

Abstract
Background and Purpose
Pulmonary vascular dysfunction is a key event in acute lung injury. We recently demonstrated that PGE2, via activation of E-prostanoid (EP)4 receptors, strongly enhances microvascular barrier function in vitro. The aim of this study was to investigate the beneficial effects of concomitant EP4 receptor activation in murine models of acute pulmonary inflammation.
Experimental Approach
Pulmonary inflammation in male BALB/c mice was induced by LPS (20 μg per mouse intranasally) or oleic acid (0.15 μL·g-1, i.v.). In-vitro, endothelial barrier function was determined by measuring electrical impedance.
Key Results
PGE2 activation of EP4 receptors reduced neutrophil infiltration, pulmonary vascular leakage and TNF-α concentration in bronchoalveolar lavage fluid from LPS-induced pulmonary inflammation. Similarly, pulmonary vascular hyperpermeability induced by oleic acid was counteracted by EP4 receptor activation. In lung function assays, the EP4 agonist ONO AE1-329 restored the increased resistance and reduced compliance upon methacholine challenge in mice treated with LPS or oleic acid. In agreement with these findings, EP4 receptor activation increased the in vitro vascular barrier function of human and mouse pulmonary microvascular endothelial cells and diminished the barrier disruption induced by LPS. The EP2 agonist ONO AE1-259 likewise reversed LPS-induced lung dysfunction without enhancing vascular barrier function.
Conclusion and Implications
Our results show that activation of the EP4 receptor strengthens the microvascular barrier function and thereby ameliorates the pathology of acute lung inflammation, including neutrophil infiltration, vascular oedema formation and airway dysfunction. This suggests a potential benefit for EP4 agonists in acute pulmonary inflammation.
Tables of Links
| TARGETS | |
|---|---|
| GPCRs | |
| EP2 receptor | |
| EP4 receptor |
| LIGANDS | |
|---|---|
| GW627368X | ONO AE1-259 |
| IL-6 | ONO AE1-329 |
| LPS | PF-04418948 |
| Misoprostol | PGE2 |
| Oleic acid | TNF-α |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013).
Introduction
Pulmonary vascular dysfunction is a key component of acute lung injury (ALI) and its more severe form, acute respiratory distress syndrome (Ware, 2006; Maniatis et al., 2008). Mortality rates of ALI patients are estimated to be 24–41% (Rubenfeld et al., 2005; Rubenfeld and Herridge, 2007). Notably, increased pulmonary microvascular permeability is associated with adverse outcome and even higher mortality rates (Bull et al., 2010). Consequently, substances promoting or protecting microvascular barrier function would seem to be promising candidates for the therapy of ALI and sepsis (Jacobson and Garcia, 2007; Birukov et al., 2013). The major COX product PGE2 has been shown to have protective effects in pulmonary inflammatory diseases (Pavord et al., 1993; Vancheri et al., 2004; Aggarwal et al., 2010). PGE2 acts via four E-type prostanoid receptors (designated as EP1–4), which show cell type-specific expression and their activation stimulates different signalling pathways leading to diverse cellular outcomes. Specifically in the lung, PGE2 reduces bronchoconstriction and blocks proinflammatory cytokine release from alveolar macrophages via EP4 receptors and prevents lung fibrosis via EP2 receptors (Konya et al., 2013a). PGE2 prevented thrombin-induced permeability of pulmonary artery endothelial cells (Birukova et al., 2007), and recently EP4 receptors were shown to mediate the endothelial barrier protecting effect of PGE2: In this study, we identified that the PGE2-EP4 signalling axis was more protective microvascular, than of macrovascular permeability in cultures of pulmonary endothelial cells (Konya et al., 2013b). Sphingosine-1 phosphate, a well-characterized protector of the endothelial barrier, attenuated adverse outcomes in murine and canine models of ALI (McVerry and Garcia, 2005). Strikingly, we observed in vitro that the microvascular barrier-enhancing effect of EP4 receptor activation was much more pronounced than that of sphingosine-1 phosphate (Konya et al., 2013b). These findings suggested a fundamental protective role for the PGE2-EP4 axis in ALI via protection of the microvascular permeability barrier.
In the present study, we have assessed the regulatory role of PGE2 and EP4 receptors in two mouse models of acute pulmonary inflammation. In LPS-induced pulmonary inflammation, PGE2 via activation of EP4 receptors reduced neutrophil infiltration, plasma extravasation and TNF-α release. Furthermore, activation of EP4 receptors inhibited pulmonary vascular permeability induced by intravenously applied oleic acid. Activation of the EP4 receptor also restored the altered airway resistance and lung compliance after methacholine challenge in mice with LPS- or oleic acid-induced lung inflammation. In vitro experiments using human and murine pulmonary microvascular endothelial cells confirmed the in vivo results, as we could show that barrier disruption following LPS was diminished by activation of EP4 receptors.
Methods
Animals
All animal care and experimental procedures used in this study complied with national and international guidelines and were approved by the Austrian Federal Ministry of Science, Research and Economy (BMWFW GZ:66.010/032-II/10b/2012; GZ:66.010/0173-II/3b/2012; GZ:66,010/0009-WF/V/3b/2015), conform with the Directive 2010/63/EU. Stuides are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath et al., 2010). A total of 392 animals were used in the experiments described here. Male BALB/c mice, 6–8 weeks old (body weight 22–25 g), were obtained from Charles River (Sulzfeld, Germany). Mice were housed in individually ventilated cages (four per cage) under controlled conditions of temperature (set point 21°C), air humidity (set point 50%) and a 12 h light/dark cycle (lights on at 06:00 h) and habituated to the environment for at least 1 week. Standard chow (altromin 1324 FORTI, Altromin, Lage, Germany) and water were provided ad libitum. Procedures were performed as humanely as possible to minimize animal suffering. Sample size calculations were performed with SigmaStat12.5 (Systat Software Inc, San Jose, CA, USA) using values obtained in pilot experiments. Mice were randomly assigned before treatment. The number (n) of animals used in the different experiments is given in the respective figure legends.
LPS-induced acute pulmonary inflammation
Mice were slightly anaesthetized with ketamine/xylazine (50 mg/5 mg·kg−1 i.p.) for intranasal applications. In preliminary experiments, we determined the LPS (Escherichia coli O55:B5) dose that induced moderate but reproducible neutrophilic inflammation in the lung which was comparable to previously published data (Haegens et al., 2009; Peters et al., 2010).
Experiment I: Mice were treated with vehicle or LPS (20 μg per mouse in PBS) or LPS in combination with PGE2 (20 μg per mouse; Goncalves de Moraes et al., 1996; Peters et al., 2010) intranasally in a volume of 25 μL to each nares at 09:00 h.
Experiment II: Mice were pretreated with vehicle or the EP4 receptor antagonist GW627368X (10 mg·kg−1) (Lee et al., 2013) s.c., 30 min before intranasal treatment as described in experiment I.
Experiment III: Mice were pretreated with vehicle or the EP4 receptor antagonist ONO AE3-208 (10 mg·kg−1) (Kabashima et al., 2002; Xu et al., 2014) s.c., 30 min before intranasal application of compounds as described in experiment I.
Experiment IV: Mice were pretreated with vehicle or the EP2 antagonist PF-04418948 (10 mg·kg−1) (af Forselles et al., 2011) s.c., 30 min before intranasal treatment as described in experiment I.
Experiment V: Mice were treated intranasally with vehicle, LPS (20 μg per mouse) or LPS in combination with the EP4 receptor agonist ONO AE1-329 (3–10–30 μg per mouse) intranasally in a volume of 25 μL to each nares at 09:00 h.
Experiment VI: Mice were treated intranasally with vehicle, LPS (20 μg per mouse) or LPS in combination with the EP2 receptor agonist ONO AE1-259 (3–10–30 μg per mouse) intranasally in a volume of 25 μL to each nares at 09:00 h.
Four hours after intranasal treatment, mice were killed with an overdose of pentobarbital (100 mg·kg−1 i.p.), followed by sampling of blood and bronchoalveolar lavage (BAL) fluid as previously described (Sturm et al., 2008). In additional experiments, mice were pretreated with the stable PGE2 analogue misoprostol (50 μg per mouse, s.c.) (Zaslona et al., 2014) 2 h before (at 08:00 h) and 10 h after intranasal application of LPS (10 μg per mouse). Mice were killed 24 h after LPS as described earlier and BAL fluid was sampled. This dose of LPS was chosen, because we found, in preliminary experiments, a higher dose (20 μg per mouse) resulted in excessive neutrophil infiltration after 24 h (data not shown). Leukocytes in blood and BAL fluid were analysed by flow cytometry as described below. TNF-α and IL-6 were determined in the BAL fluid by FlowCytomix multiplex kit (eBioscience; Vienna, Austria).
