

Abstract
Aging and bone diseases are associated with increased fracture risk. It is therefore pertinent to seek an understanding of the origins of such disease-related deterioration in bone's mechanical properties. The mechanical integrity of bone derives from its hierarchical structure, which in healthy tissue is able to resist complex physiological loading patterns and tolerate damage. Indeed, the mechanisms through which bone derives its mechanical properties make fracture mechanics an ideal framework to study bone's mechanical resistance, where crack-growth resistance curves give a measure of the intrinsic resistance to the initiation of cracks and the extrinsic resistance to the growth of cracks. Recent research on healthy cortical bone has demonstrated how this hierarchical structure can develop intrinsic toughness at the collagen fibril scale mainly through sliding and sacrificial bonding mechanisms that promote plasticity. Furthermore, the bone-matrix structure develops extrinsic toughness at much larger micrometer length-scales, where the structural features are large enough to resist crack growth through crack-tip shielding mechanisms. Although healthy bone tissue can generally resist physiological loading environments, certain conditions such as aging and disease can significantly increase fracture risk. In simple terms, the reduced mechanical integrity originates from alterations to the hierarchical structure. Here, we review how human cortical bone resists fracture in healthy bone and how changes to the bone structure due to aging, osteoporosis, vitamin D deficiency and Paget's disease can affect the mechanical integrity of bone tissue.
Introduction
The greater goals of bone research are to assess the risk of fractures in bone and to prevent their occurrence. Fracture events encompass the initiation and growth of cracks as well as the resulting complete fracture of the tissue. This makes the fracture toughness (that is, resistance to fracture) a critical aspect of bone's mechanical integrity. In healthy tissue, bone's complex structure has evolved to resist physiological forces and can further tune its mechanical resistance through remodeling. However, bone tissue is vulnerable to skeletal aging, environmental conditions and various genetic or metabolic bone diseases that increase fracture risk or affect the mechanical integrity. These diseases include osteoporosis, vitamin D deficiency and Paget's disease of bone (PDB), to name but a few. Thus, it is necessary to understand how such disease alters the bone-matrix structure leading to a higher risk of fracture and how corresponding therapeutic treatments may affect bone's mechanical properties.
Akin to most natural and engineered materials, the mechanical performance of bone derives directly from its structure. Similar to many natural materials, its structure is hierarchical, meaning that there are discrete characteristic structural features at differing length scales (Figure 1). The hierarchical structure of human cortical bone spans numerous architectural levels from the individual collagen molecules and mineral platelets at the nanoscale to the whole bone at the macroscale.1 At the smallest length scales, the key feature is the mineralized collagen fibril. Here, collagen molecules form arrays with a 67-nm offset, where nanoplatelets of hydroxyapatite mineral are embedded within the collagen array and on the surface of the fibril.2,3 At this length-scale, the intra- and extra-fibrillar matrix contains non-collagenous proteins (for example, bone sialoprotein, osteopontin, osteonectin), as well as enzymatic and non-enzymatic cross-links.4,5,6 The non-enzymatic cross-links are particularly influential to bone's mechanical integrity in disease states.7 These cross-links also known as advanced glycation end-products form through a glucose-mediated reaction and may form both intra- and interfibrillar connections.8,9 The mineralized collagen fibrils hierarchically assemble into lamellae to form osteons, which are the most prominent motif on the microstructural scale (Figure 1). The osteons are cylindrical with a nominal circular cross-section and a central vascular channel called the Haversian canal. At the outer limits of the osteon is a thin border called the cement line, which is considered to be either collagen deficient or highly mineralized relative to the surrounding tissue.10
Figure 1.
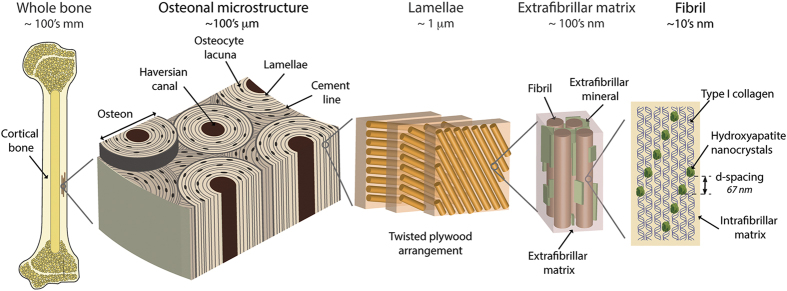
Hierarchical structure of human cortical bone. The structure of bone spans numerous length-scales from the macroscopic whole-bone structure to the nanoscale collagen and mineral components. At the microscale, the bone is composed of osteons that are 170–250 μm in diameter.60 The osteons have a central vascular channel (60–90 μm diameter60), called the Haversian canal, and a highly mineralized outer boundary, called the cement line (<5 μm thickness).10 In the osteons, each vascular channel is concentrically surrounded by lamellae (2–9 μm thickness).80 The lamellae, which are composed of bundles of collagen fibrils, have a twisted plywood arrangement, where neighboring lamellae have different fibril orientations.80 Certain lamellae may be less organized,81 and it is here that the osteocytes bone cells reside in lacunae (5–10 μm diameter) and interconnect through canaliculi (100–400 nm in diameter).82,83 The fibrils (80–100 nm diameter1) are surrounded by polycrystalline extrafibrillar mineral platelets (5 nm thickness, 50–80 nm width and 40–200 nm length3); the extrafibrillar as well as the intrafibrillar matrix may also contain molecular components, such as non-collagenous proteins or cross-links, promoting the formation of sacrificial bonds.5,9,15,16,84 In the fibrils, type I collagen molecules (1.5 nm diameter, 300 nm length) and hydroxyapatite nanocrystals (50 nm width, 25 nm height, 1.5–4 nm thickness) form a composite structure, where arrays of collagen molecules staggered at 67 nm are embedded with nanoplatelets of hydroxyapatite mineral.1 Adapted from Zimmermann et al. and Milovanovic et al.45,82
Deformation and fracture mechanisms at each level of bone's hierarchical structure contribute to its mechanical integrity, most significantly its strength and toughness. A prime benefit to the hierarchical arrangement is that strength (that is, the resistance to inelastic deformation) and toughness (that is, the resistance to crack growth or fracture) can be generated independently.11 Indeed, strength originates from intrinsic mechanisms at small length-scales that promote plasticity and determine the inherent resistance of the material to deformation or crack initiation. Intrinsic toughening mechanisms operate ahead of the crack tip to generate resistance to microstructural damage. The most prominent mechanism is that of plastic deformation, which provides a means of blunting the crack tip through the formation of ‘plastic' zones. Toughness originates from extrinsic mechanisms at micron length-scales that are large enough to shield the growth of cracks (Figure 2).12 Extrinsic toughening mechanisms, conversely, operate primarily in the wake of the crack tip to inhibit cracking by `shielding' the crack from the applied driving force, whereas intrinsic toughening mechanisms are effective in inhibiting both the initiation and the growth of cracks; extrinsic mechanisms, for example, crack bridging, are only effective in inhibiting crack growth.
Figure 2.

