

Abstract
Great success in HCV therapy was achieved by the development of direct-acting antivirals (DAA). However, the unsolved issues such as high cost and genotype dependency drive us to pursue additional therapeutic agents to be used instead or in combination with DAA. The cationic peptide p41 is one of such candidates displaying submicromolar anti-HCV potency. By electrostatic coupling of p41 with anionic poly(amino acid)-based block copolymers, antiviral peptide nanocomplexes (APN) platform was developed to improve peptide stability and to reduce cytotoxicity associated with positive charge. Herein, we developed a facile method to prepare galactosylated Gal-APN and tested their feasibility as liver-specific delivery system. In vitro, Gal-APN displayed specific internalization in hepatoma cell lines. Even though liver-targeted and non-targeted APN displayed comparable antiviral activity, Gal-APN offered prominent advantages to prevent HCV association with lipid droplets and suppress intracellular expression of HCV proteins. Moreover, in vivo preferential liver accumulation of Gal-APN was revealed in the biodistribution study. Altogether, this work illustrates the potential of Gal-APN as a novel liver-targeted therapy against HCV.
Keywords: antiviral peptide nanoparticle, liver-targeting, anionic poly(amino acid)-based block copolymer, HCV RNA, HCV proteins, biodistribution
1. Introduction
More than 185 million people worldwide are infected with the Hepatitis C Virus (HCV) [1]. This blood-borne pathogen leads to chronic hepatitis, liver cirrhosis, and hepatocellular carcinoma, and it is also the primary cause of liver transplantation in the United States [2]. In spite of being such a fatal disorder, the standard of care until 2011 was restricted to the combination of pegylated interferon-α (PEG IFN-α) and ribavirin, which exhibited limited sustained viral response and poor tolerability [3]. The emergence of direct-acting antivirals (DAA), which specifically block HCV replication, has marked a new era in HCV therapy [4]. However, the challenges such as affordability, genotype selectivity and possible long-term resistance to DAA therapy due to viral mutations [5] still necessitate the development of alternative anti-HCV drug candidates. As an option, therapeutic peptides are revolutionizing the paradigm of drug development with their prominent bioactivity [6], and, in fact, several anti-HCV peptides, targeting different stages of HCV life cycle, have been discovered [7–9]. One such, the 18-mer C5A, which is derived from the membrane anchor domain of HCV nonstructural protein NS5A and exhibits broad spectrum antiviral activity with submicromolar potency in vitro [10, 11]. The characteristics of α-helicity and amphipathicity allow this peptide to neutralize the viral particles by destabilizing the lipid composition of viral membranes without genotype dependency [12]. Its cationic derivative, with 4 residues in C5A replaced with positively charged amino acids (lysine and arginine), which we refer to as p41, also displays equivalent antiviral potency. However, the susceptibility to proteolysis and the unfavorable toxicity profile, typical for cationic peptides, remain the obstacles in their translation to clinics.
In an attempt to overcome these restraints, our group has successfully developed the well-defined antiviral peptide nanocomplexes (APN) through the immobilization of cationic p41 into nanoscale block ionomer complexes with oppositely charged poly(amino acid)-based block copolymers [13]. The p41 encapsulation into APN led to higher proteolytic stability, reduced cytotoxicity and unaltered antiviral potency of the peptides. The self-assembly behavior and preparation simplicity make the APN an extremely promising approach for peptide delivery. Using the APN platform, a more selective delivery of p41 to hepatocytes as primary sites for HCV replication can be achieved through targeting the asialoglycoprotein receptor (ASGP-R), which is a well-defined endocytotic receptor primarily expressed on parenchymal liver cells [14–16]. Therefore, we hypothesize that the decoration of the APN surface with the ligand β-D-galactose (Gal), which possesses high binding affinity to ASGP-R (Kd ~ 10−3–10−4 M) [17, 18], will target APN to the liver, thereby providing a basis for organ-specific anti-HCV therapy development.
Here, we have successfully prepared a series of APN with varying densities of the Gal ligand on the surfaces (Gal-APN) and evaluated their antiviral activities in cell culture systems. Biodistribution studies demonstrated preferential liver accumulation of Gal-decorated APN proving the effectiveness of this targeting approach for the delivery of antiviral peptides.
2. Materials and Methods
2.1 Materials
Peptide p41 (SWLRRIWRWICKVLSRFK) and Cy5-labeled p41 were custom synthesized by AnaSpec (USA). α-(9-Fluorenylmethyloxycarbonyl)amino-ω-carboxy succinimidyl ester poly(ethylene glycol) (Fmoc-PEG-NHS, MW (PEG) = 5000 g mol−1, Mw/Mn = 1.02) was purchased from Jenkem Technology (China). Amberlyst® 15 hydrogen form, molecular sieve UOP type 3 Å, L-glutamic acid γ-benzyl ester, D (+)-galactose, propargylamine, L(+)-ascorbic acid sodium salt and copper (II) sulfate pentahydrate, 98+%, A.C.S. reagent were obtained from Sigma-Aldrich (U.S.). Silica gel (for chromatography, 0.03–0.200 mm, 60 Å), 2-bromoethanol, ethyl acetate, methanol, dichloromethane (DCM), dimethylformamide (DMF), tetrahydrofuran (THF) were purchased from Acros Organics (USA). S-2-(4-Isothiocyanatobenzyl)-diethylenetriamine pentaacetic acid (SCN-DTPA) was ordered from Macrocyclics (Dallas, TX, USA). Lutetium-177 trichloride was obtained from PerkinElmer (USA).
2.2 Synthesis of Gal-terminated poly(ethylene glycol)-block-poly(L-glutamic acid) copolymer (Gal-PEG-b-PLE)
Gal-PEG-b-PLE copolymer was synthesized using the following steps (Scheme 1):
Scheme 1.

Synthesis of Gal-PEG-b-PLE via NCA-based ring-opening polymerization and subsequent click chemistry.
2.2.1 Synthesis of propargyl-PEG-NH2
Fmoc-PEG-NHS (0.5 g, 0.1 mmol) was dissolved under stirring in 5 ml of DCM at 0°C. The propargylamine (80 μl, 1.2 mmol) dissolved in 5 ml of cold DCM was added dropwise and the solution was stirred for 24 h. The solvent was removed at reduced pressure and the product was dialyzed against distilled water (MWCO 2 kDa) followed by lyophilization to give Fmoc-PEG-propargyl. Standard 20% v/v piperidine/DCM solution (3 ml) was used to remove the Fmoc-protecting group. After stirring for 2 h at room temperature, DCM was removed by rotaevaporation, reaction mixture was redissolved in water and after filtration dialyzed against distilled water (MWCO 2 kDa) for 2 days, and then lyophilized to obtain propargyl-PEG-NH2.
2.2.2 Synthesis of propargyl-PEG-b-PLE
The BLE-NCA (γ-benzyl L-glutamate-N-carboxyanhydride) monomer was prepared as described previously [19]. Benzyl-protected propargyl-PEG-b-PBLE was synthesized via ring-opening polymerization of BLE-NCA monomer using propargyl-PEG-NH2 as a macroinitiator. Propargyl-PEG-NH2 (20 mg, 0.004 mmol) was dissolved in 300 μl of anhydrous DMF with 3 μl of triethylamine (TEA) (5 equivalent of PEG) and then added into water-free and oxygen-free ampules. BLE-NCA (26.3 mg, 0.1 mmol, the feed molar ratio of propargyl-PEG-NH2 to BLE-NCA was 1:25) dissolved in 250 μl of DMF was added dropwise under nitrogen. The ampule was sealed and the reaction was allowed to proceed at 40°C for 48 h. The solvent was evaporated under vacuum and the residue was dissolved in THF followed by addition of 2 N NaOH to deprotect glutamate residues. After stirring for 4 h at 40°C, THF was removed at reduced pressure, the residual solution was neutralized by 1 M HCl, dialyzed using a dialysis membrane (MWCO 3.5 kDa) against distilled water for 48 h, and lyophilized to obtain propargyl-PEG-b-PLE.
2.2.3 Synthesis of 2′-azidoethyl-O-D-galactopyranoside
The 2′-azidoethyl-O-D-galactopyranoside was synthesized according the procedure reported by Geng et al. [20]. Briefly, the Amberlyst® 15 hydrogen form (8.0 g) and molecular sieve (4.0 g) suspended in 2-bromoethanol (50 ml) were refluxed for 30 min followed by addition of D(+)-galactose (8.0 g). After another 3 h reflux, the product formation was confirmed by TLC (ethyl acetate/methanol, 7/1, v/v, Rf: 0.3), reaction mixture was filtered, and rotaevaporated. The crude 2′-bromoethyl-D-galactopyranoside was further purified by flash column chromatography on silica gel. The obtained 2′-bromoethyl-D-galactopyranoside (3.0 g, 10.5 mmol) and sodium azide (2.1 g, 21 mmol) were further dissolved in water/acetone (7.5 ml/45 ml) and refluxed for 20 h. After removal of solvents by rotaevaporation, the residue was suspended in ethanol and filtered to remove an excess of salts (sodium azide and sodium bromide). The filtrate was concentrated under vacuum to obtain the final product as a white solid. The structure was characterized by 1H-NMR [Bruker AVANCE III instrument (400 MHz)] and mass spectrometry (Qtof Micro electrospray ionization mass spectrometer, Waters, USA).
2.2.4 Galactosylation via click reaction
The 2′-azidoethyl-O-D-galactopyranoside (3.12 mg, 20 μmol) was reacted with propargyl-PEG-b-PLE (20 mg, 4 μmol) dissolved in 100 μl of a water and methanol (1:1) mixture in the presence of CuSO4•5H2O (62.42 μg, 0.25 μmol), tris-(hydroxypropyltriazolylmethyl)amine (218.5 μg, 0.5 μmol) as the stabilizing agent and ascorbic acid (495.28 μg, 2.5 μmol) for 2 days at room temperature. The product was dialyzed against distilled water (MWCO 3.5 kDa) for another 2 days to remove all the unreacted small molecules and then lyophilized to produce Gal-PEG-b-PLE.
