

Abstract
The invertebrate cell line, Bge, from embryos of the snail Biomphalaria glabrata, remains to date the only established cell line from any species of the Phylum Mollusca. Since its establishment in 1976 by Eder Hansen, few studies have focused on profiling its cytometrics, growth characteristics or sensitivity to xenobiotics. Bge cells are reputed to be challenging to propagate and maintain. Therefore, even though this cell line is a noteworthy resource, it has not been studied widely. With growing interest in functional genomics, including genetic transfection, to elucidate molecular aspects of the snail intermediate hosts responsible for transmission of schistosomiasis, and aiming to enhance the convenience of maintenance of this molluscan cell line, we deployed the xCELLigene real time approach to study Bge cells. Doubling times for three isolates of Bge, termed CB, SL and UK, were longer than for mammalian cell lines - longer than 40 h in complete Bge medium supplemented with 7% fetal bovine serum at 25°C, ranging from ∼42 h to ∼157 h when 40,000 cells were seeded. To assess the potential of the cells for genetic transformation, antibiotic selection was explored. Bge cells were sensitive to the aminonucleoside antibiotic puromycin (from Streptomyces alboniger) from 5 μg/ml to 200 ng/ml, displaying a half maximal inhibitory concentration (IC50) of ∼1.91 μg/ml. Sensitivity to puromycin, and a relatively quick kill time (<48 h in 5 μg/ml) facilitated use of this antibiotic, together with the cognate resistance gene (puromycin N-acetyl-transferase, PAC) for selection of Bge cells transformed with the PAC gene (puroR). Bge cells transfected with a plasmid encoding puroR were partially rescued when cultured in the presence of 5 μg/ml of puromycin. These findings pave the way for the development of functional genomic tools applied to the host-parasite interaction during schistosomiasis and neglected tropical trematodiases at large.
Keywords: Biomphalaria glabrata, Molluscan embryonic cell line (Bge), xCELLigence real time cellular analysis (RTCA), Cytometrics, Genetic transformation, Antibiotic selection
Graphical abstract
1. Introduction
As the only known established molluscan cell line, the Biomphalaria glabrata embryonic cell line, Bge, represents a noteworthy resource for in vitro studies on the cellular and molecular basis of the relationship between the parasitic trematode, Schistosoma mansoni, etiological agent of the neglected tropical disease schistosomiasis, and its snail host (Knight et al., 2014). Recently there has been a rapid increase in “omics” based projects for both the parasite and snail that generated genomic and transcriptomic profiles (Raghavan and Knight, 2006; Berriman et al., 2009; Brindley et al., 2009; Chuan et al., 2010; Protasio et al., 2012; Zerlotini et al., 2013). In order to take advantage of the in vitro culture of the blood fluke intra-snail developmental stages, we considered that it was desirable to not only define cell characteristics of Bge cells, but also optimize functional genomic approaches to investigate the parasitism of the intermediate host by the parasite (Knight et al., 2014).
The snail cells or Bge conditioned culture medium have been employed to maintain in vitro intra-snail developmental stages of S. mansoni (Ivanchenko et al., 1999). Culturing miracidia in the presence of Bge cells facilitates the transformation, development and growth of sporocysts (Yoshino et al., 2013). Thus, transformed mother sporocysts, developing into daughter sporocysts in vitro, leading to the development of cercariae (the stage that is infective for the human host) has been accomplished (Coustau and Yoshino, 2000). The signaling networks operating between the snail and parasite interface can be more readily tracked by using either loss or gain of gene function tools applied to this in vitro co-culture system. Indeed, the genome sequence of B. glabrata currently being annotated will soon be released, offering an opportunity to determine targets to silence and disrupt the in vitro development of the parasite. In order to perform cytometric and genetic transformation studies using the Bge cell line, we first established the growth behavior of the three isolates of Bge, known as SL, CB and UK. The karyotypes of these isolates vary (Odoemelam et al., 2009); all exhibit aneuploidy, with modal metaphase chromosome complements of 63 and 67 for the SL and CB cell lines, respectively. These divergences confirm that these isolates have undergone substantial evolution in the laboratory since the Bge karyotype was first assessed and retained the expected 2n = 36 diploidy (Bayne et al., 1978).
Given the newly available draft genomes and the application of functional genomic tools, including RNA interference (RNAi) and transgenesis technologies, and despite the difficulties of maintaining Bge cells, there is a pressing need to establish reproducible in vitro protocols to further develop these approaches for the molluscan cells (Yoshino et al., 2013). Earlier transfection studies using lipofection to introduce plasmids bearing either the B. glabrata Heat Shock Protein (Hsp) 70 or cytomegalovirus (CMV) promotor into Bge cells, expressed the reporter gene luciferase (Lardans et al., 1996; Yoshino et al., 1998). Transgenesis in other molluscs, for example primary embryonic cells from the oyster Crassostrea gigas, has also been accomplished by using a pseudotyped pantropic retrovirus expressing luciferase (Boulo et al., 2000). With the overarching goal of applying genetic manipulation tools to the Bge cell line, here we deployed the xCELLigene Real Time Cellular Analysis (RTCA, Acea Biosciences, USA) technology to examine cell growth in real time (Smout et al., 2010; Ke et al., 2011), and performed transfections of the cells with plasmids expressing fluorescent reporters and antibiotic-resistant markers. These findings presented below pave the way for the development of functional genomic tools to investigate host-parasite interactions during schistosomiasis and neglected tropical trematodiasis at large.
2. Materials and methods
2.1. Bge cell line culture
Bge cell isolates SL (Sam Loker, University of New Mexico, USA) and CB (Christopher Bayne, Oregon State University, USA) have been described previously (Odoemelam et al., 2009). The cells maintained by one of the current authors (Joanna Bridger) at Brunel University, UK for 15 years were here termed ‘UK isolate’. All cell isolates were cultured, maintained and passaged as described previously (Odoemelam et al., 2009). In brief, Bge cells were cultured at 25°C in air in filtered sterile Bge medium (22% Schneider's Drosophila medium, 0.13% galactose, 0.45% lactalbumin hydrolysate, 14.1 μM phenol, gentamicin and 7% fetal bovine serum (FBS)). The CB isolate used for most of the analyses carried out here was passaged once each month using a cell scraper when confluence reached ∼ 90% and re-seeded at 2 × 105cells/ml in plugged T25 flasks containing 5.0 ml of fresh Bge medium at room temperature. The UK and the SL isolates were passaged at 4 and 5 - 6 weeks, respectively.
