

Abstract
Background and Purpose
The N-K-Cl cotransporters (NKCCs) mediate the coupled, electroneutral movement of Na+, K+ and Cl− ions across cell membranes. There are two isoforms of this cation co-transporter, NKCC1 and NKCC2. NKCC2 is expressed primarily in the kidney and is the target of diuretics such as bumetanide. Bumetanide was discovered by screening ∼5000 3-amino-5-sulfamoylbenzoic acid derivatives, long before NKCC2 was identified in the kidney. Therefore, structure–activity studies on effects of bumetanide derivatives on NKCC2 are not available.
Experimental Approach
In this study, the effect of a series of diuretically active bumetanide derivatives was investigated on human NKCC2 variant A (hNKCC2A) expressed in Xenopus laevis oocytes.
Key Results
Bumetanide blocked hNKCC2A transport with an IC50 of 4 μM. There was good correlation between the diuretic potency of bumetanide and its derivatives in dogs and their inhibition of hNKCC2A (r2 = 0.817; P < 0.01). Replacement of the carboxylic group of bumetanide by a non-ionic residue, for example, an anilinomethyl group, decreased inhibition of hNKCC2A, indicating that an acidic group was required for transporter inhibition. Exchange of the phenoxy group of bumetanide for a 4-chloroanilino group or the sulfamoyl group by a methylsulfonyl group resulted in compounds with higher potency to inhibit hNKCC2A than bumetanide.
Conclusions and Implications
The X. laevis oocyte expression system used in these experiments allowed analysis of the structural requirements that determine relative potency of loop diuretics on human NKCC2 splice variants, and may lead to the discovery of novel high-ceiling diuretics.
Tables of Links
| TARGETS | |
|---|---|
| Transporters | |
| NKCC1, Na-K-Cl co-transporter (SLC12A2) | |
| NKCC2 (SLC 12A1) | |
| OAT, organic anion transporter |
| LIGANDS | |
|---|---|
| Bumetanide |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013).
Introduction
Bumetanide is a widely used, highly potent loop diuretic that inhibits Na+ and Cl− reabsorption by the thick ascending limb (TAL) of the loop of Henle by blocking the Na+-K+-Cl− cotransporter (NKCC) isoform located in the apical membrane of these epithelial cells, that is, NKCC2 (Haas and Forbush, 1998). While NKCC2 is expressed primarily in the kidney, the second bumetanide-sensitive NKCC isoform, NKCC1, is expressed in many tissues and plays a major role in the regulation of intracellular Cl− concentration (Markadieu and Delpire, 2014). Although NKCC2 is encoded by a single gene, differential splicing results in the generation of three full-length splice variants (NKCC2A, NKCC2B, NKCC2F), which differ in their location along the TAL of Henle and their transport characteristics (Castrop and Schnermann, 2008; Alvarez-Leefmans, 2012). NKCC2A is the dominant isoform in humans (Carota et al., 2010) and was therefore used in the present study.
The molecular identification of NKCC1 and NKCC2 was made in 1994 (Delpire et al., 1994; Gamba et al., 1994; Xu et al., 1994), that is, long after clinical approval of bumetanide. At the time of development of bumetanide in the 1960s, its exact target in the kidney was not known. Bumetanide was discovered as a result of systematic structure–activity studies of a large series of diuretically active 3-amino-5-sulfamoylbenzoic acid derivatives (Feit, 1971) and was followed by several series of compounds related both in chemical structure and diuretic profile (Nielsen and Feit, 1978; Feit, 1981; 1990,). In other words, the synthesis of sulfamoyl benzoic acid diuretics was done long before their cellular and molecular mechanisms of action were elucidated (Feit, 1981). Rodents metabolize bumetanide and its derivatives too rapidly to allow any meaningful assessment of their diuretic potency (Olsen, 1977; Töpfer et al., 2014). Consequently, dogs were used to screen large series of diuretically active aminobenzoic acid compounds, which eventually led to discovery of bumetanide (Feit, 1971; 1981; 1990,,; Frey, 1975; Nielsen and Feit, 1978). The potency of bumetanide exceeded that of the clinically established loop diuretics furosemide and ethacrynic acid in humans as well as dogs (Cohen, 1981), indicating that the dog is a valuable translational model for structure–activity studies on loop diuretics acting via inhibition of renal NKCC2.
