

Abstract
N -Methyl-(3,4-methylenedioxyphenyl)-2-aminopropane (MDMA; ‘Ecstasy’; 1) and its β-keto analog methylone (MDMC; 2) are popular drugs of abuse. Little is known about their ring-expanded ethylenedioxy homologs. Here, we prepared N-methyl-(3,4-ethylenedioxyphenyl)-2-aminopropane (EDMA; 3), both of its optical isomers, and β-keto EDMA (i.e., EDMC; 4) to examine their effects at transporters for serotonin (SERT), dopamine (DAT), and norepinephrine (NET). In general, ring-expansion of the methylenedioxy group led to a several-fold reduction in potency at all three transporters. With respect to EDMA (3), S(+)3 was 6-fold, 50-fold, and 8-fold more potent than its R(−) enantiomer at SERT, DAT, and NET, respectively. Overall, in the absence of a β-carbonyl group, the ethylenedioxy (i.e., 1,4-dioxane) substituent seems better accommodated at SERT than at DAT and NET.
1. Introduction
The phenylalkylamine N-methyl-(3,4-methylenedioxyphenyl)-2-aminopropane (1; Figure 1), a compound commonly known as MDMA, “Ecstasy” or “Molly”, is a popular “recreational drug” and a U.S. Schedule I controlled substance. Its pharmacology and mechanism of action have been extensively investigated [1–4]. For example, MDMA (1) is considered an “empathogen” to distinguish its subjective effects in humans from those of classical phenylalkylamine hallucinogens and phenylalkylamine central stimulants [5] – even though MDMA, and its individual optical isomers, display some degree of stimulant character [e.g. 6]. MDMA’s actions are associated with its non-selective neurotransmitter releasing effects at the membrane transporters for 5-HT, dopamine, and norepinephrine (i.e., SERT, DAT, and NET, respectively) [7,8]. Accordingly, pretreatment with medications that block the transmitter-releasing effects of MDMA at SERT, DAT, and NET can significantly reduce the subjective and cardiovascular actions of MDMA (1) in human subjects under controlled laboratory conditions [9,10].
Figure 1.

Structures of MDMA (1), MDMC (2), and their ethylenedioxy counterparts EDMA (3) and EDMC (4).
The methylenedioxy substituent of MDMA (1) is common to a number of other “clandestine” and U.S. Schedule I phenylalkylamine substances, including certain “bath salts” constituents or synthetic cathinones [4,11,12]. One of the better recognized of the original bath salts constituents is the β-keto analog of MDMA (i.e., MDMC, methylone, bk-MDMA; 2) (Figure 1) [13]. MDMC (2) produces MDMA-like neurochemical and behavioral effects in rodents and acts as a neurotransmitter releaser at the three transporters mentioned above [14,15]. Despite the scheduling of MDMC (2) to ban its sale and use in the U.S., the drug continues to be confiscated by law enforcement personnel, often in tablets being sold as Ecstasy [16].
Expansion of the methylenedioxy ring of MDMA (1) and MDMC (2) to their larger ethylenedioxy (i.e., 1,4-dioxane) homologs affords N-methyl-1-(3,4-ethylenedioxyphenyl)-2-aminopropane (i.e., 3,4-ethylenedioxymethamphetamine, EDMA; 3) and 3,4-ethylenedioxymethcathinone (EDMC; 4) (Figure 1). Relatively little is known about the pharmacology of EDMA (3) and its individual optical isomers have not been reported. Likewise, EDMC (4) has not been previously investigated.
Given the growing interest in phenylalkylamine analogs as potential drugs of abuse [reviewed 11,12,17], the Drug Enforcement Administration (DEA) has solicited information on phenylalkylamines that are not yet controlled as Schedule I substances [18]. Specifically listed among these agents are some ethylenedioxy analogs, including EDMA (3). Hence, we prepared and examined EDMA (3), its individual optical isomers, and its β-keto (or methcathinone) analog 4, for comparison with MDMA (1) and MDMC (2), to function as substrates (i.e., as releasers) at SERT, DAT, and NET.
2. Chemistry
Compounds (±)1 and (±)2 as their hydrochloride (HCl) salts were on hand from previous studies in our laboratory. Compound (±)3 was synthesized from 1-(3,4-ethylenedioxyphenyl)-2-aminopropane hydrochloride [19], by converting it into its carbamate analog followed by reduction using LiAlH4. Compound (±)4 was synthesized by a nucleophilic substitution reaction using 1-(3,4-ethylenedioxyphenyl)-2-bromo-1-propanone [20] and N-methylamine. Both of these reaction sequences are shown in the Supporting Information section.