Toughening mechanisms in bone. Toughness is a competition between intrinsic and extrinsic toughening mechanisms. The intrinsic mechanisms primarily work ahead of the crack tip to develop plasticity, whereas the extrinsic mechanisms act in the crack wake (that is, after crack extension commences) to resist crack propagation through crack-tip shielding mechanisms. In human cortical bone, the extrinsic toughening mechanisms are primarily developed at large length-scales on the order of 1–100's of microns. Here, the most potent extrinsic toughening mechanisms are crack deflection and twist at cement lines and uncracked ligament bridging; however, collagen fiber bridging and microcracking could also have a role. Intrinsic toughness is developed at small length-scales, less than ∼1 μm. Here, plasticity is generated through sliding mechanisms between fibrils that are facilitated by sacrificial bonds (for example, non-collagenous proteins, cross-links) within the extrafibrillar matrix. Similarly, plasticity could also be generated within the fibril through deformation mechanisms, such as the formation of dilatational bands, which are also facilitated by sacrificial bonding in the intrafibrillar matrix. Adapted from Launey et al.85
Human cortical bone generates intrinsic resistance to fracture, similar to other materials at small length-scales where the bonds between the smallest components stretch, deform, slip and/or break. Thus, the intrinsic fracture resistance of bone is generated through plasticity at the level of the mineralized collagen fibril (see Figure 2). Here, fibrils stretch during elastic deformation and generate plasticity through intra- and extrafibrillar deformation mechanisms, such as dilatational band formation and/or fibrillar sliding. These fibrillar deformation mechanisms largely involve sacrificial bonds, such as cross-links and non-collagenous proteins that are critical to the generation of mechanical properties.5,13,14,15,16,17,18,19 Indeed, studies using animal models with deficiencies in non-collagenous proteins have found significant decreases in fracture toughness and nanoscale damage.16,17 An important distinction here is that plasticity, which is the source of bone's ductility, is generated primarily at the fibrillar level rather than solely at the collagen molecule level.20,21 The damaged caused by mechanical loading increases fracture resistance but may also trigger bone repair, where micron-scale damage (that is, linear microcracks) severing cellular connections is repaired through normal bone remodeling, and nanoscale damage (that is, diffuse damage) is repaired through a separate, as yet unknown, process.22,23,24 Further studies are required to better understand how each aspect of the complex nanostructure contributes to bone deformation, repair and overall fracture risk in health and disease.
Apart from the generation of plasticity and intrinsic toughness, extrinsic mechanisms can develop the material's resistance to the growth of cracks (Figure 2). Human cortical bone generates extrinsic resistance at the microstructural length-scale where micron-scale mechanisms are activated to resist propagation of micron-scale cracks.25 As such, the extrinsic resistance of bone shields the crack from the full mechanical driving force. In bone, crack-tip shielding is developed at the level of the osteon primarily through crack bridging and crack deflection mechanisms. Here, as a crack grows through the bone structure, collagen fibers or uncracked regions of bone matrix can remain intact in the crack wake to form ‘bridges' that span the crack opening (Figures 3a and b). These bridges can carry part of the load that would otherwise be used to further extend the crack, thereby lowering the driving force for crack propagation.26 Crack deflection and twist is another extrinsic mechanism occurring when a crack encounters an interface in the bone, such as the highly mineralized cement lines at the outer boundary of the osteons or the modulating mechanical properties of the lamellae (Figures 3c and d).27,28 If the crack deflects and/or twists away from the original crack plane, the driving force can be significantly reduced.
Figure 3.

Crack-briding and crack deflection promote extrinsic toughness. Human cortical bone develops extrinsic toughness on the microstructural scale through (a,b) uncracked ligament bridging and (c,d) crack deflection/twist mechanisms. Using (a) 3-D synchrotron tomography and (b) scanning electron microscopy, uncracked segments of bone (orange arrows) can be observed in the wake of the crack. These uncracked regions of bone tissue, commonly generated by microcracking ahead of the crack tip (red arrows), carry part of the load that would otherwise be used to extend the crack. Similarly, evidence of crack deflection can be viewed (c) on the fracture surface after testing and (d) during a test using the scanning electron microscope. Here, the crack grows from the notch (red arrows). As the crack encounters the interfaces within the bone tissue aligned with the osteons, primarily cement lines, the crack will often deflect or twist (black arrows). Adapted from Zimmermann et al.20,45
Another aspect of the bone structure that should be addressed is the porosity, which in cortical bone exists in the form of vascular canals (that is, Haversian canals) and cellular pores (that is, osteocytes lacunae and canaliculi). From a mechanics perspective, porosity can impact the mechanical properties29,30 because it reduces the load bearing area, which effectively causes a higher stress on the material. However, porosity is not the sole determinant of bone fracture risk.31,32,33,34 Indeed, compositional factors, such as the cross-linking profile, mineralization distribution, or the collagen content, also have a strong correlation with fracture risk.35,36 In terms of fracture mechanics, the precise effects of porosity on crack propagation in bone have not been isolated from other aspects of the structure that influence toughness. However, cracks are commonly observed to follow cement lines during fracture mechanics experiments in the presence of normal amounts of bone porosity.20 The effects of porosity can be corrected by normalizing the applied stress by the bone volume fracture, which is especially critical when comparing the mechanical properties of groups with significantly different porosities.19,37 Thus, although bone porosity is not the sole determinant of fracture risk in bone, it should be considered when addressing the mechanical properties of cortical bone.
Because of bone's hierarchical structure, its mechanical behavior originates from plasticity-related intrinsic mechanisms at small length-scales and crack-shielding extrinsic mechanisms at larger length-scales to create strength and toughness, respectively. Bone's intrinsic resistance can be measured by traditional characterization methods through simply a strength test or investigated more deeply through a suite of characterization methods (for example, small-angle X-ray scattering, nanoindentation, or atomic force microscopy) to assess fibrillar-level mechanical properties. Alternatively, cortical bone's intrinsic and extrinsic resistance can be quantified using fracture mechanics principles. In fracture mechanics, the toughness of a material can be measured in terms of the critical value of a parameter, such as the stress intensity K, which characterizes the stress and displacements fields in the vicinity of a crack tip; under nominally linear elastic (small-scale yielding, plane strain) conditions, one such characterizing parameter is a critical value of K known as the plane-strain fracture toughness, KIc . The fracture toughness, KIc, is defined as the critical stress intensity at fracture instability; if measured correctly, it strictly characterizes the crack-initiation toughness. However, as bone allows stable crack propagation, a single-value toughness, such as KIc, does not always fully characterize the material behavior. In this case, a crack-growth resistance curve or a R-curve can be used to more faithfully quantify material behavior during crack propagation. With an R-curve, the crack-driving force, defined in terms of K, G or J, is measured as a function of crack extension, Δa. Here the stress intensity K is a measure of the amplitude of the elastic stress and displacement fields at the crack tip. Alternatively, the toughness can be measured as a critical value of the strain-energy release rate, Gc, defined as the change in potential energy per unit increase in crack area in an elastic solid. If the presence of local plasticity is no longer small enough to be ignored, both approaches can also be expressed in terms of the J-integral, defined as the amplitude of the nonlinear elastic stress and displacement fields at the crack tip and/or as the change in potential energy per unit increase in crack area in a nonlinear elastic solid. Where nominally elastic conditions prevail, J=G=K2/E (where E is the appropriate Young's modulus). From the R-curve, the crack-initiation toughness can be measured by extrapolating the toughness to Δa→0, whereas the slope of the curve can be taken as a measure of the crack-growth toughness. An example of an R-curve is shown in Figure 4 for bone tested where cracking is perpendicular (that is, transverse orientation) and parallel (that is, longitudinal orientation) to the osteonal bone structure. The positive R-curve slope in both orientations demonstrates rising R-curve behavior stemming from the fact that the extrinsic toughening mechanisms (that is, crack deflection and bridging) increase the bone's toughness as the crack extends. However, the slope of the R-curve in the transverse orientation is significantly larger than in the longitudinal orientation, that is, the bone is easier to split than to break in two. The different R-curve slopes result from the differing extrinsic toughening mechanisms that are active in these two orientations. In the transverse orientation, the mechanism of crack deflection is paramount because once a crack deflects at the cement lines, it is extending in a direction, that is parallel to the osteons and nominally perpendicular to the path of the maximum driving force (for example, the Gmax or KII=0 path). Conversely, in the longitudinal orientation, the driving force is nominally parallel to the osteonal orientation as well as the lamellar and cement line interfaces. The preferential formation of microcracks along these interfaces activates the formation of uncracked ligaments across the crack faces, which in turn results in significant crack bridging. As is clear from the R-curves in Figure 4, crack deflection is a much more potent shielding mechanism than crack bridging in enhancing the toughness of bone, which is why bone is much tougher in the transverse orientation than in the longitudinal direction.27
Figure 4.