2.2.5 Polymer characterization
The composition of propargyl-PEG-b-PBLE was confirmed by 1H-NMR in DMSO-d6 at 80°C using a Bruker 400 MHz spectrometer. Gel permeation chromatography (GPC) measurements to determine the molecular weights and polydispersity (PDI = Mw/Mn) of the polymers were carried out at 40°C using a Shimadzu liquid chromatography system equipped with TSK-GEL® column (G4000HHR) connected to Shimadzu RI and UV/vis detectors. DMF was used as mobile phase at a flow rate of 0.6 ml/min. Poly(ethylene glycol) standards (Agilent Technologies, USA) with a molecular weight range of 106 – 34,890 were used to generate the standard curve. The Gal conjugation efficiency was determined by phenol-sulfuric acid method in microplate format [21]. Briefly, 150 μl of concentrated sulfuric acid was added to 50 μl of each sample in a series of standard aqueous solutions of D(+)-Galactose with concentrations ranging from 0.02 mM to 4 mM or Gal-PEG-b-PLE, followed by adding 30 μl of 5% phenol in water. Samples were incubated for 5 min at 90°C in a water bath. After cooling to room temperature for 5 min, the absorbance at 490 nm was measured using SpectraMaxM5 spectrophotometer (Molecular Devices Co., USA).
2.3 Gal-APN preparation and characterization
Gal-APN (Z−/+ = 1) were prepared by mixing buffered solutions (PBS, pH 7.4) of p41 and anionic block copolymers (Gal-PEG-b-PLE and propargyl-PEG-b-PLE) as described earlier [13]. The composition of the mixtures (Z−/+) was calculated as a molar ratio of carboxylic groups in the copolymers to the amino groups of lysine and arginine residues of the peptide. To obtain the APN with varying Gal densities on the surface, Gal-PEG-b-PLE and propargyl-PEG-b-PLE were combined in different molar ratios (Scheme 2). Cy5-labeled formulations were prepared in the same way by introducing 10 mol% of Cy5-labeled p41 into the APN. For all in vitro cellular studies, APN concentration was 5 μM on the basis of p41. Intensity-mean z-averaged particle diameter (Deff) and polydispersity index (PDI) of the APN were determined by dynamic light scattering (DLS) using Nano ZS Zetasizer (Malvern Instruments, UK). All measurements were performed in automatic mode at 25°C at a fixed 173° scattering angle. Software provided by manufacturer was used to calculate the Deff and PDI of APN. Data are presented as mean values ± SEM (n = 3).
Scheme 2.
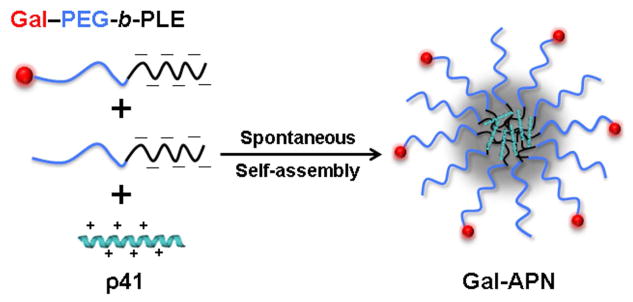
Schematic illustration of the preparation of Gal-APN.
2.4 Cell culture
Huh 7, Huh 7.5, HepG2 (hepatocarcinoma cell lines) and HeLa cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. Primary human hepatocytes isolated from a 61-year-old female with 70% viability were obtained through the Liver Tissue Cell Distribution System (Pittsburg, PA) funded by NIH (contract # HSN276201200017C). They were seeded on the coverslips coated with collagen IV and cultured in William’s E medium supplemented with nicotinamide, HEPES, sodium bicarbonate, sodium pyruvate, glucose, glutamine (Acros), L-ascorbic acid 2-phosphate sesquimagnesium salt hydrate, penicillin/streptomycin (Sigma), dexamethasone (Alexis Biochemical), ITS+Premix (BD) and 10% FBS. All cells were cultured at 37°C, 5% CO2.
2.5 Cellular uptake studies
Huh 7 or HepG2 cells (10,000 cells/well) or primary human hepatocytes (300,000 cells/well) were grown on sterile glass coverslips placed in 24-well plates and exposed to Cy5-labeled Gal-APN, APN or p41. After determined time periods, the cells were rinsed, fixed with 4% paraformaldehyde (PFA) at room temperature for 30 min, and stained with 4′,6-diamidino-2-phenylindole (DAPI). The slides were mounted with ProLong® Gold Antifade (Invitrogen, USA) and images were acquired using a LSM 710 confocal microscope (Carl Zeiss, USA). In select experiments, cells were exposed to Cy5-labeled Gal-APN with surface Gal densities ranging from 5% to 70% for 2 h, followed by washing and visualization under the confocal microscope. All the treatments were performed in duplicate. About 20 images containing 1,000–1,500 cells were collected for each time point. ImageJ (NIH) software was used for the quantification of the fluorescence from Cy5-labeled APN or p41. Mean pixel intensities in each image were normalized to the total cell number by counting the number of DAPI-labeled nuclei. For the competition assay, HepG2 (100,000 cells/well in 24-well plates) were pretreated with free D(+)-galactose (100 mM) for 0.5 h then co-incubated with APN formulations for another 1 h and imaged as described above.
For assessment of cellular uptake by flow cytometry, Huh 7 cells (400,000 cells/well) were exposed to Cy5-labeled Gal(35%)-APN or APN for 2 h, washed with PBS, trypsinized at 37°C, and harvested by centrifugation (1500 rpm, 5 min). Thereafter, the cells were resuspended in PBS, stained using LIVE/DEAD (Invitrogen, USA) to assess cell viability, fixed with 4% PFA and the median fluorescence intensity (Cy5) of each sample was measured by flow cytometry (Becton Dickinson LSR II) and analyzed using FlowJo software (Tree Star Inc.). At least 10,000 live cells from each sample were acquired in linear mode, gated to exclude debris and dead cells, and visualized in logarithmic mode. For the competition assay, D(+)-galactose (100 mM) was added to the cells for 0.5 h followed by co-incubation with Gal(35%)-APN or APN and the experiment was carried out as described above.
2.6 In vitro anti-HCV activity
To study the antiviral efficacy of Gal-APN in cell culture systems, full-length genotype 2a HCV clone JFH-1 (provided by Dr. Takaji Wakita) that replicates and produces infectious viral particles was used as a source of virus [22]. Huh 7.5 hepatoma cell line, which support HCV replication in vitro, was obtained from Dr. Charles Rice (The Rockefeller University, New York, NY) and was used in all experiments requiring viral infection. After attachment of 200,000 Huh 7.5 cells/well to 6-well plates, cells were infected with JFH-1 virus (MOI 0.1) overnight, then virus was removed and cells were replenished with fresh medium containing APN or p41 for another 48 h (post-treatment regimen). To study virocidal effects, JFH-1 virus stock solution was pre-incubated with APN or p41 for 1 h at 37°C and then these mixtures were used to infect Huh 7.5 cells at MOI 0.1 overnight. Supernatant was removed and cells were replenished with fresh media for another 48 h. HCV RNA quantification was performed using a StepOne Realtime PCR system amplifying a highly conserved sequence in the 5′ UTR of the viral genome. Total cellular RNA was extracted using the TRIzol reagent (Invitrogen, USA) and was subsequently converted to cDNA using Verso cDNA Synthesis Kit (Thermo Scientific, USA). The following primers and probe for this consensus sequence were designed using PrimerExpress Software v2.0 (Applied Biosystems): 5′UTRF GACCGGGTCCTTTCTTGGAT; 5′UTRR CCAACACTACTCGGCTAGCAGTCT; probe FAM-ATTTGGGCGTGCCCCCGC-NFQ. Positive and negative controls were included in all runs. The relative HCV RNA expression level in infected cells was quantified after normalization to cellular GAPDH mRNA levels.
2.7 Assessment of intracellular HCV localization
Huh 7.5 cells (50,000 cells/well in 24-well plates) were treated with Gal-APN, APN or p41 for 5 h, infected with JFH-1 virus (MOI 0.1) overnight, virus was removed and cells were incubated for another 48 h in fresh media. After treatment, cells were fixed with 4% PFA for 15 min at 37°C and permeabilized with 0.5% Triton X-100 for 5 min at room temperature. Goat serum in PBS (5%) was used for blocking for 30 min. For co-localization analysis of HCV core protein with lipid droplets, cells were incubated with primary HCV core antigen antibody (clone: C7-50) (Thermoscientific, Cat# MA1-080) for 1 h with dilution of 1:300. For co-localization analysis of NS5A with lipid droplets, cells were incubated with HCV NS5A antibody (1:2000) for 1 h. After washing, cells were probed with the mixture of corresponding Alexa Fluor 594-labeled secondary antibody for NS5A or HCV core and BODIPY for lipid droplets (1:100) for another 1 h. At the end, nuclei were labeled with DAPI and cells were subjected to confocal imaging analysis.
2.8 Biodistribution study
2.8.1 Synthesis of 177Lu-DTPA-p41
Prior to radiolabeling, the radiometal chelator (DTPA) was non-selectively conjugated to the amine groups on the p41 peptides. Briefly, 1 mg of p41 (0.14 μmol) and TEA (0.29 μl, 5 equivalent of p41) were dissolved in DMF. Upon stirring, SCN-DTPA (1.5 equiv of p41) in DMF was added dropwise to solution. The reaction mixture was kept stirring at room temperature for overnight. DTPA-p41 was characterized and peak-purified by RP-HPLC/MS. The UV detector connected to HPLC was set at 280 nm and 220 nm. The mobile phase was composed of water containing 0.1% formic acid (solvent A) and acetonitrile containing 0.1% formic acid (solvent B). For all purifications, the initial gradient of 62% A:38% B linearly decreased to 40% A:60% B over a 12 min time period. At the end of the run time for all HPLC experiments, the column was flushed with the gradient of 5% A:95% B and re-equilibrated to the starting gradient. To synthesize 177Lu-DTPA-p41, 30 μg of DTPA-p41 was incubated with 2.5 mCi 177LuCl3 in ammonium acetate buffer (1 M, 100 μl, pH 5.5) for 1 h at room temperature. The resulting 177Lu-DTPA-p41 was peak-purified by RP-HPLC equipped with Flow Count radiometric detector system (Bioscan, USA) and UV detector (220 nm and 280 nm). The isolated radiochemical yield was 10%–20%. L(+)-ascorbic acid sodium salt (10 mg) was added to the purified radioconjugate to reduce radiolysis. To remove the bulk of the acetonitrile and concentrate the radiolabeled peptide, dry nitrogen was applied to the mixture of 177Lu-DTPA-p41 and non-labeled p41. 177Lu-labeled APN formulations were prepared by following the same procedure as described in 2.3.