Compounds S(+)3 and R(−)3 were prepared as shown in Scheme 1; this is similar to a literature procedure reported for related phenylalkylamine optical isomers [21]. Reduction of nitroalkene 5 [19] with iron powder followed by heating an aqueous mixture gave the intermediate ketone 6. The crucial step for this synthesis was the reductive amination using either S(−)- or R(+)-α-methylbenzylamine and sodium triacetoxyborohydride; this reaction selectively afforded S,S(−)7 and R,R(+)7, whose hydrogenation at 50 p.s.i. using 10% Pd/C as catalyst led to the enantiomers S(+)8 and R(−)8, respectively. Acylation with di-tert-butyl dicarbonate to the N-Boc intermediates S(−)9 and R(+)9, followed by reduction with LiAlH4 afforded the desired enantiomers S(+)3 and R(−)3, respectively.
Scheme 1.

Reagents and conditions: (a) (i) Fe, AcOH, rt (ii) reflux (iii) H2O; (b) S-(−)-α-methylbenzylamine, NaBH(OAc)3, DCE, rt; (c) R-(+)-α-methylbenzylamine, NaBH(OAc)3, DCE, rt; (d) H2, 10% Pd/C, 50 psi; (e) (BOC)2O, Et3N, DCM, rt; (f) (i) LiAlH4, THF, 0 °C (ii) reflux (iii) HCl/Et2O.
The enantiomeric purity of the isomers of 3, determined by 1H NMR spectrometry in the presence of the chiral shift reagent S(+)-2,2,2-trifluoro-1-(9-anthryl)ethanol, was found to be >98% (detection limit) for both enantiomers. In fact, the 1H NMR spectrum of the racemate [i.e., (±)3] showed two singlets: one at δ 4.27 and another at δ 4.24 for the methylene protons of the benzodioxane nucleus, whereas only one singlet was observed for S(+)3 and R(−)3 at δ 4.27 and δ 4.24, respectively.
The S absolute configuration of the enantiomer (+)3, suggested by analogy to earlier studies [20], was also determined by comparing the sign of its optical rotation with that of S(+)3 obtained by stereoselective synthesis starting from the known S(+)1-(3,4-dihydroxyphenyl)-2-propanamine [S(+)10] [22], as shown in Scheme 2. After protecting the amine function with a Boc group, affording S(−)11, treatment with 1,2-dibromoethane furnished the 1,4-benzodioxane derivative S(−)9, whose reduction with LiAlH4 gave the desired enantiomer S(+)3.
Scheme 2.
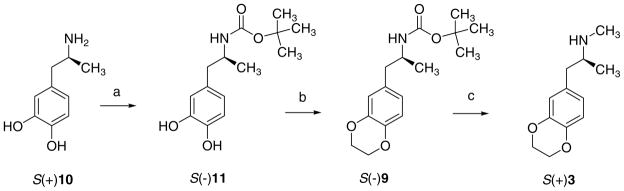
Reagents and conditions: (a) (BOC)2O, MeO−Na+, rt; (b) BrCH2CH2Br, K2CO3, DMF, rt; (c) (i) LiAlH4, THF, 0 °C (ii) reflux (iii) HCl/Et2O.
3. Results and Discussion
The potency of (±)EDMA (3) to release [3H]5-HT (EC50 = 117 nM) at SERT was approximately six times and three times greater than its potency to release [3H]MPP+ at DAT and NET (EC50 = 597 and 325 nM, respectively; Table 1). By contrast (±)MDMA (1), investigated as a comparator compound, was nearly equipotent as a releaser at all three neurotransmitter transporters (Table 1). The present findings with MDMA are consistent with our previous data [15,17] and those reported by Simmler et al. [4] and Eshleman et al. [23] who examined the effects of MDMA and related drugs in human embryonic kidney (HEK) cells transfected with human SERT, DAT and NET. Thus, the molecular mechanism of action for MDMA at monoamine transporters is similar in rats and humans. On the other hand, the potency of (±)MDMA for releasing monoamines in rat brain synaptosomes shown here (i.e., 60–70 nM) is greater than its potency in transfected HEK cells (i.e., 1–20 μM). Such discrepancies in absolute potency could be related to species differences in drug responsiveness, differences in release assay methods employed, or the absence of important neuronal membrane proteins in non-neuronal HEK cells.
Table 1.