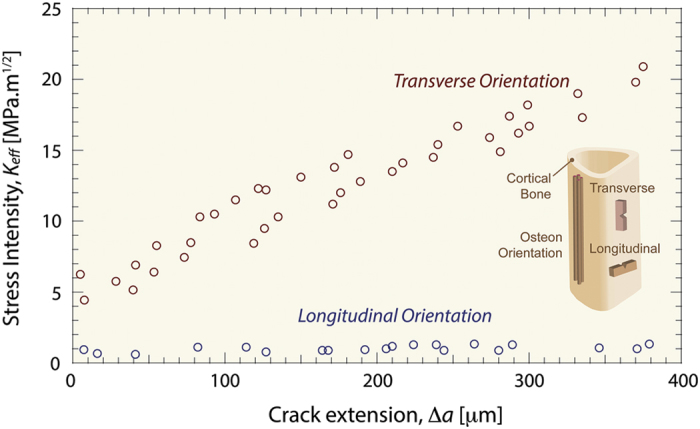
Crack-growth resistance behavior in bone. Bone is a damage tolerant material that resists the growth of cracks through its structure. As such, a single value of the toughness at instability does not completely characterize bone's behavior. Instead, the toughness must be assessed as a function of crack extension, which is called a crack-growth resistance-curve or a R-curve. Here, the amplitude of the stress and displacement fields around the crack tip (that is, K, J or G) is measured as the crack stabily extends. With R-curves, the value of the driving force as Δa→0 is a measure of the crack-initiation toughness, whereas the slope of the R-curve is a measure of the crack-growth toughness. The R-curves of human cortical bone have a positive slope, which indicates rising R-curve behavior. Essentially, the slope is able to rise because the toughness of the material increases with crack extension. Here, the microstructural extrinsic toughening mechanisms, such as crack deflection and uncracked ligament bridging, become activated with crack extension and toughen the material. In the presence of such extrinsic (shielding) mechanisms, a higher driving force is required for further crack extension. Adapted from Koester et al. and Zimmermann et al.27,39,86
As cortical bone is a damage tolerant material,11 fracture mechanics is an ideal framework to study bone's resistance to crack initiation and growth. Although our understanding of how bone resists fracture is based on experimental evidence from quasi-static loading conditions, physiological fractures occur most often in high strain rate or fatigue conditions. Recent studies have shown that fracture toughness decreases during high strain rate loading because the intrinsic viscous deformation mechanisms at the nanoscale (that is, fibrillar sliding, sacrificial bonding) are restricted. The lower plasticity at the nanoscale also influences the extrinsic crack growth behavior as the heterogeneous structure, which normally interferes with crack growth, appears more mechanically homogeneous.20,38 Although more studies are required to understand the fracture resistance of bone under physiological loading conditions, the current understanding of bone's resistance to fracture provides a starting framework to study the effects of aging and disease, where fractures can occur with minimal trauma.
Human cortical bone's complex hierarchical structure generates strength and toughness, respectively, through intrinsic plasticity-based mechanisms at small length-scales and extrinsic crack-shielding mechanisms at larger length-scales. In healthy bone, the structure is generally able to resist physiological loads and adapt to new loading environments through remodeling.20,39 However, disease states put bone at a higher risk of fracture, bending or bowing, all of which can lead to a loss in mechanical integrity. Here, we review how certain prominent bone diseases affect the structural quality of bone, specifically by examining the salient mechanisms that can cause increased fracture risk due to aging, osteoporosis, vitamin D deficiency and Paget's disease.
Aging and osteoporosis
After reaching peak bone mass in the third or fourth decade in life, both the quantity and quality of the bone tissue in the human skeleton begin to deteriorate.31,40 Here bone quality encompasses the composition and distribution of mineral and collagen, the characteristics and structural integrity of each hierarchical length scale (that is, osteon size and distribution), as well as the amount of microdamage present. This aging-related deterioration in the bone structure has a profound impact on the overall mechanical properties and fracture risk. Currently, fracture risk is mainly measured through dual-energy X-ray absorptiometry (DXA), where the degree of X-rays absorbed by the bone mineral provides a prediction of fracture risk. As the degree of bone mineralization shows a rather narrow range in health and disease,37,41 standard DXA diagnostics provide a helpful measure of areal bone mineral density, which is based on the present porosity and volume fraction of the bone (that is, bone quantity). In addition, DXA provides valuable information due to its widespread global availability and the existence of large reference populations; however, it is not necessarily an optimal indicator for predicting fracture risk. Indeed, as DXA predominantly assesses the quantity of bone mineral, many unanswered issues remain with respect to the deterioration in the quality of the tissue. Here, we briefly review experimental measurements of the structural and mechanical properties of human cortical bone due to biological aging and recent research concerning the effectiveness of bisphosphonates as a treatment for osteoporosis in human cortical bone.
Aging
Aging is associated with changes to the bone structure at small and large length-scales that deteriorate its strength (that is, intrinsic resistance) and toughness (that is, extrinsic resistance), respectively.42,43,44,45,46 The major impact to bone's intrinsic toughness is the marked aging-related increase in non-enzymatic cross-links, which has been correlated to increased fracture risk.47,48,49,50 Models and experiments measuring fibril deformation have found that cross-links restrict fibrillar sliding.45,51,52 Specifically, using small-angle X-ray scattering experiments, lower levels of fibrillar deformation in aged tissue and threefold higher levels of non-enzymatic cross-linking in comparison with young tissue suggests that the advanced glycation end-product cross-links are constraining fibrillar deformation (Figures 5a and b).45 In synchrotron X-ray scattering experiments, bone's periodic nanoscale structure scatters X-rays, with the fibril's structure scattering X-rays at small angles (SAXS) and the mineral's structure diffracting X-rays at wide angles (WAXD). Mechanical testing of the bone samples during the SAXS/WAXD experiments changes the characteristic periodicity of the nanoscale structure and thus provides a measurement of the strain within the fibril and mineral, as compared with the overall strain in the bone tissue.
Figure 5.

Aging-related changes to bone toughness. Aging of human cortical bone affects its mechanical properties at small and large length-scales. (a) In situ small-angle X-ray scattering measurements have shown that the fibrils in aged bone deform less during mechanical tensile tests, which indicates changes in the intrinsic toughness. (b) Indeed, the aged bone has a significantly higher amount of non-enzymatic cross-links. These cross-links, which form between collagen molecules and between fibrils, could constrain deformation in the aged fibrils and reduce plasticity through fibrillar sliding. (c) The aging-related deterioration in mechanical properties is also clear in fracture toughness crack-growth resistance curves. Here, decreases in the crack initiation toughness as well as the crack-growth toughness are evident with aging. Extrinsically, the decrease in the crack-growth toughness stems from the higher osteon density in aged tissue. Using 3-D synchrotron micro-computed tomography on fracture toughness samples after testing of (d) young (34–41 years) and (e) aged (85–99 years) human cortical bone, the higher osteon density and lack of uncracked ligament bridges are evident in the aged tissue. Haversian canals (green) are visible as well as the growing crack (yellow) extending from the notch (white arrow). Uncracked regions spanning the crack wake are found in young bone (orange arrows) but are less apparent and smaller in aged bone. As the osteon density is higher in the aged tissue, the crack bridges will be smaller and closer together causing a lower resistance to crack growth. Adapted from Nalla et al. and Zimmermann et al.43,45
Microdamage in the form of diffuse damage or linear microcracks has also been found to accumulate with age, with implications for both bone's intrinsic and extrinsic mechanical resistance.53,54 The lower fibrillar deformation in aged bone could account for the higher levels of damage; if the fibril is constrained by cross-links, the deformation must still occur but at a different (generally higher) length-scale.45 Mechanistically, microdamage will decrease the stiffness and strength of materials. In terms of extrinsic toughness, microcracks have also been reported to result in increased fracture toughness.26 Indeed, microcracking (that is, damage formation) ahead of the crack tip allows bridge formation and toughens the material. Therefore, an increase in microdamage in aging has consequences for the intrinsic and extrinsic mechanical resistance of bone.