2.2. Puromycin dose response curve
The puromycin dose response curves were performed by seeding 5×103 Bge cells per well into 6-well plates and 2 days later puromycin dihydrochloride (Life Technology, USA) was added to the culture medium at final concentrations of 0.05, 0.5 and 5 μg/ml. Given that puromycin is supplied at 10 mg/ml in 20 mM HEPES buffer (pH 7.2 – 7.5), cells cultured in 10 μM HEPES (but without puromycin) served as controls. The cells were observed every day under bright light using a Zeiss Axio Observer A.1 inverted microscope fitted with a digital camera (AxioCam ICc3, Zeiss, USA). Manipulation of digital images, which was limited to insertion of scale bars, adjustments of brightness and contrast, cropping and the like, was undertaken with the AxioVision release 4.6.3 software (Zeiss). The experiment was repeated three times.
2.3. Transfection of Bge cells
Bge cells cultured in 6-well plates were transfected as described (Knight et al., 2011) with either a GFP-encoding plasmid (piLenti-RNAi-GFP, Applied Biological Materials Inc. USA) (Supplementary Fig. S1A) or a murine leukemia retrovirus plasmid encoding resistance to puromycin (pLNHX_cHS4_puroR) derived from pLNHX_cHS4_650 (Suttiprapa et al., 2012) by replacing the gene for neomycin resistance (neoR) with puroR (Supplementary Fig. S1B). Briefly, using 4 μg of plasmid DNA and 3.12 μg of (N/P ratio = 6) Polyethyleneimine (PEI in vivo Plex ‘adherent’; AparnaBio, USA), nanoparticles were formed in 200 μl of Bge medium incubated at 23°C for 30 min (Knight et al., 2011; Liang et al., 2013). The cells were not washed post-transfection in PEI. Two hundred μl of plasmid DNA/PEI nanoparticle complexes were added to the monolayer of Bge cells (80 to 90% confluent) in a final volume of 2.0 ml of medium, and the cells maintained at 25 °C for at least 5 days. Transfected Bge cells were incubated at 37 °C for 30 to 60 min before examination for fluorescence. To determine the efficiency and expression of GFP using PEI-mediated transfection, we included analysis of similarly transfected HEK293T (human embryonic kidney) cells as a positive control. Following transfection, HEK293T cells were incubated at 37°C in 5% CO2. Both mammalian and Bge cells exposed to PEI only were included as controls.
2.4. Real time assessment by xCELLigence RTCA system of cell proliferation, antibiotic dose-response curve and selection of transfected cells
Cellular proliferation of Bge cells, puromycin toxicity and selection of pLNHX_cHS4_puroR-transformed Bge cells cultured in the presence of the antibiotic were assessed in real time using the xCELLigence DP system and E-plates (ACEA Biosciences, San Diego, CA, USA); see http://www.aceabio.com/main.aspx (Ke et al., 2011). This real time cellular assay (RTCA) allows the monitoring of cellular events, e.g. cell proliferation and cell toxicity (cell death), by measuring the electrical impedance across interdigitated gold micro-electrodes integrated on the bottom of tissue culture E-plates. Bge cells were seeded in E-plates and proliferation and growth in real time was assessed.
For the cellular proliferation assay of the Bge isolates termed SL, UK and CB, cells were collected, using a cell scraper, from the tissue culture flask at 90% confluence. The cells were counted using a hemocytometer and viability was measured using Trypan Blue exclusion dye (0.4% Gibco Life Technologies, USA). First, a background measurement was performed by establishing the Cell Index (CI) signal from E-plate wells containing only 100 μl of Bge medium at room temperature. Thereafter, 100 μl of cell suspension of either 5,000, 10,000, 20,000 or 40,000 was inoculated per well (in duplicate) and the cells maintained at 23 °C for 30 min to allow them to settle at the bottom of the well. The cells were plated at different densities to ensure optimum attachment at the bottom of the tissue culture E-plates since the xCELLigence system measures impedance across the microelectrodes of these plates. Cultures in E-plates were monitored in an xCELLigence DP platform for 160 h with impedance measured at intervals of 15 min. Doubling times of the different Bge isolates were estimated during a time interval of 60 h, i.e. from 20 to 80 h after the addition of the cells, with the assistance of the RTCA Software 1.2. For the puromycin dose response curve analysis, 20,000 or 30,000 CB cells were seeded and monitored as indicated above. The CB isolate was chosen for these studies because it was passaged from the original Bge cell line from the 1970s and may, therefore, be more representative of the original cell line than the SL and UK isolates. When the average CI was ∼ 0.7, the run was paused, 100 μl of cell medium was removed and replaced with 100 μl of medium containing 2× concentrated puromycin to reach the desired final concentrations, i.e. final concentrations in 200 μl per well in the E-plate, from 0.078 μg/ml to 5 μg/ml (two-fold-serial dilutions) or 10 μM HEPES buffer in Bge medium as vehicle control. The normalized CI obtained by dividing the CI value at each time point by the CI at the time of the puromycin addition, was calculated and employed to plot the dose response curves for different time points, and to estimate the half maximal inhibitory concentration (IC50) of the antibiotic (RTCA Software 1.2) (Ke et al., 2011). The IC50 was estimated based on the average of all the dose response curves for selected time points, i.e. 10 h time-windows starting from 28 h after the addition of the antibiotic. To evaluate the selection of pLNHX_cHS4_puroR-transfected Bge cells cultured in the presence of different concentrations of puromycin, 40,000 cells of the SL isolate were seeded per well and monitored as described above. When the CI reached a value between ∼0.7 to ∼1.0, the cells were transfected with (i) the plasmid pLNHX_cHS4_puroR- PEI nanocomplex, or (ii) PEI alone, following the protocol described above. A non-transfected, mock control was included. Forty-eight hours after transfection, 0, 2.5 or 5 μg/ml of puromycin were added to the culture medium and the cells were monitored for at least 84 h after the addition of the antibiotic. The cellular growth was expressed as normalized CI (above) and as the percentage of the normalized CI (% normalized CI) of experimental groups in comparison with the mock control (% normalized CI = 100).