EC50 values for test agents to release [3H]5-HT at SERT, and [3H]MPP+ at DAT and NET.
| EC50, nM (± SD)a
|
|||
|---|---|---|---|
| SERT | DAT | NET | |
| (±)EDMA (3) | 117 (± 17) [102± 3] | 597 (± 50) [108± 3] | 325 (± 61) [91± 2] |
| S(+)EDMA [(S)3] | 91 (± 20) [96± 5] | 276 (± 28) [103± 2] | 239 (± 37) [94± 3] |
| R(−)EDMA [(R)3] | 573 (± 108) [97± 5] | 14,600 (± 2850) [106± 10] | 1,952 (± 356) [85± 4] |
| (±)MDMA (1) | 61 (± 9) [98± 2] | 75 (± 5) [99± 1] | 72 (± 13) [95± 3] |
| (±)EDMC (4) | 347 (± 38) [105± 3] | 496 (± 52) [105± 4] | 327 (± 54) [84± 4] |
| (±)MDMC (2)b | 242 (± 48) | 133 (± 11) | 152 (± 33) |
EC50 values are mean (±SD) for n = 3–4 experiments performed in triplicate; efficacy values [in brackets] are % maximal release expressed as mean (±SD) as described in Methods.
EC50 values for MDMC (2) were reported earlier [15] and are included for comparison with EDMC (4).
The data depicted in Table 1 and Figure 2 demonstrate that (±)EDMA (3) exhibited approximately half the potency of (±)MDMA (1) as a releaser at SERT, but was 8-fold and 4.5-fold less potent as a releaser at DAT and NET, respectively. Thus, (±)EDMA (3) was slightly more selective for SERT over DAT and NET.
Figure 2.

Effects of (±)EDMA and its isomers on the release of [3H]5-HT at SERT and [3H]MMP+ at DAT and NET. Data are % of maximal release expressed as mean ± SD for n = 3–4 separate experiments performed in triplicate. (±)MDMA is included as a references compound.
As shown in Figure 2, the S(+) isomer of EDMA [S(+3)] was the more potent of the two optical isomers, being about 6-fold, 50-fold, and 8-fold more potent than its R-enantiomer at SERT, DAT, and NET, respectively. It has been reported previously that S(+)MDMA is more potent than R(−)MDMA at all three transporters, so the present data with EDMA support the general concept that the S-isomers of ring-substituted phenylalkylamines (i.e., amphetamine-related compounds) are more potent than their R-enantiomers as releasing agents [8]. Interestingly, R(−)EDMA was more SERT-selective when compared to its S(+)-isomer.
(±)EDMC (4) was capable of releasing [3H]substrate at SERT, DAT, and NET (EC50 = 347, 496, and 327 nM, respectively; Table 1). Unlike what was seen with (±)EDMA (3), (±)EDMC (4) displayed no transporter selectivity. Comparing the potencies of (±)EDMC (4) with those of (±)MDMC (2), the former was slightly less potent at each of the three transporters.
4. Conclusions
For the compounds examined, ring-expansion from a methylenedioxy group to an ethylenedioxy group resulted in a modest (<8-fold) decrease in release potency at SERT, DAT, and NET in the absence of the benzylic carbonyl group (i.e., comparing 1 with 3), and even less (about 2-fold) in its presence (i.e., comparing 2 with 4). Overall, the ethylenedioxy group appears to be better accommodated at SERT than at DAT or NET. The latter finding is consistent with a previous finding that SERT accepts larger substituents in this general position than DAT [24,25]. In the absence of the carbonyl group (i.e., 3), ring-expansion seems better tolerated at SERT than at DAT and NET, and the S(+) isomer of 3 is more potent than its R(−) enantiomer at all three transporters.
If the behavioral actions of EDMA (3) are related to its actions at SERT, DAT, and NET, it might be expected that its effects would be similar to MDMA (1), but that it would be less potent. Racemic MDMA is behaviorally active at a total human dose of 80–150 mg p.o. [26], and limited data indicate that racemic EDMA fails to produce similar effects in humans at 200 mg, and only a “threshold” effect at 250 mg [26].
Given the data presented here, future studies are warranted to compare the potencies of EDMA and EDMC to their methylenedioxy homologs in animal models of addiction and neurotoxicity.
5. Experimental
5.1. Chemistry
All commercially available reagents and solvents were purchased from Sigma-Aldrich Co. (St. Louis, MO) and Platte Valley Scientific Product List (Gothenburg, NE), and used as delivered. Melting points were measured in glass capillary tubes (Thomas-Hoover melting point apparatus) and are uncorrected. 1H NMR spectra were recorded with a Bruker 400 MHz spectrometer. Chemical shifts (δ) are reported in parts per million (ppm) relative to tetramethylsilane as internal standard. Optical rotations were measured using a Jasco DIP-1000 polarimeter. Reactions and product mixtures were routinely monitored by thin-layer chromatography (TLC) on silica gel precoated F254 Merck plates. Elemental analysis for C, H, and N was performed by Atlantic Microlabs (Norcross, GA) and determined values were within 0.4% of theory.