Leukocyte analysis by flow cytometry
BAL fluid and citrated whole blood were centrifuged at 400× g for 7 min at 4°C. Supernatants of BAL fluid were collected, aliquoted and stored at −70°C for further analysis. Plasma was discarded and red blood cells were lysed using BD FACS lysing solution (BD Biosciences, Vienna, Austria). Following two washing steps with Ca2+ and Mg2+ free PBS, cells were incubated for 30 min at 4°C with the following monoclonal antibodies: FITC-conjugated MHC-II (2.5 μg·mL−1), PE-conjugated CCR3 antibody (2 μg·mL−1), PE-Cy5.5-conjugated CD3e antibody (2 μg·mL−1), PE-Cy7-conjugated B220 antibody (2 μg·mL−1), APC-conjugated CD11c antibody (2 μg·mL−1) and CD16 Block (2 μg·mL−1) (all antibodies were purchased from BD Biosciences). Samples were washed once in PBS, 200 μL fixative solution was added and samples were kept on ice until analysis. Cell suspension was measured for 30 s on a FACSCalibur flow cytometer. Cell populations were analysed as events per 30 s on FlowJo software (Treestar, Costa Mesa, CA, USA; version vX.0.7). Cell populations were identified with adaptations as described elsewhere (van Rijt et al., 2004; Sturm et al., 2008). In detail, lymphocytes were identified as FSClow/SSClow CD3high/B220high cells and further divided into MHC-IIneg T- and MHC-IIhigh B-lymphocytes. FSCmed, SSCmed, CD3 neg/B220neg granulocytes were identified as CCR3neg neutrophils and CCR3high eosinophils. FSChigh/SSCmed alveolar macrophages were gated as CD11chigh/MHC-IIhigh and monocytes were identified as CD11cmed/MHC-IIneg cells.
Pulmonary vascular permeability
Mice were treated intranasally with vehicle or LPS, or LPS in combination with PGE2 or the EP4 receptor agonist ONO AE1-329 (20 μg per mouse) at 09:00 h as described earlier. Three hours post treatment, mice were injected with Evans blue (60 mg·kg−1 in saline) in the tail vein and 1 h later mice were killed with pentobarbital (100 mg·kg−1, i.p.) and exsanguinated by cutting the abdominal aorta. The lungs were perfused via the right ventricle with 10 mL of PBS containing 5 mmol·L−1 EDTA. Thereafter, lungs were excised and weighed, then homogenized in PBS (1 mL per 100 μg tissue) and incubated with two volumes of formamide (18 h, 60°C). After centrifugation (5000× g for 20 min), the absorbance at 620 nm and 740 nm was measured. The Evans blue content of tissue was calculated and corrected for the presence of heme pigments as described (Wang le et al., 2002). In order to induce pulmonary vascular leakage with oleic acid, mice were anaesthetized with ketamine (100 mg·kg−1) and xylazine (10 mg·kg−1), and oleic acid (0.15 μL·g−1) together with Evans blue (60 mg·kg−1) was injected into the tail vein at 09:00 h. Immediately thereafter, vehicle or the EP4 agonist ONO AE1-329 (20 μg per mouse) was applied intranasally. Body temperature was maintained by placing the mice on a heating pad. Ninety minutes later, mice were killed and their lungs were processed as described above.
Assessment of airway function
Mice received intranasal application of vehicle or LPS (20 μg per mouse), the EP4 receptor agonist ONO AE1-329 (20 μg per mouse) or the EP2 receptor agonist ONO AE1-259 (30 μg per mouse) at 09:00 h as described above. Four hours later, lung function was determined by the flexiVent system (flexiVent, Scireq, Montreal, QC, Canada) as described by Nemmar et al., (2011). Briefly mice were anaesthetized with ketamine (150 mg·kg−1) and xylazine (10 mg·kg−1) by i.p. injection. After surgical anaesthesia was established, tracheostomy was performed and an 18 gauge cannula was inserted into the trachea. Animals were placed on a 37°C heating pad, connected to a computer controlled small-animal ventilator and ventilated with a tidal volume of 10 mL·kg−1 at a frequency of 150 breaths per minute and a positive end-expiratory pressure of 2 cm H2O. Airway resistance (R) and compliance (C) were measured before any treatment (baseline levels) and after delivery of increasing concentrations of nebulized methacholine (1, 3, 10, 30 and 100 mg·mL−1). R and C were defined as the mean out of 12 values for each perturbation if the coefficient of determination was greater than 0.95. Assessment of lung function in mice treated with oleic acid was performed 90 min after injection of oleic acid.
Culture of endothelial cells and endothelial electrical resistance measurements
Human lung microvascular endothelial cells were purchased from Lonza (Verviers, Belgium) and maintained in EGM-2MVBulletKit medium (Lonza) with 5% FCS. Endothelial cells were cultivated as previously described (Konya et al., 2010; El-Gamal et al., 2012). Lung microvascular endothelial cells from BALB/c mice were purchased from Cell Biologics (Chicago, IL, USA) in passage 3 and were cultured until passage 9 in complete Mouse Endothelial Cell Medium Kit (Cell Biologics). Endothelial cells were seeded on 1% gelatine-coated biochips containing gold microelectrodes (ECIS 8W10E+). On the second day after seeding, cells were serum-starved for 1 h and electrical resistance was measured at 4000 Hz frequency using electric cell-substrate impedance sensing system (ECIS, Applied Biophysics, Troy, NY, USA) as described (Konya et al., 2013b). Experiments were performed in duplicate and repeated four to seven times (n) with freshly seeded cells per treatment group.
Culture of murine alveolar macrophages
Mice were killed with pentobarbital (100 mg·kg−1, i.p.) at 08:00 h and exsanguinated by cutting the abdominal aorta. BAL was performed with 12 mL PBS containing 1 mmol·L−1 EDTA and 10 mmol·L−1 HEPES, pH 7.3. BAL from three mice was pooled and centrifuged at 400× g for 7 min. Erythrocytes were lysed by adding ice-cold ammonium chloride. The washed cell pellet was resuspended in 1 mL complete RPMI medium containing 10% FCS, 1% penicillin/streptomycin and cells were counted. Cells (4 × 104 per well) were seeded in 100 μL of medium in 96-well plates. Macrophages were allowed to adhere for 1 h at 37°C in a 5% CO2 humidified incubator. Non-adherent cells and debris were removed by washing (Peters et al., 2010). The adherent macrophages were cultured in 100 μL of fresh complete medium and stimulated with LPS (10 ng·mL−1) in the absence or presence of PGE2 (30–300 nmol·L−1) or the specific EP4 agonist ONO AE1-329 (30–300 nmol·L−1). In a subset of experiments, cells were pretreated with the EP4 antagonists ONO AE3-208 (100 nmol·L−1) or GW627368X (100 nmol·L−1) for 30 min before being exposed to LPS and PGs. Cells were treated for 4 h, and thereafter the supernatant was collected for determination of TNF-α and IL-6 using FlowCytomix kit (eBioscience). These experiments were performed in triplicate and repeated five to eight times (n) with cells from different donors per group.
Lung histology and EP4 receptor immunohistochemistry
Mice were killed with pentobarbital (100 mg·kg−1) at 08:00 h, their lungs were perfused via the pulmonary artery with 0.1 mol·L−1 PBS (pH 7.4) followed by 4% buffered paraformaldehyde (Sturm et al., 2008). The lungs were dissected, postfixed for 24 h and embedded in paraffin. Five micrometre sections were deparaffinized and either haematoxylin/eosin staining was performed, or sections were immunostained for EP4 receptors.
For EP4 receptor staining, antigen retrieval was performed with antigen unmasking solution pH 6.0 (Vector, Vector Labs, Burlingame, CA, USA) and to reduce non-specific background, sections were incubated in blocking solution (Vectastain ABC Kit, Vector Labs). Thereafter, samples were incubated with a polyclonal EP4 antibody (AB133170; 1:400 diluted; Abcam, Cambridge, UK) overnight. Samples were further incubated for 45 min with a goat anti-rabbit antibody (Jackson Immunoresearch, West Grove, PA, USA; 1:200 diluted). After washing, the bound antibody was detected using the Vectastain ABC kit and 3-amino-9-ethylcarbazole as chromogen and counterstained with haematoxylin. Sections were visually examined with an Olympus BX41 microscope (Olympus Austria GmbH, Vienna, Austria) and an Olympus U Plan Apo ×40/0.8 or ×60/0.8 lens. Photographs were taken with an Olympus DP50 camera (2776 × 2074 pixels) and further processed with CELLP software (Olympus Austria GmbH) for additional white balance, contrast and brightness adjustments.
Data analysis
Statistical analysis was performed using GraphPad Prism software 6.0 (La Jolla, CA, USA). Experiments using endothelial cells and alveolar macrophages were performed in duplicate or triplicate, respectively, and were repeated n times with freshly seeded cells or with cells from different donors. Concentration–response curves were compared with each other using one-way anova for repeated measurements; the differentially treated groups of mice were tested for significant differences with one-way anova; lung function and electrical resistance measurements were analysed by two-way anova. Statistical differences between groups were determined by Tukey’s multiple comparison test. Probability values of P < 0.05 were regarded as statistically significant.
Materials
All laboratory reagents were from Sigma (Vienna, Austria) unless specified. The EP4 antagonist GW627368X was obtained from Cayman (Tallin, Estonia). ONO AE1-329, ONO AE1-259 and ONO AE3-208 were generous gifts from ONO Pharmaceutical (Osaka, Japan). CellFix and FACS-Flow from BD Bioscience. Fixative solution was prepared by adding 9 mL distilled water and 30 mL FACS-Flow to 1 mL CellFix. PGE2, ONO AE1-329 and ONO AE1-259 were dissolved in ethanol, while stock solutions of GW627368X, PF-04418948, ONO AE3-208 and misoprostol were prepared in DMSO and further diluted in PBS to produce a final concentration of the solvents of less than 0.1%.
