-induced assembly under aqueous conditions, at physiologically relevant temperatures, into well-defined vesicles with diameters of approximately 50–200 nm. The formation of nanostructures was driven by the coil/globule conformational transition of the PDEGMEMA building block above its LCST with stabilization of the nanostructures by the hydrophilic CLP. To the best of our knowledge, this is the first report on such assembled nanostructures from collagen-like peptide containing copolymers. Due to the strong propensity for CLPs to bind to natural collagen via strand invasion processes,[29] these nanosized vesicles may be used as drug carriers for targeted delivery.
Supplementary Material
Acknowledgments
This work was funded in part by the National Science Foundation (DMR 0907478 to KLK) and the National Center for Research Resources (NCRR), a component of the National Institutes of Health (P30 GM103519 for instrument resources). This work was also supported in part by the Center for Neutron Science at UD under award (U.S. department of Commerce) #70NANB7H6178. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of NCRR or NIH. The authors wish to acknowledge Chaoying Ni, Fei Deng, Yingchao Chen, and Darrin Pochan at the University of Delaware for their advice and assistance with transmission electron microscopy. We also thank Rachel Letteri at University of Massachusetts Amherst for conducting gel permeation chromatography, which was supported by the Materials Research Facilities Network (MRFN) of the UMass MRSEC on Polymers (DMR-0820506).
Footnotes
Supporting Information is available online from the Wiley Online Library or from the author.
Contributor Information
Tianzhi Luo, Department of Materials Science and Engineering, University of Delaware, Newark, Delaware, 19716, USA.
Lirong He, Institute for Technical and Macromolecular Chemistry, University of Hamburg, Bundesstrasse 45, D-20146 Hamburg, Germany.
Prof. Patrick Theato, Email: theato@chemie.uni-hamburg.de, Institute for Technical and Macromolecular Chemistry, University of Hamburg, Bundesstrasse 45, D-20146 Hamburg, Germany
Prof. Kristi L. Kiick, Email: kiick@udel.edu, Department of Materials Science and Engineering, University of Delaware, Newark, Delaware, 19716, USA; Biomedical Engineering, University of Delaware, Newark, Delaware, 19716, USA; Delaware Biotechnology Institute, Newark, Delaware, 19711, USA
References
- 1.Kadler KE, Baldock C, Bella J, Boot-Handford RP. J Cell Sci. 2007;120:1955. doi: 10.1242/jcs.03453. [DOI] [PubMed] [Google Scholar]
- 2.Ramachandran GN, Kartha G. Nature. 1955;176:593. doi: 10.1038/176593a0. [DOI] [PubMed] [Google Scholar]
- 3.Ramshaw JAM, Shah NK, Brodsky B. J Struct Biol. 1998;122:86. doi: 10.1006/jsbi.1998.3977. [DOI] [PubMed] [Google Scholar]
- 4.Watanabe K, Nishio Y, Makiura R, Nakahira A, Kojima C. Int J Pharm. 2013;446:81. doi: 10.1016/j.ijpharm.2013.02.002. [DOI] [PubMed] [Google Scholar]
- 5.Kojima C, Suehiro T, Watanabe K, Ogawa M, Fukuhara A, Nishisaka E, Harada A, Kono K, Inui T, Magata Y. Acta Biomater. 2013;9:5673. doi: 10.1016/j.actbio.2012.11.013. [DOI] [PubMed] [Google Scholar]
- 6.Ruszczak Z, Friess W. Adv Drug Deliver Rev. 2003;55:1679. doi: 10.1016/j.addr.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 7.Lee CH, Singla A, Lee Y. Int J Pharm. 2001;221:1. doi: 10.1016/s0378-5173(01)00691-3. [DOI] [PubMed] [Google Scholar]