The fracture toughness of bone generally exhibits a steady deterioration with age.42,43,44 Specifically, for crack extensions on the order of millimeters, the crack-growth toughness of young bone is some five times higher compared with aged bone (Figure 5c).43,45 Indeed, aged bone samples have a higher osteonal density compared with young bone, which can be observed using three-dimensional synchrotron computed microtomography on fracture toughness samples (Figures 5d and e). The aging-related microstructural changes correlate to lower crack-growth toughness because the size of the uncracked ligament bridges scales with the osteonal spacing.43,45 In the development of bridges, microcracks primarily form at, or near, the highly mineralized cement lines; in turn, when the osteons are closer together, the size of the bridges will decrease along with the potency of the bridging toughening mechanism. The fact that the osteonal density scales inversely with the crack-growth toughness has a large role in the fragility of aged bone.43,45
Osteoporosis treatments
Osteoporosis is associated with increased fracture risk due to changes in bone quality and bone quantity. The literature on osteoporosis-related changes in bone quality lacks studies containing large cohorts of individuals with clinically diagnosed osteoporosis and age-matched controls. Therefore, the specific changes in bone quality that differentiate osteoporosis from aging or other factors are not well known and require further investigation. However, the other side of osteoporosis, that is the loss in bone quantity, has been extensively studied and results from a remodeling imbalance where more bone is resorbed than deposited. A common treatment for osteoporosis is a class of drugs called bisphosphonates, which increase bone quantity by inhibiting bone resorption and are associated with reduced fracture risk in osteoporotic patients. Although the vast majority of bisphosphonate-related research concerns trabecular bone, the effect of bisphosphonates on cortical bone is now of interest as well due to the site-specific effects of bisphosphonates and the advent of atypical femoral fractures.55,56,57
Recent studies have highlighted changes to the multi-length-scale cortical bone structure associated with treatment. Here, we focus on reporting the results from studies investigating human cortical bone, where osteoporosis was diagnosed by osteodensitometry and measurement of the areal bone mineral density with a T-score of <2.5 s.d.s below the reference population signifying osteoporosis as measured by DXA. At small length-scales, bisphosphonates have been shown to increase collagen maturity and lower mineral crystallinity in comparison with osteoporotic cortical tissue.58 In addition, studies on bisphosphonate-treated and osteoporotic cortical bone have revealed a lower degree of mineralization in femoral mid-cortical cross-sections in comparison with young tissue;57 however, other researchers found in cylindrical cortico-cancellous bone biopsies obtained during fracture repair that bisphosphonate treatment may lead to more homogeneity in the mineralization pattern of the bone structure.59 These deviances from young tissue and treatment-naive osteoporosis cases indicate that the quality of the cortical bone structure at small length-scales is altered during bisphosphonate treatment in comparison with the osteoporotic tissue.
At larger length-scales, the reduced cortical porosity, lower number of Haversian systems, larger osteon sizes and fewer mineralized osteocyte lacunae in bisphosphonate-treated cases in comparison with osteoporosis cases may indicate trends toward counterbalancing remodeling defects with bisphosphonate treatment.57,60,61,62 The larger osteon sizes and lower amount of Haversian systems, in particular, may have implications for crack propagation and the generation of extrinsic toughness in bone.63 In the future, studies assessing the mechanical implications of bisphosphonate treatment in human cortical bone are needed, as the atypical fractures associated with long-term use have been hypothesized to be related to the accumulation of fatigue damage.55 Indeed, a recent study by Bajaj et al.64 with a canine model directly relates bisphosphonates to a deterioration in fatigue resistance.
Summary
We can conclude that, in addition to a loss in bone mass, the increase in fracture risk with aging can be related to a deterioration in the bone structure over multiple length-scales. The combined effects of a higher relative amount of non-enzymatic cross-links at the fibril level and a higher osteon density at the microstructural level reduce the mechanical integrity of aged bone: the cross-links constrain fibrillar plasticity, and the higher osteon density reduces extrinsic toughness during crack extension.45 Other aspects of the structure, such as the content of non-collagenous proteins, need to be further investigated to understand their changes with aging and osteoporosis, as well as their contribution to bone's mechanical properties.5,6
The treatment of osteoporosis with bisphosphonates (for example, zoledronic acid, alendronate, risedronate and ibandronate) may serve as an effective treatment strategy to reduce vertebral and non-vertebral fractures in post-menopausal osteoporosis.65 However, the specific effect of bisphosphonates on cortical bone's mechanical quality is still a topic of debate. Here, bisphosphonates' ability to suppress bone turnover puts the tissue at risk of developing potentially mechanistically unfavorable bone structure characteristics (for example, premature aging of the bone tissue, accumulation of damage and homogenization of mineralization63). However, experimental studies characterizing the structure of bisphosphonate-treated human cortical bone have found improvements underpinning the general effectiveness of anti-resorptive treatments.57,60 A major limitation of the studies in the literature addressing cortical bone quality in aging and osteoporosis though is the low sample sizes. Future studies characterizing the structure and mechanical properties of clinically diagnosed osteoporosis tissue using age-matched controls are still required to understand the effects of osteoporosis and bisphosphonates on the structure and mechanical properties of cortical bone.
Vitamin D deficiency
Vitamin D has an essential role in bone mineral homeostasis, where it aids the absorption of calcium and phosphate. It is synthesized in the skin as a result of sun exposure but can also be absorbed through food or vitamin supplements. Vitamin D deficiency has become a concern in certain countries, as a severe lack of vitamin D can lead to a clinical diagnosis of rickets or osteomalacia resulting in bone pain and a higher risk of fracture.66,67 Clinically, the disease is assessed through low serum 25(OH)D3 concentrations and/or high levels of unmineralized bone mass assessed through laboratory tests or a bone biopsy, respectively. Here, we briefly review the effects of vitamin D deficiency on the structure and mechanical properties of human cortical bone identified through the use of high spatial resolution techniques.
The typical characteristic of vitamin D-deficient bone is a higher amount of unmineralized bone matrix, called osteoid (Figures 6a and b). However, the quality of the remaining mineralized bone mass, which is mechanically relevant, is also of interest. In the mineralized bone of vitamin D-deficient individuals, a higher degree of mineralization, cross-link ratio (that is, mature to immature enzymatic cross-links) and carbonate-to-phosphate ratio were measured, all of which indicate an apparently higher tissue age in the vitamin D-deficient samples (Figure 6c–f).68 As a significantly greater amount of bone surfaces is covered with osteoid in vitamin D-deficient individuals, the age of the mineralized bone tissue increases because the osteoid-covered bone surfaces prevent tissue resorption.69
Figure 6.

Characteristics of vitamin D-deficient bone at small length-scales. In von Kossa stained histological sections of (a) healthy control (Nor) and (b) vitamin D-deficient human bone (D-), the vitamin D-deficient bone has a greater amount of osteoid (red) covering the mineralized tissue (black), scale bar equals 600 μm. The mineralized tissue trapped within the osteoid frame has a higher mineralization, as measured through 3-D synchrotron X-ray computed microtomography. (c) The histogram shows the distribution of mineralization values in the 3-D volume for each sample. (d) The mean calcium weight % in the histogram is significantly higher in the vitamin D-deficient samples. Fourier transform infrared spectroscopy measurements of the (e) cross-link ratio and the (f) carbonate-to-phosphate ratio were higher in the vitamin D-deficient tissue, which indicates a greater tissue age. Adapted from Busse et al.68
Mechanistically, the changes in bone quality associated with vitamin D deficiency have a measurable effect on the toughness (Figure 7a). In our study, the vitamin D-deficient tissue had a 22% decrease in initiation toughness in comparison with the healthy bone, which can be linked to the increase in tissue age of the bone matrix. The vitamin D-deficient tissue also exhibited a 31% decrease in growth toughness in comparison with healthy tissue.68 This reduction in extrinsic toughness in the vitamin D-deficient tissue was associated with a straighter crack path with fewer crack bridges (Figures 7b and c).