2.8.2 Animal studies
Biodistribution study of APN (targeted and non-targeted) and p41 were carried out on BALB/c mice (average weight, 25 g). Each animal was intravenously injected with 50 μg of 177Lu-labeled p41 or equivalent dose of 177Lu-labeled Gal-APN or APN (n = 6 per group) carrying 5 μCi radioactivity in 100 μl saline. After the designated time (15 min and 4 h), the mice were sacrificed, and tissues (liver, spleen, kidney, lung and bone) were excised, rinsed and weighed. The 177Lu radioactivity of excised organs, tail, urine + bedding, and carcasses was measured using a gamma counter, and the percentage of injected dose per gram (%ID/g) was calculated for each organ.
2.9 Statistical analysis
The data were expressed as mean ± standard deviation. For comparison between two groups, the data were analyzed using student’s t test. For multiple groups, one-way analysis of variance (ANOVA) was used. Differences were considered statistically significant at a value of p < 0.05.
3. Results
3.1 Preparation and characterization of Gal-APN
The synthesis of Gal-modified PEG-b-PLE copolymer is illustrated in Scheme 1. To this end, propargyl-terminated PEG-b-PLE block copolymer was synthesized via ring-opening polymerization of BLE-NCA monomer using propargyl-PEG-amine as a macroinitiator. The chemical composition of the resulting block copolymer was confirmed by 1H-NMR (Supporting information, Fig. S1). The degree of polymerization of the PLE block was 25 (Table 1). From the GPC analysis, the weight-average molecular weight (Mw) and the molecular weight distribution (Mw/Mn) of propargyl-PEG-b-PLE were determined to be 10,618 and 1.08, respectively (Table 1 and Fig. S1). Pursuing Gal conjugation to the copolymer using click chemistry, the azide-modified Gal was synthesized by the following two-step modification [20]: D(+)-galactose reacted with 2-bromoethanol first to give the bromo substituent, which was subsequently converted to Gal azide with sodium azide. With its molecular mass confirmed by mass spectrometry (Fig. S2), Gal azide and propargyl-PEG-b-PLE were reacted in the presence of copper (II) sulfate and sodium ascorbate in water/methanol mixture at room temperature. 1H-NMR (Fig. S1) confirmed the conversion of the alkyne groups to the triazole. The Gal conjugation yield was quantified using phenol-sulfuric acid method [21], and it showed that 67% ± 6% of the copolymer was successfully modified with Gal after click reaction. The absence of copper after 3 days’ exhaustive dialysis was confirmed by inductively coupled plasma mass spectrometry (data not shown).
Table 1.
Characteristics of propargyl-PEG-b-PBLE determined by 1H-NMR and GPC
| propargyl-PEG-b-PBLE | Feeding Ratio PEG/BLE-NCA | Degree of Polymerization | Mw (g/mol) | Mn (g/mol) | PDI |
|---|---|---|---|---|---|
| GPC | 1:25 | 26 | 10618 | 9800 | 1.08 |
| 1H-NMR | 25 | 10475 |
Gal-APN were prepared as described before [13] by simple mixing of buffered solutions of cationic p41 and anionic block copolymers (Gal-PEG-b-PLE and propargyl-PEG-b-PLE) which electrostatically bind to each other (Scheme 2). The Gal density on the surface of the Gal-APN was controlled by adjusting the molar ratio of Gal-PEG-b-PLE and propargyl-PEG-b-PLE copolymers. In this way, we prepared a series of Gal-APN with various proportions of Gal moieties ranging from 5% to 70%. APN prepared from p41 and propargyl-PEG-b-PLE was used as a non-targeted control for comparison. Gal-APN displayed a slight increase in size compared to non-targeted APN as more Gal moieties were introduced onto the APN surface (from 89 nm to 108 nm), and in all cases exhibited uniform size distributions (PDI less than 0.2) as determined by DLS (Table 2).
Table 2.
Physicochemical characteristics of Gal-APNa
| Gal Density | Deff (nm)b | PDIb |
|---|---|---|
| 0% | 89 ± 1 | 0.13 |
| 14% | 96 ± 5 | 0.14 |
| 28% | 98 ± 4 | 0.10 |
| 35% | 103 ± 5 | 0.13 |
| 42% | 98 ± 2 | 0.15 |
| 56% | 93 ± 2 | 0.13 |
| 70% | 108 ± 1 | 0.20 |
Complexes were prepared in PBS (pH 7.4, 0.14 M NaCl) at Z−/+=1. p41 concentration was 50 μM.
Effective diameter (Deff) and polydispersity indices (PDI) were determined by DLS at pH 7.4, 25°C (n = 3).
3.2 Uptake of Gal-APN in hepatoma cell lines
The internalization of Gal-APN was studied in Huh 7 cells with relatively low ASGP-R expression and in HepG2 cells, which overexpress the ASGP-R (Fig. S3). We prepared a fluorescence-labeled APN by incorporation of 10 mol% of Cy5-labeled p41 into complexes as a tracer (Table S1). The confocal microscopy suggested that Gal(35%)-APN were taken up rapidly into the Huh 7 cells, whereas the non-targeted APN were internalized more gradually (Fig. 1A). Indeed, nearly all of the cells became Cy5-positive after 2 h incubation with Gal(35%)-APN, while at least 5 h were needed for all cells to take up APN (Fig. S4). Notably, p41 alone displayed the least efficient internalization. The quantification of cell-associated fluorescence (Fig. 1B) indicated that both Gal(35%)-APN and APN displayed time-dependent cellular accumulation, but the cellular content of Gal(35%)-APN was significantly (p < 0.001) greater than of non-targeted APN. No difference in cellular uptake between Gal(35%)-APN and APN was observed in the ASGP-R negative HeLa cell line (Fig. S5). Importantly, the higher cellular uptake occurred for the Gal-APN with 5%–35% Gal moieties on their surface (Fig. 1C and Fig. S6). Above this percentage, the uptake of Gal-APN was decreased. To further elucidate whether the cellular association of Gal-APN depends on binding to ASGP-R on the hepatocytes surface, Huh 7 cells were treated with Gal(35%)-APN or APN in the presence of excess free galactose (100 mM). Flow cytometric analysis revealed that the uptake of Gal(35%)-APN was suppressed to the level of the non-targeted APN in the presence of free galactose (Fig. 1D). This clearly demonstrates the involvement of a specific receptor-mediated uptake mechanism.
Figure 1.

Uptake of Gal-APN by Huh 7 cells. (A) Representative confocal microscopy images of Huh 7 incubated with Cy5-labeled Gal(35%)-APN, APN or p41 for 2 h at 37°C. The merged images of Cy5-labeled p41 (red) and DAPI-stained nuclei (blue) (objective 40x). (B) Quantification of internalization kinetics of Gal(35%)-APN, APN and p41 in Huh 7 cells. Mean fluorescence intensity per cell was calculated from images taken at indicated time points. Asterisks show statistically significant differences between Gal(35%)-APN and APN at the corresponding time points. (C) Gal-APN uptake as a function of Gal density following 2 h incubation. *** indicates significantly higher uptake vs. non-targeted APN. (D) Inhibitory effect of free Gal on the uptake of Gal(35%)-APN as analyzed by flow cytometry. Huh 7 cells were treated with free Gal (100 mM) for 0.5 h, followed by co-incubation with either Gal(35%)-APN (blue) or APN (black) for another 2 h. Cells treated with Gal(35%)-APN (grey) or APN (red) were used as controls. In all experiments, cells were treated with APN and peptide at a dose equivalent to 5 μM of p41. Data are presented as mean ± SEM (n = 15–20); *p < 0.05, ***p < 0.001, NS - not significant.
Contrary to our expectation, the internalization kinetics and cellular uptake of Gal(35%)-APN and APN in HepG2 cells that abundantly express ASGP-R (Fig. S3) were not significantly different during continuous incubation with the complexes (Fig. 2A). Similar behavior of Gal(35%)-APN was also observed in primary human hepatocytes (Fig. S7). Nevertheless, the competition experiments demonstrated that the uptake of Gal-APN into HepG2 cells was inhibited by the presence of soluble Gal (Fig. 2B and Fig. S8). By contrast, no attenuation in internalization was observed in the cells treated with non-targeted APN under the same experimental conditions. These findings indicate that uptake of targeted Gal-APN is at least partially mediated by interaction with ASGP-R on cultured HepG2 cells.
Figure 2.

Uptake of Gal-APN into HepG2 cells. (A). Time-dependent uptake of Gal(35%)-APN and APN at 37°C as quantified from confocal images (objective 20x). Data are presented as mean ± SEM (n = 15–20). (B). Representative confocal images of HepG2 cells incubated with Cy5-labeled Gal(35%)-APN and APN in the absence or presence of free galactose. For competition experiments, cells were pre-treated for 0.5 h with 100 mM of galactose followed by co-incubation with corresponding APN for 1 h. The merged images of Cy5-labeled p41 (red) and cell nuclei stained with DAPI (blue) (objective 20x). In all experiments, cells were treated with APN at a dose equivalent to 5 μM peptide at 37°C.
