

Abstract
Systemic autoimmune diseases such as lupus affect multiple organs, usually in a diverse fashion where only certain organs are affected in individual patients. It is unclear whether the ‘local’ immune cells play a role in regulating tissue specificity in relation to disease heterogeneity in systemic autoimmune diseases. Here, we used skin as a model to determine the role of tissue-resident dendritic cells in local and systemic involvement within a systemic lupus disease model. Skin-resident dendritic cells, namely Langerhans cells (LC), have been implicated in regulating tolerance or autoimmunity using elegant transgenic models, however, their role in local versus systemic immune regulation is unknown. We demonstrate that while lymphocytes from skin-draining lymph nodes of autoimmune-prone MRL/MpJ-Faslpr/lpr mice react spontaneously to a physiological skin self-Ag desmoglein-3, epicutaneous applications of desmoglein-3 induced tolerance that is dependent on LCs. Inducible ablation of LCs in adult, preclinical MRL/MpJ-Faslpr/lpr and MRL/MpJ-Fas+/+ mice resulted in increased autoantibodies against skin Ags and markedly accelerated lupus dermatitis with increased local macrophage infiltration, but had no effect on systemic autoantibodies such as anti-dsDNA Abs or disease in other organs such as kidneys, lung, and liver. Furthermore, skin-draining lymph nodes of LC-ablated MRL/MpJ-Faslpr/lpr mice had significantly fewer CD4+ T-cells producing anti-inflammatory cytokine IL-10 than LC-intact controls. These results indicate that a skin-resident dendritic cell population regulates local tolerance in systemic lupus and emphasize the importance of the local immune milieu in preventing tissue-specific autoimmunity yet have no effect on systemic autoimmunity.
INTRODUCTION
Systemic autoimmune diseases, such as systemic lupus erythematosus (SLE)3, involve various combinations of multiple organ autoimmunity and tissue injury. Thus, patients present with a wide variety of clinical manifestations. Dysregulation of immune cells, such as dendritic cells (DCs), and B and T cells, has been implicated in the systemic autoimmunity best reflected by the presence of autoAbs against systemic autoantigens, such as dsDNA (1). However, since specific organ involvement varies from patient to patient, organ-specific tolerance may also play a role in regulating tissue-specific injury. Evidence supports a pivotal role of DCs in the pathogenesis of systemic autoimmune diseases. In fact, a constitutive deletion of all DCs (CD11c+cells) in autoimmune-prone MRL/MpJ-Faslpr/lpr (MRL-lpr) mice ameliorates lupus (2). However, DCs comprise heterogeneous subsets of cells with different origins, organ of residence, and potentially differential roles. While many studies implicate myeloid and plasmacytoid DC populations in the pathogenesis of SLE (3), the role of tissue-resident DCs in the maintenance of organ-specific tolerance in autoimmunity and development of disease is not well defined.
Langerhans cells (LC) are epidermis-resident DCs that express langerin and migrate through dermal lymphatics to skin-draining lymph nodes (dLN). LCs are the prototype tissue-resident DCs studied to gain insights into tissue-specific DC function. LCs act as sentinels to orchestrate the immune response against foreign Ags including pathogens in skin. In the steady state, LCs have been implicated to induce peripheral tolerance to skin self-Ags (4, 5). However, transgenically-expressed epidermal Ag can be cross-presented by migratory DC to CD8+ T cells in dLN under steady state conditions resulting in autoimmunity (6, 7). Such immune stimulatory versus regulatory roles of LCs have been well elucidated using elegant transgenic models, however, little is known about the role of LCs in chronic autoimmune conditions such as SLE. Analyses of LCs in skin biopsies from humans and mice with SLE have revealed conflicting information. For example, skin biopsies from patients with SLE show increased numbers (8) or an irregular distribution (9) of LCs in some studies, but reduced numbers of LCs in skin lesions in other studies (10). Another study reported the migration of LCs into lupus skin lesions preceding the infiltration with lymphocytes (11). In order to explore the contribution of LCs in the maintenance of skin tolerance prior to the onset of clinical disease, we utilized murine strains that spontaneously develop an autoimmune disease resembling human SLE.
MRL-lpr and MRL/MpJ-Fas+/+ (MRL+/+) mice spontaneously develop a syndrome with anti-dsDNA Abs, nephritis, vasculitis, and dermatitis that histopathologically mimics chronic cutaneous lupus in humans (12). As in a human autoimmune bullous disease, pemphigus vulgaris (13), MRL-lpr mice develop autoantibodies to the cadherin-type desmosomal glycoprotein, desmoglein 3 (Dsg3) (14) that is expressed in stratified squamous epithelium in skin, oral mucosa and esophagus but not in other tissues such as kidney, liver, intestine, and muscle (14, 15). Furthermore, immunization with Dsg3 induces lupus-like skin disease in mice (14), and the adoptive transfer of Dsg3-reactive CD4+ T cell clones into immune-deficient mice induces interface dermatitis that is a feature of cutaneous lupus and a few other inflammatory skin diseases (15). Importantly, the adoptively transferred Dsg3-reactive T cells infiltrate skin and mucous membranes, but not other organs that do not express Dsg3 (15). Taken together, these observations suggest a role for tissue-specific autoimmunity in the development of lupus skin disease, which may be regulated differently from the systemic disease process. Hence, we utilized Dsg3 as a model skin autoantigen to investigate local tolerance induction in the murine lupus model.
Previously we demonstrated that lupus-prone MRL-lpr mice have impaired migration of langerin-expressing skin-DCs from skin to dLN, which is detected early in life and correlates with the onset and severity of autoimmune skin disease (16). This migration defect leading to the paucity of langerin-expressing DCs in dLN might jeopardize the maintenance of tolerance to skin Ags, thus contributing to the development of lupus dermatitis. Using an endogenous skin self-Ag and an inducible LC ablation model, we specifically examined the role of LCs in maintaining local skin tolerance by ablating LCs pre-clinically to exacerbate the LC deficiency in MRL mice. While several langerin+ DC ablative models exist (17–19), we chose an inducible model (20) to ablate LCs in young lupus mice to obviate any potential compensatory pathways arising as a result of constitutive LC ablation from early development as well as to mimic the loss of peripheral tolerance observed clinically in young human adults who develop lupus. Using this model, we for the first time demonstrate a role of LCs in protecting against the breakdown of skin tolerance in genetically autoimmune-prone mice and preventing the development of autoimmune dermatitis in murine lupus.
MATERIALS AND METHODS
Mice
MRL-lpr, MRL+/+, C3H/HeOuJ (C3H), C57BL/6 (B6), and C57BL-6 Faslpr/lpr (B6-lpr) mice were purchased (Jackson Laboratory) and/or bred locally. B6 Langerin-DTR-eGFP knock-in (Lang-DTR) mice that express human diphtheria toxin receptor (DTR) and enhanced green fluorescent protein (eGFP) downstream of the internal stop codon of the Cd207 gene (langerin [Lang]) were provided by Bernard Malissen (20). Lang-DTR B6 mice were backcrossed onto the MRL-lpr and MRL+/+ backgrounds for more than 10 generations. To generate Lang-DTR B6-lpr mice, B6 knock-in mice were crossed with B6-lpr mice, and the F1 offspring intercrossed. Mice were maintained in specific pathogen-free conditions.
Antibodies
Abs against CD4 (GK1.5), CD8 (53–6.7), CD11b (M1/70), CD16/32 (93), CD11c (N418), CD19 (1D3), CD44 (IM7), CD62L (MEL-1), CD69 (H1.2F3), FoxP3 (FJK-16s), I-A/E (M5/114.15.2), IL-17 (eBio17B7), INFγ (XMG1.2), Langerin (RMUL.2), TCRβ (H57-597), and TNFα (MP6-XT22) were purchased from eBioscience (San Diego, CA). Abs for Ki-67 (B56), IL-2 (JES6-5H4), and IL-10 (JES5-16E3) were purchased from BD Biosciences (San Jose, CA).
Flow cytometry
Fc receptors were blocked by incubating single cell suspensions on ice for at least 30 min with CD16/32 before staining. For cytokine assays, cells were cultured for 5–6 h with PMA/ionomycin in RPMI with 10% FCS at 37°C and 5% CO2. At the last 2–3 h of culture, BD GolgiStop was added for IL-10 staining and BD GolgiPlug was added for TNFα, INFγ, IL-2, and IL-17 staining. Samples were acquired on FACSCalibur or LSR-II flow cytometers (BD Biosciences). Data were analyzed with FlowJo software (TreeStar, Ashland, OR).
Epicutaneous tolerization assay
To administer the Ag in a steady state environment, we avoided the use of tape stripping and other traumatic procedures that are generally used to facilitate the skin penetrance of Ag. In initial experiments, we used acetone/olive oil as a carrier for Ags, followed by a patch (Duo-Derm extra thin) to keep in place the emulsion. This approach led to inconsistent results, as the patch was torn by many animals soon after its application. After trials of several carriers and application methods, we selected to emulsify the Ag (500 μg) with an equal volume of incomplete Freund’s adjuvant (Sigma). Mouse ears were painted epicutaneously with 50 μl (25 μl per ear) of an emulsion containing Dsg3 or hen egg lysozyme (HEL; Fisher Scientific, Pittsburg, PA) or PBS alone. Painted ears were blow air-dried to ensure consistent application of the Ag. Cervical lymph nodes were harvested 7 d later and their single cell suspensions cultured with increasing volumes of the indicated Ag in complete RPMI (10% FCS, L-glutamate, penicillin, streptavidin, non-essential amino acids, HEPES and β-mercaptoethanol) at 37°C and 5% CO2. On day 10, [3H]thymidine (Perkin Elmer, Waltham, MA) was added to each well and incorporation measured on a beta scintillation counter 18–24 h thereafter.