Figure 7.

Toughness of bone with vitamin D deficiency. (a) Fracture toughness measurements in the form of crack-growth resistance curves were measured in a set of healthy/control (Nor) and vitamin D-deficient (D-) human bone biopsies. The toughness of the vitamin D-deficient samples was lower compared with controls; specifically, a 22% decrease in the crack-initiation toughness and a 31% decrease in the crack-growth toughness were measured. The samples used for toughness testing also exhibited different trends in crack extension observable with 3-D synchrotron computed microtomography. (b) The control samples had a deflected crack path consistent with the behavior of healthy tissue, whereas (c) the vitamin D-deficient samples had a significantly straighter crack path with a 30% smaller deflection angle than the control samples. These changes accounted for the decrease in the fracture toughness in the vitamin D-deficient bone. Adapted from Busse et al.68
Summary
Vitamin D deficiency causes an imbalance in bone calcium homeostasis, which creates a scenario where mineralized tissue is trapped within an osteoid frame. The trapped tissue cannot be resorbed and takes on the characteristics of aged tissue with a decrease in bone quality. Therefore, the unmineralized osteoid traps the mineralized tissue, leading to premature aging and a decreased fracture resistance, which in turn can reduce toughness, in part by inhibiting deflected crack paths.68 Vitamin D deficiency can clearly have a significant impact on the mechanical integrity and toughness of human cortical bone.
Paget's disease of bone (PDB)
PDB is the second most common bone disease after osteoporosis, generally occurring in individuals over the age of 50 years.70,71 Paget's disease affects localized skeletal sites, most often the pelvis, spine, femur or tibia.70,71 The disease is most often diagnosed through X-rays, by the bone's strikingly high increase in bone volume due to high levels of bone turnover.71 Individuals with Paget's disease commonly suffer from bone enlargement, bowing/deformity and pain. Incomplete fractures, termed pseudo or fissure fractures, can be found at sites with severe bowing and deformity.72 Thus, Paget's disease clearly compromises the mechanical integrity of bone tissue. However, despite the bowing and cracking, the overall fracture risk to patients is low, and fracture events at pathological skeletal sites are uncommon (occurring in ∼2% of patients).73,74
For Paget's disease to produce the marked changes in bone's mechanical integrity (that is, bowing and deformities) at the macroscale, the disease must be changing the bone structure and in turn the mechanical mechanisms through which the bone resists deformation and fracture. Indeed, the overall mineral content is significantly lower in Paget's disease, which has also a greater heterogeneity of mineralized bone packets (Figure 8a–c). These compositional changes in the tissue are related to bone's intrinsic resistance, which governs the overall stiffness and hardness. The diseased tissue has a significantly lower stiffness and hardness, that is, more plasticity (Figures 8d and e), as measured through nanoindentation. Thus, the compositional changes in the diseased tissue resulting in a lower mineralization affect both the elastic (that is, stretching of bonds generating stiffness) and the plastic (that is, permanent deformation promoting ductility and energy absorption) mechanical properties, resulting in a lower stiffness and more plasticity.37
Figure 8.

Characteristics of Paget's disease at small length-scales. Paget's disease of bone affected the composition of the bone structure at small length-scales. The mineral distribution in (a) control and (b) Paget's disease samples was measured with quantitative back-scattered electron imaging. (c) The Paget's disease of bone samples had a lower average Ca-Wt% value and a higher degree of low mineralized bone, as seen from the histogram, which shows the distribution of Ca-Wt% values in the images. These changes in composition at the nanoscale have a direct result on the mechanical properties. Nanoindentation was used to measure the (d) Young's modulus and (e) hardness of the control and diseased cases. Here, the disease samples have a lower stiffness, due to the lower mineralization, as well as a lower hardness, which measures the resistance to plastic deformation. Adapted from Zimmermann et al.37
Paget's disease also affects the higher level structural organization in bone. In cortical regions, instead of the structural patterns characteristic of healthy tissue (that is, parallel aligned Haversian canals), the pathological tissue is a patchwork of lamellar and woven bone, with less organized collagen fiber orientation.75 Interestingly, fracture toughness measurements on healthy and diseased tissue from the iliac crest show little difference in toughness (Figure 9a); however, the mechanism for resisting crack propagation changes.37 Despite the limited number of toughness samples restricting statistical comparisons, there was still clear evidence of higher fracture toughness in the transversely oriented control and PDB bone samples, in comparison with the longitudinally oriented bone, which has a comparatively weak resistance to crack growth.27 Indeed, the healthy (control) bone resists crack propagation and derives toughness in the usual way through crack deflection mechanisms (Figure 9b).27 Conversely, human iliac crest bone from patients with Paget's disease exhibits a straighter crack path (Figure 9c). In these diseased cases, the straighter crack path arises due to the disorganized microstructure, which cannot effectively deflect cracks. Although the ability for the diseased tissue to generate extrinsic toughening is severely degraded, the bone structure compensates through a more pronounced mechanical resistance through plastic deformation, that is, Paget's diseased bone shows higher ductility (intrinsic toughness).37 Indeed, the Paget's tissue has a lower mineralization and a correspondingly lower hardness, which both indicate that the tissue has the capacity for higher levels of plasticity similar to other tissues with low mineralization levels and significant plastic deformation.76,77 Thus, although toughness is generated through crack deflection in healthy bone (extrinsic toughening), the diseased tissue may generate toughness primarily through intrinsic plasticity mechanisms that absorb energy during deformation and fracture. However, further studies on the fracture toughness in bowed/deformed long bones would add further detail on the mechanisms affecting the resistance to fracture in PDB.
Figure 9.

Toughness of bone with Paget's disease. (a) The toughness of control and Paget's disease of bone samples was measured as a function of crack extension. Despite the marked changes in the bone structure at small and large length-scales in Paget's disease of bone, the diseased tissue was not significantly different from healthy/control samples. 3-D synchrotron X-ray computed microtomography was used to investigate the crack path (yellow) through the Haversian canal structure (blue) after toughness testing. (b) The control samples exhibited crack deflection, which is known to extrinsically increase the toughness in bone. (c) However, the diseased bone displayed much straighter crack paths. As the diseased bone did not exhibit much crack deflection, we believe that the comparable toughness in the Paget's diseased bone is derived from the increase in plasticity and possibly due to its lower mineralization, which is consistent with the occurrence of stable fractures found in clinical cases. Adapted from Zimmermann et al.37
Treatment of PDB
Paget's disease is primarily treated with anti-resorptive agents, such as bisphosphonates. Anti-resorptive treatments act by suppressing bone remodeling. In individuals treated for PDB, anti-resorptive agents have been shown to normalize the serum alkaline phosphatase levels, which is a marker of bone formation.78 In addition, in many cases significant improvements in osteolytic lesions are visible radiologically, and histomorphometric values return to normal with the formation of lamellar bone.79
Summary
Characterizing the multi-scale structure and mechanical properties in Paget's disease provides a basis for explaining some of the clinical symptoms and the overall mechanical integrity of the diseased bone tissue. In particular, the lower mineral content of the diseased tissue with a corresponding lower stiffness and lower resistance to deformation could directly account for the occurrence of harmful bowing and deformities in affected skeletal sites. In addition, the presence of subcritical (that is, stable) cracks, so called fissure fractures, in Paget's disease most likely occur due to the extreme deformations associated with the deformities/bowing in long bones. However, the fact that these fractures remain in the tissue and do not cause complete bone failure is in line with the same propensity for the altered structure to resist crack growth through plastic deformation. Indeed, the structural changes associated with Paget's disease provide an interesting view into the interplay and connections between intrinsic toughening mechanisms at small length-scales, which are primarily plasticity related, and extrinsic toughening at larger length-scales, which primarily shield the growing crack.