3.3 Gal-APN anti-HCV activity in cell culture systems
Anti-HCV activity of Gal(35%)-APN and APN was evaluated against JFH1 virus (MOI 0.1) in the Huh 7.5 cell line. MOI 0.1 has been demonstrated as the most efficient for JFH-1 virus in Huh7.5 cells by others [10]. As was previously demonstrated, both p41 and APN displayed dose-dependent antiviral activity [13]. To rule out any confounding effects due to cytotoxicity of free p41, all formulations (Gal-APN, APN and p41 alone) were used at a dose equivalent to 5 μM peptide which was shown to be sufficient to suppress HCV infection without inducing cytotoxicity [13]. The antiviral potency of formulations was determined by quantifying the intracellular HCV RNA replication using RT-qPCR. To assess whether APN can suppress an ongoing HCV infection and block cell-to-cell viral spread, the cells were first infected with virus and then treated with formulations (at a dose equivalent to 5 μM p41). As shown in Fig. 3A, both Gal(35%)-APN and APN were able to suppress viral replication after 48 h of cell treatment. It is likely that the lack of the differences in magnitude of the antiviral response between targeted and non-targeted APN might be due to the strong virocidal activity of p41 peptide, which is not affected by targeted delivery of the peptide. To test this hypothesis, virus was pre-incubated with formulations for 1 h and then Huh 7.5 cells were infected with this mixture. As shown in Fig. 3B, in contrast to the viral replication observed in control cultures, the pre-incubation of both Gal(35%)-APN and APN with virus stock induced 99% inhibition of intracellular viral RNA accumulation. Overall, it appeared that in terms of reducing viral RNA replication in cell culture systems, Gal-APN is as effective as APN.
Figure 3.
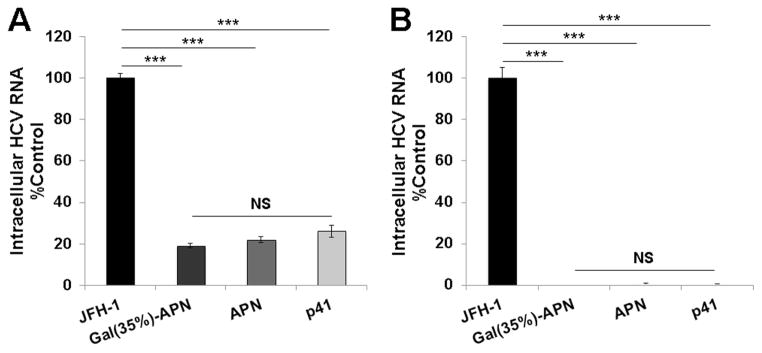
In vitro anti-HCV activity of Gal-APN. (A) APN suppress the ongoing HCV infection. Cells were infected with JFH-1 virus (MOI 0.1) overnight and then treated with Gal(35%)-APN, APN or p41 for 48 h. (B) Virocidal effect: JFH-1 virus (MOI 0.1) was pre-incubated with respective formulations for 1 h at 37°C, then cells were infected with these mixtures overnight, washed, and cultured for another 48 h. For all experiments, intracellular HCV RNA levels were determined by RT-qPCR and normalized to GAPDH mRNA levels. Data are presented as %control ± SEM (n = 3) in comparison to HCV RNA level in non-treated infected cells (100%); *** p < 0.001, NS - not significant.
We attempted to further elucidate the antiviral mechanism of p41 that is beyond the virocidal effect and interferes with the intracellular viral life cycle. Since amphipathic α-helical p41 is derived from the membrane anchor domain of NS5A [10], we hypothesized that p41 competes with NS5A and HCV core proteins for binding to lipid droplets, thereby preventing the assembly and release of mature viral particles. As expected, infected cells exhibited high co-localization of HCV core and NS5A proteins with lipid droplets (Fig. 4A and 4C). When cells were treated with APN or free peptide for 5 h before exposure to HCV, the expression of HCV core protein and NS5A, as well as their localization to lipid droplets, were substantially reduced. Notably, in both cases the strongest inhibition of co-localization with lipid droplets was observed after treatment with Gal(35%)-APN, which significantly (p < 0.001) exceeded the inhibition effects of APN and p41 (Fig. 4B and 4D).
Figure 4.

Gal-APN suppress association of HCV proteins with lipid droplets in infected Huh 7.5 cells. (A, C) Representative confocal microscopy images of the co-localization of HCV core (A) or NS5A (C) proteins with lipid droplets. Cells were pretreated with Gal(35%)-APN, APN or p41 (at a dose equivalent to 5 μM of p41) for 5 h before overnight infection (MOI 0.1). In 48 h, cells were analyzed using BODIPY to stain lipid droplets (green) and immunofluorescence with Alexa Fluor 594-labeled antibodies either to HCV core or NS5A proteins (red). Cell nuclei were stained with DAPI (blue). Co-localization of lipid droplets with HCV core or NS5A proteins defined as yellow overlap of green and red fluorescence (objective 63x and 20x). (B, D) Quantitative analysis of co-localization of lipid droplets with HCV core (B) or NS5A (D) proteins from the images (objective 20x). Data are presented as mean ± SEM (n = 15–20), **p < 0.01, ***p < 0.001, NS - not significant.
In addition to the direct antiviral effect of Gal-APN, the indirect aspect of antiviral action was also investigated based on the activation of interferon-sensitive genes (ISGs) (Fig. S9). ISGs were induced by 4 h exposure to low dose of interferon alpha (IFN-α, 100 U/ml), which itself did not affect HCV RNA levels. Meanwhile, cells were treated with Gal(35%)-APN 48 h post-infection with or without IFN-α exposure. Gal-APN itself had no effect on ISGs activation, however, the accumulation of ISGs (viperin and 2′5′ oligoadenylate synthase-1) was significantly enhanced by Gal-APN after triggering of IFN-α signaling.
3.4 Biodistribution studies
As a proof of concept, the in vivo biodistribution of Gal(14%)-APN, APN, and p41 was tested in BALB/c mice using 177Lu-DTPA-p41 as a probe. Since no significant differences were observed in the uptake of Gal-APN within 5%–35% range of ligand surface densities, Gal(14%)-APN with an average level of galactosylation were selected for further in vivo studies. Following intravenous administration of the radiolabeled compounds, biodistribution studies showed that all formulations were rapidly captured by the liver (~13 %ID/g) within 15 min (Fig. S10A) with no significant differences observed during this short time frame. However, at 4 h after injection, significantly (p < 0.001) higher hepatic disposition of Gal(14%)-APN (6.0 ± 1.6 %ID/g) was observed compared to APN (2.6 ± 1.7 %ID/g) and p41 (1.4 ± 0.3 %ID/g) (Fig. 5A). The trend remained the same after the liver was perfused. The slight drop of radioactivity after perfusion implied that the majority of the drugs were associated with the liver tissue. The liver-to-blood ratios in animals injected with the liver-targeted Gal-APN was calculated to be 6.1 ± 2.1 and was significantly (p < 0.001) higher compared to non-targeted APN (2.1 ± 0.8) and free p41 (1.3 ± 0.6). Our preliminary data showed that radiolabeled formulations were associated with both liver hepatocytes and non-parenchymal cells (consisting primarily of Kupffer cells and sinusoidal endothelial cells). However, preferential accumulation in hepatocytes was observed for Gal(14%)-APN (9.2% ± 2.3% of total dose accumulated in the liver), while for APN and free p41, these values were 5.8% ± 1.4% and 3.7% ± 0.1%, respectively (Fig. S10B). When compared to APN and p41, a slightly higher extent of Gal(14%)-APN accumulation was also observed in the spleen and lung, due to these organs contain high numbers of macrophages expressing lectin receptors (Fig. 5B). Overall, the in vivo data demonstrates the increased hepatic localization of Gal-APN, most likely as a result of Gal-APN binding to ASGP-R expressed on the surface of hepatocytes.
Figure 5.

Biodistribution of radiolabeled Gal-APN in vivo at 4 h post i.v. injection. (A) Quantification of the liver (before and after perfusion) and blood concentrations of the Gal(14%)-APN, APN and p41 (50 μg p41 or its APN format per animal). The values in parenthesis indicate the mean liver (perfused)-to-blood ratio. (B). Tissue distribution of Gal(14%)-APN, APN and p41. Apart from liver, a higher accumulation of Gal(14%)-APN was found in lung and kidney. All results were interpreted as mean percentage of injected dose per gram of tissue (%ID/g) ± SEM (n = 6); *p < 0.05, **p < 0.01, ***p < 0.001, NS - not significant.
4. Discussion
The targeting of bioactive molecules to specific organs is an attractive strategy to increase local concentration of therapeutics at the anticipated site of action, while reducing off-target effects. Viral replication in HCV infection occurs predominantly in the hepatocytes. The ASGP-R, a C-type lectin receptor presented in abundance on the sinusoidal surface of hepatic parenchymal cells, mediates the binding and removal of glycoconjugates containing exposed terminal galactosyl or N-acetylgalactosaminyl sugars. Despite that ASGP-R has been also detected on extra-hepatic cells such as peritoneal and liver residential macrophages, in human kidney, thoroid and activated T cells, the ASGP-R has been intensely validated as a potential target for drug and gene delivery to the liver [23–25]. For example, it was demonstrated that N-glycosylated human interferon beta directed to ASGP-R efficiently inhibited hepatitis B virus infection in vivo [26]. Coulstock et al. [27] demonstrated increased targeting to the liver of interferon genetically fused with a ASGP-R-specific antibody following systemic intravenous administration.