5.1.1. (±)-1-(3,4-Ethylenedioxyphenyl)-N-methyl-2-aminopropane hydrochloride [(±)3]
Triethylamine in anhydrous Et2O (0.4 mL) and ethyl chloroformate (0.14 mL) in anhydrous Et2O (1 mL) were added to a suspension of 1-(3,4-ethylenedioxyphenyl)-2-aminopropane hydrochloride [19] (0.35 g) in anhydrous Et2O (10 mL) and stirred at room temperature under an N2 atmosphere for 1 h. Reaction mixture was filtered and solvent removed under reduced pressure to afford the corresponding carbamate as a yellow oil (0.3 g, 79%); IR (Diamond, cm−1) 3066 (NH), 1754 (N-CO-O-), 1577 (CH2 CH2O). A solution of the above carbamate (0.30 g, 1.13 mmol) in anhydrous THF (12 mL) was added in a dropwise manner at 0 °C to a stirred suspension of LiAlH4 (0.13 g, 3.40 mmol) in anhydrous THF (12 mL). The reaction mixture was heated at reflux under an N2 atmosphere for 3 h, and then cooled at 0 °C and quenched by the dropwise addition of Et2O (50 mL), H2O (3 mL) and 15% NaOH (0.5 mL). The white precipitate was removed by filtration and the filtrate was dried (Na2SO4). The solvent was evaporated under reduced pressure to give a yellow oily residue that was dissolved in absolute EtOH (5 mL) and converted to the hydrochloride salt by the addition of saturated solution of HCl/Et2O. The precipitate was collected by filtration and recrystallized from iPrOH to afford 0.03 g (11%) of the product as a white solid: mp 150 – 151°C; 1H NMR (DMSO-d6) δ 1.25 (d, 3H, CH3), 2.54 (s, 3H, CH3), 2.52–2.58 (m, 1H, CH), 3.08–3.13 (m, 1H CH), 3.26–3.30 (m, 1H, CH), 4.22 (s, 4H, CH2), 6.66 (dd, J = 8.2, 1.9 Hz, 1H, ArH), 6.74 (d, J = 1.9 Hz, 1H, ArH), 6.80 (d, J = 8.2 Hz, 1H, ArH), 7.86 (s, 3H, NH3+). Anal. calcd for (C12H17NO2 x HCl) C, 59.14; H, 7.44; N, 5.74. Found: C, 58.91; H, 7.42; N, 5.53.
5.1.2. S(+)1-(3,4-Ethylenedioxyphenyl)-N-methyl-2-aminopropane hydrochloride [S(+)3]
A solution of S(−)9 (0.05 g, 0.17 mmol) in dry THF (2 mL) was added in a dropwise manner at 0 ° C to a stirred suspension of LiAlH4 (0.02 g, 0.52 mmol) in dry THF (5 mL). The mixture was heated at reflux under an N2 atmosphere for 4 h, allowed to cool, and EtOAc (5 mL) was added, followed by the addition of aqueous 20% NaOH (0.5 mL). The white precipitate was removed by filtration and the residue was washed with EtOAc (3 × 5 mL). The combined organic portion was dried (Na2SO4) and the solvent was evaporated under reduced pressure to give a yellow oil (0.03 g, 84%), which was converted to the hydrochloride salt in anhydrous Et2O by addition of HCl-saturated anhydrous Et2O. The precipitate was collected by filtration and recrystallized from iPrOH/Et2O to give S(+)3 as white crystals (72% yield); mp 192–194 ° C; [α]25D = +14.5° (c 0.5, H2O); 1H NMR (CDCl3) δ 1.34 (d, 3H, CH3), 2.71 (s, 3H, CH3) 2.74 (dd, 1H, CH2), 3.25 (dd, 1H, CH2), 3.36 (m, 1H, CH), 4.25 (s, 4H, CH2O), 6.68–6.79 (m, 3H, ArH), 9.62 (bs, 2H, NH2+). Anal. calcd for (C12H17NO2 x HCl) C, 59.14; H, 7.44; N, 5.74. Found: C, 58.97; H, 7.41; N, 5.72.
5.1.3. R(−)1-(3,4-Ethylenedioxyphenyl)-N-methyl-2-aminopropane hydrochloride [R(−)3]
The compound was prepared from R(−)9 in the same manner as S(+)3: 67% yield; [α]25D = −14.2° (c 0.5, H2O). The 1H NMR spectrum was identical to that of S(+)3. Anal. calcd for (C12H17NO2 x HCl) C, 59.14; H, 7.44; N, 5.74. Found: C, 58.96; H, 7.46; N, 5.66.