2.5 Statistical analyses
Levels of statistical significance among and between treatments were determined using ANOVA and a Student's t-test. P ≤ 0.05 was considered significant. At least two biological replicates of each experiment were performed, each of which included two to four technical replicates, i.e. number of wells per group.
3. Results
3.1. Bge cells display a doubling time much longer than mammalian cells
In order to evaluate cell proliferation and quantify the doubling time of the isolates, RTCA (xCELLigence) was employed. Different numbers of cells of each Bge isolate ranging from 5,000 to 40,000 cells were seeded per well in the E-plates. The three isolates, SL, UK and CB proliferated in the E-plates, displaying increasing CI values over time (Fig. 1). However, ostensible differences in the proliferative behavior, i.e. the shape of the cell proliferation CI curve over time, among the isolates were evident. Interestingly, the increasing CI values were evident when more than 10,000 cells were seeded per well, and when 5,000 cells were seeded per well, no incremental CI signals were detected in any of the Bge isolates. In addition, remarkably, differences in the estimated doubling times were detected among not only the three isolates, but also among the different numbers of seeded cells per well within each isolate. Bge isolates SL, UK and CB displayed doubling times longer than 40 h ranging from either ∼42 h to ∼157 h or from ∼ 41 h to ∼ 121 h, when 40,000 or 20,000 cells were seeded per well, respectively. Despite these differences, the UK isolate exhibited the shortest doubling time ranging from 42 h to 56 h, when 40,000 to 10,000 cells were seeded per well (Fig. 2). Concerning the cell size and morphology of the Bge cells, at ∼60 % confluence, the average length of the three isolates were similar, ranging from 14 to 24 μm; significant differences in the morphology were not apparent (Fig. 1 and Supplementary Fig. S2).
Fig. 1.
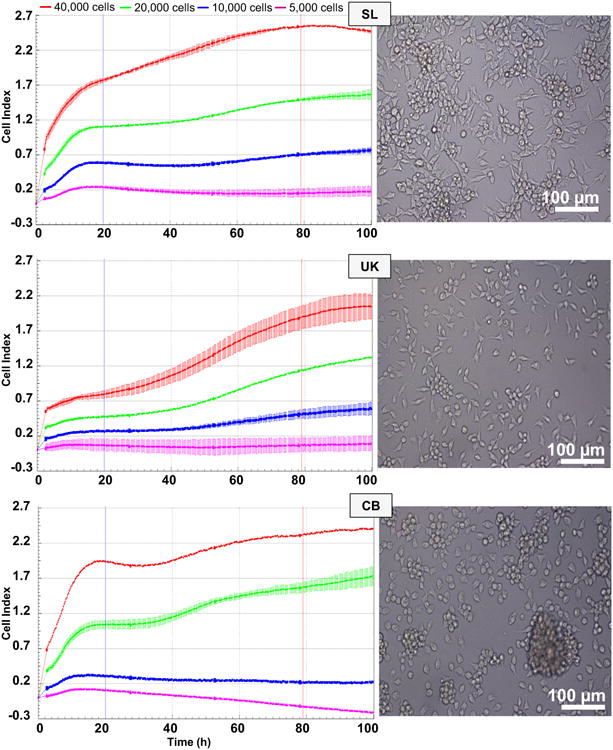
Cytometric analysis of the three Bge (Biomphalaria glabrata embryonic) cell isolates. Cellular proliferation of Bge cell isolates SL, UK and CB measured in real time by xCELLigence. The time interval employed to estimate the doubling times is indicated by the vertical bars (∼20 h and ∼ 80 h). Error bars: ±1 S.D. of the mean. Representative images of Bge cell isolates SL, UK and CB. Magnification 20×, scale bars: 100 μm The number of cells seeded per well is indicated above the graphs.
Fig. 2.
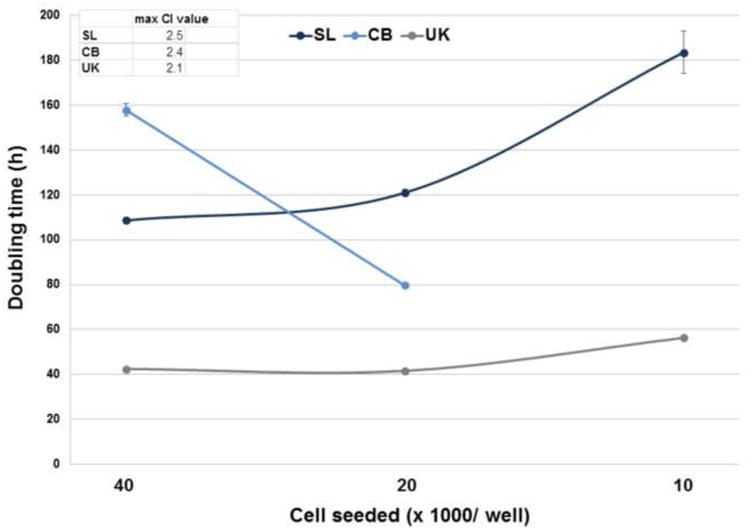
Doubling times estimated for the three Bge (Biomphalaria glabrata embryonic) isolates. Doubling times of the Bge isolates were calculated during a time interval of 60 h (from 20 to 80 h after the addition of the cells, see Fig. 1) for increasing numbers of cells seeded per well, with the assistance of the real time cellular analysis (RTCA) Software 1.2. Error bars: ±1 S.D. of the mean. The maximum Cell Index (CI) values reached by the three Bge isolates are indicated.