In our previous in vitro studies, we demonstrated that incorporation of cationic p41, an antiviral peptide derived from the membrane anchor domain of HCV nonstructural protein NS5A, into nanosized APN substantially increases peptide proteolytic stability, reduces its intrinsic cytotoxicity, while preserving antiviral activity against HCV. Here, exploiting a concept of the liver-targeted delivery, we designed ASGP-R-directed APN coated with Gal ligands to deliver an antiviral HCV-specific peptide to hepatocytes, which is anticipated to eventually benefit treatment outcomes. A series of Gal-decorated APNs with various densities of Gal ligands was prepared by controlling the molar ratio of Gal-PEG-b-PLE/propargyl-PEG-b-PLE copolymers. The targeting properties of Gal-decorated APN were evaluated in hepatic cells with differential expression of ASGP-R. Interestingly, the uptake efficiency of Gal-APN did not correlate with ASGP-R expression level. Rapid and preferential accumulation of Gal-APN over APN was observed in Huh 7 cells with relatively low receptor expression. Moreover, an efficient uptake of Gal-APN was observed when the density of the Gal ligand did not exceed 35%. The affinity of the ASGP-R is a consequence of oligovalent interactions with its physiological ligands, so-called “cluster glycoside effect” [28]. Studies using natural and synthetic ligands have illustrated the importance of the special arrangements of the terminal Gal residues for the binding affinity to ASGP-R. Based on affinity studies of several asialoglycopeptides and synthetic cluster glycosides with defined sugar arrangements and geometries it was concluded that appropriate sugar spacing (at least 15Å) is required for optimal receptor recognition [28–30]. It is possible that increase in ligand density on the Gal-APN above 35% can lead to formation of structures with shorter intergalactose distances and results in lower biding affinity of the corresponding Gal-APN. Furthermore, observed ligand density effects can be also related but not limited to improper orientation of the ligand, steric hindrance of neighboring molecules or competitive behaviors for the binding of the receptor. Similar negative cooperative binding effects were previously reported for other ligand-decorated nanoformulations such as liposomes [31, 32], nanogels [33], polymeric micelles and dendrimers [34], and superparamagnetic iron oxide nanoparticles [35].
Surprisingly, there were nearly no differences in the uptake of targeted and non-targeted APN in cells with higher density of ASGP-R (~76,000 and 3,000 ASGP-R/cell for HepG2 and Huh 7, respectively [36]). Nevertheless, the uptake of Gal-APN but not APN was significantly inhibited by co-incubation with free Gal in both cell lines. The latter indicates that uptake of galactosylated APN into hepatic cells involves a specific ASGP-R-mediated mechanism, while non-targeted APN utilize some alternative endocytotic pathway. This can possibly be attributed to the difference in composition of the outer shell of the APN. However, the functional relationship between APN composition, ligand density and ASGP-R expression on hepatic cells remains to be established.
Huh 7.5 cells, as an in vitro model for HCV infection, were used to evaluate the antiviral activity of Gal-APN. The life cycle of HCV requires its penetration into hepatocytes via receptors for viral entry, replication, and degradation of a viral polypeptide into smaller structural and non-structural viral proteins, which then are assembled on lipid droplets in the vicinity of endoplasmic reticulum (ER) into full infectious particles. These particles then leave the cells in the complexes with very low density lipoproteins, which are “sensed” by new intact hepatocytes leading to spread of infection [37, 38]. Thus, in the liver, HCV may persist extracellularly as well as intracellularly. In our study, we demonstrated that antiviral activity of p41 (as a part of APN and Gal-APN) also consists of two components: extracellular virocidal component related to viral membranolytic activity of the peptide and intracellular component, which presumably affects viral assembly on lipid droplets. No difference was found between the capability of Gal-APN and APN to reduce intracellular HCV RNA replication when the formulations were either added to the cells after preincubation with virus or were applied post-infection. This may be attributed to the dominant virocidal effect of p41 before its penetration into hepatocytes, which corroborates the results reported by Cheng et al. [10] that amphipathic α-helical peptides derived from the membrane anchor domain of the HCV NS5A protein, including p41, destabilize the viral lipid membranes. This extracellular virocidal component may not depend on targeted APN delivery via ASGP-R in the context of in vitro experiments. However, in vivo, targeted delivery of APN to hepatocytes would provide additional benefit of specific accumulation of Gal-APN and its antiviral payload in the ASGP-R-expressing hepatocytes that are a primary site of HCV replication. In fact, based on our biodistribution experiments, p41 peptide is mainly delivered to liver and hepatocytes via Gal-APN, where, as expected, it will ultimately provide the desired antiviral effect and prevent the spread of extracellularly secreted virus to the neighboring non-infected cells. Besides, the co-staining of HCV core and NS5A proteins with lipid droplets after the treatment with targeted or non-targeted APN sheds light on the additional intracellular antiviral mechanism of p41 that is beyond its virocidal action. Specifically, when the formulations were delivered to the cells before viral entry (avoiding preliminary extracellular contact of APNs with HCV), it prevented the binding of both core and NS5A proteins to lipid droplets via the common amphipathic α-helical domain. This binding of HCV proteins to lipid droplets is necessary for assembly and release of fully assembled infectious viral particles [37, 38]. The decreased association between viral proteins and lipid droplets may be vital for the virus assembly. Here, we show that Gal-APN demonstrated the advantages over APN and free p41 to inhibit this step as a prerequisite for virion production. Furthermore, we also observed the reduced expression of HCV core and NS5A proteins after Gal-APN treatment. Currently, we have not identified the exact intracellular compartment, where HCV interacts with p41. Since both the viral particles and Gal-APN enter hepatocytes via receptor-mediated endocytosis [39, 40], and the destabilization of APN and p41 release can occur probably due to the protonation of carboxylic groups of PLE chains in the APN after acidification in endosomes, we cannot exclude that, in part, inactivation of virus may also be attributed to virocidal effects of APN in this compartment. In addition, arginine-rich α–helical structure of p41 as well as its membranolytic properties may facilitate the endosomal release of these peptides into the cytosol [41]. We also cannot exclude the possibility that p41 may access the HCV replication complex on the ER as a result of multiple fusion of APN/p41-containing vesicles, using lipid droplets or heat shock proteins, or other endocytosis tools as trafficking vehicles. These mechanisms definitely require more detailed investigations.
The effects of antiviral small molecules (including recently discovered DAA) are potentiated by activation of host innate immunity, namely, activation of ISGs that encode generation of endogenous antiviral proteins. This activation of innate immunity diminishes the chance of relapses of chronic HCV infection. It is likely that in addition to disturbance of HCV assembly on lipid droplets, Gal-APN might provide an indirect effect on IFN-α-induced activation of ISGs that encode the expression of antiviral proteins. In the infected cells, HCV core and NS5A proteins suppress IFN-α signaling and prevent activation of ISGs, thereby abolishing the synthesis of intracellular antiviral proteins and diminishing anti-HCV protection [42–44]. While the major antiviral mechanism of Gal-APN is distinct from exogenous IFN-α, we demonstrated that Gal-APN indirectly potentiate the endogenous IFN-α signaling, thereby increasing its downstream antiviral ISGs activation in liver cells (Fig. S9). We strongly believe that apart from the potent extracellular virocidal action, the combination of advanced intracellular effects of Gal-APN and subsequent ISG activation may potentiate in vivo antiviral activity of Gal-APN.
In vivo, using radiolabeled peptides, we subsequently showed that the galactosylation of APN offered certain advantages including preferential liver uptake as well as accumulation in hepatic parenchymal cells (hepatocytes). It appeared that the biodistribution profile of Gal-APN and APN is highly dose-dependent: if lower dose were administered due to inaccurate injection, the distribution of p41 in all excised organs (%ID/g) was correspondingly decreased (data not shown). This is reconciled with previous findings that the lower plasma concentration of protein-based block ionomer complexes makes them more susceptible to dissociation due to the dilution in the bloodstream and interaction with charged serum proteins [45]. For the Gal-APN, the levels of radioactivity in all other tissues assayed were lower than the liver, which was in line with what was obtained for both the APN and p41. The relatively high level of radioactivity detected in the kidney can be attributed to renal excretion of the peptides that was already apparent 15 min following injection of p41 or its formulations.
Interestingly, slightly but significantly higher uptake of Gal-APN compared to non-targeted APN or free p41 was detected in the spleen and lungs (Fig. 5). Since galactosylation did not alter the physicochemical properties of APN, such as size and charge, this indicates the increased binding of Gal-APN to galactose-specific C-type lectins expressed on macrophages in these organs rather than enhanced opsonization [46–48]. Collectively, our findings suggest that preferential hepatic uptake of Gal-APN was a result of ASGP-R binding on the surface of hepatocytes and that galactosylated APN can be used to affect the biodistribution of therapeutic antiviral peptides. However, we should acknowledge that ASGP-R function and expression level in the liver of HCV patients are influenced by many factors including oxidative stress, toxic substances (such as alcohol), viral infection (Fig. S3), etc. [49]. Therefore, hepatic distribution of Gal-APN under pathological conditions should be evaluated, which will be crucial for treatment outcome. To serve this purpose, currently, our group is in the process of establishing a HCV animal model using urokinase-type plasminogen activator (uPA)-NOG mice with reconstituted humanized liver [50].
In the frame of this study, we have not aimed to compare the efficiency of Gal-APN with DAA that already demonstrated their clinical specificity and efficiency. However, apart from the predominant liver accumulation, another advantage of Gal-APN (due to the strong virocidal effects of the peptide) might be the lack of dependence on high rate of HCV mutations, which with time may limit viral sensitivity to DAA. Potentially, Gal-APN can be used for the development of drug combinations that target different aspects of the viral life cycle. APN could complement the DAA potency, interfere with multiple viral genotypes, overcome resistance development and, eventually, reduce treatment duration.
5. Conclusion
In the present study, we designed and synthesized the galactosylated nanocomplexes of antiviral peptides as a platform for targeted therapy against HCV. In vitro, Gal-APN displayed favorable ASGP-R-mediated uptake by hepatoma cell lines and primary human hepatocytes. Even though both targeted and non-targeted APN efficiently suppress HCV RNA replication in cell culture systems, Gal-APN displayed a significantly stronger ability than APN to prevent intracellular binding of HCV core and NS5A proteins to lipid droplets, which is known to be an essential step for viral assembly and release. The preferential hepatic accumulation of Gal-APN in animals further indicates the possibility of improved therapeutic outcomes in vivo. Overall, the results presented here demonstrate the potential of Gal-decorated APN as a specific therapeutic strategy against HCV.
Supplementary Material
Figure S1. 1H-NMR spectra for (A) propargyl-PEG-NH2, (B) propargyl-PEG-b-PBLE, (C) propargyl-PEG-b-PLE, and (D) Gal-PEG-b-PLE. Peak assignments are as follows:
(A) 1H-NMR (DMSO-d6, 25°C): δ ppm = 8.1 (1H, s, -NHCO-), 3.8–4.0 (2H, m, -CH2-), 3.2–3.8 (4H, m, -CH2CH2O-), 3.07 (1H, s, CH≡C-).