Preparation of Dsg3
For bacterial expression of Dsg3, the extracellular portion of Dsg3 was cloned in to BamHI and NotI sites in pGEX4T1 (GE Healthcare Life Sciences, Pittsburg, PA). Primers were chosen to PCR amplify from the signal sequence up to the transmembrane domain (5′-gcgc GGATCC GAACTGCATGTGAAGCCG-3′ and 5′-cgcgc GCGGCCGC TCACAGCCTCCAGGATGACT-3′) using cDNA as a template (clone 40130335, Open Biosystems). The plasmid was confirmed by sequencing. For expression, the plasmid was transformed into BL21 and grown to a density of 0.8. Protein expression was induced using 100 μM IPTG overnight at room temperature. Inclusion body purification protocol was from Pamela Bjorkman (http://www.its.caltech.edu/~bjorker/protocols.html). Cells were pelleted and resuspended in 13 ml of solution buffer (50mM Tris pH 8, 25% sucrose, 1mM EDTA, 0.1% sodium azide, 10mM DTT). After sonication for 30 pulses of 10 seconds each, 12.5 ml of lysis buffer (50mM Tris pH 8, 1% Triton X-100, 1% sodium deoxycholate, 100mM sodium chloride, 0.1% sodium azide, 10mM DTT) supplemented with lysozyme, DNAse I, and magnesium chloride to final concentrations of 200 μg/ml, 10 μg/ml, and 1mM respectively. After one hour incubation at room temperature, EDTA was added to a final concentration of 7mM and the lysate was snap frozen in liquid nitrogen and thawed at 37 °C for 30 minutes. Magnesium chloride was increased to 5 mM and incubation allowed to continue for one hour at room temperature. EDTA was increased to 14 mM. Inclusion bodies were pelleted by centrifugation at 11,000g at 4°C. The pellet was washed with wash buffer (50mM Tris pH 8, 100 mM sodium chloride, 1mM EDTA, 0.1% sodium azide, 1mM DTT, 0.5% Triton X-100) at 4°C. Another wash was performed without Triton X-100. LPS in rDsg3 preparations was measured using Limulus Amebocyte Lysate (LAL) chromogenic endotoxin quantitation kit (Pierce), which showed trace amounts of LPS in most preparations. rDsg3 was pre-incubated with polymyxin B (Sigma) at 10μg/ml for 30 min at 37°C to control for endotoxin contamination prior to use for in vitro culture studies.
Isolating cells from skin
To isolate DCs from skin, ears were first split into dorsal and ventral halves with the cartilage removed from the ventral sheet. To split epidermis from dermis, ear half was floated dermal side down in PBS containing 2.5 mg/ml dispase II (Roche, Nutley, NJ) at 4°C for 16–20 h. Epidermis and dermis were separated using forceps and cut into small pieces in RPMI containing 1mg/ml collagenase IV (Worthington, Lakewood, NJ) and 1mg/ml DNase (Sigma, St. Louis, MO). Tissue was incubated with the cocktail for 1 h at 37°C to obtain homogenous cell suspensions. 25mM of ETDA was added during the last 5 min of incubation.
Immunofluorescence
Ears were first split into dorsal and ventral halves using forceps. With the dermal side down, ear halves were floated on top PBS containing 3.8% NH4SCN (Sigma) at 37°C for 30 mins to split epidermis from dermis. After the epidermis and dermis were separated using forceps, tissue was fixed immediately in acetone at −20°C. Tissue was washed 3 times with PBS and blocked with 1% BSA and CD16/32 in PBS for at least 30 min. Tissues were stained with Alexa488-Langerin (eBioRMUL.2, eBiosciences) and Alexa647-CD11c (Invitrogen labeling kit). Images were acquired on a Leica SP1-Inverted confocal microscope and processed using Leica imaging software.
Preparation of whole-tissue skin lysate
Nair was applied to MRL-lpr mouse skin to remove hair and then washed off thoroughly. Skin was harvested and homogenized on ice with an electric homogenizer in lysis buffer (150mM NaCl, 20mM Tris-HCl, 0.5% Triton x100) containing protease inhibitor cocktail (Sigma). Homogenized skin was transferred to Eppendorf tubes and centrifuged for 20 min at 12000 rpm at 4°C. Supernatants were collected and stored at −20°C. Protein concentration was determined by BCA assay (Pierce, Rockford, IL).
Detection of autoantibodies
Autoantibodies were detected by Western blot or ELISA, as described previously (21, 22). To detect skin-reactive autoantibodies in mouse serum, skin lysate (50μg per lane) was incubated with NuPage LDS Sample Buffer (Invitrogen) and DTT at 70°C for 10mins and then run in a 4–12% gradient bis/tris polyacrylamide gel (Invitrogen, Grand Island, NY) in MOPS buffer. Precision Plus Protein Kaleidoscope Standards (Bio-rad, Hercules, CA) were run in every third lane. Gels were blotted to 0.45μm PVDF membranes (Invitrogen). Blots were then incubated with Ponceau S (0.1% Ponceau Red, 5% Acetic Acid) to visualize protein. After washing blots with 0.05% PBS-Tween 20 (PBS-T) and reactivated in methanol, membranes were blocked with 5% milk in PBS-T overnight at 4°C. Blots were then cut into strips so that each strip had one lane of lysate and a half lane of the molecular weight ladder. Blot strips were incubated with 200x diluted mouse serum for 1.5 h. On each blot, one strip was incubated with a standard of pooled serum samples from 24 MRL-lpr mice as a control. After washing with PBS-T, membranes were incubated with 1:25,000 diluted secondary Ab (Mouse TrueBlot ULTRA: anti-mouse IgG HRP, eBioscience) for 1 h at room temperature. Blots were visualized using SuperSignal West Femto Maximum Sensitivity Substrate Kit (Thermo Scientific, Waltham, MA). Band densities were calculated using ImageJ software. The 63kDa band visualized by Ponceau S staining was used to adjust for loading volume variances.
To detect autoantibodies to dsDNA by ELISA, 5μg/ml of calf thymus DNA (Sigma) was plated at 50μl per well in 96-well EIA/RIA plates and incubated overnight at 4°C. After washing plates thrice with 0.05% PBS-Tween 20 (PBS-T), wells were blocked with 2% BSA in PBS for 2 h at room temperature. The plates were then washed thrice with PBS-T, incubated with 50μl of 100x diluted mouse serum for 2 h at room temperature, washed 3-times again, and incubated with goat anti-mouse IgG conjugated to HRP (Santa Cruz Biotechnology, Dallas, TX) for 1 h at room temperature. After a final triple wash, plates were developed using the TMB Substrate Reagent Kit (Biolegend, San Diego, CA). Plates were read at 450 nm absorption. Serum from an old NZB/W mouse with severe nephritis was used as a positive control and to calculate the ELISA unit value [(Absorbance of sample – Absorbance of blank)/(Absorbance of positive control – Absorbance of blank)]. Anti-Dsg3 Abs were detected in serum diluted at 1:20 using mouse desmoglein3 Ab ELISA kit (MBL International, Woburn, MA).
Evaluation of disease severity
To assess clinical skin disease, blinded observers scored skin lesions on a scale of 0 (none) to 3 (severe), as previously described (23, 24). A score ≥ 2 (marked alopecia with or without hemorrhage) reflected mice with skin disease, as shown in Fig. 4 and 5. Histopathological scoring of H&E-stained sections of ear skin, nape skin, lung, liver, and kidney was performed by two independent reviewers trained by a dermatopathologist, using criteria described in Supplemental Table S1. Tissues were fixed in 10% formalin for 24 h and then stored in 70% ethanol until processed by paraffin embedding, sectioning, and standard H&E staining techniques. Proteinuria was measured using Siemens Albustix reagent strips.
FIGURE 4. Inducible LC ablation in adult, preclinical MRL-lpr mice accelerates and exacerbates autoimmune skin disease, but does not affect disease in other organ systems.

A, B. 6–8-wk-old female wild-type or knock-in (DTR) MRL-lpr littermates received 6–8 i.p. injections of DT (Depleted) or PBS (Intact) every 10–14 d, and animals were monitored for lupus skin disease. A, Representative images of clinical lesions of LC-intact and LC-depleted mice taken at 20 wks of age. B, Cumulative prevalence of animals without skin lesions (< 2+). Results demonstrate accelerated skin disease in LC ablated mice (n=14) compared to the three control groups (n=12–26 per group; *** p < 0.001 for overall log rank test; p=0.004 for red vs. green; p<0.001 for red vs. black; p=0.002 for red vs. blue). Control groups were not significantly different among themselves (p > 0.2). The median age of skin disease was 18 wks in the LC-depleted mice and 28–33 wks in the control groups. C, D, Ear and nape skin tissues were harvested at 20 wks of age, and skin sections stained with H&E. Representative photomicrographs are shown in C (scale bar indicates 200μm). D, H&E-stained skin sections were scored, as described in Supplemental Table S1. Results are expressed as the mean ± SEM composite histological scores (**p < 0.01, n = 10 and 13; ***p< 0.001, n = 10 and 11). E, H&E stained sections of lung, liver, and kidney from littermate LC-intact and LC-depleted MRL-lpr mice were scored, as described in Supplemental Table S1. Results are expressed as composite histology scores from individual mice; n=9–12 mice/group). F, Proteinuria was estimated using dipstick urinalysis, and read on a scale of trace (0.5) and 1–3 (10–11 mice/group).
FIGURE 5. Acceleration of skin autoimmunity by LC ablation occurs only in the autoimmune background and is independent of the Faslpr mutation.

A, Lang-DTR knock-in mice in B6, B6-lpr, MRL+/+, and MRL-lpr backgrounds were injected with DT or vehicle for 8 wks, and monitored for skin disease, as described in Figure 3A. Results are shown as the frequency of moderate to severe skin lesions (score ≥ 2+) at 26 wks of age in LC-ablated B6 and B6-lpr mice, and LC-ablated and LC-intact MRL+/+ and MRL-lpr mice. None of the LC-ablated B6 (n = 11) and B6-lpr (n=9) developed any skin lesions until their termination at one year of age. LC ablation in both MRL strains increased skin disease (**p = 0.007, 15 vehicle and 18 DT-injected MRL+/+ mice; ***p<0.001, 15 vehicle and 14 DT-injected MRL-lpr mice; Fisher’s exact test). B, Cumulative prevalence of mice without skin disease (< 2+ lesions). 9–15-wk-old Lang-DTR MRL+/+ mice were treated with PBS or DT every 10–14 d for 6–8 injections, and monitored for skin lesions (*p = 0.04, log rank test, n=16 and 19 mice respectively).