3.2. Bge cells are sensitive to the antibiotic aminonucleoside puromycin
The toxicity of the aminonucleoside puromycin (from Streptomyces alboniger), routinely employed for antibiotic selection of puromycin resistance marker-expressing mammalian cells (Ni et al., 2014), was investigated. Bge cells were sensitive to puromycin from 50 ng/ml to 5 μg/ml for all three isolates, i.e. most of the cells (> ∼90%) were dead between 2 and 5 days after the addition of the drug at 5 μg/ml and 50 ng/ml, respectively (Fig. 3).
Fig. 3.
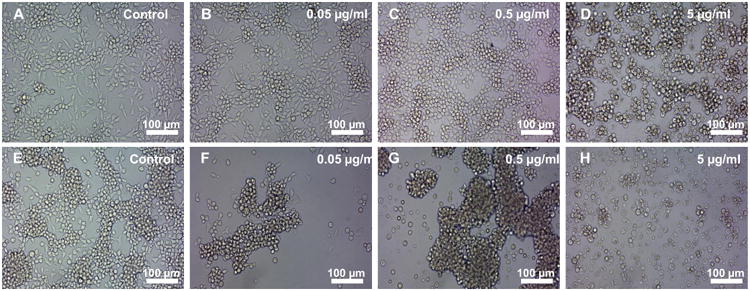
Bge (Biomphalaria glabrata embryonic) cells are sensitive to the antibiotic puromycin. Representative images of Bge cells cultured in the presence of the indicated concentrations of puromycin, 2 days (A - D) and 5 days (E - H) after addition of the antibiotic. Scale bars: 100 μ m.
In order to quantify the cellular toxicity and the IC50 of the antibiotic puromycin, dose response curves were generated using xCELLigence. Within less than 5 h after the addition of 5 μg/ml of puromycin, the cell toxicity was evident and irreversible (Fig. 4A). Additionally, at lower concentrations of the antibiotic, i.e. 0.2 and 1 μg/ml, the mortality, based on the decreasing value of the normalized CI over time, was evident. However, the Bge cells clearly recovered ∼20 h after the addition of low concentrations of puromycin (Fig. 4A). The IC50 of the antibiotic was calculated with the assistance of the xCELLigence RTCA Software 1.2. Dose response curves were performed with concentrations of puromycin ranging from 0.078 to 5 μg/ml (two-fold serial dilutions), and the normalization CI time point was chosen immediately before the addition of the antibiotic. Fig. 4B shows the dose response curves of Bge cells cultured in the presence of increasing concentrations of the antibiotic (i.e. normalized CI plotted against the log of puromycin concentrations) at different time points after the addition of puromycin. The IC50 of the antibiotic was 10-5.72 g/ml, i.e. ∼1.91 μg/ml, for Bge cells under the conditions tested here. The time-dependent IC50 was stable from 24 h after puromycin addition and remained almost constant for 3 more days (Fig. 4C).
Fig. 4.

Dose response of Bge (Biomphalaria glabrata embryonic) cells to puromycin. (A) Cell proliferation measured by xCELLigence and shown as the normalized Cell Index (CI) over time of the Bge cells cultured in the presence of increasing concentrations of puromycin as indicated. Vertical bar represents the time point when the normalization was performed immediately before the addition of puromycin (arrow), i.e. ∼79 h after the cells were seeded. (B) Puromycin dose response curves generated at indicated time points after the addition of the antibiotic and employed to estimate the IC50 values with the assistance of real time cellular analysis (RTCA) Software 1.2. The mean IC50 is indicated, i.e. 10-5.72 g/ml = ∼1.91 μg/ml. (C) Time-dependent IC50 values from 24 to 72 h after the addition of the antibiotic. The mean IC50 is indicated at ∼1.91 μg/ml as the horizontal red dashed line.
3.3. A PEI-based transfection protocol was effective to deliver reporter genes into BGE cells
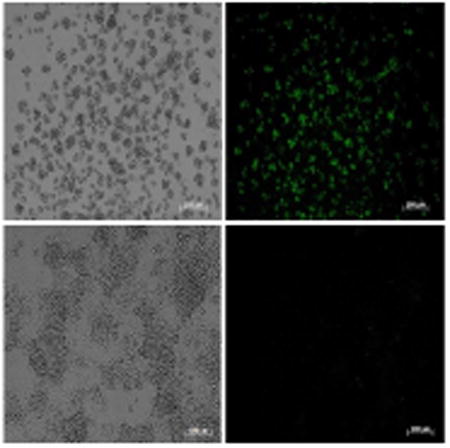
Transfection protocols for Bge cells have been previously reported (Lardans et al., 1996; Yoshino et al., 1998). However, in the current study we employed for the first known time, a recently described PEI-based protocol for transfection of snails and S. mansoni schistosomules (Knight et al., 2011; Liang et al., 2013). Firstly, as proof-of-concept, mammalian cells were transfected following this protocol. The human kidney cell line HEK293T was transfected with the piLenti-RNAi-GFP plasmid encoding GFP driven by the CMV promoter (Supplementary Fig. S1A), and 18 h after transfection a strong GFP signal was evident in comparison with control cells (Fig. 5A - F). The GFP signal increased over time and more cells displayed green fluorescence 48 h after transfection (not shown). Secondly, we proceeded to transfect Bge cells with the same plasmid encoding GFP and following the protocol successfully employed to transfect snails (Knight et al., 2011). GFP-positive-Bge cells were evident 5 days after transfection in comparison with control cells exposed to PEI alone (Fig. 5G - L).
Fig. 5.