Results
PGE2 via EP4 receptor activation inhibits LPS-induced neutrophil infiltration in the lung
Intranasal treatment of mice with LPS (20 μg per mouse) caused a pronounced increase in neutrophil counts in the BAL fluid (Figure 1A) after 4 h, with only a moderate increase in monocyte and T-lymphocyte count (see Supporting Information Fig. S1). In parallel, an increase of neutrophils was also observed in blood samples (see Supporting Information Fig. S2). In additional experiments, treatment with the stable PGE2 analogue misoprostol (50 μg per mouse), which was used here because of the short half-life of PGE2, abolished neutrophilia in the BAL fluid 24 h after intranasal application of LPS (10 μg per mouse; see Supporting Information Fig. S3). All further experiments were performed 4 h after LPS challenge. The LPS-induced neutrophilia in the airways was abolished by co-administration with PGE2 (20 μg per mouse) (Figure 1A). Notably, pretreatment of the animals with the EP4 receptor antagonist GW627368X or ONO AE3-208 (both at 10 mg·kg−1 s.c.), 30 min before LPS/PGE2 co-administration reversed the inhibitory effect of PGE2 on LPS-induced pulmonary neutrophil infiltration, but had no effect on neutrophil counts in the absence of PGE2 (Figure 1A). To further substantiate this finding, LPS (20 μg per mouse) was intranasally applied in combination with the selective EP4 receptor agonist ONO AE1-329 (3–30 μg per mouse), which likewise caused a reduction of BAL neutrophils (Figure 1B). These observations were reflected at the tissue level where the dense leukocyte infiltration seen after LPS (Figure 1D) treatment was markedly reduced by PGE2 and ONO AE1-329 (Figure 1E and F respectively). In addition, we found that pretreatment of mice with the EP2 receptor antagonist PF-04418947 (10 mg·kg−1 s.c.) counteracted the effect of PGE2 while intranasal co-administration with the EP2 receptor agonist ONO AE1-259 (3–30 μg per mouse) exerted only minor effects on LPS-induced neutrophil infiltration (see Supporting Information Fig. S4).
Figure 1.
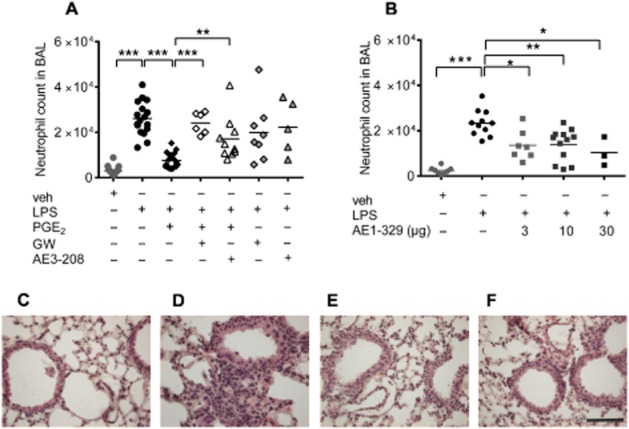
Neutrophil counts in BAL fluid of (A) mice treated intranasally with vehicle (veh; n = 18), LPS (20 μg per mouse; n = 17), PGE2 (20 μg per mouse) concomitantly with LPS (n = 22) or pretreated with the EP4 receptor antagonists GW627368X (GW; 10 mg·kg−1 s.c.; n = 8) or ONO AE3-208 (10 mg·kg−1 s.c.; n = 5) 30 min before intranasal LPS or LPS concomitantly applied with PGE2 (n = 6 or 10 respectively). (B) Intranasal application of vehicle (veh, n = 8) or LPS (20 μg per mouse, n = 11) alone or in combination with the EP4 receptor agonist ONO AE1-329 (n = 3–10). Each point shows the neutrophil count in BAL fluid from one animal. Data were analysed using one-way anova and multiple comparisons were calculated with Tukey’s post test. *P < 0.05, **P < 0.01, ***P < 0.001. (C–F) Haematoxylin/eosin staining of lungs with intranasal application of (C) vehicle, (D) LPS (20 μg per mouse), (E) PGE2 (20 μg per mouse) given concomitantly with LPS and (F) ONO AE1-329 (20 μg per mouse) concomitantly applied with LPS. Lung histology micrographs are representative of three independent experiments; scale bar = 50 μm.
PGE2 via EP4 receptor activation inhibits LPS-induced pulmonary vascular leakage
Intranasal application of LPS (20 μg per mouse) caused significant vascular leakage after 4 h, which could be inhibited by co-administration of PGE2 (20 μg per mouse) (Figure 2A), as well as by the EP4 receptor agonist ONO AE1-329 (20 μg per mouse) (Figure 2B). These data suggest that LPS-induced pulmonary vascular leakage is inhibited by PGE2 via EP4 receptor activation.
Figure 2.
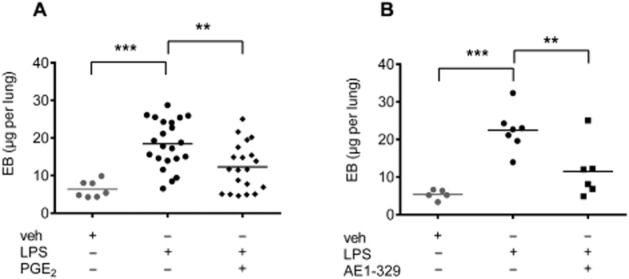
Evans blue extravasation in the lung of mice intranasally treated (A) with vehicle (veh, n = 7), LPS (20 μg per mouse, n = 22) or LPS in combination with PGE2 (20 μg per mouse, n = 20). (B) Intranasal treatment with vehicle (veh, n = 5), LPS (20 μg per mouse, n = 7) or in combination with the EP4 receptor agonist ONO AE1-329 (20 μg per mouse, n = 6). Each point shows μg Evans blue (EB) per lung from one animal. Data were analysed using one-way anova and multiple comparisons were calculated with Tukey’s post test. **P < 0.01, ***P < 0.001.
Effect of PGE2 and EP4 receptor activation on LPS-induced cytokine release
Cytokine concentrations were determined in BAL fluid which had been obtained from the experiments described earlier. Low levels of IL-1α, IL-2, IL-5, IL-10 and GM-CSF were found while IFN-γ and IL-17 were undetectable in the BAL fluid (data not shown). LPS (20 μg per mouse) caused an increase of TNF-α and IL-6 that was significantly inhibited by PGE2 (20 μg per mouse; Figure 3A and B). Unexpectedly, this inhibitory effect of PGE2 on cytokine release was not influenced by pretreatment of mice with the EP4 receptor antagonists GW627368X (10 mg·kg−1 s.c.) or ONO AE3-208 (10 mg·kg−1 s.c.; Figure 3A and B), or by the EP2 receptor antagonist PF-04418948 (10 mg·kg−1 s.c.; Supporting Information Fig. S5). Interestingly, concomitant application of the specific EP4 receptor agonist ONO AE1-329 or the EP2 receptor agonist ONO AE1-259 (at a dose of 30 μg per mouse) along with LPS abolished the increase of TNF-α, without affecting the increase in IL-6 (Figure 3C and D, and Supporting Information Fig. S5).
Figure 3.
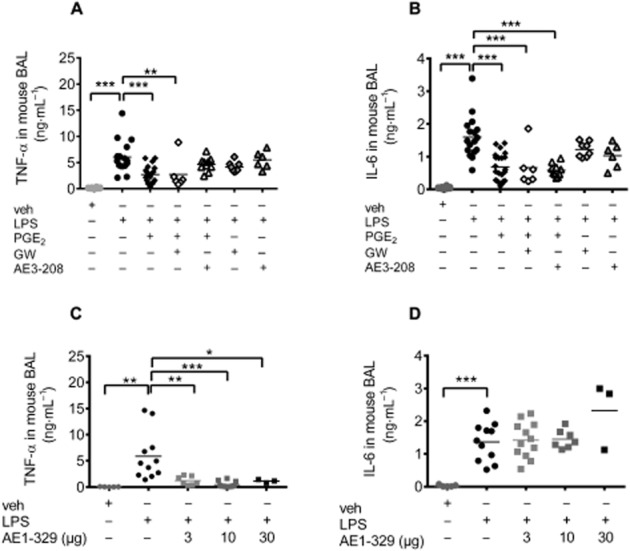
TNF-α (A,C) and IL-6 (B,D) levels in the BAL fluid of (A,B) mice treated intranasally with vehicle (veh; n = 18), LPS (20 μg per mouse; n = 17), PGE2 (20 μg per mouse) concomitantly with LPS (n = 22) or pretreated with the EP4 receptor antagonists GW627368X (GW; 10 mg·kg−1 s.c.; n = 8) or ONO AE3-208 (10 mg·kg−1 s.c.; n = 5) 30 min before intranasal LPS or LPS concomitantly applied with PGE2 (n = 6 or 10 respectively). (C,D) Intranasal application of vehicle (veh, n = 8) or LPS (20 μg per mouse, n = 11) alone or in combination with the EP4 receptor agonist ONO AE1-329 (n = 3–10). Each point shows the cytokine concentration (ng mL−1) from one animal. Data were analysed using one-way anova and multiple comparisons were calculated with Tukey’s post test. *P < 0.05, **P < 0.01, ***P < 0.001.
Because TNF-α is derived predominantly from activated macrophages, we isolated bronchoalveolar macrophages from mice. In the supernatants of unstimulated macrophages, we were unable to detect any TNF-α; however, treatment with LPS (10 ng·mL−1) for 4 h caused an increase in TNF-α that could be significantly inhibited by PGE2 (100–300 nmol·L−1; Figure 4A). Pretreatment of the cells with the EP4 receptor antagonists ONO AE3-208 (300 nmol·L−1) or GW627368X (1 μmol·L−1) blocked the inhibitory effect of PGE2 (30 nmol·L−1) on TNF-α release (Figure 4B). In addition, the EP4 receptor agonist ONO AE1-329 mimicked the effect of PGE2 (Figure 4C). Interestingly, both PGE2 (30 nmol·L−1) and ONO AE1-329 (100 nmol·L−1) augmented the secretion of IL-6 from alveolar macrophages (Figure 4D), and this was inhibited by ONO AE3-208 (300 nmol·L−1), which contrasted with the reduction by PGE2 of the IL-6 content of lungs in vivo (Figure 3B). These observations suggested that EP4 receptor activation might be involved in the regulation of TNF-α secretion, while additional PG receptors, and possibly cells other than macrophages, were likely to mediate the effect of PGE2 on cytokine release, in particular IL-6.