Detection of immune cell infiltrates in skin by immunohistochemistry
Five-micron frozen tissue sections were fixed in 4°C acetone for 10 min, incubated with 0.3% hydrogen peroxide for 30 min to block endogenous peroxidase activity, rinsed with PBS, and blocked with PBS containing 1% BSA and 5% normal rabbit serum for 30 min at room temperature. Avidin/biotin blocking kit (Vector Laboratories, Burlingame, CA) was used to block endogenous avidin and biotin. Sections were then incubated at 4°C with primary mAbs CD4 (1:20 dilution; BD PharMingen), CD8 (1:20 dilution; eBioscience), CD68 (1:200 dilution; Serotec), and CD11c (1:20 dilution; BD PharMingen), rinsed with PBS, and then incubated with biotinylated secondary Ab anti-Rat IgG (1:100; Vector Laboratories) for 1 h at room temperature. Sections were then incubated with Avidin-peroxidase complex (Vectastatin ABC kit, Vector Laboratories) for 30 min at room temperature, rinsed, and then exposed to 3,3′-diaminobenzidine for a chromogenic reaction and counterstained with hematoxylin. Negative control slide sections were stained in the same way without the primary Abs. Images were viewed on an Olympus BX51 microscope and acquired using MicroSuite (V) software. All images were prepared using Adobe Photoshop software.
Statistics
The cumulative prevalence of animals without skin lesions was analyzed using log rank tests. For MRL-lpr mice, first an overall log rank test was used and then pairwise log rank tests to compare two curves individually. The Fisher’s exact test was conducted to compare the frequency of mice with skin disease at 20-wks of age. Data on anti-dsDNA antibody levels, proteinuria, histology scores, and Western blot band density were analyzed using Wilcoxon’s rank sum test. Data on immune cell subsets, cytokine producing cells, and skin infiltrates were analyzed using two-tailed Student’s t test. Results of the epicutaneous tolerance assay were analyzed using two-way ANOVA for dose response assays, and one-way ANOVA with a Tukey post test for single dose assays.
RESULTS
Spontaneous loss of tolerance to skin autoantigens in mice prone to develop lupus dermatitis
MRL-lpr mice have a marked reduction in langerin-expressing DCs in their dLNs at an early age (16). Such deficiency of skin-DCs in dLN might lead to the loss of tolerance to skin. To test this, we first analyzed sera from MRL-lpr mice against skin lysate (Supplemental Fig. S1), which revealed Abs against multiple Ags, including a 110kDa band that is consistent with the molecular weight of Dsg3, a known skin autoantigen (14). To determine if the reactivity to skin-autoantigens arises prior to disease onset, dLN cells from naive preclinical MRL-lpr and age-matched MRL+/+ and C3H mice were cultured with Dsg3 or irrelevant, foreign Ags namely hen egg lysozyme (HEL) and OVA. Whereas the proliferative response of dLN cells to Con A was equivalent in the three strains of mice, dLN cells mounted a significant proliferative response to Dsg3 in MRL-lpr mice as compared to age-matched control animals (Fig. 1A, B). There was no difference in Ki67+ cells in dLN cells cultured with medium alone, HEL (Fig. 1A) or OVA (data not shown). CD4+ and CD8+ T cells as well as TCRβ+CD4−CD8− (double negative [DN]) cells proliferated in response to Dsg3, and 75% of Dsg3-reactive CD4+ T cells expressed memory marker CD44, with 38% cells bearing effector memory T cell markers (CD62L−/loCD44+) (Fig. 1C). These data suggest a spontaneous loss of tolerance to skin autoantigens in MRL-lpr mice prior to the onset of disease.
FIGURE 1. Spontaneous proliferation to a skin autoantigen in the dLN of MRL-lpr mice.
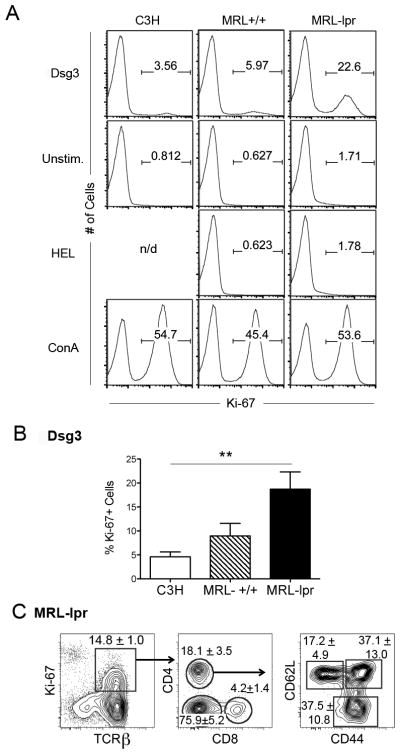
Freshly isolated cells from the dLNs of 5–8-wk-old female naïve MRL-lpr mice, MRL+/+ mice, and MHCII-matched C3H control mice were cultured with Dsg3 (Dsg3, 500 μg/ml) or hen egg lysozyme (HEL, 500 μg/ml) as a non-autoantigen control (n = 4 mice/group). ConA was used as a positive control, and medium alone cultures (Unstim.) served as a negative control. After 72 h, dLN cell proliferation was measured using Ki-67 expression. A, Representative histograms show Ki-67 expression on lymphocytes. Numbers on the histogram represent the % of proliferating cells. n/d = not done. B, Percentage of proliferating cells (Ki-67+ as gated in A) in response to Dsg3 from three independent experiments are shown as the mean ± SEM (**p <0.01; n = 4 per group). C, Proliferating T cells from MRL-lpr cultures containing Dsg3 were analyzed for T cell subsets and activation/memory markers. Representative FACS plots of proliferating T cells (TCRβ+ Ki-67+, left panel) were gated off live cells, and subsequently gated to show percentages of proliferating T cell subsets (CD4, CD8 and double negative, middle panel), and activation/memory CD4+ T cells (CD44 and CD62L, right panel) in MRL-lpr mice. Gate frequencies indicate the mean ± SD (n = 3 per group) from three independent experiments.
Tolerance to a skin autoantigen can be induced by epicutaneous applications of the autoantigen in lupus-prone mice
To directly test the role of langerin-expressing DCs in the breakdown of tolerance to skin autoantigens in MRL-lpr mice in vivo, we utilized an inducible tolerization model using high-dose autoantigen Dsg3 in young, pre-dermatitis mice. On day 0 and 1, high-dose Dsg3 (500 μg) or PBS mixed with equal proportions of vehicle emulsion were applied epicutaneously onto mouse ears and dried thoroughly with fan to ensure efficient application. After 7 days, dLN cells were harvested to assess a recall proliferative response to Dsg3 (Fig. 2A). Consistent with data in Figure 1, LC-intact MRL-lpr mice applied with only vehicle emulsion had a robust in vitro proliferative response to Dsg3 autoantigen in a dose-dependent manner. In contrast, MRL-lpr mice that received the tolerizing regimen of Dsg3 administered epicutaneously on ear skin showed a significant reduction in the proliferation of dLN cells to Dsg3 (p = 0.003) (Fig. 2B). The stimulation index (SI, ratio of cpm in cultures containing Dsg3 : cpm in wells containing media alone) at the highest recall concentration was 8-fold lower in Dsg3-painted animals than in control vehicle-painted animals (mean SI of 3 and 24, respectively). As a control for the in vivo immunogenicity of Dsg3, a s.c. injection of Dsg3 emulsified in complete Freund’s adjuvant at the hock site induced a strong in vitro recall proliferative response (Supplemental Fig. S2). To highlight that Dsg3-induced epicutaneous tolerance is self-Ag specific, epicutaneous applications of foreign Ags (HEL or OVA) did not elicit an in vitro proliferative response in spontaneous or tolerizing conditions (Fig. 2C [HEL] and data not shown [OVA]). The epicutaneous applications of the self-Ag Dsg3 also did not affect the in vitro response to the control foreign Ag (Fig. 2E, left two columns), and vice versa (Fig. 2F). The SI in Dsg3 recall response in vehicle-painted animals was comparable between Fig. 2B and Fig. 2F (mean SI of 24 and 27, respectively). Thus, although MRL-lpr mice develop spontaneous loss of tolerance to skin autoantigens, they are still amenable to tolerance induction as the topical administration of high doses of a tissue autoantigen can induce tolerance in an Ag-specific manner in MRL-lpr mice.
FIGURE 2. LCs are needed for the induction of tolerance to epicutaneously applied skin autoantigens.

A, Tolerization schema. Mice received a single PBS or DT i.p. injection 10 d prior to skin application of an Ag-containing emulsion. On day 0 and 1, emulsions containing vehicle (PBS) or a high dose of Ags (500 μg of a self [Dsg3] or foreign Ag [HEL or OVA]) were applied twice epicutaneously on preclinical (7–11-wk-old) female MRL-lpr mouse ears bilaterally. Seven days later, dLNs were harvested and dLN cells cultured in vitro with varying doses of the applied Ag (0–500 μg/ml) in triplicate. After 72 h, Ag-recall response was assessed using [3H]-Thymidine proliferation assay. Results are expressed in panels B–F as the mean ± SD triplicate CPM values. A dotted horizontal line in each panel represents [3H]-thymidine incorporation in cells cultured with medium alone (Media). B, In vitro recall response to Dsg3 was assessed in PBS-injected mice (LC-intact) that received a vehicle or Dsg3-containing emulsion (**, p=0.003). C, In vitro recall response to a foreign Ag (HEL) after its epicutaneous applications in PBS-injected mice (LC-intact). D, In vitro recall response to Dsg3 was assessed in DT-injected mice (LC-depleted). E, Epicutaneous application of a self Ag does not affect the in vitro responsiveness to a control foreign Ag. LC intact or depleted mice were epicutaneously (ECI) treated with Dsg3 or vehicle, and dLN cells cultured with control Ag. Mean ± SD triplicate CPM values from cultures containing HEL (500 μg/ml) are shown. F, Epicutaneous application of a foreign Ag does not affect the in vitro responsiveness to a self Ag. LC intact mice were epicutaneously treated with a foreign Ag (HEL) or vehicle, and dLN cells cultured with increasing doses of Dsg3. Results are shown as the mean ± SD triplicate CPM values. G, Representative FACS plots show frequency of Treg cells in LC-intact and LC-depleted mice 7 d after the epicutaneous application of vehicle or Dsg3. Plots were gated from TCRβ+, CD4+ lymphocytes. Gate frequencies indicate mean ± SD of 3 independent experiments (** p < 0.01; one-way ANOVA with a Tukey post-test; n = 3 mice). Results shown are representative of three independent experiments, each using 3–5 mice per condition.