- 8.Luo TZ, Kiick KL. Eur Polym J. 2013;49:2998. doi: 10.1016/j.eurpolymj.2013.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yu SM, Li Y, Kim D. Soft Matter. 2011;7:7927. doi: 10.1039/C1SM05329A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shoulders MD, Raines RT. Annu Rev Biochem. Vol. 78. Palo Alto: 2009. p. 929. Annual Reviews. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fallas JA, Dong JH, Tao YZJ, Hartgerink JD. J Biol Chem. 2012;287:8039. doi: 10.1074/jbc.M111.296574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Persikov AV, Ramshaw JAM, Brodsky B. J Biol Chem. 2005;280:19343. doi: 10.1074/jbc.M501657200. [DOI] [PubMed] [Google Scholar]
- 13.Boudko SP, Engel J. J Mol Biol. 2004;335:1289. doi: 10.1016/j.jmb.2003.11.054. [DOI] [PubMed] [Google Scholar]
- 14.Barth D, Kyrieleis O, Frank S, Renner C, Moroder L. Chem-Eur J. 2003;9:3703. doi: 10.1002/chem.200304918. [DOI] [PubMed] [Google Scholar]
- 15.Yang W, Chan VC, Kirkpatrick A, Ramshaw JAM, Brodsky B. J Biol Chem. 1997;272:28837. doi: 10.1074/jbc.272.46.28837. [DOI] [PubMed] [Google Scholar]
- 16.He LR, Theato P. Eur Polym J. 2013;49:2986. [Google Scholar]
- 17.O’Leary LER, Fallas JA, Bakota EL, Kang MK, Hartgerink JD. Nat Chem. 2011;3:821. doi: 10.1038/nchem.1123. [DOI] [PubMed] [Google Scholar]
- 18.Przybyla DE, Chmielewski J. J Am Chem Soc. 2008;130:12610. doi: 10.1021/ja804942w. [DOI] [PubMed] [Google Scholar]
- 19.Krishna OD, Kiick KL. Biomacromolecules. 2009;10:2626. doi: 10.1021/bm900551c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cejas MA, Kinnney WA, Chen C, Vinter JG, Almond HR, Balss KM, Maryanoff CA, Schmidt U, Breslav M, Mahan A, Lacy E, Maryanoff BE. P Natl Acad Sci USA. 2008;105:8513. doi: 10.1073/pnas.0800291105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cejas MA, Kinney WA, Chen C, Leo GC, Tounge BA, Vinter JG, Joshi PP, Maryanoff BE. J Am Chem Soc. 2007;129:2202. doi: 10.1021/ja066986f. [DOI] [PubMed] [Google Scholar]
- 22.Rele S, Song YH, Apkarian RP, Qu Z, Conticello VP, Chaikof EL. J Am Chem Soc. 2007;129:14780. doi: 10.1021/ja0758990. [DOI] [PubMed] [Google Scholar]
- 23.Pires MM, Przybyla DE, Perez CMR, Chmielewski J. J Am Chem Soc. 2011;133:14469. doi: 10.1021/ja2042645. [DOI] [PubMed] [Google Scholar]
- 24.Pires MM, Lee J, Ernenwein D, Chmielewski J. Langmuir. 2012;28:1993. doi: 10.1021/la203848r. [DOI] [PubMed] [Google Scholar]
- 25.Hsu W, Chen YL, Horng JC. Langmuir. 2012;28:3194. doi: 10.1021/la204351w. [DOI] [PubMed] [Google Scholar]
- 26.Perez CMR, Rank LA, Chmielewski J. Chem Commun. 2014;50:8174. doi: 10.1039/c4cc03171g. [DOI] [PubMed] [Google Scholar]
- 27.Perez CMR, Panitch A, Chmielewski J. Macromol Biosci. 2011;11:1426. doi: 10.1002/mabi.201100230. [DOI] [PubMed] [Google Scholar]
- 28.Hernandez-Gordillo V, Chmielewski J. Biomaterials. 2014;35:7363. doi: 10.1016/j.biomaterials.2014.05.019. [DOI] [PubMed] [Google Scholar]
- 29.Wang AY, Mo X, Chen CS, Yu SM. J Am Chem Soc. 2005;127:4130. doi: 10.1021/ja0431915. [DOI] [PubMed] [Google Scholar]