Figure 4.
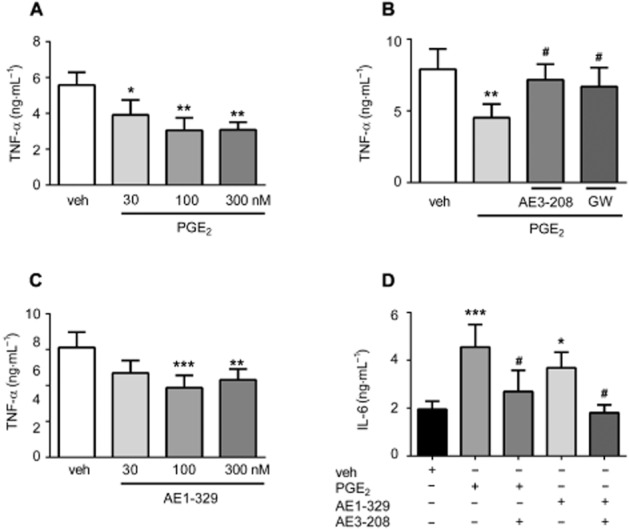
TNF-α (A–C) and IL-6 (D) concentrations in the supernatants of isolated murine alveolar macrophages. Cells were treated with LPS (10 ng·mL−1) for 4 h in the presence of (A) vehicle (veh) or PGE2 (concentrations as indicated), n = 5, (B) vehicle (veh) or PGE2 (30 nmol·L−1) in the absence or presence of the EP4 receptor antagonists ONO AE3-208 (300 nmol·L−1) or GW627368X (GW, 1 μmol·L−1) for 30 min, n = 6, (C) vehicle (veh) or the EP4 receptor agonist ONO AE1-329 (concentrations as indicated), n = 8, (D) vehicle (veh) or PGE2 (30 nmol·L−1) or ONO AE1-329 (100 nmol·L−1) in the absence or presence of the EP4 receptor antagonist ONO AE3-208 (300 nmol·L−1), n = 7. Data are shown as mean + SEM of n independent experiments performed in triplicate. Data were analysed using one-way anova for repeated measurements and multiple comparisons were calculated with Tukey’s post test. *P < 0.05, **P < 0.01, ***P < 0.001 as compared with LPS treatment, #P < 0.05 as compared with PGE2 or EP4 agonist treatment.
Activation of EP4 receptors promotes pulmonary microvascular endothelial barrier function in vitro
In cultures of murine lung endothelial cells, PGE2 (3–300 nmol·L−1) and the selective EP4 receptor agonist ONO AE1-329 (0.1–30 nmol·L−1) (Figure 5A and B) increased endothelial barrier function, and ONO AE3-208 prevented the endothelial barrier enhancement induced by the EP4 receptor agonist (30 nmol·L−1) and PGE2 (30 nmol·L−1) (Figure 5C and D). In agreement with these functional observations, flow cytometric analysis confirmed that murine pulmonary microvascular endothelial cells in culture expressed EP4 receptors (see Figure 5E). These observations extend our previous findings made in human endothelial cells (Konya et al., 2013b). In contrast, the EP2 receptor agonist ONO AE1-259 (up to 100 nmol·L−1) did not affect the barrier function (data not shown; n = 3).
Figure 5.
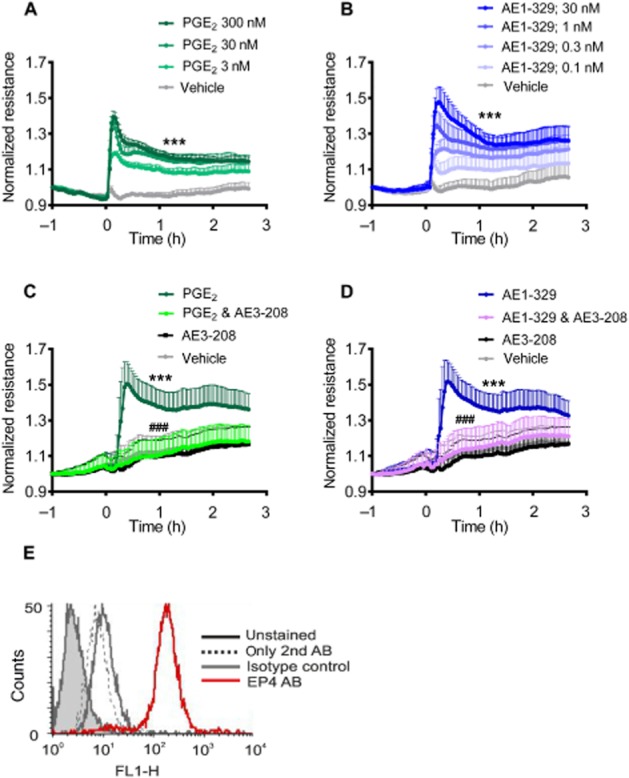
Electrical resistance of murine endothelial cell monolayers treated with either vehicle or (A) PGE2 or the (B) EP4 agonist ONO AE1-329 at the indicated concentrations. (C). Cells were treated with PGE2 (30 nmol·L−1) or (D) the EP4 agonist ONO AE1-329 (30 nmol·L−1) alone or in the presence of the EP4 antagonist ONO AE3-208 (1 μmol·L−1, 15 min pretreatment). Data are shown as mean normalized resistance + SEM of four independent experiments performed in duplicate. Data were analysed by two-way anova for repeated measurements and multiple comparisons were calculated with Tukey’s post test. ***P < 0.001 as compared with vehicle; ###P < 0.001 as compared with PGE2 or the EP4 agonist. (E) EP4 receptor expression on murine pulmonary microvascular cells using flow cytometry (typical example of n = 3). AB, antibody.
We have recently reported that activation of EP4 receptors on human endothelial cells strengthens the barrier function of the monolayer (Konya et al., 2013b). Here, we showed that LPS (1 μg·mL−1) 1.6 h after addition, decreased endothelial barrier function in vitro, as demonstrated by reduced electrical resistance. The EP4 receptor agonist ONO AE1-329 (100 nmol·L−1) or PGE2 (30 nmol·L−1) in the presence of LPS caused an increase in barrier function for up to 2.1 and 2.9 h, respectively, and attenuated the LPS effect for up to 7 h (Figure 6A and B). Pre-incubation with the EP4 receptor-selective antagonist ONO AE3-208 (300 nmol·L−1) abolished the endothelial barrier enhancement induced by the EP4 receptor agonist and PGE2 (30 nmol·L−1) in the presence of LPS (Figure 6C and D). This was the first time that EP4 receptor activation had been shown to prevent the barrier disruption induced by LPS.
Figure 6.
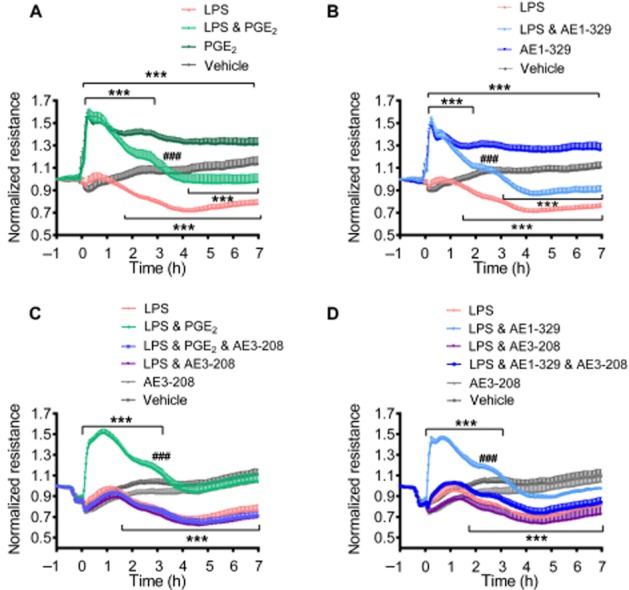
Electrical resistance of human endothelial cell monolayers treated with vehicle or LPS (1 μg·mL−1) in the absence or presence of (A,C) PGE2 (30 nmol·L−1), (B,D) the EP4 receptor agonist ONO AE1-329 (100 nmol·L−1). (C,D) Cells were pretreated with the EP4 receptor antagonist ONO AE3-208 (300 nmol·L−1) for 15 min. Data are shown as mean normalized resistance + SEM of 4–7 independent experiments performed in duplicate. Data were analysed by two-way anova for repeated measurements and multiple comparisons were calculated with Tukey’s post test. ***P < 0.001 as compared with vehicle; ###P < 0.001 as compared with LPS.
Moreover, immunohistochemistry of murine lung tissue demonstrated pulmonary endothelial cells to stain positively for EP4 receptors (Figure 7) while no signal was detected with isotype control (see Supporting Information Fig. S6).
Figure 7.