LCs are necessary for the epicutaneous tolerance induction to a skin autoantigen
To evaluate the in vivo contribution of LCs in the autoantigen-specific tolerance in autoimmune background, we generated Lang-DTR MRL-lpr mice by introgressing the knock-in mutation from the stock B6 (20) onto the MRL-lpr background for more than 10 generations. In this model, a single i.p. injection of diphtheria toxin (DT) temporally deletes all langerin+ DCs that comprise of LCs and a recently discovered subset of langerin+ DCs that inhabit the dermis, called Lang+dDC (25–27). Recent studies accounting for both langerin+ subsets suggest that LCs and Lang+dDCs appear to elicit diverse roles in regulating immunity and tolerance (5, 18, 28). In the Lang-DTR model, Lang+dDCs begin to repopulate by day 3 but epidermal LCs begin to repopulate after day 14 of DT injection (Supplemental Fig. S3), as is consistent with the published literature (25, 29). We utilized this differential repopulation time kinetics between LC and Lang+dDC to assess the effect of epidermal LC-selective depletion in the skin tissue upon tolerance induction. Dsg3 emulsion was epicutaneously applied 10 d after DT injection, a time-point when only LCs were absent from the skin. In contrast to LC-intact mice (Fig. 2B), LC-depleted mice did not show any reduction in the recall proliferative response of dLN cells to Dsg3 when pre-treated with Dsg3 as compared to animals receiving vehicle emulsion application (Fig. 2D; p = 0.59). There was no difference in proliferation between vehicle-treated LC-intact vs. LC-depleted mice (Fig. 2B [vehicle] vs. Fig. 2D [vehicle]; p = 0.13). However, the recall proliferative responses of dLN cells to Dsg3 after epicutaneous tolerization to Dsg3 were different (p = 0.007) between LC-intact (Fig. 2B [Dsg3]) and LC-depleted mice (Fig. 2D [Dsg3]). As a control, the in vitro proliferation of dLN cells to an irrelevant Ag (HEL) after epicutaneous application of Dsg3 was not different between LC-intact and LC-depleted mice (Fig. 2E, grey vs. black columns). Furthermore, the Ag-specific in vitro proliferation of cells from non-skin draining (mesenteric) lymph nodes was unaffected by the epicutaneous application of Dsg3 in both LC-intact and LC-depleted mice (data not shown). These data indicate that LCs are necessary for in vivo tolerance induction by epicutaneous application of a skin autoantigen at the tissue level.
To investigate if LC depletion affects the activation, differentiation, and apoptosis of T cells upon high-dose tolerance to a self-Ag, we analyzed the respective markers, including annexin V, CD25, CD44, CD62L, CD69 and FoxP3, on local dLN T cells from vehicle or Dsg3 treated LC-intact and LC-depleted mice. Whereas the frequency of activated, memory and apoptotic CD4+ T cells were not significantly different between the groups (data not shown), the frequency of conventional Treg cells (CD4+CD25+Foxp3+ T cells) was significantly increased in LC-intact mice treated with epicutaneous Dsg3 as compared to vehicle-treated mice (Fig. 2G, LC Intact). Such increase in Treg cells upon Dsg3 tolerance induction was not observed in LC-depleted mice. The epicutaneous application of a foreign Ag, HEL, did not affect the proportions of Treg cells as compared to vehicle-treated animals (data not shown). These data suggest a role of LCs in the generation of Treg cells during epicutaneous tolerance induction to self-Ag.
Inducible LC depletion in adult, preclinical MRL-lpr mice promotes skin-specific autoantibody response, but not systemic B cell hyperactivation
Since LCs are required for tolerance induction to a test skin autoantigen (Fig. 2), we surmised that LC ablation will also result in increased autoantibody responses to skin Ags. To test this in vivo in a chronic autoimmune disease model, we used a long-term LC ablation strategy by administering repeated injections of DT in young Lang-DTR MRL-lpr mice and measured serum autoantibody responses. Specifically, young 6–8-wk-old female mice were given DT injections every 10–14 d for up to 8 total injections. This injection regimen allowed for continual depletion of LCs during the time prior to dermatitis onset, whereas Lang+dDCs have repopulated and are present (Fig. 3A, B) during this period. Results show that serum autoantibodies against homogenized whole-skin lysate were increased in LC-depleted mice (Fig. 3C) compared to LC-intact mice. Specifically, the 26kDa, 40kDA, 73kDA, and cumulative (indicated as “All”) band densities were comparatively higher in LC-depleted mice compared to LC-intact controls (p = 0.05–0.02). Although not statistically significant, 24kDa, 48kDa, 53kDa, 63kDa, and 110kDa (representing Dsg3) bands also showed trends of increased densities in LC-depleted mice compared to controls (Fig. 3D). In contrast, serum autoantibody levels to the systemic autoantigen dsDNA were similar between LC-depleted and LC-intact mice (Fig. 3E), thus demonstrating tissue specificity with depletion of tissue specific DCs. Therefore, the depletion of the local DCs, the LCs in skin, led to increased autoantibodies to skin but had no impact on Abs to a systemic autoantigen.
FIGURE 3. Inducible LC ablation in adult, pre-clinical MRL-lpr mice promotes autoantibody response to skin autoantigens, but does not increase systemic autoantibody levels.

A, LC ablation schema. Lang-DTR knock-in MRL-lpr mice are given repeated DT injections (initial dose 1μg starting at 6–8-wks of age, followed by subsequent 100ng every 10–14 d) to continuously deplete epidermal LCs in pre-clinical period. White cells represent absent DCs. B, LCs and Lang+dDCs in the skin of MRL-lpr mice after 8 repeated weekly injections. FACS plots of epidermal cells were gated off large cells, and dermal cells were gated off MCHII+ cells. Gate frequencies represent mean ± SD, with dermal migratory LCs (Group I) and Lang+dDC (Group II) gate frequencies located under their corresponding FACS plot (n=3–6 mice/plot; *** p < 0.001, ** p < 0.01, * p < 0.05). C, Western Blots were used to assess levels of serum IgG autoantibodies against whole-skin lysate in LC-intact and LC-depleted Lang-DTR (DTR) MRL-lpr mice (n=8–11 mice/group). Serum samples from BALB/c and B6 mice were used as negative controls, and a pooled serum sample from inbred MRL-lpr mice was used as a reference serum between different Western blot experiments. Representative blots are shown. Numbers indicate molecular sizes in kDa. Ponceau S staining (PR) of skin lysate was performed for loading controls, with the corresponding 63 kDa PR band shown. D, Band densities of the indicated protein sizes were calculated relative to the corresponding band in the reference serum as well as normalized against Ponceau S staining. (All bands *p=0.033; 26kDa *p=0.041; 40kDa *p=0.050; 63kDa #p=0.061; 73kDa *p=0.012; n=8–11 mice/group). E, Serum anti-dsDNA autoantibodies were measured using ELISA. Results are expressed as the U/ml from individual mice. Results are pooled from three independent experiments.
LC depletion in adult, preclinical MRL-lpr mice accelerates dermatitis, but not other systemic manifestations of lupus
Since LC ablation in adult, pre-clinical MRL-lpr mice results in impaired epicutaneous tolerance induction to skin autoantigen (Fig. 2) and increased skin autoantibody production (Fig. 3), we surmised that continual LC ablation in pre-clinical mice will accelerate autoimmune dermatitis. Indeed, LC-depleted mice had a marked acceleration of clinically relevant lupus dermatitis lesions (score ≥ 2) compared to their littermate controls (Fig. 4A), with a median onset of 18 versus 28 wks of age, respectively (p < 0.001) (Fig. 4B).
To confirm that the acceleration of clinical manifestations of dermatitis in LC-ablated mice is consistent with lupus skin histopathology, we scored skin sections for acanthosis, vacuolar changes, lymphocytic infiltrates, apoptotic keratinocytes, and fibrosis, as described in Supplemental Table S1. LC-ablated mice had markedly increased histology scores in both ear and nape skin (Fig. 4C, D). However, LC-ablated mice had no increased pathology in non-cutaneous organs such as the lungs, liver or kidneys compared to LC-intact mice (Fig. 4E, F). Thus, LC ablation exacerbates lupus dermatitis, without affecting other organ systems.
Acceleration of dermatitis by LC ablation occurs in the autoimmune MRL background and is independent of the Faslpr mutation
To determine if the long-term LC depletion regimen that accelerates dermatitis in young MRL-lpr mice will induce skin autoimmune disease in non-autoimmune mouse strains, we depleted LCs in Lang-DTR B6 mice. There was no incidence of skin disease up to over one year of follow up in these mice (Fig. 5A). This suggests a requirement for an autoimmune genetic background or the lpr mutation for LC ablation-induced trigger in skin inflammation.
To assess whether the dermatitis exacerbating effect of LC ablation in MRL-lpr mice is owing to the lpr mutation, we generated Lang-DTR B6-Faslpr/lpr (B6-lpr) and Lang-DTR MRL+/+ mice by introgressing the knock-in mutation in these backgrounds. Whereas LC ablation did not elicit autoimmune dermatitis in B6-lpr mice up to one year of follow up (Fig. 5A), LC ablated MRL+/+ mice experienced a significant acceleration of autoimmune skin disease compared to littermate LC-intact controls (Fig. 5A, B). LC-ablated MRL+/+ mice had a median onset of skin disease at 41 wks compared to only 17% of LC-intact mice that developed skin disease at this age (p=0.04). Thus, LC ablation accelerates dermatitis in a genetically autoimmune-prone background, regardless of the presence of the lpr mutation.