- 30.Li Y, Ho D, Meng H, Chan TR, An B, Yu H, Brodsky B, Jun AS, Yu SM. Bioconjugate Chem. 2013;24:9. doi: 10.1021/bc3005842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang AY, Foss CA, Leong S, Mo X, Pomper MG, Yu SM. Biomacromolecules. 2008;9:1755. doi: 10.1021/bm701378k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li Y, Foss CA, Summerfield DD, Doyle JJ, Torok CM, Dietz HC, Pomper MG, Yu SM. P Natl Acad Sci USA. 2012;109:14767. doi: 10.1073/pnas.1209721109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Krishna OD, Jha AK, Jia XQ, Kiick KL. Biomaterials. 2011;32:6412. doi: 10.1016/j.biomaterials.2011.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang Z, Lai YX, Yu L, Ding JD. Biomaterials. 2010;31:7873. doi: 10.1016/j.biomaterials.2010.07.014. [DOI] [PubMed] [Google Scholar]
- 35.Humtsoe JO, Kim JK, Xu Y, Keene DR, Hook M, Lukomski S, Wary KK. J Biol Chem. 2005;280:13848. doi: 10.1074/jbc.M410605200. [DOI] [PubMed] [Google Scholar]
- 36.Harris JM, Chess RB. Nat Rev Drug Discov. 2003;2:214. doi: 10.1038/nrd1033. [DOI] [PubMed] [Google Scholar]
- 37.Klok HA. J Polym Sci Pol Chem. 2005;43:1. [Google Scholar]
- 38.Ding JX, Xiao CS, Tang ZH, Zhuang XL, Chen XS. Macromol Biosci. 2011;11:192. doi: 10.1002/mabi.201000238. [DOI] [PubMed] [Google Scholar]
- 39.Wigenius J, Bjork P, Hamedi M, Aili D. Macromol Biosci. 2010;10:836. doi: 10.1002/mabi.200900463. [DOI] [PubMed] [Google Scholar]
- 40.Shu JY, Tan C, DeGrado WF, Xu T. Biomacromolecules. 2008;9:2111. doi: 10.1021/bm800113g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sahin E, Kiick KL. Biomacromolecules. 2009;10:2740. doi: 10.1021/bm900474k. [DOI] [PubMed] [Google Scholar]
- 42.Kopecek J, Yang JY. Abstr Pap Am Chem Soc. 2011;241 [Google Scholar]
- 43.Elder AN, Dangelo NM, Kim SC, Washburn NR. Biomacromolecules. 2011;12:2610. doi: 10.1021/bm200393k. [DOI] [PubMed] [Google Scholar]
- 44.Lee OS, Liu YM, Schatz GC. J Nanopart Res. 2012;14 [Google Scholar]
- 45.Aluri S, Pastuszka MK, Moses AS, MacKay JA. Biomacromolecules. 2012;13:2645. doi: 10.1021/bm300472y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Grieshaber SE, Farran AJE, Lin-Gibson S, Kiick KL, Jia XQ. Macromolecules. 2009;42:2532. doi: 10.1021/ma802791z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Grieshaber SE, Farran AJE, Bai S, Kiick KL, Jia XQ. Biomacromolecules. 2012;13:1774. doi: 10.1021/bm3002705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen Y, Youn P, Furgeson DY. J Control Release. 2011;155:175. doi: 10.1016/j.jconrel.2011.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yu YC, Berndt P, Tirrell M, Fields GB. J Am Chem Soc. 1996;118:12515. [Google Scholar]
- 50.Gore T, Dori Y, Talmon Y, Tirrell M, Bianco-Peled H. Langmuir. 2001;17:5352. [Google Scholar]
- 51.Luo JN, Tong YW. Acs Nano. 2011;5:7739. doi: 10.1021/nn202822f. [DOI] [PubMed] [Google Scholar]
- 52.Krishna OD, Wiss KT, Luo TZ, Pochan DJ, Theato P, Kiick KL. Soft Matter. 2012;8:3832. doi: 10.1039/C2SM07025A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wiss KT, Krishna OD, Roth PJ, Kiick KL, Theato P. Macromolecules. 2009;42:3860. [Google Scholar]
- 54.Roth PJ, Wiss KT, Zentel R, Theato P. Macromolecules. 2008;41:8513. [Google Scholar]
- 55.Persikov AV, Ramshaw JAM, Kirkpatrick A, Brodsky B. Biochemistry-Us. 2005;44:1414. doi: 10.1021/bi048216r. [DOI] [PubMed] [Google Scholar]
- 56.Sakakiba S, Tanaka N, Kakudo M, Okuyama K, Ashida T, Kishida Y. J Mol Biol. 1972;65:371. doi: 10.1016/0022-2836(72)90288-4. [DOI] [PubMed] [Google Scholar]