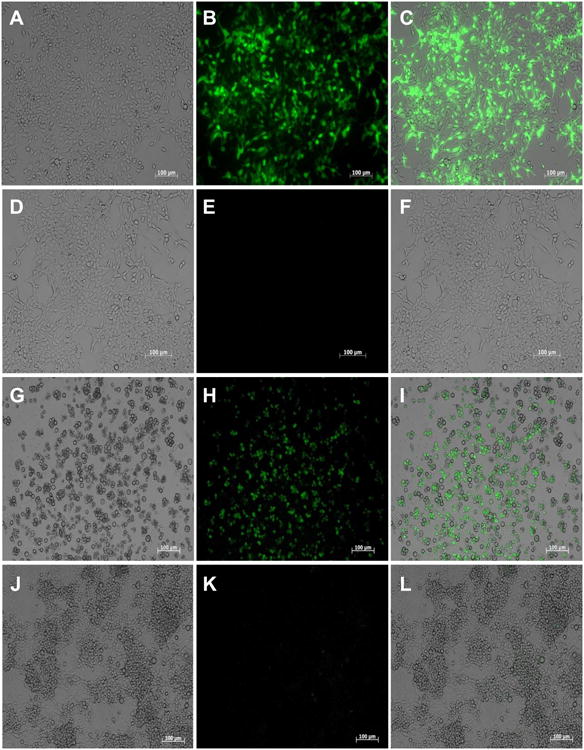
GFP expression in cells transfected with the plasmid piLenti-RNAi-GFP and polyethylenimine (PEI). (A – F) Representative images of transfected human cells (HEK 293T) examined under bright field (A), fluorescent field (B) and merged (C). PEI only-transfected control human cells examined either under bright (D), or fluorescence field (E) and merged (F). Pictures of HEK 293T cells were captured 18 h after transfection, (G – L) Representative images of transfected Bge (Biomphalaria glabrata embryonic) cells examined under either bright (G), or fluorescence field (H) and merged (I). PEI only-transfected Bge cells examined under either bright (J), or fluorescence field (K) and merged (L). Pictures of Bge cells were captured 5 days after transfection. Scale bar: 100 μm.
3.4. Bge cells transfected with a puromycin resistance marker were partially rescued in the presence of antibiotic
Once the sensitivity profile of Bge cells to puromycin was established, together with an effective protocol to transfect the cells with plasmid DNA, we decided to investigate antibiotic selection and enrichment of Bge cells transfected with a puromycin resistant marker-encoding plasmid. Bge cells were seeded and grown in E-plates employing xCELLigence that allows the quantification of the effect (Rakers et al., 2014). The Bge cells were transfected with the plasmid pLNHX_cHS4_puroR (Supplementary Fig. S1B) after they had reached CI values between ∼0.7 and ∼1.0. Two experimental groups were included; (i) cells transfected with the plasmid pLNHX_cHS4_puroR - PEI nanocomplex, or (ii) PEI alone, and a third non-transfected, mock control. Two days after the transfection puromycin was added to the culture medium at 0, 2.5 or 5 μg/ml and the cells were further monitored for at least 84 h. The percentage of the normalized CI of experimental groups compared with the mock control (% normalized CI = 100) was calculated for the different puromycin concentrations and plotted over time (Fig. 6). No significant differences among the three groups, i.e. the two experimental transfected groups and the control, were observed when the cells were cultured with 0 or 2.5 μg/ml of puromycin. However, a partial but significant rescue of ∼40% was detected for cells transfected with both PEI and pLNHX_cHS4_puroR and cultured for ∼80 h in the presence of 5 μg/ml of the antibiotic in comparison with control cells (Fig. 6A). The partial rescue of the transfected cells compared with control cells became apparent from ∼2 days after the addition of 5 μg/ml of puromycin and continued until the end of monitoring (Fig. 6B).
Fig. 6.

Partial rescue of Bge (Biomphalaria glabrata embryonic) cells transfected with a puromycin-resistant marker and cultured in the presence of the antibiotic. Cell growth is expressed as a percentage of the normalized Cell Index of transfected cells compared with mock control cells (growth rate = 100%). All cells were cultured in the presence of puromycin. (A) Cell growth in mock control, cells transfected only with PEI and cells transfected with both PEI and plasmid pLNHX_cHS4_puroR and cultured for 80 h after the addition of 0, 2.5 or 5 μg/ml of puromycin as indicated. (B) Cell growth overtime after the addition of 5 μg/ml of puromycin in mock control, cells transfected only with PEI and cells transfected with both PEI and plasmid pLNHX_cHS4_puroR. Error bars: ±1 S.D. of the mean. * P ≤ 0.05.
4. Discussion
From the time of its establishment in 1976 (Hansen, 1976), few laboratories have been able to routinely maintain the Bge cell line for experimental use. Notwithstanding this complication, advances have been reported including the culture from miracidium to cercaria in the presence of Bge cells (Ivanchenko et al., 1999) and demonstration that Bge cells behave like hemocytes, the cellular mediators of the snail innate defense system (Humphries and Yoshino, 2006).
Three different isolates of Bge cells were analyzed in the current study; SL and CB that were previously described (Odoemelam et al., 2009), and a third isolate maintained by us (UK). Significant differences were not evident among the isolates in terms of the morphology and size of the cells. However, marked variability in the doubling times among the isolates occurred, as quantified by the xCELLigence assay. The UK isolate displayed the shortest doubling time, ranging between ∼40 h to ∼56 h, and was the most independent of the number of seeded cells. Remarkably, the doubling times of both SL and CB isolates were not only longer than the UK isolate but also variable, depending on the number of cells seeded. CB displayed the longest doubling time (∼157 h) and, inversely, SL displayed the shortest doubling time (∼108 h) when more cells were seeded. These differences are likely due to genetic changes to the three isolates that have independently evolved in culture. Indeed, karyotypes of CB and SL isolates had altered dramatically, revealing pronounced aneuploidy, from when this was first determined by Bayne et al. (1978). The karyotype remains to be determined for the UK cell isolate. However, based on these data showing that this isolate was the fastest growing, we could assume that its karyotype may also have changed in culture.