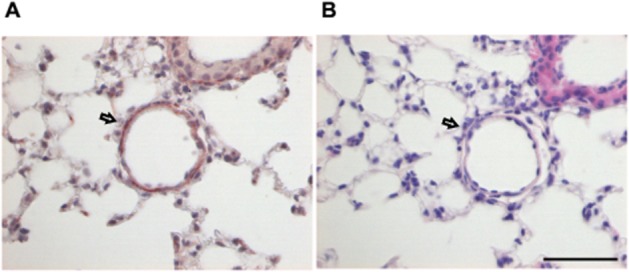
Immunohistochemical staining of EP4 receptors in murine lung tissue. (A) The endothelial layer of a lung vessel as indicated by the arrow is stained brown with the EP4 receptor antibody. (B) Haematoxylin–eosin staining of a consecutive section which shows the same vessel. Scale bar = 50 μm. Micrographs are representative for five lungs.
EP4 receptor activation attenuates oleic acid-induced vascular leakage in the lung
These observations indicated that stimulation of EP4 receptors on endothelial cells might be involved in protection against acute pulmonary inflammation. To further substantiate this notion, we used another pulmonary inflammation model that is primarily based on pulmonary vascular injury and investigated the involvement of EP4 receptors in oleic acid-induced vascular leakage. Intravenous administration of oleic acid (0.15 μL·g−1 body weight) caused a pronounced increase in Evans blue extravasation in the lung after 90 min. Intranasal treatment of the mice with the selective EP4 receptor agonist ONO AE1-329 (20 μg) significantly diminished this response (Figure 8), arguing for an important role of EP4 receptors in endothelial protection.
Figure 8.
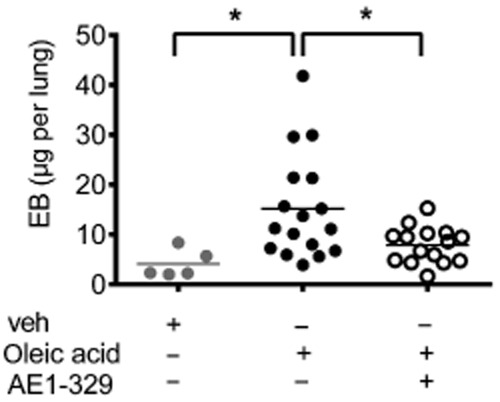
Evans blue (EB) extravasation in the lung of mice that received vehicle (veh, n = 5) or oleic acid (0.15 μL·g−1, n = 17) injected into the tail vein. The EP4 receptor agonist ONO AE1-329 (20 μg per mouse, n = 15) was given intranasally. Each point shows microgram EB per lung of one animal 90 min after oleic acid injection. Data were analysed by one-way anova and multiple comparisons were calculated with Tukey’s post test. *P < 0.05.
EP4 receptor activation attenuates LPS- and oleic acid-induced airway hyperresponsiveness to methacholine
Finally, we investigated whether EP4 receptor-mediated lung protection also translates to regulation of respiratory function. Four hours after intranasal application of LPS (20 μg per mouse) and 90 min after i.v. injection of oleic acid (0.15 μL·g−1 body weight), no changes in baseline resistance and compliance were seen in mechanically ventilated mice; however, significantly increased responses to aerosolized methacholine (10–100 ng·mL−1) were observed with respect to resistance and compliance as compared with vehicle-treated control mice (Figure 9). While concomitant treatment with PGE2 did not significantly affect resistance, it showed a minor improvement of compliance. The EP4 receptor agonist ONO AE1-329 (20 μg per mouse) reversed the airway hyperresponsiveness induced by LPS and by oleic acid (Figure 9). As it is known that the EP2 receptor mediates the bronchodilator effect of PGE2 in mice, we investigated this in our model. The EP2 receptor agonist ONO AE1-259 (30 μg per mouse) given concomitantly with LPS intranasally, also significantly inhibited the LPS-induced increase in resistance and decrease in compliance (see Supporting Information Fig. S7).
Figure 9.
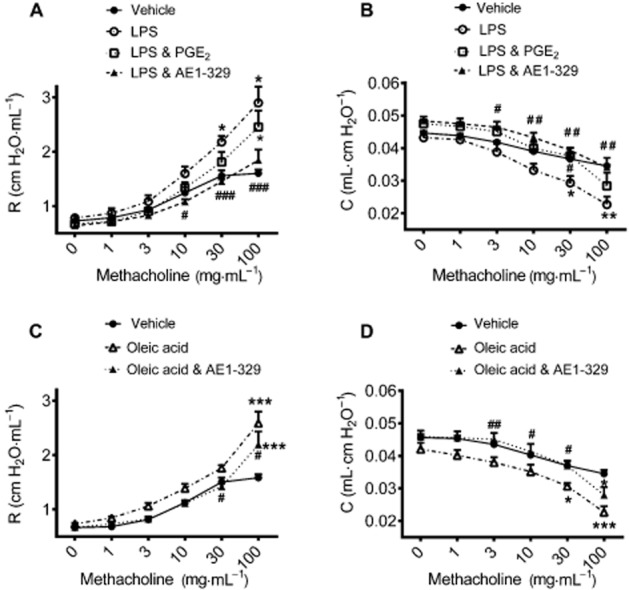
Lung function measured by the flexiVent system. Resistance (A,C) and compliance (B,D) induced by inhalation of increasing concentrations of methacholine was recorded 4 h after intranasal application of LPS (20 μg per mouse; A,B) and 90 min after oleic acid (0.15 μL·g−1 body weight; C,D) injection into the tail vein. Concomitant intranasal application of (A,B) PGE2 (20 μg per mouse) or (A–D) the EP4 receptor agonist ONO AE1-329 (20 μg per mouse). Data are shown as mean + SEM (n = 6 individual experiments per group). Data were analysed by two-way anova for repeated measurements and multiple comparisons were calculated with Tukey’s post test *P < 0.05, **P < 0.01, ***P < 0.001, compared with control; #P < 0.05, ##P < 0.01, compared with LPS or oleic acid.
Discussion
In this study, we have demonstrated that concomitant activation of the PGE2 receptor, EP4, ameliorates LPS-induced lung pathology, including recruitment of neutrophils, plasma extravasation and airway dysfunction, which are key clinical features of ALI (Abraham, 2003). We provide evidence that one major mechanism behind these beneficial PGE2 effects is via facilitation of endothelial barrier function through activation of endothelial EP4 receptors. A second mechanism appears to be the reduction of cytokine secretion observed after PGE2 administration. The limiting role of PGE2 in LPS-induced neutrophil accumulation has been recognized previously (Goncalves de Moraes et al., 1996; Peters et al., 2010). Our data corroborate these findings. The crucial finding of our study, however, is that activation of EP4 receptors is largely responsible for this, as the protective PGE2 effect was prevented by the EP4 receptor antagonists GW627368X and ONO AE3-208, and was mimicked by the selective EP4 receptor agonist ONO AE1-329. In addition, we also found an involvement of the EP2 receptor, as the EP2 receptor antagonist PF-04418948 blocked the inhibitory effect of PGE2 on neutrophil accumulation. However, the inhibitory effect of the highly selective EP2 receptor agonist ONO AE1-259 (Maruyame and Ohuchida, 2000) did not reach statistical significance. The EP4 and EP2 receptor antagonists by themselves did not change neutrophil infiltration induced by LPS. This was unexpected, as LPS is known to increase COX-2 expression and increase PG synthesis (Lin et al., 1996; Amann et al., 1999; Murakami et al., 2000). Nevertheless, the endogenous levels of PGE2 may be too low in our experimental model to affect neutrophil infiltration. Accordingly, we observed only a minor (twofold) increase in the levels of PGE2 metabolites in the BAL fluid after LPS treatment (data not shown). In a similar model, however, Birrell et al. (2015) found that EP4 receptor knockout mice showed exaggerated responses to LPS and other inflammatory stimuli in the lung. These data were published while our current study was under revision.
Endothelial barrier dysfunction in the lung leads to increased vascular permeability to fluid and macromolecules, resulting in pulmonary oedema. In the present study, we obtained evidence in two in vivo models, which are LPS and oleic acid-induced pulmonary vascular dysfunction, that PGE2 and an EP4 receptor-selective analogue were capable of strengthening the endothelial barrier, thereby preventing increased vascular permeability in the lungs.
These results are compatible with our in vitro data obtained recently with human microvascular endothelial cells, in which neutrophil adhesion to, and migration through, endothelial monolayers in vitro was blocked by EP4 receptor activation, which we could link to reduced E-selectin expression and enhanced barrier function respectively (Konya et al., 2013b). The latter effect was reflected by enhanced electrical resistance and increased formation of adherens junctions by vascular endothelial-cadherin. In extension of this idea, we have demonstrated in the current study that the LPS-induced disruption of endothelial barrier function in human pulmonary microvascular endothelial cells was prevented by PGE2 via EP4 receptor activation.
As already observed with human microvascular lung endothelial cells (Konya et al., 2013b), our present data demonstrate that PGE2 was likewise able to increase basal vascular barrier function in mouse pulmonary microvascular endothelial cells in vitro, reflected by an increase in electrical resistance. This effect was mimicked by the EP4 receptor agonist and was counteracted by the EP4 receptor antagonist. Our immunohistochemical data also showed that the EP4 receptor was expressed on endothelial layers of lung parenchymal vessels in the mouse lung. In contrast, we did not find the EP2 receptor to be involved in barrier function.
Recent studies have shown that the stable PGI2 analogue iloprost increases endothelial barrier function, which is mediated by PKA/Epac/Rap1/Rac, and leads to improvement of ventilation-induced ALI as well as LPS-induced ALI, with regard to neutrophil infiltration, increased protein content in BAL fluid and Evans blue extravasation (Birukova et al., 2010; 2013,). In vitro, iloprost treatment suppressed the LPS-induced cytoskeletal alterations and inflammatory signalling in endothelial cells (Birukova et al., 2013). In addition, Peters et al. (2010) reported on the role of the EP2 receptor in LPS-induced ALI, as butaprost mimicked the effect of PGE2 in inhibiting neutrophil accumulation in BAL fluid. Here, we confirmed the effect of EP2 receptor activation on neutrophil infiltration in our model, but this latter effect seems to be unrelated to a direct effect on the endothelial barrier.