5.1.4. (±)-1-(3,4-Ethylenedioxyphenyl)-2-(methylamino)-1-propanone hydro- chloride [(±)4]
Methylamine in EtOH (0.60 mL, 13.08 mmol) was added to a stirred solution of 1-(3,4-ethylenedioxyphenyl)-2-bromopropanone [20] (0.88 g, 3.27 mmol) in anhydrous benzene (12 mL) at room temperature under an N2 atmosphere in a sealed tube. The reaction mixture was allowed to stir at room temperature for 36 h, filtered, and the solvent was evaporated under reduced pressure. The residue was dissolved in Et2O (25 mL) and washed with H2O (3 × 5 mL). The organic portions were combined and acidified with 2M HCl (10 mL). The aqueous portion was basified with saturated NaHCO3 (25 mL) and extracted with Et2O (3 × 10 mL). The combined organic portions were washed with brine (3 × 5 mL), dried (Na2SO4), and evaporated to dryness under reduced pressure to yield a residue (0.15 g) as a free base that was converted to the hydrochloride salt and purified by recrystallization to afford 0.04 g (6%) of the product as a buff-colored powder; mp 218–219 °C, absolute EtOH/Et2O; 1H NMR (DMSO-d6) δ 1.43 (d, 3H, CH3), 2.57 (s, 3H, CH3), 4.33 (m, 4H, CH2O), 5.07 (m, 1H, CH), 7.05 (d, 1H, ArH), 7.56 (dd, J =5.7, 1.9 Hz, 2H, ArH), 9.28 (s, 1H, NH+). Anal. calcd for (C12H15NO2 x HCl) C, 55.93; H, 6.26; N, 5.43. Found: C, 56.01; H, 6.12; N, 5.33.
5.1.5. 1-(3,4-Ethylenedioxyphenyl)propan-2-one (6)
A solution of 5 [19] (1.40 g, 6.33 mmol) in glacial AcOH (18 mL) was added in a dropwise manner at room temperature to a stirred suspension of Fe powder (4.80 g, 86.40 mmol) in glacial AcOH (18 mL). The resulting mixture was heated at reflux for 3 h, and then cooled to room temperature. Excess iron was removed by filtration, and the residue was diluted with H2O (50 mL) and extracted with DCM (3 × 30 mL). The combined organic portion was washed with aqueous 2N NaOH and dried (Na2SO4). Solvent was removed under reduced pressure and the residual oil was chromatographed on a silica gel column (Aldrich silica gel 60) using hexanes:EtOAc (9:1) as eluent to give 6 as a yellow oil (78% yield); 1H NMR (CDCl3) δ 2.23 (s, 3H, CH3), 3.68 (s, 2H, CH2), 4.27 (s, 4H, CH2O), 6.68–6.72 (m, 2H, ArH), 6.85 (s, 1H, ArH).
5.1.6. S,S(−)N-(1-Phenylethyl)-1-(3,4-ethylenedioxyphenyl)-2-aminopropane hydrochloride [S,S(−)7]
Glacial AcOH (0.22 g, 3.64 mmol) and NaBH(OAc)3 (1.08 g, 5.10 mmol) were added to a stirred solution of 6 (0.70 g, 3.64 mmol) and S(−)α-methylbenzylamine (0.44 g, 3.64 mmol) in anhydrous DCE (15 ml). The reaction mixture was allowed to stir at room temperature under an N2 atmosphere for 24 h and then basified with aqueous 1N NaOH to pH 9. The mixture was extracted with Et2O (3 × 20 mL), the combined organic portion was dried (Na2SO4) and solvent was evaporated to give a yellow oil that was converted to the hydrochloride salt in anhydrous Et2O by addition of HCl-saturated anhydrous Et2O. The precipitate was collected by filtration and recrystallized from iPrOH to afford S,S(−)7 as white crystals in 50% yield; mp 180–182 °C; [α]25D = −34.4° (c 1.0, MeOH); 1H NMR (CDCl3) δ 1.39 (d, 3H, CH3), 1.95 (d, 3H, CH3), 2.73 (dd, 1H, CH2), 2.95 (m, 1H, CH), 3.27 (dd, 1H, CH2), 4.20 (s, 4H, CH2O), 4.39 (m, 1H, CH), 6.45 (s, 1H, ArH), 6.47 (d, 1H, ArH) 6.70 (d, 1H, ArH), 7.47 (m, 3H, ArH), 7.68 (d, 2H, ArH), 9.77 (bs, 1H, NH2+), 10.25 (bs, 1H, NH2+).
5.1.7. R,R(+)N-(1-Phenylethyl)-1-(3,4-ethylenedioxyphenyl)-2-aminopropane hydrochloride [R,R(+)7]
The compound was prepared in the same manner as S,S(−)7 using R(+)α-methylbenzylamine; [α]25D = +33.6° (c 1.0, MeOH). The 1H NMR spectrum was identical to that of S,S(−)7.