Concluding remarks
Fracture-mechanics-based toughness measurements provide a valuable framework for assessing the mechanical integrity of human cortical bone. The bone is able to generate substantial strength and toughness through the hierarchical assembly of two components with very different mechanical properties: ductile collagen molecules and brittle nanoplatelets of hydroxyapatite mineral. Here, deformation mechanisms at the smallest length-scales generate bone's intrinsic resistance (that is, strength and crack-initiation toughness) mainly through plasticity provided by sliding mechanisms at the fibrillar level. At larger length-scales, the bone structure generates extrinsic toughness (that is, crack-growth toughness) through crack-tip shielding mechanisms, such as crack deflection, principally at cement lines, and uncracked ligament bridging. The intrinsic and extrinsic toughening mechanisms work in concert to resist the initiation and propagation of cracks in healthy cortical tissue.
Assessing fracture risk and preventing fractures are critical issues in bone health. Many factors affect the fracture toughness of human cortical bone, which can become more prone to fracture due to aging, disease or other environmental conditions (for example, vitamin deficiencies or irradiation). The fracture risk or mechanical integrity of bone changes because aging, disease or other factors affect the bone structure at multiple length-scales, which in turn affects the potency of the intrinsic and extrinsic toughening mechanisms. We have reviewed here recent studies to investigate the effects of aging, vitamin D deficiency and PDB on the fracture toughness of human cortical bone. In many cases, changes to the mineralization or cross-linking profile at small length-scales directly affect the level of plasticity (that is, intrinsic toughness), whereas changes to the distribution or character of the osteons at the microstructural scale affect the bone's ability to resist the growth of cracks (that is, extrinsic toughness). Studying these disease conditions aids further progress in the diagnosis, prevention and treatment of diseases that increase the fracture risk or affect the mechanical integrity of human cortical bone.
Acknowledgments
We thank numerous individuals who have provided research or input to this review, including Drs Claire Acevedo, Joel Ager, Tamara Alliston, Michael Amling, Hrishikesh Bale, Holly Barth, Neil Dave, Bernd Gludovatz, Michael Hahn, Sophi Ionova-Martin, Till Köhne, Kurt Koester, Jay Kruzic, Nancy Lane, Max Launey, Ravi Nalla, Diana Olvera, Brian Panganiban, Klaus Püschel, Eric Schaible, Simon Tang, Tony Tomsia, Wei Yao and Jozef Zustin. Several interactions/collaborations with Drs Markus Buehler, David Burr, Paul Hansma and Deepak Vashishth are also gratefully acknowledged. Elizabeth A. Zimmermann is supported by the Alexander von Humboldt Foundation, Robert O. Ritchie by the Air Force Office of Scientific Research, Multi-University Research Initiative grant AFOSR-FA9550-15-1-0009, and Björn Busse by the Emmy Noether program under grant no. BU 2562-2/1 from the German Research Foundation (DFG).
Footnotes
The authors declare no conflict of interest.
References
- Weiner S, Wagner HD. The material bone: structure mechanical function relations. Annu Rev Mater Sci 1998; 28: 271–298. [Google Scholar]
- Arsenault A. Image-analysis of collagen-associated mineral distribution in cryogenically prepared turkey leg tendons. Calcif Tissue Int 1991; 48: 56–62. [DOI] [PubMed] [Google Scholar]
- McNally EA, Schwarcz HP, Botton GA, Arsenault AL. A model for the ultrastructure of bone based on electron microscopy of ion-milled sections. PLoS One 2012; 7: e29258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robins SP. Biochemistry and functional significance of collagen cross-linking. Biochem Soc Trans 2007; 35: 849–852. [DOI] [PubMed] [Google Scholar]
- Thurner PJ, Katsamenis OL. The role of nanoscale toughening mechanisms in osteoporosis. Curr Osteoporos Rep 2014; 12: 351–356. [DOI] [PubMed] [Google Scholar]
- Grynpas MD, Tupy JH, Sodek J. The distribution of soluble, mineral-bound, and matrix-bound proteins in osteoporotic and normal bones. Bone 1994; 15: 505–513. [DOI] [PubMed] [Google Scholar]
- Bailey AJ. Molecular mechanisms of ageing in connective tissues. Mech Ageing Dev 2001; 122: 735–755. [DOI] [PubMed] [Google Scholar]
- Knott L, Bailey AJ. Collagen cross-links in mineralizing tissues: a review of their chemistry, function, and clinical relevance. Bone 1998; 22: 181–187. [DOI] [PubMed] [Google Scholar]
- Avery NC, Bailey AJ. Enzymic and non-enzymic cross-linking mechanisms in relation to turnover of collagen: relevance to aging and exercise. Scand J Med Sci Sports 2005; 15: 231–240. [DOI] [PubMed] [Google Scholar]
- Skedros JG, Holmes JL, Vajda EG, Bloebaum RD. Cement lines of secondary osteons in human bone are not mineral-deficient: new data in a historical perspective. Anat Rec 2005; 286A: 781–803. [DOI] [PubMed] [Google Scholar]
- Ritchie RO. The conflicts between strength and toughness. Nat Mater 2011; 10: 817–822. [DOI] [PubMed] [Google Scholar]
- Zimmermann EA, Barth HD, Ritchie RO. The multiscale origins of fracture resistance in human bone and its biological degradation. JOM 2012; 64: 486–493. [Google Scholar]
- Gupta HS, Wagermaier W, Zickler GA, Raz-Ben Aroush D, Funari SS, Roschger P et al. Nanoscale deformation mechanisms in bone. Nano Lett 2005; 5: 2108–2111. [DOI] [PubMed] [Google Scholar]
- Nair AK, Gautieri A, Chang S-W, Buehler MJ. Molecular mechanics of mineralized collagen fibrils in bone. Nat Commun 2013; 4: 1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fantner GE, Hassenkam T, Kindt JH, Weaver JC, Birkedal H, Pechenik L et al. Sacrificial bonds and hidden length dissipate energy as mineralized fibrils separate during bone fracture. Nat Mater 2005; 4: 612–616. [DOI] [PubMed] [Google Scholar]
- Poundarik AA, Diab T, Sroga GE, Ural A, Boskey AL, Gundberg CM et al. Dilatational band formation in bone. Proc Natl Acad Sci USA 2012; 109: 19178–19183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurner PJ, Chen CG, Ionova-Martin S, Sun L, Harman A, Porter A et al. Osteopontin deficiency increases bone fragility but preserves bone mass. Bone 2010; 46: 1564–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta HS, Krauss S, Kerschnitzki M, Karunaratne A, Dunlop JWC, Barber AH et al. Intrafibrillar plasticity through mineral/collagen sliding is the dominant mechanism for the extreme toughness of antler bone. J Mech Behav Biomed Mater 2013; 28: 366–382. [DOI] [PubMed] [Google Scholar]
- Karunaratne A, Esapa CR, Hiller J, Boyde A, Head R, Bassett JD et al. Significant deterioration in nanomechanical quality occurs through incomplete extrafibrillar mineralization in rachitic bone: evidence from in-situ synchrotron X-ray scattering and backscattered electron imaging. J Bone Miner Res 2012; 27: 876–890. [DOI] [PubMed] [Google Scholar]