In addition to increased vascular permeability and influx of neutrophils into the lung, ALI is also characterized by increased levels of proinflammatory cytokines (Abraham, 2003; Lesur et al., 2010). TNF-α and IL-6 are elevated in ALI and cause vascular barrier dysfunction, oedema formation as well as orchestrating neutrophil recruitment into lung tissue (Horvath et al., 1988; Millar et al., 1989; Hocking et al., 1990; Birukova et al., 2012). However, whether IL-6 is actually a proinflammatory or anti-inflammatory cytokine in ALI is not clear as, in LPS-induced lung injury, IL-6 plays an anti-inflammatory role (Xing et al., 1998; Bhargava et al., 2013), while in a ‘two hit’ murine model of LPS and ventilator-induced lung injury it showed an overall proinflammatory effect (Goldman et al., 2014). We found increased TNF-α and IL-6 in the BAL fluid of mice which had been treated intranasally with LPS. As expected, exogenous PGE2 prevented the increase in TNF-α. Treatment of mice with the EP4 receptor antagonists GW627368X and ONO AE3-208 as well as the EP2 receptor antagonist PF-04418948 did not prevent the inhibitory effect of PGE2. However, intranasal administration of the EP4 receptor agonist ONO AE1-329 as well as the EP2 receptor agonist ONO AE1-259, albeit at a higher dose, mimicked the effect of PGE2. Therefore, the inhibitory effect of PGE2 on TNF-α in vivo seems to be mediated by both receptors and this could explain why blockade of either receptor alone was not effective.
By sharp contrast to TNF-α release, PGE2 and EP4 receptor activation were found to increase IL-6 production in macrophages as well as in neutrophils, smooth muscle cells and fibroblasts (Yokoyama et al., 2013). While we corroborated this observation in isolated alveolar macrophages, we also found that exogenous PGE2 treatment of mice attenuated the LPS-induced increase of IL-6 in BAL fluid. Even more strikingly, EP4 or EP2 receptor blockade did not reverse the inhibition of IL-6 secretion by PGE2, just as EP4 and EP2 agonists did not influence IL-6 secretion in vivo, while reversing the inhibitory effect of PGE2 on neutrophil recruitment. Interestingly, a recent study showed that PGE2 inhibits IL-6 release in mouse and human monocytic cell lines (Birrell et al., 2015). In the lung, in addition to macrophages, a variety of cells produce IL-6, such as epithelial cells, endothelial cells, interstitial fibroblasts and other inflammatory cells (Rincon and Irvin, 2012). Therefore, we assume that the inhibition of IL-6 release by PGE2 in vivo involves other cell types than macrophages. Due to the lack of effect of the EP4 and EP2 agonists and antagonists in vivo, it remains to be established which EP receptor (EP1 or EP3 or IP) and which cell type is involved in the PGE2 regulation of IL-6 secretion in the lungs.
Proinflammatory cytokines in the lungs can lead to airway hyperresponsiveness (Brightling et al., 2008; Rincon and Irvin, 2012) which we measured in ventilated mice by invasive spirometry. LPS- and oleic acid-induced pulmonary inflammation was accompanied by enhanced bronchoconstrictor responses and decreased compliance after methacholine inhalation. Importantly, treatment of mice with the EP4 receptor agonist completely normalized LPS- and oleic acid-induced airway dysfunction to levels observed in vehicle-treated mice. In this respect, the EP4 receptor agonist had a greater effect than PGE2 at the given dose, as PGE2 largely corrected the enhanced fall in compliance elicited by methacholine, but had little effect on bronchoconstrictor responses. This discrepancy might be explained by the fact that PGE2 acts on three additional receptors, that is EP1–3, of which EP1 and EP3 receptors have been described as mediating bronchoconstriction in mice, by activating cholinergic reflexes (Tilley et al., 2003; Buckley et al., 2011). In contrast, the EP4 receptor has been implicated to directly relax airway smooth muscle in humans, while the EP2 receptor mediates the bronchodilator effect of PGE2 in mice (Tilley et al., 2003; Buckley et al., 2011; Benyahia et al., 2012; Konya et al., 2013a), which was shown in our experiments using an EP2 agonist. Therefore, it is likely that the beneficial effect of the EP4 receptor agonist on LPS- and oleic acid-induced lung dysfunction was due to its anti-inflammatory properties, rather than direct bronchodilation.
In conclusion, the current study provides ample evidence that EP4 receptor activation, concomitant with an inflammatory stimulus, strongly induces anti-inflammatory mechanisms, protects pulmonary microvascular endothelial barrier function and thereby ameliorates the pathology of pulmonary inflammation, such as neutrophil infiltration, plasma extravasation and deterioration of lung function in mice. Therefore, our data support a potential benefit for selective EP4 receptor agonists in acute pulmonary inflammation.
Acknowledgments
This work was supported by the Austrian Science Fund FWF (Grant P22521-B18 to A. H., P25531-B23 to V. K., P26185-B19 to R. S. and P21004-B02, P22976-B18 to G. M.) and the Jubiläumsfonds of the Austrian National Bank (14263 to A. H. and 14853 to G. M.). K. J., I. A. and J. M. were funded by the PhD Program DK-MOLIN (FWF – W1241). The authors appreciate the excellent technical assistance of Martina Ofner and Iris Red.
Glossary
- ALI
acute lung injury
- BAL
bronchoalveolar lavage
Author contributions
J. M., K. J., W. P., A. T. and T. B. performed and analysed the in vitro experiments. I. A. performed the immunohistochemistry. P. L., R. F. and I. L. performed the in vivo experiments and analysed the data. V. K., G. M., L. M. M., A. O., I. T. L., R. S. and A. H. provided intellectual input into study design, data interpretation and contributed to the manuscript preparation. V. K., R. S. and A. H. wrote the manuscript.
Conflict of interest
A. H. has received consultancy fees from Bayer Pharma AG (Berlin, Germany).
Supporting Information
Figure S1 Bronchoalveolar (BAL) fluid was collected 4 h post intranasal LPS (20 μg per mouse) or vehicle administration, and the numbers of (A) eosinophils, (B) monocytes, (C) macrophages, (D) T-lymphocytes and (E) B-lymphocytes were determined by flow cytometry. Each point shows the cell count from one animal. Data were analysed using one-way anova and multiple comparisons were calculated with Tukey’s post test. *P < 0.05, **P < 0.01.
Figure S2 Blood was collected 4 h post intranasal LPS or vehicle administration, and the numbers of neutrophils were determined by flow cytometry. (A) Intranasal administration of vehicle (veh), LPS (20 μg per mouse) alone or together with PGE2 (20 μg per mouse), and in mice pretreated the EP4 receptor antagonist GW627368X (GW; 10 mg·kg−1 s.c.; 30 min before PGE2). (B) Intranasal application of vehicle (veh), LPS (20 μg per mouse) alone or together with the EP4 receptor agonist ONO AE1-329 (concentrations are indicated). Each point shows the neutrophil count from one animal. Data were analysed using one-way anova and multiple comparisons were calculated with Tukey’s post test. **P < 0.01, ***P < 0.001.
Figure S3 Neutrophil count in BAL fluid of mice treated intranasally with vehicle (veh), LPS (10 μg per mouse) or misoprostol (50 μg per mouse, s.c.) 2 h before and 10 h after intranasal application of LPS. Twenty-four hours after LPS application, mice were killed and neutrophils in the BAL fluid were determined by flow cytometry. Each point shows the neutrophil count from one animal. Data were analysed using one-way anova and multiple comparisons were calculated with Tukey’s post test. ***P < 0.001.
Figure S4 Neutrophil counts in BAL fluid of (A) mice treated intranasally with vehicle (veh), LPS (20 μg per mouse), PGE2 (20 μg per mouse) concomitantly with LPS, or pretreated with the EP2 antagonist PF-04418948 (PF; 10 mg·kg−1 s.c.). (B) Intranasal application of vehicle (veh), LPS (20 μg per mouse) and the EP2 receptor agonist ONO AE1-259 (concentrations as indicated) concomitantly with LPS. Each point shows the neutrophil count in BAL fluid from one animal. Data were analysed using one-way anova and multiple comparisons were calculated with Tukey’s post test. *P < 0.05, **P < 0.01, ***P < 0.0.
Figure S5 TNF-α (A) and IL-6 (B) levels in the bronchoalveolar lavage (BAL) fluid of mice (A,B) treated intranasally with vehicle (veh), LPS (20 μg per mouse) or the EP2 receptor agonist ONO AE1-259 (concentrations as indicated) concomitantly with LPS. Each point shows the cytokine concentration (ng·mL−1) from one animal. Data were analysed using one-way anova and multiple comparisons were performed with Tukey’s post test. *P < 0.05, **P < 0.01, ***P < 0.001.
Figure S6 Isotype control for EP4 staining. The endothelial layer of a lung vessel is indicated by the arrow. Scale bar = 50 μm. Micrograph is representative for five lungs.