(B) 1H-NMR (DMSO-d6, 80°C): δ ppm = 8.1–8.3 (1H, br, CH≡C-CH2-NHCO-), 7.8–8.0 (2H, m, -NHCO-), 7.1–7.4 (5H, m, PhH), 4.9–5.2 (2H, m, PhCH2-), 4.3 (2H, s, CH≡C-CH2-), 3.8–4.0 (1H, m, -CH-), 3.3–3.8 (4H, m, -OCH2CH2O-), 2.2–2.4 (2H, br, -COCH2-), 1.9–2.2 (2H, br, -CH2CH-).
(C) 1H-NMR (D2O, 25 °C): δ ppm = 4.2–4.6 (1H, s, -CH-), 4.1–4.2 (2H, s, -COCH2O-), 4.0–4.1 (2H, s, CH≡C-CH2-), 3.5–3.9 (4H, m, -OCH2CH2O-), 2.49 (2H, s, -COCH2-), 1.8–2.3 (2H, m, -CH2CH-).
(D) 1H-NMR (D2O, 25°C): δ ppm = 8.1 (1H, s, -CH2N(N=)CH=C(N=)CH2O-), 4.9 (1H, m, saccharide α-H), 4.2–4.6 (1H, br, -CH-), 4.1–4.2 (2H, s, -COCH2O-), 4.0–4.1 (2H, s, -CH2N(N=)CH=C(N=)CH2O-), 3.5–4.0 (4H, m, -OCH2CH2O-), 2.4–3.0 (2H, br, -COCH2-), 2.0–2.4 (2H, br, -CH2CH-).
(E) Typical GPC elution curves of precursor propargyl-PEG-NH2 (magenta) and propargyl-PEG-b-PBLE (black).
Figure S2. Mass spectra of 2′-Azidoethyl-O-D-galactopyranoside. The theoretical molecular weight is 249 Da. The main peak shown in the spectra is 249 + 23 (Na) = 272 Da.
Figure S3. Western blot analysis of ASGP-R expression level in hepatoma cells (non-infected and infected) and primary human hepatocytes. Lanes: (1) HepG2 cells; (2) Huh 7 cells; (3) Huh 7.5 cells; (4) Huh 7.5 cells after 1 day infection; (5) Huh 7.5 cells after 2 days infection; (6) Huh 7.5 cells after 3 days infection; (7) primary human hepatocytes; (8) HeLa cells (negative control).
Figure S4. Representative confocal microscopy images (40x) of Huh 7 cells incubated with Cy5-labeled Gal(35%)-APN, APN or p41 (equivalent to 5 μM of p41) for 5 h at 37°C. The merged images of Cy5-labeled p41 (red) and cell nuclei stained with DAPI (blue).
Figure S5. Representative confocal images (20x) of HeLa cells (ASGP-R negative) after 1 h incubation with Gal(35%)-APN and APN at 37°C. The peptide concentration is 5 μM. The cellular nuclei were stained with DAPI (blue), and p41 was labeled with Cy5 (red).
Figure S6. Gal-APN uptake as a function of Gal density following 2 h incubation. Mean fluorescence intensity per cell was calculated from confocal microscopy images containing 1,000–1,500 cells (n = 15–20). Data are presented as mean ± SEM. *p < 0.05, **p < 0.01, and ***p < 0.001 indicate significantly higher uptake vs. non-targeted APN. No significant difference in uptake of Gal-APN was observed within 5%–35% range of ligand density.
Figure S7. Uptake of Gal-APN into primary human hepatocytes. (A). Representative confocal images (40x) of freshly isolated primary human hepatocytes after 2 h and 5 h incubation with Cy5-labeled Gal(35%)-APN and APN (red) at a dose equivalent to 5 μM of peptide. The cellular nuclei were stained with DAPI (blue). (B). Internalization kinetics of Gal(35%)-APN and APN as quantified by ImageJ from confocal images. Data are presented as mean fluorescence per cell ± SEM (n = 15–20); **p < 0.01, ***p < 0.001.
Figure S8. In HepG2 cells, the internalization of Gal(35%)-APN was effectively blocked by free Gal (blue) compared to the cells treated only with Gal(35%)-APN (grey). In contrast, APN uptake remained the same in the absence (red) or the presence (black) of free galactose. ASGP-R were blocked by free galactose (100 mM) for 0.5 h before 1 h co-incubation with Gal(35%)-APN or APN (at a dose equivalent to 5 μM p41). The median fluorescence intensity (Cy5) of each sample was acquired by Becton Dickinson LSRII cytometer (BD Biosciences, USA) and analyzed by FlowJo (Tree Star Inc., USA). At least 10,000 cells from each sample were acquired in linear mode, gated to exclude debris and dead cells, and visualized in logarithmic mode.
Figure S9. The effect of Gal-APN on ISGs expression. After incubation with Gal(35%)-APN (at a dose equivalent to 5 μM of peptide), cells were treated with IFN-α (100 U/ml) for 4 h before harvesting to activate ISGs expression. ISGs [2′-5′-oligoadenylate synthase 1 (OAS-1) and viperin] activation and HCV RNA were presented as %control ± SEM (n=3), where 100% were ISGs or HCV RNA expression in infected cells only exposed to IFN-α; ***p < 0.001.
Figure S10. Biodistribution study of radiolabeled Gal(14%)-APN, APN and p41 in BALB/c mice. (A) Liver accumulation of drugs (before and after perfusion) at 15 min post i.v. injection. Data are presented as mean percentage of injected dose per gram of tissue (%ID/g) ± SEM (n = 3). (B) Intrahepatic cellular localization at 4 h post-injection. *p < 0.05, NS – not significant.
Table S1. Physicochemical characteristics of Cy5-labeled APNa for confocal imaging analysis
a10 mol% Cy5-p41 were introduced into complexes. p41 concentration was 5 μM.
bEffective diameter (Deff) and polydispersity indices (PDI) were determined by DLS at pH 7.4, 25°C (n = 3).
1. Western Blot analysis
The cell lysates of HepG2, Huh 7, primary human hepatocytes, Huh 7.5, and JFH-1-infected Huh 7.5 were prepared by using 1x SDS sample buffer and were quantified with a Micro BCA™ protein assay kit (Pierce, USA). After cell lysates were mixed with β-mercaptoethanol with the ratio of 9:1, an equal amount of protein from each lysate was subjected to SDS-PAGE and then transferred to PVDF membrane at 80 mA and 4°C for overnight. Next day, membranes were blocked with 10% nonfat milk in Tris-buffered saline containing 0.1% Tween 20 (TBST) for 2 h at room temperature. The membranes were then incubated with rabbit polyclonal ASGP-R1 antibody (Thermo Scientific, USA) diluted 1:1000 for overnight at 4°C and then with goat anti-rabbit horseradish peroxidase-conjugated secondary antibody (Thermo scientific, USA) with 1:5000 dilution for 1 h. Both primary and secondary antibodies were diluted with TBST containing 10% nonfat milk. The membranes were washed in TBST for 30 min before the immunoblots were developed with ECL (Pierce, USA). An equal protein load was confirmed by immunoblot with β-actin.
2. The effect of Gal-APN on ISGs expression
Huh 7.5 cells (200,000 cells/well) were seeded on 6-well plate for overnight. Cells were treated with Gal(35%)-APN (5 μM) for 48 h after overnight viral infection (MOI 0.1), and 4 h before harvesting, ISGs were activated by IFN-α (100 units/ml). Total cellular RNA was isolated using TRIzol reagent and subsequently converted to cDNA. Antiviral ISGs [2′-5′-oligoadenylate synthetase 1 (OAS-1) and viperin] abundance and HCV RNA replication were quantified by real-time qPCR.
3. Liver distribution study
Perfused liver was dissociated in the presence of collagenase using a gentle MACS dissociator (Mitenyi Biotec, Germany). The dissociated liver was centrifuged at 700 rpm for 5 min to obtain the pellet of hepatocytes. Subsequently, non-parenchymal cells (NPCs) were harvested by further centrifuging supernatant for 5 min at 1500 rpm. The 177Lu radioactivity of hepatocytes and NPCs was measured using a gamma counter. The cell number and viability were determined by the trypan blue exclusion method.