LC depletion in adult, preclinical MRL-lpr mice reduces IL-10 producing CD4+ T cells in dLN, and increases macrophage infiltration in skin
To understand the mechanism underlying accelerated skin disease in LC-depleted MRL mice, we first asked if the depletion of LCs chronically results in an impaired tolerance induction to a skin autoantigen. To address this, we epicutaneously applied high doses of Dsg3 or vehicle to MRL-lpr mice after 6 DT or PBS injections. Similar to results in mice with acute LC depletion after one DT injection (Fig. 2D), chronically LC-depleted MRL-lpr mice did not develop high dose tolerance to Dsg3, and exhibited significantly increased recall proliferative responses to Dsg3 as compared to their age-matched LC-intact control littermates (data not shown). Such lack of local skin-specific immune tolerance in LC depleted mice may account for the accelerated skin disease in autoimmune-prone mice.
To test the hypothesis that the loss of tolerance with increased skin autoimmunity in chronically LC-ablated MRL-lpr mice results from an altered homeostasis of effector versus regulatory cells in the dLN, we continuously depleted LCs during the pre-clinical period in young mice (as in Fig. 3A) and examined its effect on immune cell subsets and cytokine production in the dLN (Fig. 6A–F). There was no difference in the frequency of CD4+, CD8+ and DN T cells (Fig. 6A), nor naïve, memory, effector and activated CD4+ T cells (Fig. 6B) in the dLN of LC ablated mice compared to LC-intact mice. There was a trend towards increased B cell numbers in the dLN of LC-ablated mice, but this difference was not statistically significant (Fig. 6C, p = 0.06). Interestingly, in contrast to the acute LC depletion that prevented the FoxP3+ Treg cell expansion that is observed upon epicutaneous tolerance induction to Dsg3 in LC-intact mice (Fig. 2G), we found an increase in the frequency of FoxP3+ Tregs in chronically LC-depleted mice compared to LC-intact mice (p = 0.02) (Fig. 6D).
FIGURE 6. Effect of sustained LC ablation in adult, preclinical MRL-lpr mice on immune cell phenotype in skin and dLN.

6–8-wk-old Lang-DTR MRL-lpr mice were treated with vehicle or DT every 10–14 d, dLNs and skin tissue were harvested after 6–8 injections, and analyzed for immune cell subsets and cytokine production. A–C, Frequency of T cell subsets in dLN, including CD4, CD8 and DN (TCRβ+B220+CD4−CD8−) T cells (A), CD4+ naïve, memory, and effector activated T cells (B), and B cells (C) in LC-intact versus LC-depleted mice. Error bars show SEM (n = 5–13 mice/group). D, dLN cells were analyzed for Treg cells (CD4+CD25+FoxP3+). Representative flow plots depict FoxP3/CD25 staining in gated CD4+ T cells. Results are expressed on the left as the mean ± SEM of CD25+FoxP3+ Treg cells (*, p = 0.02, n = 6 LC-intact and 5 LC-depleted mice). E, F, Single cell suspensions from dLNs were stimulated with PMA/ionomycin, and analyzed for intracellular cytokines in gated TCRβ+CD4+ cells. Representative flow plots of IL-10 producing cells among Treg cells (FoxP3+ CD4+) and FoxP3−CD4+ T cells are shown in E. Results are expressed in F as the mean (horizontal bars) and individual data symbols of cytokine producing FoxP3−CD4+ T cells (*, p =0.04; n = 9 LC-intact and 10 LC-depleted mice). G, H, Nape skin from LC-intact and LC-depleted MRL-lpr mice was stained for the presence of macrophages (CD68), T cell subsets (CD4 and CD8), and CD11c by immunohistochemistry. Representative sections of nape skin are shown in G. Scale bar indicates 100μm (at original 20x magnification). H, Infiltrative cells were quantified using HistoQuest Software (TissueGnostics) (***p = 0.001; 4 mice/group). Differences were not statistically significant for CD4, CD8, and CD11c staining. Results represent at least three independent experiments.
To determine the effect of LC depletion on T cell functions, we stimulated dLN cells with PMA/ionomycin and analyzed intracellular cytokine production (Fig. 6E, F). While LC ablation had no effect on the frequency of CD4+ T cells producing IFN-γ, IL-2, TNF-α and IL-17, the frequency of IL-10 producing CD4+ T cells was reduced in the dLN of LC-ablated mice compared to LC-intact controls (p = 0.04) (Fig. 6F). Since FoxP3+ Tregs as well as CD4+Foxp3− IL-10-producing (Tr1) cells can suppress inflammation in an IL-10-dependent manner (30), we analyzed IL-10 production in CD4+FoxP3+ vs. CD4+FoxP3− T cells (Fig. 6E). There was no difference in IL-10+CD4+FoxP3+ T cells between the groups (p=0.28; n=4 mice/group). Thus, LC ablation reduces the frequency of IL-10–producing CD4+ T cells in dLN of MRL-lpr mice, but had no significant effects on pro-inflammatory cytokine-producing CD4+ T cells.
In order to determine which cell types may be affected locally at the site of disease, we performed immunohistochemical analysis in skin tissues from LC-ablated vs. control animals. We found that LC-ablated MRL-lpr mice had a significant increase in CD68+ macrophages in the skin as compared to LC-intact controls (Fig. 6G, H). The number of infiltrating CD4+ and CD8+ T cells, and CD11c+ cells were not statistically different between the groups at 20 wks of age. To detect cytokines in skin, we separated epidermis and dermis using dispase and isolated leukocytes using collagenase IV. Isolated leukocytes from the epidermis and dermis were analyzed for intracellular cytokines including TNFα, IL-1β, IL-17, IFNγ and IL-10 (data not shown). We have thus far found no significant differences in these cytokines in the epidermis and dermis of chronically LC-depleted MRL-lpr mice as compared to control mice.
DISCUSSION
In this article, we report a novel protective role of LCs in lupus dermatitis. We demonstrate that LC ablation in adult MRL-lpr mice led to inability to induce tolerance to a skin autoantigen, elevated Abs to skin Ags, and accelerated dermatitis, with no effect on disease in other organs tested or on anti-dsDNA autoantibodies. The increased lupus dermatitis in LC-ablated mice was associated with reduced IL-10-producing CD4+ T cells in dLNs, and increased local dermal macrophage infiltration. Thus, the tissue-resident immune cells regulate local immune tolerance without affecting systemic responses. These observations offer a potential explanation that immune regulation at the local level may underlie the heterogeneity of multiple organ involvement in lupus.
An important function of skin migratory DC is to acquire and transport skin Ags to the dLN in the steady state to induce peripheral tolerance (4, 28). The role of tolerance in tissues has been elegantly examined using ectopic expression of transgenes encoding neoantigens in combination with mice expressing transgenic TCRs specific to the neoantigen. For example, in mice where OVA is expressed by keratinocytes adoptively transferred OVA-specific transgenic CD8+ T cells undergo tolerance (31). It is important, however, to understand whether peripheral tolerance is impaired in autoimmune-prone conditions, such as cutaneous SLE. In order to investigate the role of LCs in the local skin versus systemic organ tolerance, we reasoned to use a physiological antigen system that is specific for skin and is not cross-reactive with other systemic autoantigens. Although, antibodies against laminin have been reported in patients with cutaneous LE, these autoantibodies have also been implicated in lupus nephritis (32–34). Previous studies have also shown cross-reactivity between antibodies against laminin or α-actinin and dsDNA (34, 35). In animals, immunizations with laminin or a passive transfer of anti-laminin antibodies either had no effect or induced limited nephritis, myositis, abortion, reproductive failure or subepidermal splits but no lupus dermatitis (36). A previous study showed that immunizations with a skin-associated autoantigen, Dsg3, induced lupus-like skin disease in FcγRIIb−/− mice. Genetically lupus-prone MRL-lpr mice also spontaneously develop autoantibodies to Dsg3, which correlate with skin disease severity (14). Furthermore, IgG deposits at the dermal-epidermal junction co-localized with Dsg3 in the ultraviolet B-induced NZB/W cutaneous lupus model (37). Importantly, the adoptive transfer of Dsg3-reactive CD4+ T cells into immune-deficient mice induced skin inflammation that mimics interface dermatitis that is a distinct T cell-mediated inflammation at the dermal-epidermal junction seen in cutaneous lupus, graft-versus-host disease and other diseases (15). In this model, the adoptively transferred Dsg3-reactive T cells infiltrate the stratified squamous epithelium in skin, oral mucosa, and esophagus which express Dsg3, but not the other organs, such as liver, small and large intestine, spleen and muscle, which do not express Dsg3 (14, 15). Taken together, the Dsg3 provides a suitable model antigen system for our studies to investigate the local immune tolerance in skin.
Using the Dsg3 model antigen system, we demonstrate that MRL-lpr mice that have reduced numbers of LCs in their dLN exhibit spontaneous loss of tolerance to Dsg3. This spontaneous breakdown in tolerance to Dsg3 observed in MRL-lpr mice can be corrected by epicutaneous applications of high doses of Dsg3, for which LCs are necessary. This role of LCs in inducing skin tolerance is in resonance with previous bone marrow chimera studies, where only host LCs expressed the MHCII I-E that could present the Ag to the transferred transgenic CD4+ T cells and tolerize them (38). We further show that despite a reduced LC migration and tolerance breakdown, skin-reactive T cells in MRL-lpr mice can be tolerized. This has important therapeutic implications such that epicutaneous applications of skin autoantigens might restore tolerance to skin and prevent relapses of lupus dermatitis. Such induced tolerance can be boosted further by improving the migration of LCs so they can more efficiently transport the epicutaneously applied Ags to dLN. Taken together, these observations implicate the LC migration defect in causing the breakdown of tolerance to skin autoantigens in autoimmune-prone mice.
The mechanisms by which LC depletion prevents the high dose tolerance induction to self-Ags remain to be determined. We did not observe a significant effect of high dose tolerance to Dsg3 on CD4+ T cell activation and activation-induced cell death in dLN of MRL-lpr mice, but the frequency of CD4+CD25+Foxp3+ Treg cells was significantly increased 7 d after an epicutaneous application of Dsg3 in LC-intact mice. Such increase in Treg cells upon Dsg3 tolerance induction was not observed in LC-depleted MRL-lpr mice. These data are consistent with a previous study that showed that the generation of Tregs that is observed upon ultraviolet radiation is prevented in LC-depleted mice (39). Additionally, we found a reduced frequency of IL-10 producing FoxP3− CD4+ T cells in the dLN of LC-ablated MRL-lpr mice as compared to LC-intact mice. Such IL-10 producing FoxP3− CD4+ T cells can inhibit T cell expansion in vivo, hence referred to as IL-10 Tregs (40). IL-10 has also been shown to mediate T cell tolerance (41), and to skew DCs toward a tolerogenic phenotype (42). Thus, our data suggest a role of LCs in the generation of both FoxP3+ and IL-10 Treg cells in dLN. It remains to be determined whether the impaired generation of FoxP3+ Treg cells in response to high dose skin antigen or the reduced IL-10 Treg population in the dLN of LC-ablated MRL-lpr mice contributes to loss of tolerance to skin Ags.