Figure S7 Lung function was measured by the flexiVent system. Resistance (A) and compliance (B) towards inhalation of increasing concentrations of methacholine was recorded 4 h after intranasal application of vehicle, LPS (20 μg per mouse) or LPS concomitantly applied with the EP2 receptor agonist ONO AE1-259 (30 μg per mouse). Data are shown as mean + SEM (n = 6 individual experiments per group). Data were analysed by two-way anova for repeated measurements and multiple comparisons were calculated with Tukey’s post test. **P < 0.01, ***P < 0.001 as compared with vehicle; ##P < 0.01, ###P < 0.001 as compared with LPS.
References
- Abraham E. Neutrophils and acute lung injury. Crit Care Med. 2003;31:S195–S199. doi: 10.1097/01.CCM.0000057843.47705.E8. [DOI] [PubMed] [Google Scholar]
- Aggarwal S, Moodley YP, Thompson PJ, Misso NL. Prostaglandin E2 and cysteinyl leukotriene concentrations in sputum: association with asthma severity and eosinophilic inflammation. Clin Exp Allergy. 2010;40:85–93. doi: 10.1111/j.1365-2222.2009.03386.x. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Benson He, Faccenda E, Pawson AJ, Sharman JL. Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: G Protein-Coupled Receptors. Br J Pharmacol. 2013;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amann R, Schuligoi R, Peskar BA. Eicosanoid release in the endotoxin-primed isolated perfused rat lung and its pharmacological modification. Inflamm Res. 1999;48:632–636. doi: 10.1007/s000110050514. [DOI] [PubMed] [Google Scholar]
- Benyahia C, Gomez I, Kanyinda L, Boukais K, Danel C, Leseche G, et al. PGE(2) receptor (EP(4)) agonists: potent dilators of human bronchi and future asthma therapy? Pulm Pharmacol Ther. 2012;25:115–118. doi: 10.1016/j.pupt.2011.12.012. [DOI] [PubMed] [Google Scholar]
- Bhargava R, Janssen W, Altmann C, Andrés-Hernando A, Okamura K, Vandivier RW, et al. Intratracheal IL-6 protects against lung inflammation in direct, but not indirect, causes of acute lung injury in mice. PLoS ONE. 2013;8:e61405. doi: 10.1371/journal.pone.0061405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birrell MA, Maher SA, Dekkak B, Jones V, Wong S, Brook P, et al. Anti-inflammatory effects of PGE2 in the lung: role of the EP4 receptor subtype. Thorax. 2015;70:740–747. doi: 10.1136/thoraxjnl-2014-206592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birukov KG, Zebda N, Birukova AA. Barrier enhancing signals in pulmonary edema. Compr Physiol. 2013;3:429–484. doi: 10.1002/cphy.c100066. [DOI] [PubMed] [Google Scholar]
- Birukova AA, Zagranichnaya T, Fu P, Alekseeva E, Chen W, Jacobson JR, et al. Prostaglandins PGE(2) and PGI(2) promote endothelial barrier enhancement via PKA- and Epac1/Rap1-dependent Rac activation. Exp Cell Res. 2007;313:2504–2520. doi: 10.1016/j.yexcr.2007.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birukova AA, Fu P, Xing J, Cokic I, Birukov KG. Lung endothelial barrier protection by iloprost in the 2-hit models of ventilator-induced lung injury (VILI) involves inhibition of Rho signaling. Transl Res. 2010;155:44–54. doi: 10.1016/j.trsl.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birukova AA, Tian Y, Meliton A, Leff A, Wu T, Birukov KG. Stimulation of Rho signaling by pathologic mechanical stretch is a ‘second hit’ to Rho-independent lung injury induced by IL-6. Am J Physiol Lung Cell Mol Physiol. 2012;302:L965–L975. doi: 10.1152/ajplung.00292.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birukova AA, Wu T, Tian Y, Meliton A, Sarich N, Tian X, et al. Iloprost improves endothelial barrier function in lipopolysaccharide-induced lung injury. Eur Respir J. 2013;41:165–176. doi: 10.1183/09031936.00148311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brightling C, Berry M, Amrani Y. Targeting TNF-alpha: a novel therapeutic approach for asthma. J Allerg Clin Immunol. 2008;121:5–12. doi: 10.1016/j.jaci.2007.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley J, Birrell MA, Maher SA, Nials AT, Clarke DL, Belvisi MG. EP4 receptor as a new target for bronchodilator therapy. Thorax. 2011;66:1029–1035. doi: 10.1136/thx.2010.158568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bull TM, Clark B, McFann K, Moss M National Institutes of Health/National Heart, Lung, and Blood Institute ARDS Network. Pulmonary vascular dysfunction is associated with poor outcomes in patients with acute lung injury. Am J Respir Crit Care Med. 2010;182:1123–1128. doi: 10.1164/rccm.201002-0250OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Gamal D, Holzer M, Gauster M, Schicho R, Binder V, Konya V, et al. Cyanate is a novel inducer of endothelial icam-1 expression. Antioxid Redox Signal. 2012;16:129–137. doi: 10.1089/ars.2011.4090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- af Forselles KJ, Root J, Clarke T, Davey D, Aughton K, Dack K, et al. In vitro and in vivo characterization of PF-04418948, a novel, potent and selective prostaglandin EP2 receptor antagonist. Br J Pharmacol. 2011;164:1847–1856. doi: 10.1111/j.1476-5381.2011.01495.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman JL, Sammani S, Kempf C, Saadat L, Letsiou E, Wang T, et al. Pleiotropic effects of interleukin-6 in a ‘two-hit’ murine model of acute respiratory distress syndrome. Pulm Circ. 2014;4:280–288. doi: 10.1086/675991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goncalves de Moraes VL, Boris Vargaftig B, Lefort J, Meager A, Chignard M. Effect of cyclo-oxygenase inhibitors and modulators of cyclic AMP formation on lipopolysaccharide-induced neutrophil infiltration in mouse lung. Br J Pharmacol. 1996;117:1792–1796. doi: 10.1111/j.1476-5381.1996.tb15356.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haegens A, Heeringa P, van Suylen RJ, Steele C, Aratani Y, O’Donoghue RJ, et al. Myeloperoxidase deficiency attenuates lipopolysaccharide-induced acute lung inflammation and subsequent cytokine and chemokine production. J Immunol. 2009;182:7990–7996. doi: 10.4049/jimmunol.0800377. [DOI] [PubMed] [Google Scholar]
- Hocking DC, Phillips PG, Ferro TJ, Johnson A. Mechanisms of pulmonary edema induced by tumor necrosis factor-alpha. Circ Res. 1990;67:68–77. doi: 10.1161/01.res.67.1.68. [DOI] [PubMed] [Google Scholar]
- Horvath CJ, Ferro TJ, Jesmok G, Malik AB. Recombinant tumor necrosis factor increases pulmonary vascular permeability independent of neutrophils. Proc Natl Acad Sci U S A. 1988;85:9219–9223. doi: 10.1073/pnas.85.23.9219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson JR, Garcia JG. Novel therapies for microvascular permeability in sepsis. Curr Drug Targets. 2007;8:509–514. doi: 10.2174/138945007780362719. [DOI] [PubMed] [Google Scholar]
- Kabashima K, Saji T, Murata T, Nagamachi M, Matsuoka T, Segi E, et al. The prostaglandin receptor EP4 suppresses colitis, mucosal damage and CD4 cell activation in the gut. J Clin Invest. 2002;109:883–893. doi: 10.1172/JCI14459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konya V, Sturm EM, Schratl P, Beubler E, Marsche G, Schuligoi R, et al. Endothelium-derived prostaglandin I2 controls the migration of eosinophils. J Allergy Clin Immunol. 2010;125:1105–1113. doi: 10.1016/j.jaci.2009.12.002. [DOI] [PubMed] [Google Scholar]
- Konya V, Marsche G, Schuligoi R, Heinemann A. E-type prostanoid receptor 4 (EP4) in disease and therapy. Pharmacol Ther. 2013a;138:485–502. doi: 10.1016/j.pharmthera.2013.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konya V, Ullen A, Kampitsch N, Theiler A, Philipose S, Parzmair GP, et al. Endothelial E-type prostanoid 4 receptors promote barrier function and inhibit neutrophil trafficking. J Allergy Clin Immunol. 2013b;131:532–540. doi: 10.1016/j.jaci.2012.05.008. e531–e532. [DOI] [PubMed] [Google Scholar]
- Lee IT, Lin C-C, Cheng S-E, Hsiao L-D, Hsiao Y-C, Yang C-M. TNF-alpha induces cytosolic phospholipase A2 expression in human lung epithelial cells via JNK1/2- and p38 MAPK-dependent AP-1 activation. PLoS ONE. 2013;8:e72783. doi: 10.1371/journal.pone.0072783. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Lesur I, Textoris J, Loriod B, Courbon C, Garcia S, Leone M, et al. Gene expression profiles characterize inflammation stages in the acute lung injury in mice. PLoS ONE. 2010;5:e11485. doi: 10.1371/journal.pone.0011485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JY, Wang LF, Lin RH. The association between lung innate immunity and differential airway antigen-specific immune responses. Int Immunol. 1996;8:499–507. doi: 10.1093/intimm/8.4.499. [DOI] [PubMed] [Google Scholar]
- Maniatis NA, Kotanidou A, Catravas JD, Orfanos SE. Endothelial pathomechanisms in acute lung injury. Vascul Pharmacol. 2008;49:119–133. doi: 10.1016/j.vph.2008.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama T, Ohuchida S. Selective agonists and antagonists for prostaglandin E2 receptor subtypes. Tanpakushitsu Kakusan Koso. 2000;45:1001–1007. [PubMed] [Google Scholar]