5.1.8. S(+)1-(3,4-Ethylenedioxyphenyl)-2-aminopropane hydrochloride [S(+)8]
Pd/C catalyst (10%, 0.35 g) was added to a solution of S,S(−)7 (0.58 g, 1,74 mmol) in MeOH (30 mL). The reaction mixture was hydrogenated at ca. 50 psi for 40 h and then filtered. The solvent was evaporated under reduced pressure and the residue was recrystallized from acetone to give S(+)8 as white crystals in 73% yield; mp 166–167 °C; [α]25D = +30.1° (c 0.5, H2O); 1H NMR (CDCl3) δ 1.42 (d, 3H, CH3), 2.79 (dd, 1H, CH2), 3.10 (dd, 1H, CH2), 3.53 (m, 1H, CH), 4.27 (s, 4H, CH2O), 6.74–6.86 (m, 3H, ArH), 8.43 (bs, 3H, NH3+).
5.1.9. S(+)1-(3,4-Ethylenedioxyphenyl)-2-aminopropane hydrochloride [R(−)8]
The compound was prepared from R,R(+)7 in the same manner as S(+)8; [α]25D = −30.6° (c 0.5, H2O). The 1H NMR spectrum was identical to that of S(+)8.
5.1.10. S(−)1-(3,4-Ethylenedioxyphenyl)propan-2-yl carbamic acid tert-butyl ester [S(−)9]
Method 1 (Scheme 1): A solution of S(+)8 (0.15 g, 0.65 mmol) in aqueous 1N NaOH (15 mL) was extracted with Et2O (3 × 10 mL). The combined organic portion was dried (Na2SO4) and the solvent was evaporated to give the free base as a yellow oil (0.12 g, 0.62 mmol). Di-tert-butyl dicarbonate (0.14 g, 0.62 mmol) was added to a solution of the free base and NEt3 (0.10 mL, 0.62 mmol) in anhydrous DCM (5 mL) at 0 ° C under an N2 atmosphere. The reaction mixture was stirred at room temperature for 1 h and then quenched with H2O (10 mL). The mixture was extracted with DCM (3 × 10 mL) and the combined organic portion was dried (Na2SO4). Solvent was removed under reduced pressure and the residual oil was then chromatographed on a silica gel column (Aldrich silica gel 60) using hexanes:EtOAc (8:2) as eluent to give S(−)9 as a colorless oil in 83% yield.
Method 2 (Scheme 2): 1,2-Dibromoethane (0.04 mL, 0.45 mmol) was added to a stirred mixture of S(−)11 (0.10 g, 0.38 mmol), K2CO3 (0.16 g, 1.14 mmol) and dry DMF (5 mL). The reaction mixture was allowed to stir at room temperature under an N2 atmosphere for 24 h. Water (10 mL) was added and the resulting mixture was extracted with EtOAc (3 × 10 mL). The organic extracts were combined and dried (Na2SO4). Solvent was evaporated under reduced pressure and the residual oil was chromatographed on a silica gel column (Aldrich silica gel 60) using hexanes:EtOAc (8:2) as eluent to give S(−)9 as a colorless oil in 45% yield; [α]25D = −13.3° (c 0.5, CHCl3); 1H NMR (CDCl3) δ 1.09 (d, 3H, CH3), 1.44 (s, 9H, CH3) 2.56 (dd, 1H, CH2), 2.72 (dd, 1H, CH2), 3.84 (m, 1H, CH), 4.25 (s, 4H, CH2O), 6.66–6.77 (m, 3H, ArH).
5.1.11. R(+)1-(3,4-Ethylenedioxyphenyl)propan-2-ylcarbamic acid tert-butyl ester [R(+)9]
This compound was also prepared from R(−)8 in the same manner employed for the preparation of S(−)9 (Scheme 1); [α]25D = +13.0° (c 0.5, CHCl3). The 1H NMR spectrum was identical to that of S(−)9.
5.1.12. S(−)1-(3,4-Dihydroxyphenyl)-2-ylcarbamic acid tert-butyl ester [S(−)11]
Sodium methoxide (0.04 g, 0.74 mmol) in dry MeOH (0.5 mL) was added to a stirred solution of S(+)10 [21] (0.15 g, 0.61 mmol) and di-tert-butyl dicarbonate (0.15 g, 0.69 mmol) in MeOH (10 mL). The reaction mixture was allowed to stir at room temperature under an N2 atmosphere for 0.5 h. Solvent was evaporated under reduced pressure, and the residue was diluted with EtOAc (20 mL). The inorganic salt was removed by filtration and solvent was removed under reduced pressure. The residual oil was chromatographed on a silica gel column (Aldrich silica gel 60) using hexanes:EtOAc (7:3) as eluent to give S(−)11 as a colorless oil in 62% yield; [α]25D = −3.0° (c 1.0, CHCl3); 1H NMR (CDCl3) δ 1.08 (d, 3H, CH3), 1.44 (s, 9H, CH3) 2.54 (dd, 1H, CH2), 2.65 (dd, 1H, CH2), 3.82 (m, 1H, CH), 6.58 (dd, 1H, ArH), 6.77 (m, 2H, ArH).