The continuous in vivo ablation of LCs in adult, preclinical MRL-lpr mice led to increased serum autoantibodies to endogenous skin Ags. A previous investigation that used a patch immunization method where the dorsal mouse ears were tape-stripped five times with Scotch tape prior to applying a filter paper wetted with Staphylococcus aureus–derived exfoliative toxin or OVA showed reduced levels of antibodies against these foreign antigens in LC-depleted mice as compared to LC-intact controls (43). It is likely that the repeated tape stripping used in this study caused local trauma leading to a marked increase in the activation and migration of Ag-loaded LCs to dLN, where they facilitated antibody production. To be able to administer the Ag in a steady state environment, we avoided the use of tape stripping and applied the Ag in an emulsion with incomplete Freund’s adjuvant followed by blow air drying of the painted emulsion to ensure the consistent application. Use of acetone/olive oil and other carriers for Ags followed by a Duo-Derm patch led to inconsistent results on epicutaneous tolerance (data not shown). Nevertheless, these observations suggest that LCs may be competent to support the production of antibodies against foreign Ags, whereas they regulate T–B cell tolerance to self-Ags at the steady state in lupus. These observations also suggest a plasticity in the role of LCs on immune responses, which may depend on the nature of the Ag (e.g., foreign or self), route of Ag exposure, and presence or absence of environmental cues, including trauma, pathogens, and inflammation (44).
Skin biopsies from patients with SLE show increased numbers (8) or an irregular distribution (9) of LCs in some studies, but reduced LC numbers in skin lesions by others (10, 45). An irregular distribution with clusters of large LCs is also seen in the epidermis of preclinical MRL-lpr mice (16), which may represent activated LCs that are unable to migrate to dLNs. To directly investigate the role of LCs in lupus dermatitis, a previous study generated transgenic MRL-lpr mice that use the human langerin promoter to express attenuated DT subunit A. These mice have a constitutive LC ablation from early development (2). In order to obviate the potential compensatory pathways arising as a result of life-long LC deficiency, we chose an inducible model and an ablation schema that continuously deletes LCs only during the pre-clinical young adult life of MRL-lpr and MRL+/+ mice. This LC ablation regimen resulted in a profound acceleration of dermatitis, such that MRL+/+ mice that normally do not display any clinical or pathological evidence of dermatitis until 8–10 months of age begin to exhibit severe dermatitis upon LC depletion at an earlier age (0% vs. 39% in LC-intact vs. LC-depleted mice, respectively, by 26 weeks of age). A similar acceleration of dermatitis was seen upon LC depletion in MRL-lpr mice (33% vs. 92% in LC-intact vs. LC-depleted mice, respectively, by 26 weeks of age). In contrast, the constitutive ablation of LCs using the Langerin-DTA transgenic model did not result in increased skin disease in MRL-lpr mice (2). Thus, it would be important to identify mechanisms that might compensate for the loss of LCs from birth in the constitutive LC ablation model. Nevertheless, our data clearly demonstrate a protective role of LCs in young adult mice prone to develop lupus dermatitis.
The ability of LCs to confer tolerance to skin Ags may underlie their protective role in lupus dermatitis. Increased dermatitis in LC-depleted mice might also be due to the reduced frequency of CD4+ T cells that produce IL-10. Previous studies have shown that IL-10 deficiency can exacerbate dermatitis in MRL-lpr mice (46), and T cell-derived IL-10 has been shown to play a role in curtailing skin inflammation in contact hypersensitivity (CHS) (47, 48). IL-10 may also exert its effect in skin inflammation via its ability to induce the apoptosis and inhibit the activation of pDCs (49) that infiltrate the lesions of cutaneous SLE (50). IL-10 can control DC function during the effector phase of inflammatory skin reaction (42), and to inhibit macrophage functions (51). Thus, the reduced IL-10 Tregs in dLN of LC-ablated MRL-lpr mice might contribute to the increased macrophage infiltration in skin that we observed. While further studies are needed to understand how LCs regulate the homeostasis of IL-10 producing cells, it is possible that LCs mediate their effects at least in part via IL-10 producing CD4+ T cells.
The elucidation of immune stimulatory versus regulatory role of LCs has been complicated by the discovery of Lang+dDC (25–27), prompting re-examination of prior studies using langerin as an exclusive marker for LCs alone. Recent studies accounting for both langerin+ subsets suggest that LCs and Lang+dDCs appear to elicit diverse and complex roles in regulating immunity and tolerance (5, 18, 28), which are not fully elucidated yet. Like epidermal LCs, Lang+dDCs that arise from bone marrow precursors to populate dermis migrate to dLNs both in steady state and in response to inflammation (18). Unlike LCs, Lang+dDCs arrive at dLNs quickly and are continually replaced by new recruits from the blood, such that only ~50% Lang+dDCs are reduced on day 2 after a 3rd to 8th DT injection in Lang-DTR MRL-lpr mice. This differential kinetics allowed us to demonstrate that the continuous depletion of LCs, with Lang+dDCs mostly present, in preclinical Lang-DTR MRL-lpr mice injected with DT every 10–14 d exacerbates dermatitis. It is possible, however, that a functional alteration in repopulating Lang+dDCs confers the observed phenotype in this model. This issue can be addressed by using Lang-DTA mice, where Lang+dDCs are continually present. Roles of LC and Lang+dDC have been extensively investigated in CHS. Results have been variable under different experimental conditions. Under conditions where only LCs are ablated, LCs appear to negatively regulate CHS (17, 39, 52, 53). However, depletion of LCs and Lang+dDCs both resulted in the diminished (19) or unaffected (20) CHS. Other studies suggest a compensatory role of these two skin DCs in the sensitization phase of CHS (54). Consensus is emerging that LCs may generally regulate CHS, whereas Lang+dDCs may promote CHS (5, 18, 28). Our ongoing studies will determine the role of Lang+dDCs and other heterogeneous populations of Lang− dermal DCs that remain intact in Lang-DTR mice in the development of lupus dermatitis.
Epicutaneous tolerance with Dsg3 led to increased CD4+CD25+FoxP3+ Treg cells in LC-intact, but not in acutely LC-depleted mice, indicating a requirement of LC in generating Treg cells in response to high dose epicutaneous Ag. These data are consistent with previous reports indicating a role of LCs in the induction of Treg cells (39, 44). However, we were surprised to find an increase in the frequency of Tregs in the dLN of chronically LC-depleted mice compared to LC-intact mice. The reason for this discrepancy in the role of LCs in Treg generation is unclear. Recent studies using the OVA transgenic model suggest that migratory Lang+dDCs, not LCs, mediated tolerance in CD4+ T cells, where adoptively transferred naive OVA-specific transgenic CD4+ T cells were converted into FoxP3+ Treg cells (28). Thus, it is possible to speculate that Lang+dDCs that remain mostly intact and re-populate rather quickly after multiple DT injections contribute to the accumulation of Tregs in the dLN of chronically LC-ablated MRL-lpr mice. However, despite the presence of Lang+dDCs and increased Tregs, the skin autoimmunity is accelerated in chronically LC-ablated MRL-lpr mice. It is also possible that Lang+dDCs that re-populate the dermis after repeated LC ablations are inefficient in inducing functionally competent FoxP3+ Tregs, especially under autoimmune conditions. In fact, Tregs are functionally impaired in MRL-lpr mice (55). Thus, an increase in Treg cells might still not be commensurate with a profound increase in activated T cells in these mice.
Our observations offer a potential explanation that immune regulation at the local level may underlie the heterogeneity of multiple organ involvement in lupus. There are other examples of differential regulation of inflammation in different organs in a systemic autoimmune disease. For example, MRL-lpr mice rendered deficient in β2-microglobulin (56), CD1d (23), and CD40L (57) exhibit increased lupus dermatitis, but less or unaffected renal disease. In another example, nephritis is accelerated by multiple pregnancies in MRL-lpr mice while skin disease is prevented (58), presumably due to opposite effects of IL-10 in skin versus kidneys. Higher renal IL-10 levels were associated with more nephritis, whereas higher IL-10 levels in skin were associated with lower skin disease. The latter observation is akin to our findings showing reduced IL-10-producing CD4+ T cells in dLN of mice with increased dermatitis. We show a specific role for LCs in the epidermis in young adult mice to regulate tolerance in skin, but not other organs. The localized worsening of skin disease with epidermal LC ablation may be due to the unique location of this DC subset.
This study highlights the role of local immune tissue regulation as a key regulatory layer for tolerance induction in systemic autoimmune disorders. These data have implications for therapy at the local organ level, providing a target for therapy to correct a local breakdown in tolerance rather than attempting correction at a systemic level. This has potential advantages in patients with persistent cutaneous lupus when systemic symptoms are absent or have subsided. Further studies should continue to shed light on predictors of organ specificity in the context of systemic multi-organ autoimmune diseases.
Supplementary Material
Acknowledgments
We thank Dr. Bernard Malissen (Marseille, France) for providing Lang-DTR knock-in B6 mice, Mr. Miguel-Angel Gutierrez for performing LAL assay, Ms. Dora Acuna for immunohistochemical staining of skin section, Dr. Robert Modlin for reading the manuscript, TissueGnostics for help with analyses of stained skin sections, Julia Pinkhasov for helpful discussions, and David Elashoff, Department of Medicine Biostatistics Core, for statistical analysis.