- Zimmermann EA, Gludovatz B, Schaible E, Busse B, Ritchie RO. Fracture resistance of human cortical bone across multiple length-scales at physiological strain rates. Biomaterials 2014; 35: 5472–5481. [DOI] [PubMed] [Google Scholar]
- Gautieri A, Vesentini S, Redaelli A, Buehler MJ. Viscoelastic properties of model segments of collagen molecules. Matrix Biol 2012; 31: 141–149. [DOI] [PubMed] [Google Scholar]
- Herman BC, Cardoso L, Majeska RJ, Jepsen KJ, Schaffler M. Activation of bone remodeling after fatigue: Differential response to linear microcracks and diffuse damage. Bone 2010; 47: 766–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dooley C, Tisbo P, Lee TC, Taylor D. Rupture of osteocyte processes across microcracks: the effect of crack length and stress. Biomech Model Mechanobiol 2012; 11: 759–766. [DOI] [PubMed] [Google Scholar]
- Seref-Ferlengez Z, Basta-Pljakic J, Kennedy OD, Philemon CJ, Schaffler MB. Structural and mechanical repair of diffuse damage in cortical bone in vivo. J Bone Miner Res 2014; 29: 2537–2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie RO. Mechanisms of fatigue-crack propagation in metals, ceramics and composites: role of crack tip shielding. Mater Sci Eng A 1988; 103: 15–28. [Google Scholar]
- Nalla RK, Kruzic JJ, Ritchie RO. On the origin of the toughness of mineralized tissue: microcracking or crack bridging? Bone 2004; 34: 790–798. [DOI] [PubMed] [Google Scholar]
- Koester KJ, Ager JW, Ritchie RO. The true toughness of human cortical bone measured with realistically short cracks. Nat Mater 2008; 7: 672–677. [DOI] [PubMed] [Google Scholar]
- Katsamenis OL, Jenkins T, Thurner PJ. Toughness and damage susceptibility in human cortical bone is proportional to mechanical inhomogeneity at the osteonal-level. Bone 2015; 76: 158–168. [DOI] [PubMed] [Google Scholar]
- Currey JD. The effect of porosity and mineral content on the Young's modulus of elasticity of compact bone. J Biomech 1988; 21: 131–139. [DOI] [PubMed] [Google Scholar]
- Schaffler MB, Burr DB. Stiffness of compact bone: effects of porosity and density. J Biomech 1988; 21: 13–16. [DOI] [PubMed] [Google Scholar]
- Hui S, Slemenda C, Johnston C. Age and bone mass as predictors of fracture in a prospective-study. J Clin Invest 1988; 81: 1804–1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aspray TJ, Prentice A, Cole TJ, Sawo Y, Reeve J, Francis RM. Low bone mineral content is common but osteoporotic fractures are rare in elderly rural Gambian women. J Bone Miner Res 1996; 11: 1019–1025. [DOI] [PubMed] [Google Scholar]
- Xiaoge D, Eryuan L, Xianping W, Zhiguang Z, Gan H, Zaijing J et al. Bone mineral density differences at the femoral neck and Ward's triangle: a comparison study on the reference data between Chinese and Caucasian women. Calcif Tissue Int 2000; 67: 195–198. [DOI] [PubMed] [Google Scholar]
- Kanis JA, Johnell O, Oden A, Dawson A, De Laet C, Jonsson B. Ten year probabilities of osteoporotic fractures according to BMD and diagnostic thresholds. Osteoporo Int 2001; 12: 989–995. [DOI] [PubMed] [Google Scholar]
- Yeni YN, Brown CU, Norman TL. Influence of bone composition and apparent density on fracture toughness of the human femur and tibia. Bone 1998; 22: 79–84. [DOI] [PubMed] [Google Scholar]
- Nyman JS, Roy A, Tyler JH, Acuna RL, Gayle HJ, Wang X. Age-related factors affecting the postyield energy dissipation of human cortical bone. J Orthop Res 2007; 25: 646–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann EA, Köhne T, Bale HA, Panganiban B, Gludovatz B, Zustin J et al. Modifications to nano- and microstructural quality and the effects on mechanical integrity in Paget's disease of bone. J Bone Miner Res 2015; 30: 264–273. [DOI] [PubMed] [Google Scholar]
- Kulin RM, Jiang F, Vecchio KS. Loading rate effects on the R-curve behavior of cortical bone. Acta Biomater 2011; 7: 724–732. [DOI] [PubMed] [Google Scholar]
- Zimmermann EA, Launey ME, Barth HD, Ritchie RO. Mixed-mode fracture of human cortical bone. Biomaterials 2009; 30: 5877–5884. [DOI] [PubMed] [Google Scholar]
- Heaney RP, Abrams S, Dawson-Hughes B, Looker A, Marcus R, Matkovic V et al. Peak bone mass. Osteoporosis Int 2000; 11: 985–1009. [DOI] [PubMed] [Google Scholar]
- Koehne T, Vettorazzi E, Küsters N, Lüneburg R, Kahl-Nieke B, Püschel K et al. Trends in trabecular architecture and bone mineral density distribution in 152 individuals aged 30–90 years. Bone 2014; 66: 31–38. [DOI] [PubMed] [Google Scholar]
- Zioupos P, Currey JD. Changes in the stiffness, strength, and toughness of human cortical bone with age. Bone 1998; 22: 57–66. [DOI] [PubMed] [Google Scholar]
- Nalla RK, Kruzic JJ, Kinney JH, Balooch M, Ager JW, Ritchie RO. Role of microstructure in the aging-related deterioration of the toughness of human cortical bone. Mater Sci Eng C 2006; 26: 1251–1260. [Google Scholar]
- Wang X, Shen X, Li X, Mauli Agrawal C. Age-related changes in the collagen network and toughness of bone. Bone 2002; 31: 1–7. [DOI] [PubMed] [Google Scholar]
- Zimmermann EA, Schaible E, Bale H, Barth HD, Tang SY, Reichert P et al. Age-related changes in the plasticity and toughness of human cortical bone at multiple length scales. Proc Natl Acad Sci USA 2011; 108: 14416–14421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granke M, Makowski AJ, Uppuganti S, Does MD, Nyman JS. Identifying novel clinical surrogates to assess human bone fracture toughness. J Bone Miner Res 2015; 30: 1290–1300. doi: 10.1002/jbmr.2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odetti P, Rossi S, Monacelli F, Poggi A, Cirnigliaro M, Federici M et al. Advanced glycation end products and bone loss during aging. In: Baynes JW, Monnier VM, Ames JM, Thorpe SR (eds) Maillard Reaction: Chemistry at the Interface of Nutrition, Aging, and Disease New York Academy of Sciences: NY, USA, 2005, pp 710–717. [DOI] [PubMed] [Google Scholar]
- Saito M, Marumo K, Fujii K, Ishioka N. Single-column high-performance liquid chromatographic fluorescence detection of immature, mature, and senescent cross-links of collagen. Anal Biochem 1997; 253: 26–32. [DOI] [PubMed] [Google Scholar]
- Sell D, Monnier V. Structure elucidation of a senescence cross-link from human extracellular-matrix - Implication of pentoses in the aging process. J Biol Chem 1989; 264: 21597–21602. [PubMed] [Google Scholar]
- Vashishth D, Gibson GJ, Khoury JI, Schaffler MB, Kimura J, Fyhrie DP. Influence of nonenzymatic glycation on biomechanical properties of cortical bone. Bone 2001; 28: 195–201. [DOI] [PubMed] [Google Scholar]
- Siegmund T, Allen MR, Burr DB. Failure of mineralized collagen fibrils: Modeling the role of collagen cross-linking. J Biomech 2008; 41: 1427–1435. [DOI] [PubMed] [Google Scholar]
- Buehler MJ. Nanomechanics of collagen fibrils under varying cross-link densities: atomistic and continuum studies. J Mech Behav Biomed Mater 2008; 1: 59–67. [DOI] [PubMed] [Google Scholar]
- Schaffler MB, Choi K, Milgrom C. Aging and matrix microdamage accumulation in human compact bone. Bone 1995; 17: 521–525. [DOI] [PubMed] [Google Scholar]