- McGrath JC, Drummond GB, McLachlan EM, Kilkenny C, Wainwright CL. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVerry BJ, Garcia JG. In vitro and in vivo modulation of vascular barrier integrity by sphingosine 1-phosphate: mechanistic insights. Cell Signal. 2005;17:131–139. doi: 10.1016/j.cellsig.2004.08.006. [DOI] [PubMed] [Google Scholar]
- Millar AB, Foley NM, Singer M, Johnson NM, Meager A, Rook GA. Tumour necrosis factor in bronchopulmonary secretions of patients with adult respiratory distress syndrome. Lancet. 1989;2:712–714. doi: 10.1016/s0140-6736(89)90772-1. [DOI] [PubMed] [Google Scholar]
- Murakami M, Naraba H, Tanioka T, Semmyo N, Nakatani Y, Kojima F, et al. Regulation of prostaglandin E2 biosynthesis by inducible membrane-associated prostaglandin E2 synthase that acts in concert with cyclooxygenase-2. J Biol Chem. 2000;275:32783–32792. doi: 10.1074/jbc.M003505200. [DOI] [PubMed] [Google Scholar]
- Nemmar A, Al-Salam S, Zia S, Marzouqi F, Al-Dhaheri A, Subramaniyan D, et al. Contrasting actions of diesel exhaust particles on the pulmonary and cardiovascular systems and the effects of thymoquinone. Br J Pharmacol. 2011;164:1871–1882. doi: 10.1111/j.1476-5381.2011.01442.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavord ID, Wong CS, Williams J, Tattersfield AE. Effect of inhaled prostaglandin E2 on allergen-induced asthma. Am Rev Respir Dis. 1993;148:87–90. doi: 10.1164/ajrccm/148.1.87. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledge base of drug targets and their ligands. Nucl. Acids Res. 2014;42(Database Issue):D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters T, Mann TS, Henry PJ. Inhibitory influence of protease-activated receptor 2 and E-prostanoid receptor stimulants in lipopolysaccharide models of acute airway inflammation. J Pharmacol Exp Ther. 2010;335:424–433. doi: 10.1124/jpet.109.163253. [DOI] [PubMed] [Google Scholar]
- van Rijt LS, Kuipers H, Vos N, Hijdra D, Hoogsteden HC, Lambrecht BN. A rapid flow cytometric method for determining the cellular composition of bronchoalveolar lavage fluid cells in mouse models of asthma. J Immunol Methods. 2004;288:111–121. doi: 10.1016/j.jim.2004.03.004. [DOI] [PubMed] [Google Scholar]
- Rincon M, Irvin CG. Role of IL-6 in asthma and other inflammatory pulmonary diseases. Int J Biol Sci. 2012;8:1281–1290. doi: 10.7150/ijbs.4874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubenfeld GD, Herridge MS. Epidemiology and outcomes of acute lung injury. Chest. 2007;131:554–562. doi: 10.1378/chest.06-1976. [DOI] [PubMed] [Google Scholar]
- Rubenfeld GD, Caldwell E, Peabody E, Weaver J, Martin DP, Neff M, et al. Incidence and outcomes of acute lung injury. N Engl J Med. 2005;353:1685–1693. doi: 10.1056/NEJMoa050333. [DOI] [PubMed] [Google Scholar]
- Sturm EM, Schratl P, Schuligoi R, Konya V, Sturm GJ, Lippe IT, et al. Prostaglandin E2 inhibits eosinophil trafficking through E-prostanoid 2 receptors. J Immunol. 2008;181:7273–7283. doi: 10.4049/jimmunol.181.10.7273. [DOI] [PubMed] [Google Scholar]
- Tilley SL, Hartney JM, Erikson CJ, Jania C, Nguyen M, Stock J, et al. Receptors and pathways mediating the effects of prostaglandin E2 on airway tone. Am J Physiol Lung Cell Mol Physiol. 2003;284:L599–L606. doi: 10.1152/ajplung.00324.2002. [DOI] [PubMed] [Google Scholar]
- Vancheri C, Mastruzzo C, Sortino MA, Crimi N. The lung as a privileged site for the beneficial actions of PGE2. Trends Immunol. 2004;25:40–46. doi: 10.1016/j.it.2003.11.001. [DOI] [PubMed] [Google Scholar]
- Wang le F, Patel M, Razavi HM, Weicker S, Joseph MG, McCormack DG, et al. Role of inducible nitric oxide synthase in pulmonary microvascular protein leak in murine sepsis. Am J Respir Crit Care Med. 2002;165:1634–1639. doi: 10.1164/rccm.2110017. [DOI] [PubMed] [Google Scholar]
- Ware LB. Pathophysiology of acute lung injury and the acute respiratory distress syndrome. Semin Respir Crit Care Med. 2006;27:337–349. doi: 10.1055/s-2006-948288. [DOI] [PubMed] [Google Scholar]
- Xing Z, Gauldie J, Cox G, Baumann H, Jordana M, Lei XF, et al. IL-6 is an antiinflammatory cytokine required for controlling local or systemic acute inflammatory responses. J Clin Invest. 1998;101:311–320. doi: 10.1172/JCI1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S, Zhang Z, Ogawa O, Yoshikawa T, Sakamoto H, Shibasaki N, et al. An EP4 antagonist ONO-AE3-208 suppresses cell invasion, migration, and metastasis of prostate cancer. Cell Biochem Biophys. 2014;70:521–527. doi: 10.1007/s12013-014-9951-2. [DOI] [PubMed] [Google Scholar]
- Yokoyama U, Iwatsubo K, Umemura M, Fujita T, Ishikawa Y. The prostanoid EP4 receptor and its signaling pathway. Pharmacol Rev. 2013;65:1010–1052. doi: 10.1124/pr.112.007195. [DOI] [PubMed] [Google Scholar]
- Zaslona Z, Okunishi K, Bourdonnay E, Domingo-Gonzalez R, Moore BB, Lukacs NW, et al. Prostaglandin E2 suppresses allergic sensitization and lung inflammation by targeting the E prostanoid 2 receptor on T cells. J Allerg Clin Immunol. 2014;133:379–387. doi: 10.1016/j.jaci.2013.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Bronchoalveolar (BAL) fluid was collected 4 h post intranasal LPS (20 μg per mouse) or vehicle administration, and the numbers of (A) eosinophils, (B) monocytes, (C) macrophages, (D) T-lymphocytes and (E) B-lymphocytes were determined by flow cytometry. Each point shows the cell count from one animal. Data were analysed using one-way anova and multiple comparisons were calculated with Tukey’s post test. *P < 0.05, **P < 0.01.
Figure S2 Blood was collected 4 h post intranasal LPS or vehicle administration, and the numbers of neutrophils were determined by flow cytometry. (A) Intranasal administration of vehicle (veh), LPS (20 μg per mouse) alone or together with PGE2 (20 μg per mouse), and in mice pretreated the EP4 receptor antagonist GW627368X (GW; 10 mg·kg−1 s.c.; 30 min before PGE2). (B) Intranasal application of vehicle (veh), LPS (20 μg per mouse) alone or together with the EP4 receptor agonist ONO AE1-329 (concentrations are indicated). Each point shows the neutrophil count from one animal. Data were analysed using one-way anova and multiple comparisons were calculated with Tukey’s post test. **P < 0.01, ***P < 0.001.
Figure S3 Neutrophil count in BAL fluid of mice treated intranasally with vehicle (veh), LPS (10 μg per mouse) or misoprostol (50 μg per mouse, s.c.) 2 h before and 10 h after intranasal application of LPS. Twenty-four hours after LPS application, mice were killed and neutrophils in the BAL fluid were determined by flow cytometry. Each point shows the neutrophil count from one animal. Data were analysed using one-way anova and multiple comparisons were calculated with Tukey’s post test. ***P < 0.001.
Figure S4 Neutrophil counts in BAL fluid of (A) mice treated intranasally with vehicle (veh), LPS (20 μg per mouse), PGE2 (20 μg per mouse) concomitantly with LPS, or pretreated with the EP2 antagonist PF-04418948 (PF; 10 mg·kg−1 s.c.). (B) Intranasal application of vehicle (veh), LPS (20 μg per mouse) and the EP2 receptor agonist ONO AE1-259 (concentrations as indicated) concomitantly with LPS. Each point shows the neutrophil count in BAL fluid from one animal. Data were analysed using one-way anova and multiple comparisons were calculated with Tukey’s post test. *P < 0.05, **P < 0.01, ***P < 0.0.
Figure S5 TNF-α (A) and IL-6 (B) levels in the bronchoalveolar lavage (BAL) fluid of mice (A,B) treated intranasally with vehicle (veh), LPS (20 μg per mouse) or the EP2 receptor agonist ONO AE1-259 (concentrations as indicated) concomitantly with LPS. Each point shows the cytokine concentration (ng·mL−1) from one animal. Data were analysed using one-way anova and multiple comparisons were performed with Tukey’s post test. *P < 0.05, **P < 0.01, ***P < 0.001.
Figure S6 Isotype control for EP4 staining. The endothelial layer of a lung vessel is indicated by the arrow. Scale bar = 50 μm. Micrograph is representative for five lungs.
Figure S7 Lung function was measured by the flexiVent system. Resistance (A) and compliance (B) towards inhalation of increasing concentrations of methacholine was recorded 4 h after intranasal application of vehicle, LPS (20 μg per mouse) or LPS concomitantly applied with the EP2 receptor agonist ONO AE1-259 (30 μg per mouse). Data are shown as mean + SEM (n = 6 individual experiments per group). Data were analysed by two-way anova for repeated measurements and multiple comparisons were calculated with Tukey’s post test. **P < 0.01, ***P < 0.001 as compared with vehicle; ##P < 0.01, ###P < 0.001 as compared with LPS.