To date, considerable progress developing functional genomics tools for schistosomes has been reported (Beckmann and Grevelding, 2012; Mann et al., 2014; Rinaldi et al., 2012). However, few if any reports have addressed functional genomics for B. glabrata or other intermediate host snails of schistosomes. Therefore, our results showing that Bge cells are sensitive to puromycin from 5 μg/ml to 200 ng/ml, for all three isolates, and the fast kill time (2 days at 5 μg) is particularly encouraging. Also noteworthy is that this sensitivity enabled the moderate selection of cells harboring a plasmid expressing the puromycin N-acetyl-transferase (PAC) gene while cultured in the antibiotic. Transfection assisted with nanoparticles of PEI has been reported to deliver double-stranded RNA and naked plasmid in snails and schistosomes (Knight et al., 2011; Liang et al., 2013), respectively, but is reported here for the first known time for transient transfection of the snail cell line. Although a partial selection of the cells transfected with the plasmid bearing the PAC gene, driven by the 5′ long terminal repeat (LTR) of the Moloney Leukaemia Virus (MLV) was consistently observed under drug pressure, there was only a minor difference between survival of transfected cells and control non-transfected cells (rescue of ∼40% was observed for cells transfected with both PEI and pLNHX_cHS4_puroR in comparison with control cells). Reasons for this lower than expected selection could be that optimum function of the 5′ LTR promoter occurs at 37°C whereas Bge cells were maintained at 23°C (room temperature). Furthermore, we used a heterologous virus promoter in these studies. To date, only one snail promoter, HSP 70, has been characterized and shown to drive the expression of the reporter luciferase gene (Yoshino et al., 1998). To improve the selection, we are currently testing the use of a pantropic virus delivery system (Mann et al., 2014), and preliminary results are encouraging. Following the availability of the genome sequence of this snail, which is pending (unpublished data) more snail promoters and cis-regulatory sequences likely will be characterized and optimized for genetic transformation studies.
With the findings presented here, characterizing the cytometrics, analyzing Bge cell sensitivity to puromycin, and subsequent use of the antibiotic to partially select the transfected cells, we have advanced prospects for facile efficient transfection of this cell line from the snail host of S. mansoni. We aim to transfer this technology to the intact snail, studying the sensitivity of this gastropod at large to antibiotics, including puromycin, to facilitate the eventual manipulation of the snail genome to dissect the molecular pathways that enable schistosomes (sporocyts) to develop in vivo, and hence shine a light on the host-parasite interaction. In this regard, primary cell lines derived from the gill tissue and hemocytes of the abalone mollusc, Haliotis tuberculata have been used to examine host pathogen interactions and to test the effects of toxins, respectively (Minguez et al., 2014; Pichon et al., 2013). Investigators in this field would benefit from access to techniques and protocols that optimize co-culture of schistosomes with Bge cells.
Molluscs and flatworms constitute the majority of lophotrocozoan taxa, yet only a single established cell line, the Bge cell line, exists from this super-phylum. This is in contrast to the wide application of insect cell lines, including from the lepidopteran Spodoptera frugiperda. The model Sf9 cell line from this moth, and baculovirus system, is used routinely for the production of functional recombinant proteins (Fogal et al., 2014; Schneider and Seifert, 2010). Cell lines from the fruit fly, Drosophila melanogaster, are also widely employed for in vitro cellular and molecular studies, reviewed by Ceriani (2007), for example, mutagenesis of the S2 cell line using the CRISPR/Cas9 gene editing approach has been recently accomplished (Bassett and Liu, 2014; Bassett et al., 2014). There is also a clear need for an established cell line in the schistosome molecular tool-kit and progress in this endeavor is being made, especially for Schistosoma japonicum (see review by Ye et al. (2013)). Finally, the introduction here of the xCELLigence approach to analyze non-model, less conventional cell lines such as Bge of B. glabrata, is notable and predicts follow-up studies to examine host-parasite interaction in more detail such as in Transwell/co-culture experiments (Roshan Moniri et al., 2014). With the increasing availability of sequence information of the genome and transcriptome of both the snail host and the schistosome, it is opportune and timely to optimize Bge cells as companion ‘tools’ to provide more fundamental information on the schistosome/snail host-parasite relationship, vector biology and indeed novel interventions. Since the snail is the natural obligate intermediate host for the schistosome parasite, discovery of mediators in the development of the in vitro co-culture of the parasite with the Bge cell line that we can interfere with can identify potential candidates for novel control strategies. The three Bge cell isolates described here are available, as frozen stocks or in tissue culture from the National Institutes of Health (NIH) -National Institute of Allergy and Infectious Diseases (NIAID), Schistosomiasis Repository (SR3) at Biodefense and Emerging Infections (BEI) resources (http://www.beiresources.org). Manassas, Virginia, USA.
Supplementary Material
Supplementary Fig. S1. Constructs employed in the current study. (A) Schematic of the plasmid piLenti-RNAi-GFP (Applied Biological Materials, U.S.A). (B) Schematic of the murine leukemia virus plasmid pLNHX_cHS4_650 encoding the neomycin-resistant marker (Suttiprapa et al., 2012) that was replaced with the puromycin resistant marker to engineer pLNHX_cHS4_puroR (C). MCS, multiple cloning site. Maps are not to scale.
Supplementary Fig. S2. Representative micrograph of Bge (Biomphalaria glabrata embryonic) cells in culture. The diameter of the cells was estimated with the assistance of AxioVision release 4.6.3 software (Zeiss, Germany). Scale bar: 50μm
Highlights.
The xCELLigene system was employed to study cytometrics, growth characteristics, and sensitivity of Bge cells to xenobiotics.
Bge cells were sensitive to puromycin, displaying a half maximal inhibitory concentration (IC50) of ∼1.91 μg/ml.
Bge cells transfected by PEI/ puroR encoding plasmid nanoparticles were partially rescued when cultured with puromycin.
Results pave the way for developing functional genomic tools to investigate the host-parasite interaction.