- Diab T, Vashishth D. Morphology, localization and accumulation of in vivo microdamage in human cortical bone. Bone 2007; 40: 612–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shane E, Burr D, Ebeling PR, Abrahamsen B, Adler RA, Brown TD et al. Atypical subtrochanteric and diaphyseal femoral fractures: report of a task force of the American Society for Bone and Mineral Research. J Bone Miner Res 2010; 25: 2267–2294. [DOI] [PubMed] [Google Scholar]
- Allen MR, Kubek DJ, Burr DB. Cancer treatment dosing regimens of zoledronic acid result in near-complete suppression of mandible intracortical bone remodeling in beagle dogs. J Bone Miner Res 2010; 25: 98–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milovanovic P, Zimmermann EA, Riedel C, vom Scheidt A, Herzog L, Krause M et al. Multi-level characterization of human femoral cortices and their underlying osteocyte network reveal trends in quality of young, aged, osteoporotic, and antiresorptive-treated bone. Biomaterials 2015; 45: 46–55. [DOI] [PubMed] [Google Scholar]
- Bala Y, Depalle B, Farlay D, Douillard T, Meille S, Follet H et al. Bone micromechanical properties are compromised during long-term alendronate therapy independently of mineralization. J Bone Miner Res 2012; 27: 825–834. [DOI] [PubMed] [Google Scholar]
- Donnelly E, Meredith DS, Nguyen JT, Gladnick BP, Rebolledo BJ, Shaffer AD et al. Reduced cortical bone compositional heterogeneity with bisphosphonate treatment in postmenopausal women with intertrochanteric and subtrochanteric fractures. J Bone Miner Res 2012; 27: 672–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernhard A, Milovanovic P, Zimmermann EA, Hahn M, Djonic D, Krause M et al. Micro-morphological properties of osteons reveal changes in cortical bone stability during aging, osteoporosis, and bisphosphonate treatment in women. Osteoporos Int 2013; 24: 2671–2680. [DOI] [PubMed] [Google Scholar]
- Roschger P, Rinnerthaler S, Yates J, Rodan GA, Fratzl P, Klaushofer K. Alendronate increases degree and uniformity of mineralization in cancellous bone and decreases the porosity in cortical bone of osteoporotic women. Bone 2001; 29: 185–191. [DOI] [PubMed] [Google Scholar]
- Borah B, Dufresne T, Nurre J, Phipps R, Chmielewski P, Wagner L et al. Risedronate reduces intracortical porosity in women with osteoporosis. J Bone Miner Res 2010; 25: 41–47. [DOI] [PubMed] [Google Scholar]
- Ettinger B, Burr DB, Ritchie RO. Proposed pathogenesis for atypical femoral fractures: lessons from materials research. Bone 2013; 55: 495–500. [DOI] [PubMed] [Google Scholar]
- Bajaj D, Geissler JR, Allen MR, Burr DB, Fritton JC. The resistance of cortical bone tissue to failure under cyclic loading is reduced with alendronate. Bone 2014; 64: 57–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pazianas M, Epstein S, Zaidi M. Evaluating the antifracture efficacy of bisphosphonates. Rev Recent Clin Trials 2009; 4: 122–130. [DOI] [PubMed] [Google Scholar]
- Lips P. Vitamin D deficiency and secondary hyperparathyroidism in the elderly: consequences for bone loss and fractures and therapeutic implications. Endocr Rev 2001; 22: 477–501. [DOI] [PubMed] [Google Scholar]
- Whyte MP, Thakker RV. Rickets and osteomalacia. Medicine (Baltimore) 2005; 33: 70–74. [Google Scholar]
- Busse B, Bale HA, Zimmermann EA, Panganiban B, Barth HD, Carriero A et al. Vitamin D deficiency induces early signs of aging in human bone, increasing the risk of fracture. Sci Transl Med 2013; 5: 193ra88. [DOI] [PubMed] [Google Scholar]
- Teitelbaum SL. Renal osteodystrophy. Hum Pathol 1984; 15: 306–323. [DOI] [PubMed] [Google Scholar]
- Davie M, Davies M, Francis R, Fraser W, Hosking D, Tansley R. Paget's disease of bone: a review of 889 patients. Bone 1999; 24: 11S–12S. [DOI] [PubMed] [Google Scholar]
- Seitz S, Priemel M, Zustin J, Beil FT, Semler J, Minne H et al. Paget's disease of bone: histologic analysis of 754 patients. J Bone Miner Res 2009; 24: 62–69. [DOI] [PubMed] [Google Scholar]
- Cushing FR, Bone HG. Radiographic diagnosis and laboratory evaluation of Paget's disease of bone. Clin Rev Bone Miner Metab 2002; 1: 115–134. [Google Scholar]
- Melton LJ, Tiegs RD, Atkinson EJ, O'Fallon WM. Fracture risk among patients with Paget's disease: a population-based cohort study. J Bone Miner Res 2000; 15: 2123–2128. [DOI] [PubMed] [Google Scholar]
- Van Staa TP, Selby P, Leufkens HGM, Lyles K, Sprafka JM, Cooper C. Incidence and natural history of Paget's disease of bone in England and Wales. J Bone Miner Res 2002; 17: 465–471. [DOI] [PubMed] [Google Scholar]
- Meunier PJ, Coindre JM, Edouard CM, Arlot ME. Bone histomorphometry in Paget's disease. Quantitative and dynamic analysis of pagetic and nonpagetic bone tissue. Arthritis Rheum 1980; 23: 1095–1103. [DOI] [PubMed] [Google Scholar]
- Launey ME, Chen P-Y, McKittrick J, Ritchie RO. Mechanistic aspects of the fracture toughness of elk antler bone. Acta Biomater 2010; 6: 1505–1514. [DOI] [PubMed] [Google Scholar]
- Krauss S, Fratzl P, Seto J, Currey JD, Estevez JA, Funari SS et al. Inhomogeneous fibril stretching in antler starts after macroscopic yielding: indication for a nanoscale toughening mechanism. Bone 2009; 44: 1105–1110. [DOI] [PubMed] [Google Scholar]
- Miller PD, Brown JP, Siris ES, Hoseyni MS, Axelrod DW, Bekker PJ. A randomized, double-blind comparison of risedronate and etidronate in the treatment of Paget's disease of bone. Am J Med 1999; 106: 513–520. [DOI] [PubMed] [Google Scholar]
- Reid IR, Nicholson GC, Weinstein RS, Hosking DJ, Cundy T, Kotowicz MA et al. Biochemical and radiologic improvement in Paget's disease of bone treated with alendronate: A randomized, placebo-controlled trial. Am J Med 1996; 101: 341–348. [DOI] [PubMed] [Google Scholar]
- Ascenzi M-G, Ascenzi A, Benvenuti A, Burghammer M, Panzavolta S, Bigi A. Structural differences between ‘dark' and ‘bright' isolated human osteonic lamellae. J Struct Biol 2003; 141: 22–33. [DOI] [PubMed] [Google Scholar]
- Reznikov N, Shahar R, Weiner S. Three-dimensional structure of human lamellar bone: the presence of two different materials and new insights into the hierarchical organization. Bone 2014; 59: 93–104. [DOI] [PubMed] [Google Scholar]
- Milovanovic P, Zimmermann EA, Hahn M, Djonic D, Püschel K, Djuric M et al. Osteocytic canalicular networks: morphological implications for altered mechanosensitivity. ACS Nano 2013; 7: 7542–7551. [DOI] [PubMed] [Google Scholar]
- You L-D, Weinbaum S, Cowin SC, Schaffler MB. Ultrastructure of the osteocyte process and its pericellular matrix. Anat Rec 2004; 278: 505–513. [DOI] [PubMed] [Google Scholar]
- Knott L, Whitehead CC, Fleming RH, Bailey AJ. Biochemical changes in the collagenous matrix of osteoporotic avian bone. Biochem J 1995; 310: 1045–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Launey ME, Buehler MJ, Ritchie RO. On the mechanistic origins of toughness in bone. Annu Rev Mater Res 2010; 40: 25–53. [Google Scholar]
- Koester KJ, Barth HD, Ritchie RO. Effect of aging on the transverse toughness of human cortical bone: evaluation by R-curves. J Mech Behav Biomed Mater 2011; 4: 1504–1513. [DOI] [PubMed] [Google Scholar]