Acknowledgments
This study was supported by the National Institute of General Medical Sciences Center of Biomedical Research Excellence (Grant P20GM103480 to L.Y.P. and T.K.B). HCV cell culture studies were performed at Nebraska-Western Iowa Health Care System, Omaha, NE (N.A.O.). The authors would like to thank Dr. Samuel Sanderson for his help with peptide synthesis and purification and Swapnil Desale for the synthesis of monomer as well as Susan Brusnahan, Dr. Wei Fan, Dr. Zhengyuan Zhou, Dr. Wen Shi, Wenting Zhang, Yinnong Jia, and Weizhe Li for assistance with the animal studies. We also thank the Advanced Microscopy, Flow Cytometry and NMR Core facilities at the University of Nebraska Medical Center for their excellent technical assistance and the Nebraska Research Initiative and Fred and Pamela Buffet Cancer Center support grant (P30CA036717) for their support of the Core Facilities.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Thomas DL. Global control of hepatitis C: where challenge meets opportunity. Nat Med. 2013;19:850–8. doi: 10.1038/nm.3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alter MJ. Hepatitis C virus infection in the United States. J Hepatol. 1999;31:88–91. doi: 10.1016/s0168-8278(99)80381-x. [DOI] [PubMed] [Google Scholar]
- 3.Fried MW. Side effects of therapy of hepatitis C and their management. Hepatology. 2002;36:S237–S44. doi: 10.1053/jhep.2002.36810. [DOI] [PubMed] [Google Scholar]
- 4.Pockros PJ. Review: New direct-acting antivirals in the development for hepatitis C virus infection. Therap Adv Gastroenterol. 2010;3:191–202. doi: 10.1177/1756283X10363055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pawlotsky JM. New hepatitis C therapies: the toolbox, strategies, and challenges. Gastroenterology. 2014;146:1176–92. doi: 10.1053/j.gastro.2014.03.003. [DOI] [PubMed] [Google Scholar]
- 6.Latham PW. Therapeutic peptides revisited. Nat Biotechnol. 1999;17:755–8. doi: 10.1038/11686. [DOI] [PubMed] [Google Scholar]
- 7.Liu X, Huang Y, Cheng M, Pan L, Si Y, Li G, et al. Screening and Rational Design of Hepatitis C Virus Entry Inhibitory Peptides Derived from GB Virus A NS5A. J Virol. 2013;87:1649–57. doi: 10.1128/JVI.02201-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kota S, Coito C, Mousseau G, Lavergne J-P, Strosberg AD. Peptide inhibitors of hepatitis C virus core oligomerization and virus production. J Gen Virol. 2009;90:1319–28. doi: 10.1099/vir.0.008565-0. [DOI] [PubMed] [Google Scholar]
- 9.Liu S, McCormick KD, Zhao W, Zhao T, Fan D, Wang T. Human apolipoprotein E peptides inhibit hepatitis C virus entry by blocking virus binding. Hepatology. 2012;56:484–91. doi: 10.1002/hep.25665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheng G, Montero A, Gastaminza P, Whitten-Bauer C, Wieland SF, Isogawa M, et al. A virocidal amphipathic α-helical peptide that inhibits hepatitis C virus infection in vitro. Proc Natl Acad Sci U S A. 2008;105:3088–93. doi: 10.1073/pnas.0712380105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bobardt MD, Cheng G, de Witte L, Selvarajah S, Chatterji U, Sanders-Beer BE, et al. Hepatitis C virus NS5A anchor peptide disrupts human immunodeficiency virus. Proc Natl Acad Sci U S A. 2008;105:5525–30. doi: 10.1073/pnas.0801388105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cho N-J, Dvory-Sobol H, Xiong A, Cho S-J, Frank CW, Glenn JS. Mechanism of an Amphipathic α-Helical Peptide’s Antiviral Activity Involves Size-Dependent Virus Particle Lysis. ACS Chem Biol. 2009;4:1061–7. doi: 10.1021/cb900149b. [DOI] [PubMed] [Google Scholar]
- 13.Zhang J, Mulvenon A, Makarov E, Wagoner J, Knibbe J, Kim JO, et al. Antiviral peptide nanocomplexes as a potential therapeutic modality for HIV/HCV co-infection. Biomaterials. 2013;34:3846–57. doi: 10.1016/j.biomaterials.2013.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stockert RJ. The asialoglycoprotein receptor: relationships between structure, function, and expression. Physiol Rev. 1995;75:591–609. doi: 10.1152/physrev.1995.75.3.591. [DOI] [PubMed] [Google Scholar]
- 15.Pricer WE, Ashwell G. The binding of desialylated glycoproteins by plasma membranes of rat liver. J Biol Chem. 1971;246:4825–33. [PubMed] [Google Scholar]
- 16.Hudgin RL, Pricer WE, Ashwell G, Stockert RJ, Morell AG. The isolation and properties of a rabbit liver binding protein specific for asialoglycoproteins. J Biol Chem. 1974;249:5536–43. [PubMed] [Google Scholar]
- 17.Stokmaier D, Khorev O, Cutting B, Born R, Ricklin D, Ernst TO, et al. Design, synthesis and evaluation of monovalent ligands for the asialoglycoprotein receptor (ASGP-R) Bioorg Med Chem. 2009;17:7254–64. doi: 10.1016/j.bmc.2009.08.049. [DOI] [PubMed] [Google Scholar]
- 18.D’Souza AA, Devarajan PV. Asialoglycoprotein receptor mediated hepatocyte targeting — Strategies and applications. J Control Release. 2015;203:126–39. doi: 10.1016/j.jconrel.2015.02.022. [DOI] [PubMed] [Google Scholar]
- 19.Desale SS, Cohen SM, Zhao Y, Kabanov AV, Bronich TK. Biodegradable hybrid polymer micelles for combination drug therapy in ovarian cancer. J Control Release. 2013;171:339–48. doi: 10.1016/j.jconrel.2013.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Geng J, Mantovani G, Tao L, Nicolas J, Chen G, Wallis R, et al. Site-directed conjugation of “clicked” glycopolymers to form glycoprotein mimics: binding to mammalian lectin and induction of immunological function. J Am Chem Soc. 2007;129:15156–63. doi: 10.1021/ja072999x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Masuko T, Minami A, Iwasaki N, Majima T, Nishimura S-I, Lee YC. Carbohydrate analysis by a phenol–sulfuric acid method in microplate format. Anal Biochem. 2005;339:69–72. doi: 10.1016/j.ab.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 22.Lindenbach BD, Evans MJ, Syder AJ, Wölk B, Tellinghuisen TL, Liu CC, et al. Complete replication of hepatitis C virus in cell culture. Science. 2005;309:623–6. doi: 10.1126/science.1114016. [DOI] [PubMed] [Google Scholar]
- 23.Murao A, Nishikawa M, Managit C, Wong J, Kawakami S, Yamashita F, et al. Targeting efficiency of galactosylated liposomes to hepatocytes in vivo: effect of lipid composition. Pharm Res. 2002;19:1808–14. doi: 10.1023/a:1021433206081. [DOI] [PubMed] [Google Scholar]
- 24.Managit C, Kawakami S, Nishikawa M, Yamashita F, Hashida M. Targeted and sustained drug delivery using PEGylated galactosylated liposomes. Int J Pharm. 2003;266:77–84. doi: 10.1016/s0378-5173(03)00383-1. [DOI] [PubMed] [Google Scholar]
- 25.Ishida E, Managit C, Kawakami S, Nishikawa M, Yamashita F, Hashida M. Biodistribution characteristics of galactosylated emulsions and incorporated probucol for hepatocyte-selective targeting of lipophilic drugs in mice. Pharm Res. 2004;21:932–9. doi: 10.1023/b:pham.0000029280.39882.ff. [DOI] [PubMed] [Google Scholar]
- 26.Eto T, Takahashi H. Enhanced inhibition of hepatitis B virus production by asialoglycoprotein receptor-directed interferon. Nat Med. 1999;5:577–81. doi: 10.1038/8462. [DOI] [PubMed] [Google Scholar]
- 27.Coulstock E, Sosabowski J, Ovečka M, Prince R, Goodall L, Mudd C, et al. Liver-targeting of interferon-alpha with tissue-specific domain antibodies. PLoS One. 2013;8:e57263. doi: 10.1371/journal.pone.0057263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee YC, Townsend R, Hardy MR, Lönngren J, Arnarp J, Haraldsson M, et al. Binding of synthetic oligosaccharides to the hepatic Gal/GalNAc lectin. Dependence on fine structural features. J Biol Chem. 1983;258:199–202. [PubMed] [Google Scholar]
- 29.Biessen EA, Beuting DM, Roelen HC, van de Marel GA, Van Boom JH, Van Berkel TJ. Synthesis of cluster galactosides with high affinity for the hepatic asialoglycoprotein receptor. J Med Chem. 1995;38:1538–46. doi: 10.1021/jm00009a014. [DOI] [PubMed] [Google Scholar]
- 30.Rensen PC, Sliedregt LA, Ferns M, Kieviet E, van Rossenberg SM, van Leeuwen SH, et al. Determination of the upper size limit for uptake and processing of ligands by the asialoglycoprotein receptor on hepatocytesin vitro and in vivo. J Biol Chem. 2001;276:37577–84. doi: 10.1074/jbc.M101786200. [DOI] [PubMed] [Google Scholar]
- 31.Reddy J, Abburi C, Hofland H, Howard S, Vlahov I, Wils P, et al. Folate-targeted, cationic liposome-mediated gene transfer into disseminated peritoneal tumors. Gene Ther. 2002;9:1542–50. doi: 10.1038/sj.gt.3301833. [DOI] [PubMed] [Google Scholar]
- 32.Kawano K, Maitani Y. Effects of polyethylene glycol spacer length and ligand density on folate receptor targeting of liposomal Doxorubicin in vitro. J Drug Deliv. 2010;2011:160967. doi: 10.1155/2011/160967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nukolova NV, Oberoi HS, Cohen SM, Kabanov AV, Bronich TK. Folate-decorated nanogels for targeted therapy of ovarian cancer. Biomaterials. 2011;32:5417–26. doi: 10.1016/j.biomaterials.2011.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Poon Z, Chen S, Engler AC, Lee Hi, Atas E, von Maltzahn G, et al. Ligand-Clustered “Patchy” Nanoparticles for Modulated Cellular Uptake and In Vivo Tumor Targeting. Angew Chem Int Ed Engl. 2010;49:7266–70. doi: 10.1002/anie.201003445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Elias DR, Poloukhtine A, Popik V, Tsourkas A. Effect of ligand density, receptor density, and nanoparticle size on cell targeting. Nanomedicine. 2013;9:194–201. doi: 10.1016/j.nano.2012.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li Y, Huang G, Diakur J, Wiebe LI. Targeted delivery of macromolecular drugs: asialoglycoprotein receptor (ASGPR) expression by selected hepatoma cell lines used in antiviral drug development. Curr Drug Deliv. 2008;5:299–302. doi: 10.2174/156720108785915069. [DOI] [PubMed] [Google Scholar]