Acknowledgments
We thank Dr. Irene Riz, who generously provided the plasmid piLenti-RNAi-GFP and the HEK293T cells. These studies were supported by awards 1R21AI109532-01A1 (GR, VHM, PJB) and 1R01AI072773 (PJB, VHM, GR) from the National Institute of Allergy & Infectious Diseases, National Institutes of Health, USA.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bassett A, Liu JL. CRISPR/Cas9 mediated genome engineering in Drosophila. Methods. 2014;69:128–136. doi: 10.1016/j.ymeth.2014.02.019. [DOI] [PubMed] [Google Scholar]
- Bassett AR, Tibbit C, Ponting CP, Liu JL. Mutagenesis and homologous recombination in Drosophila cell lines using CRISPR/Cas9. Biol Open. 2014;3:42–49. doi: 10.1242/bio.20137120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayne CJ, Owczarzak A, Allen JR. Molluscan (Biomphalaria) Cell line: serology, karyotype, behavioral, and enzyme electrophoretic characterization. J of Inv Path. 1978;32:35–39. [Google Scholar]
- Beckmann S, Grevelding CG. Paving the way for transgenic schistosomes. Parasitol. 2012;139:651–668. doi: 10.1017/S0031182011001466. [DOI] [PubMed] [Google Scholar]
- Berriman M, Haas BJ, LoVerde PT, Wilson RA, Dillon GP, Cerqueira GC, Mashiyama ST, Al-Lazikani B, Andrade LF, Ashton PD, Aslett MA, Bartholomeu DC, Blandin G, Caffrey CR, Coghlan A, Coulson R, Day TA, Delcher A, DeMarco R, Djikeng A, Eyre T, Gamble JA, Ghedin E, Gu Y, Hertz-Fowler C, Hirai H, Hirai Y, Houston R, Ivens A, Johnston DA, Lacerda D, Macedo CD, McVeigh P, Ning Z, Oliveira G, Overington JP, Parkhill J, Pertea M, Pierce RJ, Protasio AV, Quail MA, Rajandream MA, Rogers J, Sajid M, Salzberg SL, Stanke M, Tivey AR, White O, Williams DL, Wortman J, Wu W, Zamanian M, Zerlotini A, Fraser-Liggett CM, Barrell BG, El-Sayed NM. The genome of the blood fluke Schistosoma mansoni. Nature. 2009;460:352–358. doi: 10.1038/nature08160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulo V, Cadoret JP, Shike H, Shimizu C, Miyanohara A, Burns JC. Infection of cultured embryo cells of the pacific oyster, Crassostrea gigas, by pantropic retroviral vectors. In Vitro Cell Dev Biol Anim. 2000;36:395–399. doi: 10.1290/1071-2690(2000)036<0395:IOCECO>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- Brindley PJ, Mitreva M, Ghedin E, Lustigman S. Helminth genomics: The implications for human health. PLoS Negl Trop Dis. 2009;3:e538. doi: 10.1371/journal.pntd.0000538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceriani MF. Basic protocols for Drosophila S2 cell line: maintenance and transfection. Methods Mol Biol. 2007;362:415–422. doi: 10.1007/978-1-59745-257-1_33. [DOI] [PubMed] [Google Scholar]
- Chuan J, Feng Z, Brindley PJ, McManus DP, Han Z, Jianxin P, Hu W. Our wormy world: genomics, proteomics and transcriptomics in East and southeast Asia. Adv Parasitol. 2010;73:327–371. doi: 10.1016/S0065-308X(10)73011-6. [DOI] [PubMed] [Google Scholar]
- Coustau C, Yoshino TP. Flukes without snails: advances in the in vitro cultivation of intramolluscan stages of trematodes. Exp Parasitol. 2000;94:62–66. doi: 10.1006/expr.1999.4462. [DOI] [PubMed] [Google Scholar]
- Fogal S, Carotti M, Giaretta L, Lanciai F, Nogara L, Bubacco L, Bergantino E. Human Tyrosinase Produced in Insect Cells: A Landmark for the Screening of New Drugs Addressing its Activity. Mol Biotechnol. 2015;57:45–57. doi: 10.1007/s12033-014-9800-y. [DOI] [PubMed] [Google Scholar]
- Hansen E. A cell line from embryos of Biomphalaria glabrata (Pulmonata): Establishment and characteristics. In: Maranorosche K, editor. Invertebrate tissue culture: Research applications. Academic Press; New York: 1976. pp. 75–97. [Google Scholar]
- Humphries JE, Yoshino TP. Schistosoma mansoni excretory-secretory products stimulate a p38 signalling pathway in Biomphalaria glabrata embryonic cells. Int J Parasitol. 2006;36:37–46. doi: 10.1016/j.ijpara.2005.08.009. [DOI] [PubMed] [Google Scholar]
- Ivanchenko MG, Lerner JP, McCormick RS, Toumadje A, Allen B, Fischer K, Hedstrom O, Helmrich A, Barnes DW, Bayne CJ. Continuous in vitro propagation and differentiation of cultures of the intramolluscan stages of the human parasite Schistosoma mansoni. Proc Natl Acad Sci U S A. 1999;96:4965–4970. doi: 10.1073/pnas.96.9.4965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke N, Wang X, Xu X, Abassi YA. The xCELLigence system for real-time and label-free monitoring of cell viability. Methods Mol Biol. 2011;740:33–43. doi: 10.1007/978-1-61779-108-6_6. [DOI] [PubMed] [Google Scholar]
- Knight M, Arican-Goktas HD, Ittiprasert W, Odoemelam EC, Miller AN, Bridger JM. Schistosomes and snails: a molecular encounter. Front Genet. 2014;5:230. doi: 10.3389/fgene.2014.00230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight M, Miller A, Liu Y, Scaria P, Woodle M, Ittiprasert W. Polyethyleneimine (PEI) mediated siRNA gene silencing in the Schistosoma mansoni snail host, Biomphalaria glabrata. PLoS Negl Trop Dis. 2011;5:e1212. doi: 10.1371/journal.pntd.0001212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lardans V, Boulo V, Duclermortier P, Serra E, Mialhe E, Capron A, Dissous C. DNA transfer in a Biomphalaria glabrata embryonic cell line by DOTAP lipofection. Parasitol Res. 1996;82:574–576. doi: 10.1007/s004360050166. [DOI] [PubMed] [Google Scholar]