5.2 In Vitro Release Assays
Subjects
Male Sprague-Dawley rats (Charles River, Wilmington, MA, USA) weighing 250–350 g were housed three per cage with free access to food and water and maintained on a 12 h light/dark cycle with lights on from 7:00 a.m. to 7:00 p.m. Animal facilities were accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care, and procedures were carried out in accordance with the Institutional Animal Care and Use Committee and the National Institutes of Health guidelines on care and use of animal subjects in research (National Research Council, 2011).
Procedure
Rats were euthanized by CO2 narcosis, and brains were processed to yield synaptosomes as previously described [27]. Whole brain minus caudate and cerebellum was used to prepare synaptosomes for SERT and NET assays, whereas caudate was used for DAT assays. One whole brain minus caudate and cerebellum (for SERT and NET assays) or one pair of caudates (for DAT assays) was diluted in 10 mL of ice-cold 10% sucrose containing 1 μM reserpine. Tissue was homogenized using a Potter-Elvehjem homogenizer, centrifuged at 1000 g for 10 min at 4 °C, and supernatants (i.e., synaptosomal preparations) were retained on ice. Supernatants were diluted with sucrose solution to yield protein concentrations of 900 μg/mL for SERT and NET assays and 90 μg/mL for DAT assays. In the release procedure, 5 nM [3H]5-HT was used as a radioligand substrate for SERT whereas 9 nM [3H]-1-methyl-4-phenylpyridinium ([3H]MPP+) was used as the radiolabeled substrate for NET and DAT. All buffers used in the release assay methods contained 1 μM reserpine to block vesicular uptake of substrates. The selectivity of release assays was optimized for a single transporter by including unlabeled blockers to prevent the uptake of [3H]5-HT or [3H]MPP+ by competing transporters. Synaptosomes were preloaded with radiolabeled substrate in Krebs-phosphate buffer for 1 h (steady state). Release assays were initiated by adding 850 μL of preloaded synaptosomes to 150 μL of test drug. Eight point dilution curves, with doses ranging from 10 to 10,000 nM, were performed in triplicate on three separate occasions for each test drug. Release was terminated by vacuum filtration, and retained radioactivity was quantified by liquid scintillation counting. The specific counts per minute (cpm) for each assay (i.e., total cpm – nonspecififc cpm) were 7000, 1700, and 2500 for SERT, NET, and DAT, respectively.
Data Analysis
Statistical analyses were carried out using GraphPad Prism (v. 6.0; GraphPad Scientific, San Diego, CA, USA). EC50 values and corresponding SDs for stimulation of release were calculated based on non-linear regression analysis. Efficacy for test compounds was expressed as a percentage of maximal release (i.e., % Emax), which was defined as the release produced by 100 μM tyramine for SERT and 10 μM tyramine for DAT and NET. These concentrations of tyramine induce the efflux of all ‘releasable’ tritium from synaptosomes under the assay conditions described.
Reagents
[3H]5-HT (specific activity = 30 Ci mmol−1) was purchased from Perkin Elmer (Shelton, CT, USA). [3H]MPP+ (specific activity = 85 Ci mmol−1) was purchased from American Radiolabeled Chemicals (St. Louis, MO, USA). All other chemicals and reagents were acquired from Sigma-Aldrich (St. Louis, MO, USA).