- 37.Shavinskaya A, Boulant S, Penin F, McLauchlan J, Bartenschlager R. The lipid droplet binding domain of hepatitis C virus core protein is a major determinant for efficient virus assembly. J Biol Chem. 2007;282:37158–69. doi: 10.1074/jbc.M707329200. [DOI] [PubMed] [Google Scholar]
- 38.Shi ST, Polyak SJ, Tu H, Taylor DR, Gretch DR, Lai M. Hepatitis C virus NS5A colocalizes with the core protein on lipid droplets and interacts with apolipoproteins. Virology. 2002;292:198–210. doi: 10.1006/viro.2001.1225. [DOI] [PubMed] [Google Scholar]
- 39.Meertens L, Bertaux C, Dragic T. Hepatitis C virus entry requires a critical postinternalization step and delivery to early endosomes via clathrin-coated vesicles. J Virol. 2006;80:11571–8. doi: 10.1128/JVI.01717-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Geffen I, Spiess M. Asialoglycoprotein receptor. Int Rev Cytol. 1992;137B:181–219. doi: 10.1016/s0074-7696(08)62605-4. [DOI] [PubMed] [Google Scholar]
- 41.Appelbaum JS, LaRochelle JR, Smith BA, Balkin DM, Holub JM, Schepartz A. Arginine topology controls escape of minimally cationic proteins from early endosomes to the cytoplasm. Chem Biol. 2012;19:819–30. doi: 10.1016/j.chembiol.2012.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lin W, Choe WH, Hiasa Y, Kamegaya Y, Blackard JT, Schmidt EV, et al. Hepatitis C virus expression suppresses interferon signaling by degrading STAT1. Gastroenterology. 2005;128:1034–41. doi: 10.1053/j.gastro.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 43.Duong FH, Filipowicz M, Tripodi M, La Monica N, Heim MH. Hepatitis C virus inhibits interferon signaling through up-regulation of protein phosphatase 2A. Gastroenterology. 2004;126:263–77. doi: 10.1053/j.gastro.2003.10.076. [DOI] [PubMed] [Google Scholar]
- 44.Zhang T, Lin RT, Li Y, Douglas SD, Maxcey C, Ho C, et al. Hepatitis C virus inhibits intracellular interferon alpha expression in human hepatic cell lines. Hepatology. 2005;42:819–27. doi: 10.1002/hep.20854. [DOI] [PubMed] [Google Scholar]
- 45.Klyachko NL, Manickam DS, Brynskikh AM, Uglanova SV, Li S, Higginbotham SM, et al. Cross-linked antioxidant nanozymes for improved delivery to CNS. Nanomedicine. 2012;8:119–29. doi: 10.1016/j.nano.2011.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Imai Y, Akimoto Y, Mizuochi S, Kimura T, Hirano H, Irimura T. Restricted expression of galactose/N-acetylgalactosamine-specific macrophage C-type lectin to connective tissue and to metastatic lesions in mouse lung. Immunology. 1995;86:591–8. [PMC free article] [PubMed] [Google Scholar]
- 47.Mizuochi S, Akimoto Y, Imai Y, Hirano H, Irimura T. Unique tissue distribution of a mouse macrophage C-type lectin. Glycobiology. 1997;7:137–46. doi: 10.1093/glycob/7.1.137. [DOI] [PubMed] [Google Scholar]
- 48.Onami TM, Lin M-Y, Page DM, Reynolds SA, Katayama CD, Marth JD, et al. Generation of mice deficient for macrophage galactose-and N-acetylgalactosamine-specific lectin: limited role in lymphoid and erythroid homeostasis and evidence for multiple lectins. Mol Cell Biol. 2002;22:5173–81. doi: 10.1128/MCB.22.14.5173-5181.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McVicker BL, Tuma DJ, Kubik JA, Hindemith AM, Baldwin CR, Casey CA. The effect of ethanol on asialoglycoprotein receptor—mediated phagocytosis of apoptotic cells by rat hepatocytes. Hepatology. 2002;36:1478–87. doi: 10.1053/jhep.2002.37137. [DOI] [PubMed] [Google Scholar]
- 50.Gutti TL, Knibbe JS, Makarov E, Zhang J, Yannam GR, Gorantla S, et al. Human hepatocytes and hematolymphoid dual reconstitution in treosulfan-conditioned uPA-NOG mice. Am J Pathol. 2014;184:101–9. doi: 10.1016/j.ajpath.2013.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. 1H-NMR spectra for (A) propargyl-PEG-NH2, (B) propargyl-PEG-b-PBLE, (C) propargyl-PEG-b-PLE, and (D) Gal-PEG-b-PLE. Peak assignments are as follows:
(A) 1H-NMR (DMSO-d6, 25°C): δ ppm = 8.1 (1H, s, -NHCO-), 3.8–4.0 (2H, m, -CH2-), 3.2–3.8 (4H, m, -CH2CH2O-), 3.07 (1H, s, CH≡C-).
(B) 1H-NMR (DMSO-d6, 80°C): δ ppm = 8.1–8.3 (1H, br, CH≡C-CH2-NHCO-), 7.8–8.0 (2H, m, -NHCO-), 7.1–7.4 (5H, m, PhH), 4.9–5.2 (2H, m, PhCH2-), 4.3 (2H, s, CH≡C-CH2-), 3.8–4.0 (1H, m, -CH-), 3.3–3.8 (4H, m, -OCH2CH2O-), 2.2–2.4 (2H, br, -COCH2-), 1.9–2.2 (2H, br, -CH2CH-).
(C) 1H-NMR (D2O, 25 °C): δ ppm = 4.2–4.6 (1H, s, -CH-), 4.1–4.2 (2H, s, -COCH2O-), 4.0–4.1 (2H, s, CH≡C-CH2-), 3.5–3.9 (4H, m, -OCH2CH2O-), 2.49 (2H, s, -COCH2-), 1.8–2.3 (2H, m, -CH2CH-).
(D) 1H-NMR (D2O, 25°C): δ ppm = 8.1 (1H, s, -CH2N(N=)CH=C(N=)CH2O-), 4.9 (1H, m, saccharide α-H), 4.2–4.6 (1H, br, -CH-), 4.1–4.2 (2H, s, -COCH2O-), 4.0–4.1 (2H, s, -CH2N(N=)CH=C(N=)CH2O-), 3.5–4.0 (4H, m, -OCH2CH2O-), 2.4–3.0 (2H, br, -COCH2-), 2.0–2.4 (2H, br, -CH2CH-).
(E) Typical GPC elution curves of precursor propargyl-PEG-NH2 (magenta) and propargyl-PEG-b-PBLE (black).
Figure S2. Mass spectra of 2′-Azidoethyl-O-D-galactopyranoside. The theoretical molecular weight is 249 Da. The main peak shown in the spectra is 249 + 23 (Na) = 272 Da.
Figure S3. Western blot analysis of ASGP-R expression level in hepatoma cells (non-infected and infected) and primary human hepatocytes. Lanes: (1) HepG2 cells; (2) Huh 7 cells; (3) Huh 7.5 cells; (4) Huh 7.5 cells after 1 day infection; (5) Huh 7.5 cells after 2 days infection; (6) Huh 7.5 cells after 3 days infection; (7) primary human hepatocytes; (8) HeLa cells (negative control).
Figure S4. Representative confocal microscopy images (40x) of Huh 7 cells incubated with Cy5-labeled Gal(35%)-APN, APN or p41 (equivalent to 5 μM of p41) for 5 h at 37°C. The merged images of Cy5-labeled p41 (red) and cell nuclei stained with DAPI (blue).
Figure S5. Representative confocal images (20x) of HeLa cells (ASGP-R negative) after 1 h incubation with Gal(35%)-APN and APN at 37°C. The peptide concentration is 5 μM. The cellular nuclei were stained with DAPI (blue), and p41 was labeled with Cy5 (red).
Figure S6. Gal-APN uptake as a function of Gal density following 2 h incubation. Mean fluorescence intensity per cell was calculated from confocal microscopy images containing 1,000–1,500 cells (n = 15–20). Data are presented as mean ± SEM. *p < 0.05, **p < 0.01, and ***p < 0.001 indicate significantly higher uptake vs. non-targeted APN. No significant difference in uptake of Gal-APN was observed within 5%–35% range of ligand density.
Figure S7. Uptake of Gal-APN into primary human hepatocytes. (A). Representative confocal images (40x) of freshly isolated primary human hepatocytes after 2 h and 5 h incubation with Cy5-labeled Gal(35%)-APN and APN (red) at a dose equivalent to 5 μM of peptide. The cellular nuclei were stained with DAPI (blue). (B). Internalization kinetics of Gal(35%)-APN and APN as quantified by ImageJ from confocal images. Data are presented as mean fluorescence per cell ± SEM (n = 15–20); **p < 0.01, ***p < 0.001.
Figure S8. In HepG2 cells, the internalization of Gal(35%)-APN was effectively blocked by free Gal (blue) compared to the cells treated only with Gal(35%)-APN (grey). In contrast, APN uptake remained the same in the absence (red) or the presence (black) of free galactose. ASGP-R were blocked by free galactose (100 mM) for 0.5 h before 1 h co-incubation with Gal(35%)-APN or APN (at a dose equivalent to 5 μM p41). The median fluorescence intensity (Cy5) of each sample was acquired by Becton Dickinson LSRII cytometer (BD Biosciences, USA) and analyzed by FlowJo (Tree Star Inc., USA). At least 10,000 cells from each sample were acquired in linear mode, gated to exclude debris and dead cells, and visualized in logarithmic mode.
Figure S9. The effect of Gal-APN on ISGs expression. After incubation with Gal(35%)-APN (at a dose equivalent to 5 μM of peptide), cells were treated with IFN-α (100 U/ml) for 4 h before harvesting to activate ISGs expression. ISGs [2′-5′-oligoadenylate synthase 1 (OAS-1) and viperin] activation and HCV RNA were presented as %control ± SEM (n=3), where 100% were ISGs or HCV RNA expression in infected cells only exposed to IFN-α; ***p < 0.001.
Figure S10. Biodistribution study of radiolabeled Gal(14%)-APN, APN and p41 in BALB/c mice. (A) Liver accumulation of drugs (before and after perfusion) at 15 min post i.v. injection. Data are presented as mean percentage of injected dose per gram of tissue (%ID/g) ± SEM (n = 3). (B) Intrahepatic cellular localization at 4 h post-injection. *p < 0.05, NS – not significant.
Table S1. Physicochemical characteristics of Cy5-labeled APNa for confocal imaging analysis
a10 mol% Cy5-p41 were introduced into complexes. p41 concentration was 5 μM.
bEffective diameter (Deff) and polydispersity indices (PDI) were determined by DLS at pH 7.4, 25°C (n = 3).