- Liang S, Knight M, Jolly ER. Polyethyleneimine mediated DNA transfection in schistosome parasites and regulation of the WNT signaling pathway by a dominant-negative SmMef2. PLoS Negl Trop Dis. 2013;7:e2332. doi: 10.1371/journal.pntd.0002332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann VH, Suttiprapa S, Skinner DE, Brindley PJ, Rinaldi G. Pseudotyped murine leukemia virus for schistosome transgenesis: approaches, methods and perspectives. Transgenic Res. 2014;23:539–556. doi: 10.1007/s11248-013-9779-3. [DOI] [PubMed] [Google Scholar]
- Minguez L, Halm-Lemeille MP, Costil K, Bureau R, Lebel JM, Serpentini A. Assessment of cytotoxic and immunomodulatory properties of four antidepressants on primary cultures of abalone hemocytes (Haliotis tuberculata) Aquat Toxicol. 2014;153:3–11. doi: 10.1016/j.aquatox.2013.10.020. [DOI] [PubMed] [Google Scholar]
- Ni P, Zhang Q, Chen H, Chen L. Inactivation of an integrated antibiotic resistance gene in mammalian cells to re-enable antibiotic selection. Biotechniques. 2014;56:198–201. doi: 10.2144/000114160. [DOI] [PubMed] [Google Scholar]
- Odoemelam E, Raghavan N, Miller A, Bridger JM, Knight M. Revised karyotyping and gene mapping of the Biomphalaria glabrata embryonic (Bge) cell line. Int J Parasitol. 2009;39:675–681. doi: 10.1016/j.ijpara.2008.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichon D, Cudennec B, Huchette S, Djediat C, Renault T, Paillard C, Auzoux-Bordenave S. Characterization of abalone Haliotis tuberculata-Vibrio harveyi interactions in gill primary cultures. Cytotechnology. 2013;65:759–772. doi: 10.1007/s10616-013-9583-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Protasio AV, Tsai IJ, Babbage A, Nichol S, Hunt M, Aslett MA, De Silva N, Velarde GS, Anderson TJ, Clark RC, Davidson C, Dillon GP, Holroyd NE, LoVerde PT, Lloyd C, McQuillan J, Oliveira G, Otto TD, Parker-Manuel SJ, Quail MA, Wilson RA, Zerlotini A, Dunne DW, Berriman M. A systematically improved high quality genome and transcriptome of the human blood fluke Schistosoma mansoni. PLoS Negl Trop Dis. 2012;6:e1455. doi: 10.1371/journal.pntd.0001455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghavan N, Knight M. The snail (Biomphalaria glabrata) genome project. Trends Parasitol. 2006;22:148–151. doi: 10.1016/j.pt.2006.02.008. [DOI] [PubMed] [Google Scholar]
- Rakers S, Imse F, Gebert M. Real-time cell analysis: sensitivity of different vertebrate cell cultures to copper sulfate measured by xCELLigence((R)) Ecotoxicol. 2014;23:1582–1591. doi: 10.1007/s10646-014-1279-6. [DOI] [PubMed] [Google Scholar]
- Rinaldi G, Eckert SE, Tsai IJ, Suttiprapa S, Kines KJ, Tort JF, Mann VH, Turner DJ, Berriman M, Brindley PJ. Germline transgenesis and insertional mutagenesis in Schistosoma mansoni mediated by murine leukemia virus. PLoS Pathog. 2012;8:e1002820. doi: 10.1371/journal.ppat.1002820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roshan Moniri M, Young A, Reinheimer K, Rayat J, Dai LJ, Warnock GL. Dynamic assessment of cell viability, proliferation and migration using real time cell analyzer system (RTCA) Cytotechnology. 2014;67:379–386. doi: 10.1007/s10616-014-9692-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider EH, Seifert R. Sf9 cells: a versatile model system to investigate the pharmacological properties of G protein-coupled receptors. Pharmacol Ther. 2010;128:387–418. doi: 10.1016/j.pharmthera.2010.07.005. [DOI] [PubMed] [Google Scholar]
- Smout MJ, Kotze AC, McCarthy JS, Loukas A. A novel high throughput assay for anthelmintic drug screening and resistance diagnosis by real-time monitoring of parasite motility. PLoS Negl Trop Dis. 2010;4:e885. doi: 10.1371/journal.pntd.0000885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suttiprapa S, Rinaldi G, Brindley PJ. Prototypic chromatin insulator cHS4 protects retroviral transgene from silencing in Schistosoma mansoni. Transgenic Res. 2012;21:555–566. doi: 10.1007/s11248-011-9556-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Q, Dong HF, Grevelding CG, Hu M. In vitro cultivation of Schistosoma japonicum-parasites and cells. Biotechnol Adv. 2013;31:1722–1737. doi: 10.1016/j.biotechadv.2013.09.003. [DOI] [PubMed] [Google Scholar]
- Yoshino TP, Bickham U, Bayne CJ. Molluscan cells in culture: primary cell cultures and cell lines. Can J Zool. 2013;91 doi: 10.1139/cjz-2012-0258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshino TP, Wu XJ, Liu HD. Transfection and heat-inducible expression of molluscan promoter-luciferase reporter gene constructs in the Biomphalaria glabrata embryonic snail cell line. Am J Trop Med Hyg. 1998;59:414–420. doi: 10.4269/ajtmh.1998.59.414. [DOI] [PubMed] [Google Scholar]
- Zerlotini A, Aguiar ER, Yu F, Xu H, Li Y, Young ND, Gasser RB, Protasio AV, Berriman M, Roos DS, Kissinger JC, Oliveira G. SchistoDB: an updated genome resource for the three key schistosomes of humans. Nucleic Acids Res. 2013;41:D728–D731. doi: 10.1093/nar/gks1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Fig. S1. Constructs employed in the current study. (A) Schematic of the plasmid piLenti-RNAi-GFP (Applied Biological Materials, U.S.A). (B) Schematic of the murine leukemia virus plasmid pLNHX_cHS4_650 encoding the neomycin-resistant marker (Suttiprapa et al., 2012) that was replaced with the puromycin resistant marker to engineer pLNHX_cHS4_puroR (C). MCS, multiple cloning site. Maps are not to scale.
Supplementary Fig. S2. Representative micrograph of Bge (Biomphalaria glabrata embryonic) cells in culture. The diameter of the cells was estimated with the assistance of AxioVision release 4.6.3 software (Zeiss, Germany). Scale bar: 50μm