Supplementary Material
Acknowledgments
This work was supported in part by PHS grant DA033930 and by the Intramural Program of the National Institute on Drug Abuse.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Green AR, Cross AJ, Goodwin GM. Psychopharmacology. 1993;119:247. doi: 10.1007/BF02246288. [DOI] [PubMed] [Google Scholar]
- 2.Parrott AC. Hum Psychopharmacol Clin Exp. 2001;16:557. doi: 10.1002/hup.351. [DOI] [PubMed] [Google Scholar]
- 3.Baylen CA, Rosenberg H. Addiction. 2006;101:933. doi: 10.1111/j.1360-0443.2006.01423.x. [DOI] [PubMed] [Google Scholar]
- 4.Simmler LD, Buser TA, Donzelli M, Schramm Y, Dieu LH, Huwyler J, Chaboz S, Hoener MC, Liechti ME. Br J Pharmacol. 2013;168:458. doi: 10.1111/j.1476-5381.2012.02145.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Metzner R. In: Through the Gateway of the Heart. Accounts of Experiences with MDMA and Other Empathogenic Substances. Adamson S, editor. Four Trees Publications; San Francisco: 1985. pp. 1–6. [Google Scholar]
- 6.Young R, Glennon RA. Pharmacol Biochem Behav. 2008;88:318. doi: 10.1016/j.pbb.2007.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rudnick G, Wall SC. Proc Nat Acad Sci USA. 1992;89:1817. doi: 10.1073/pnas.89.5.1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Setola V, Hufeisen SJ, Grande-Allen KJ, Vesely I, Glennon RA, Blough B, Rothman RB, Roth BL. Mol Pharmacol. 2003;63:1223. doi: 10.1124/mol.63.6.1223. [DOI] [PubMed] [Google Scholar]
- 9.Liechti ME, Vollenweider FX. Hum Psychopharmacol. 2001;16:589. doi: 10.1002/hup.348. [DOI] [PubMed] [Google Scholar]
- 10.Hysek CM, Simmler LD, Ineichen M, Grouzmann E, Hoener MC, Brenneisen R, Huwyler J, Liechti ME. Clin Pharmacol Ther. 2011;90:246. doi: 10.1038/clpt.2011.78. [DOI] [PubMed] [Google Scholar]
- 11.Iversen LE. ‘Consideration of the cathinones’. Advisory Council on the Misuse of Drugs. A report submitted to the Home Secretary of the UK. 2010 Mar 31; [Google Scholar]
- 12.Glennon RA. Adv Pharmacol. 2014;69:581. doi: 10.1016/B978-0-12-420118-7.00015-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Methylone (bk-MDMA) Critical Review Report. Expert Committee on Drug Dependence 36th Meeting, World Health Organization, Agenda item 4.14; 2014. [Google Scholar]
- 14.Cozzi NV, Sievert MK, Shulgin AT, Jacob P, Ruoho AE. Eur J Pharmacol. 1999;381:63. doi: 10.1016/s0014-2999(99)00538-5. [DOI] [PubMed] [Google Scholar]
- 15.Baumann MH, Ayestas MA, Partilla JS, Sink JR, Shulgin AT, Brandt SD, Rothman RB, Ruoho AE, Cozzi NV. Neuropsychopharmacology. 2012;37:1192. doi: 10.1038/npp.2011.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.U. S. Drug Enforcement Administration, Office of Diversion Control. National Forensic Laboratory Information System Report. 2014 Jun; https://www.nflis.deadiversion.usdoj.gov.
- 17.Baumann MH, Partilla JS, Lehner KR. Eur J Pharmacol. 2013;698:1. doi: 10.1016/j.ejphar.2012.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rannazzisi JT. Federal Register. 2006 Oct 20;71:203. 62017. [Google Scholar]
- 19.Vallejos G, Fierro A, Rezende MC, Sepulveda-Boza S, Reyes-Parada M. Bioorg Med Chem. 2005;13:4450. doi: 10.1016/j.bmc.2005.04.045. [DOI] [PubMed] [Google Scholar]
- 20.Bockmühl M, Erhart G. 1964973A. US Patent. 1934 Jul 3;
- 21.Nichols DE, Barfknecht CF, Rusterholz DB, Benington F, Morin RD. J Med Chem. 1973;16:480. doi: 10.1021/jm00263a013. [DOI] [PubMed] [Google Scholar]
- 22.Milhazes N, Cunha-Oliveira T, Martins P, Garrido J, Oliveira C, Rego AC, Borges F. Chem Res Toxicol. 2006;19:1294. doi: 10.1021/tx060123i. [DOI] [PubMed] [Google Scholar]
- 23.Eshleman AJ, Wofrum KM, Hatfield MG, Johnson RA, Murphy KV, Janowsky A. Biochem Pharmacol. 2013;85:1803. doi: 10.1016/j.bcp.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sakloth F, Kolanos R, Mosier PD, Bonano JS, Banks ML, Partilla JS, Baumann MH, Negus SS, Glennon RA. Br J Pharmacol. 2015;172:2210. doi: 10.1111/bph.13043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bonano JS, Banks ML, Kolanos R, Sakloth F, Barnier ML, Glennon RA, Cozzi NV, Partilla JS, Baumann MH, Negus SS. Br J Pharmacol. 2015;172:2433. doi: 10.1111/bph.13030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shulgin A, Shulgin A. Pihkal. Transform Press; Berkeley, CA: 1991. [Google Scholar]
- 27.Baumann MH, Dersch CM, Romero DV, Rice KC, Carroll FI, Partilla JS. Synapse. 2001;39:32. doi: 10.1002/1098-2396(20010101)39:1<32::AID-SYN5>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.