Footnotes
Grant Support. This work was supported by NIH R01 AI080778 (RRS) and a Pilot Award from the UCLA Office of the Vice Chancellor for Research (RRS). JKK was supported by NIH T32 AR053463 (RRS), NIH K12 UCLA Building Interdisciplinary Research Careers in Women’s Health (BIRCWH, PI, Gautam Chaudhuri), Rheumatology Research Foundation Physician Scientist Development Award and Young Investigator Award, and Arthritis Foundation Postdoctoral Fellowship; RLP was supported by Lupus Foundation of America Gina Finzi Memorial Student Fellowship; AUE was supported by Arthritis Foundation Meyer Young Investigator Award; PJK was supported by NIH (K08 AR062593) and Rheumatology Research Foundation Bridge Award; and DJL was supported by NIH (AR59126) and the Joseph B. Gould Foundation. Confocal microscopy was performed at UCLA CNSI Advanced Microscopy Facility, supported with NIH-NCRR CJX1-443835-WS-29646 and NSF CHE-0722519. Flow cytometry was performed in the UCLA Flow Cytometry Core Facility, supported by NIH CA-16042 and AI-28697. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Abbreviations used in this article: B6, C57BL/6; B6-lpr, C57BL-6 Faslpr/lpr; C3H, C3H/HeOuJ; DC, dendritic cell; dDC, dermal dendritic cell; dLN, skin-draining lymph nodes; Dsg3, desmoglein-3; DT, diphtheria toxin; DTR, diphtheria toxin receptor; eGFP, enhanced green fluorescent protein; HEL, hen egg lysozyme; Lang, Langerin; Lang-DTR, Langerin-DTR-eGFP; LC, Langerhans cell; MRL+/+, MRL/MpJ-Fas+/+; MRL-lpr, MRL/MpJ-Faslpr/lpr; SLE, systemic lupus erythematosus.
References
- 1.Singh RR, Dubey S, Pinkhasov J. Immune tolerance defects in lupus. In: Wallace DJ, Hahn BH, editors. Dubois’ Lupus Erythematosus and Related Syndromes. 8. Elsevier Saunders; Philadelphia: 2013. pp. 256–272. [Google Scholar]
- 2.Teichmann LL, Ols ML, Kashgarian M, Reizis B, Kaplan DH, Shlomchik MJ. Dendritic cells in lupus are not required for activation of T and B cells but promote their expansion, resulting in tissue damage. Immunity. 2010;33:967–978. doi: 10.1016/j.immuni.2010.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ganguly D, Haak S, Sisirak V, Reizis B. The role of dendritic cells in autoimmunity. Nature reviews Immunology. 2013;13:566–577. doi: 10.1038/nri3477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Steinman RM, Nussenzweig MC. Avoiding horror autotoxicus: the importance of dendritic cells in peripheral T cell tolerance. Proc Natl Acad Sci U S A. 2002;99:351–358. doi: 10.1073/pnas.231606698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clausen BE, Kel JM. Langerhans cells: critical regulators of skin immunity? Immunology and cell biology. 2010;88:351–360. doi: 10.1038/icb.2010.40. [DOI] [PubMed] [Google Scholar]
- 6.Azukizawa H, Kosaka H, Sano S, Heath WR, Takahashi I, Gao XH, Sumikawa Y, Okabe M, Yoshikawa K, Itami S. Induction of T-cell-mediated skin disease specific for antigen transgenically expressed in keratinocytes. Eur J Immunol. 2003;33:1879–1888. doi: 10.1002/eji.200323630. [DOI] [PubMed] [Google Scholar]
- 7.Mayerova D, Parke EA, Bursch LS, Odumade OA, Hogquist KA. Langerhans cells activate naive self-antigen-specific CD8 T cells in the steady state. Immunity. 2004;21:391–400. doi: 10.1016/j.immuni.2004.07.019. [DOI] [PubMed] [Google Scholar]
- 8.Kanauchi H, Furukawa F, Imamura S. Evaluation of ATPase-positive Langerhans’ cells in skin lesions of lupus erythematosus and experimentally induced inflammations. Arch Dermatol Res. 1989;281:327–332. doi: 10.1007/BF00412976. [DOI] [PubMed] [Google Scholar]
- 9.Sontheimer RD, Bergstresser PR. Epidermal Langerhans cell involvement in cutaneous lupus erythematosus. J Invest Dermatol. 1982;79:237–243. doi: 10.1111/1523-1747.ep12500069. [DOI] [PubMed] [Google Scholar]
- 10.Andrews BS, Schenk A, Barr R, Friou G, Mirick G, Ross P. Immunopathology of cutaneous human lupus erythematosus defined by murine monoclonal antibodies. J Am Acad Dermatol. 1986;15:474–481. doi: 10.1016/s0190-9622(86)70196-5. [DOI] [PubMed] [Google Scholar]
- 11.Bergroth V, Konttinen YT, Johansson E. Langerhans cells in SLE skin. A role in lymphocyte migration and activation in situ. Scand J Rheumatol. 1985;14:411–416. doi: 10.3109/03009748509102046. [DOI] [PubMed] [Google Scholar]
- 12.Hahn BH, Singh RR. Animal models of systemic lupus erythematosus. In: Wallace DJ, Hahn BH, editors. Dubois’ Lupus Erythematosus. 7. Lippincott, Williams and Wilkins; Philadelphia: 2007. pp. 299–355. [Google Scholar]
- 13.Amagai M, Klaus-Kovtun V, Stanley JR. Autoantibodies against a novel epithelial cadherin in pemphigus vulgaris, a disease of cell adhesion. Cell. 1991;67:869–877. doi: 10.1016/0092-8674(91)90360-b. [DOI] [PubMed] [Google Scholar]
- 14.Nishimura H, Strominger JL. Involvement of a tissue-specific autoantibody in skin disorders of murine systemic lupus erythematosus and autoinflammatory diseases. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:3292–3297. doi: 10.1073/pnas.0510756103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Takahashi H, Kouno M, Nagao K, Wada N, Hata T, Nishimoto S, Iwakura Y, Yoshimura A, Yamada T, Kuwana M, Fujii H, Koyasu S, Amagai M. Desmoglein 3-specific CD4+ T cells induce pemphigus vulgaris and interface dermatitis in mice. J Clin Invest. 2011;121:3677–3688. doi: 10.1172/JCI57379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eriksson AU, Singh RR. Cutting edge: migration of langerhans dendritic cells is impaired in autoimmune dermatitis. Journal of immunology. 2008;181:7468–7472. doi: 10.4049/jimmunol.181.11.7468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaplan DH, Jenison MC, Saeland S, Shlomchik WD, Shlomchik MJ. Epidermal langerhans cell-deficient mice develop enhanced contact hypersensitivity. Immunity. 2005;23:611–620. doi: 10.1016/j.immuni.2005.10.008. [DOI] [PubMed] [Google Scholar]
- 18.Igyarto BZ, Kaplan DH. Antigen presentation by Langerhans cells. Curr Opin Immunol. 2013;25:115–119. doi: 10.1016/j.coi.2012.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bennett CL, van Rijn E, Jung S, Inaba K, Steinman RM, Kapsenberg ML, Clausen BE. Inducible ablation of mouse Langerhans cells diminishes but fails to abrogate contact hypersensitivity. J Cell Biol. 2005;169:569–576. doi: 10.1083/jcb.200501071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kissenpfennig A, Henri S, Dubois B, Laplace-Builhe C, Perrin P, Romani N, Tripp CH, Douillard P, Leserman L, Kaiserlian D, Saeland S, Davoust J, Malissen B. Dynamics and function of Langerhans cells in vivo: dermal dendritic cells colonize lymph node areas distinct from slower migrating Langerhans cells. Immunity. 2005;22:643–654. doi: 10.1016/j.immuni.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 21.Singh RR, Ebling FM, Sercarz EE, Hahn BH. Immune tolerance to autoantibody-derived peptides delays development of autoimmunity in murine lupus. J Clin Invest. 1995;96:2990–2996. doi: 10.1172/JCI118371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang JQ, Singh AK, Wilson MT, Satoh M, Stanic AK, Park JJ, Hong S, Gadola SD, Mizutani A, Kakumanu SR, Reeves WH, Cerundolo V, Joyce S, Van Kaer L, Singh RR. Immunoregulatory role of CD1d in the hydrocarbon oil-induced model of lupus nephritis. J Immunol. 2003;171:2142–2153. doi: 10.4049/jimmunol.171.4.2142. [DOI] [PubMed] [Google Scholar]
- 23.Yang JQ, Chun T, Liu H, Hong S, Bui H, Van Kaer L, Wang CR, Singh RR. CD1d deficiency exacerbates inflammatory dermatitis in MRL-lpr/lpr mice. Eur J Immunol. 2004;34:1723–1732. doi: 10.1002/eji.200324099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang JQ, Saxena V, Xu H, Van Kaer L, Wang CR, Singh RR. Repeated alpha-galactosylceramide administration results in expansion of NK T cells and alleviates inflammatory dermatitis in MRL-lpr/lpr mice. J Immunol. 2003;171:4439–4446. doi: 10.4049/jimmunol.171.8.4439. [DOI] [PubMed] [Google Scholar]
- 25.Bursch LS, Wang L, Igyarto B, Kissenpfennig A, Malissen B, Kaplan DH, Hogquist KA. Identification of a novel population of Langerin+ dendritic cells. J Exp Med. 2007;204:3147–3156. doi: 10.1084/jem.20071966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ginhoux F, Collin MP, Bogunovic M, Abel M, Leboeuf M, Helft J, Ochando J, Kissenpfennig A, Malissen B, Grisotto M, Snoeck H, Randolph G, Merad M. Blood-derived dermal langerin+ dendritic cells survey the skin in the steady state. J Exp Med. 2007;204:3133–3146. doi: 10.1084/jem.20071733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Poulin LF, Henri S, de Bovis B, Devilard E, Kissenpfennig A, Malissen B. The dermis contains langerin+ dendritic cells that develop and function independently of epidermal Langerhans cells. J Exp Med. 2007;204:3119–3131. doi: 10.1084/jem.20071724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lutz MB, Dohler A, Azukizawa H. Revisiting the tolerogenicity of epidermal Langerhans cells. Immunol Cell Biol. 2010;88:381–386. doi: 10.1038/icb.2010.17. [DOI] [PubMed] [Google Scholar]