- 57.Persikov AV, Ramshaw JAM, Kirkpatrick A, Brodsky B. Biochemistry-Us. 2000;39:14960. doi: 10.1021/bi001560d. [DOI] [PubMed] [Google Scholar]
- 58.Kar K, Amin P, Bryan MA, Persikov AV, Mohs A, Wang YH, Brodsky B. J Biol Chem. 2006;281:33283. doi: 10.1074/jbc.M605747200. [DOI] [PubMed] [Google Scholar]
- 59.Miles CA. Biopolymers. 2007;87:51. doi: 10.1002/bip.20787. [DOI] [PubMed] [Google Scholar]
- 60.Cheon JB, Kim BC, Park YH, Park JS, Moon JY, Nahm JH, Cho CS. Macromol Chem Phys. 2001;202:395. [Google Scholar]
- 61.Qin SH, Geng Y, Discher DE, Yang S. Adv Mater. 2006;18:2905. [Google Scholar]
- 62.Topp MDC, Dijkstra PJ, Talsma H, Feijen J. Macromolecules. 1997;30:8518. [Google Scholar]
- 63.Zhang WQ, Shi LQ, Wu K, An YG. Macromolecules. 2005;38:5743. [Google Scholar]
- 64.Isoda K, Kanayama N, Miyamoto D, Takarada T, Maeda M. React Funct Polym. 2011;71:367. [Google Scholar]
- 65.Zhao CW, Zhuang XL, He CL, Chen XS, Jing XB. Macromol Rapid Commun. 2008;29:1810. [Google Scholar]
- 66.Nuhn H, Klok HA. Biomacromolecules. 2008;9:2755. doi: 10.1021/bm800784y. [DOI] [PubMed] [Google Scholar]
- 67.McDaniel JR, Radford DC, Chilkoti A. Biomacromolecules. 2013;14:2866. doi: 10.1021/bm4007166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cacace MG, Landau EM, Ramsden JJ. Q Rev Biophys. 1997;30:241. doi: 10.1017/s0033583597003363. [DOI] [PubMed] [Google Scholar]
- 69.Zhang YJ, Cremer PS. Curr Opin Chem Biol. 2006;10:658. doi: 10.1016/j.cbpa.2006.09.020. [DOI] [PubMed] [Google Scholar]
- 70.Ravoo BJ, Stuart MCA, Brisson ADR, Weringa WD, Engberts J. Chem Phys Lipids. 2001;109:63. doi: 10.1016/s0009-3084(00)00203-6. [DOI] [PubMed] [Google Scholar]
- 71.Wei H, Cheng SX, Zhang XZ, Zhuo RX. Prog Polym Sci. 2009;34:893. [Google Scholar]
- 72.Otsuka I, Fuchise K, Halila S, Fort S, Aissou K, Pignot-Paintrand I, Chen YG, Narumi A, Kakuchi T, Borsali R. Langmuir. 2010;26:2325. doi: 10.1021/la902743y. [DOI] [PubMed] [Google Scholar]
- 73.Zhao Y, Gao HY, Liang GD, Zhu FM, Wu Q. Polym Chem. 2014;5:962. [Google Scholar]
- 74.Marsden HR, Handgraaf JW, Nudelman F, Sommerdijk N, Kros A. J Am Chem Soc. 2010;132:2370. doi: 10.1021/ja909540a. [DOI] [PubMed] [Google Scholar]
- 75.Koga T, Kamiwatari S, Higashi N. Langmuir. 2013;29:15477. doi: 10.1021/la404328b. [DOI] [PubMed] [Google Scholar]
- 76.Bacinello D, Garanger E, Taton D, Tam KC, Lecommandoux S. Biomacromolecules. 2014;15:1882. doi: 10.1021/bm500296n. [DOI] [PubMed] [Google Scholar]
- 77.Pires MM, Ernenwein D, Chmielewski J. Biomacromolecules. 2011;12:2429. doi: 10.1021/bm2004934. [DOI] [PubMed] [Google Scholar]
- 78.Li Y, Yu SM. Curr Opin Chem Biol. 2013;17:968. doi: 10.1016/j.cbpa.2013.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Meuer S, Braun L, Schilling T, Zentel R. Polymer. 2009;50:154. [Google Scholar]
- 80.Brodsky B, Persikov AV. In: Fibrous Proteins: Coiled-Coils, Collagen and Elastomers. Parry DAD, Squire JM, editors. Vol. 70. Elsevier Academic Press Inc; San Diego: 2005. p. 301. [Google Scholar]
- 81.Olsen BD, Segalman RA. Mater Sci Eng R-Rep. 2008;62:37. [Google Scholar]
- 82.Holmdahl R, Bockermann R, Backlund J, Yamada H. Ageing Res Rev. 2002;1:135. doi: 10.1016/s0047-6374(01)00371-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.