- 29.Noordegraaf M, Flacher V, Stoitzner P, Clausen BE. Functional redundancy of Langerhans cells and Langerin+ dermal dendritic cells in contact hypersensitivity. The Journal of investigative dermatology. 2010;130:2752–2759. doi: 10.1038/jid.2010.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huber S, Gagliani N, Esplugues E, O’Connor W, Jr, Huber FJ, Chaudhry A, Kamanaka M, Kobayashi Y, Booth CJ, Rudensky AY, Roncarolo MG, Battaglia M, Flavell RA. Th17 cells express interleukin-10 receptor and are controlled by Foxp3(−) and Foxp3+ regulatory CD4+ T cells in an interleukin-10-dependent manner. Immunity. 2011;34:554–565. doi: 10.1016/j.immuni.2011.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Waithman J, Allan RS, Kosaka H, Azukizawa H, Shortman K, Lutz MB, Heath WR, Carbone FR, Belz GT. Skin-derived dendritic cells can mediate deletional tolerance of class I-restricted self-reactive T cells. J Immunol. 2007;179:4535–4541. doi: 10.4049/jimmunol.179.7.4535. [DOI] [PubMed] [Google Scholar]
- 32.Groth S, Vafia K, Recke A, Dahnrich C, Zillikens D, Stocker W, Kuhn A, Schmidt E. Antibodies to the C-terminus of laminin gamma1 are present in a distinct subgroup of patients with systemic and cutaneous lupus erythematosus. Lupus. 2012;21:1482–1483. doi: 10.1177/0961203312460113. [DOI] [PubMed] [Google Scholar]
- 33.Hanrotel-Saliou C, Segalen I, Le Meur Y, Youinou P, Renaudineau Y. Glomerular antibodies in lupus nephritis. Clinical reviews in allergy & immunology. 2011;40:151–158. doi: 10.1007/s12016-010-8204-4. [DOI] [PubMed] [Google Scholar]
- 34.Renaudineau Y, Deocharan B, Jousse S, Renaudineau E, Putterman C, Youinou P. Anti-alpha-actinin antibodies: a new marker of lupus nephritis. Autoimmun Rev. 2007;6:464–468. doi: 10.1016/j.autrev.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 35.Ben-Yehuda A, Rasooly L, Bar-Tana R, Breuer G, Tadmor B, Ulmansky R, Naparstek Y. The urine of SLE patients contains antibodies that bind to the laminin component of the extracellular matrix. J Autoimmun. 1995;8:279–291. doi: 10.1006/jaut.1995.0021. [DOI] [PubMed] [Google Scholar]
- 36.Florea F, Bernards C, Caproni M, Kleindienst J, Hashimoto T, Koch M, Sitaru C. Ex vivo pathogenicity of anti-laminin gamma1 autoantibodies. The American journal of pathology. 2014;184:494–506. doi: 10.1016/j.ajpath.2013.10.019. [DOI] [PubMed] [Google Scholar]
- 37.Lee YF, Cheng CC, Lan JL, Hsieh TY, Lin NN, Lin HY, Chiu YT. Effects of mycophenolate mofetil on cutaneous lupus erythematosus in (NZB x NZW) F1 mice. Journal of the Chinese Medical Association : JCMA. 2013;76:615–623. doi: 10.1016/j.jcma.2013.07.010. [DOI] [PubMed] [Google Scholar]
- 38.Shklovskaya E, O’Sullivan BJ, Ng LG, Roediger B, Thomas R, Weninger W, Fazekas de St Groth B. Langerhans cells are precommitted to immune tolerance induction. Proc Natl Acad Sci U S A. 2011;108:18049–18054. doi: 10.1073/pnas.1110076108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schwarz A, Noordegraaf M, Maeda A, Torii K, Clausen BE, Schwarz T. Langerhans cells are required for UVR-induced immunosuppression. J Invest Dermatol. 2010;130:1419–1427. doi: 10.1038/jid.2009.429. [DOI] [PubMed] [Google Scholar]
- 40.O’Garra A, Vieira PL, Vieira P, Goldfeld AE. IL-10-producing and naturally occurring CD4+ Tregs: limiting collateral damage. J Clin Invest. 2004;114:1372–1378. doi: 10.1172/JCI23215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Singh RR, Hahn BH, Sercarz EE. Neonatal peptide exposure can prime T cells and, upon subsequent immunization, induce their immune deviation: implications for antibody vs. T cell-mediated autoimmunity. J Exp Med. 1996;183:1613–1621. doi: 10.1084/jem.183.4.1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Clausen BE, Girard-Madoux MJ. IL-10 control of dendritic cells in the skin. Oncoimmunology. 2013;2:e23186. doi: 10.4161/onci.23186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ouchi T, Kubo A, Yokouchi M, Adachi T, Kobayashi T, Kitashima DY, Fujii H, Clausen BE, Koyasu S, Amagai M, Nagao K. Langerhans cell antigen capture through tight junctions confers preemptive immunity in experimental staphylococcal scalded skin syndrome. J Exp Med. 2011;208:2607–2613. doi: 10.1084/jem.20111718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kautz-Neu K, Noordegraaf M, Dinges S, Bennett CL, John D, Clausen BE, von Stebut E. Langerhans cells are negative regulators of the anti-Leishmania response. J Exp Med. 2011;208:885–891. doi: 10.1084/jem.20102318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bos JD, I, van Garderen D, Krieg SR, Poulter LW. Different in situ distribution patterns of dendritic cells having Langerhans (T6+) and interdigitating (RFD1+) cell immunophenotype in psoriasis, atopic dermatitis, and other inflammatory dermatoses. J Invest Dermatol. 1986;87:358–361. doi: 10.1111/1523-1747.ep12524811. [DOI] [PubMed] [Google Scholar]
- 46.Yin Z, Bahtiyar G, Zhang N, Liu L, Zhu P, Robert ME, McNiff J, Madaio MP, Craft J. IL-10 regulates murine lupus. Journal of immunology. 2002;169:2148–2155. doi: 10.4049/jimmunol.169.4.2148. [DOI] [PubMed] [Google Scholar]
- 47.Roers A, Siewe L, Strittmatter E, Deckert M, Schluter D, Stenzel W, Gruber AD, Krieg T, Rajewsky K, Muller W. T cell-specific inactivation of the interleukin 10 gene in mice results in enhanced T cell responses but normal innate responses to lipopolysaccharide or skin irritation. J Exp Med. 2004;200:1289–1297. doi: 10.1084/jem.20041789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rubtsov YP, Rasmussen JP, Chi EY, Fontenot J, Castelli L, Ye X, Treuting P, Siewe L, Roers A, Henderson WR, Jr, Muller W, Rudensky AY. Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity. 2008;28:546–558. doi: 10.1016/j.immuni.2008.02.017. [DOI] [PubMed] [Google Scholar]
- 49.Duramad O, Fearon KL, Chan JH, Kanzler H, Marshall JD, Coffman RL, Barrat FJ. IL-10 regulates plasmacytoid dendritic cell response to CpG-containing immunostimulatory sequences. Blood. 2003;102:4487–4492. doi: 10.1182/blood-2003-07-2465. [DOI] [PubMed] [Google Scholar]
- 50.Vermi W, Lonardi S, Morassi M, Rossini C, Tardanico R, Venturini M, Sala R, Tincani A, Poliani PL, Calzavara-Pinton PG, Cerroni L, Santoro A, Facchetti F. Cutaneous distribution of plasmacytoid dendritic cells in lupus erythematosus. Selective tropism at the site of epithelial apoptotic damage. Immunobiology. 2009;214:877–886. doi: 10.1016/j.imbio.2009.06.013. [DOI] [PubMed] [Google Scholar]
- 51.Bogdan C, Vodovotz Y, Nathan C. Macrophage deactivation by interleukin 10. J Exp Med. 1991;174:1549–1555. doi: 10.1084/jem.174.6.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bobr A, Olvera-Gomez I, Igyarto BZ, Haley KM, Hogquist KA, Kaplan DH. Acute ablation of Langerhans cells enhances skin immune responses. J Immunol. 2010;185:4724–4728. doi: 10.4049/jimmunol.1001802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Igyarto BZ, Jenison MC, Dudda JC, Roers A, Muller W, Koni PA, Campbell DJ, Shlomchik MJ, Kaplan DH. Langerhans cells suppress contact hypersensitivity responses via cognate CD4 interaction and langerhans cell-derived IL-10. J Immunol. 2009;183:5085–5093. doi: 10.4049/jimmunol.0901884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Honda T, Nakajima S, Egawa G, Ogasawara K, Malissen B, Miyachi Y, Kabashima K. Compensatory role of Langerhans cells and langerin-positive dermal dendritic cells in the sensitization phase of murine contact hypersensitivity. The Journal of allergy and clinical immunology. 2010;125:1154–1156. e1152. doi: 10.1016/j.jaci.2009.12.005. [DOI] [PubMed] [Google Scholar]
- 55.Divekar AA, Dubey S, Gangalum PR, Singh RR. Dicer insufficiency and microRNA-155 overexpression in lupus regulatory T cells: an apparent paradox in the setting of an inflammatory milieu. J Immunol. 2011;186:924–930. doi: 10.4049/jimmunol.1002218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chan OT, Paliwal V, McNiff JM, Park SH, Bendelac A, Shlomchik MJ. Deficiency in beta(2)-microglobulin, but not CD1, accelerates spontaneous lupus skin disease while inhibiting nephritis in MRL-Fas(lpr) nice: an example of disease regulation at the organ level. J Immunol. 2001;167:2985–2990. doi: 10.4049/jimmunol.167.5.2985. [DOI] [PubMed] [Google Scholar]
- 57.Peng SL, McNiff JM, Madaio MP, Ma J, Owen MJ, Flavell RA, Hayday AC, Craft J. alpha beta T cell regulation and CD40 ligand dependence in murine systemic autoimmunity. J Immunol. 1997;158:2464–2470. [PubMed] [Google Scholar]
- 58.Kokeny G, Godo M, Nagy E, Kardos M, Kotsch K, Casalis P, Bodor C, Rosivall L, Volk HD, Zenclussen AC, Hamar P. Skin disease is prevented but nephritis is accelerated by multiple pregnancies in autoimmune MRL/LPR mice. Lupus. 2007;16:465–477. doi: 10.1177/0961203307079456. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.