In 1979, Frizzell et al.1979 proposed that the diuretic effect of loop diuretics was mediated by inhibition of a co-transport of Na+ plus Cl−. This prompted Palfrey et al. (1980) to study the effect of various aminobenzoic acid derivatives, including bumetanide and some of its derivatives, on a cation co-transport system in avian erythrocytes, resulting in a good correlation between diuretic potency in the dog assay and their inhibition of cation cotransport in this model system. This cation co-transport in erythrocytes was later shown to be mediated by NKCC1 (Flatman and Creanor, 1999). To our knowledge, a similar correlation analysis is not available for mammalian NKCC2 transporters, the molecular targets of bumetanide and other loop diuretics in the apical membrane of TAL of Henle epithelial cells. This prompted us to perform such an analysis with bumetanide and several of its derivatives previously synthesized by Peter W. Feit. Bumetanide derivatives were chosen on the basis of their diuretic activity in dogs, covering a wide range of diuretic potencies and structural modifications (Figure 1). To determine the inhibitory profile of bumetanide and its derivatives on human NKCC2 splice variant A (hNKCC2A) in an isolated system, NKCC2 activity was assayed in Xenopus oocytes heterologously expressing hNKCC2A.
Figure 1.
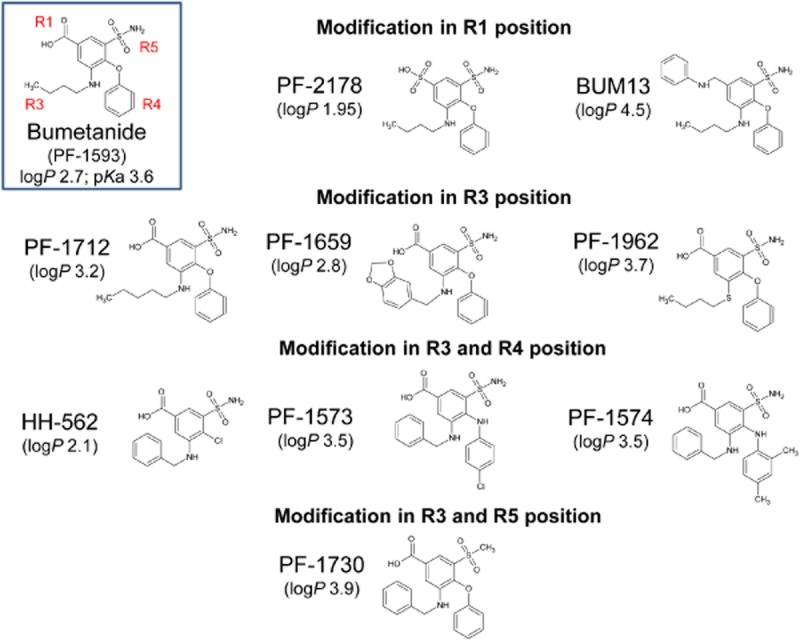
Structures of bumetanide derivatives and their code numbers and, for comparison, bumetanide (PF-1593). For all compounds, lipophilicity (logP) is indicated. Furthermore, the acidic dissociation constant, pKa, of the carboxylic group is shown for bumetanide. Bumetanide derivatives had similar pKa values, except PF-2178 (0.7) and BUM13 (7.0).
Methods
Chemistry
The bumetanide derivatives tested in this study are illustrated in Figure 1. With the exception of BUM13 and BUM13Ox, all samples of bumetanide derivatives used for the present in vitro experiments were from the batches originally used for determining diuretic activity in the dog assay (see below). The synthesis of these compounds has been described previously by one of us (PWF): PF-2178, Palfrey et al. (1980); PF-1712, Feit (1971); PF-1659, Feit (1971); PF-1962, Feit et al. (1974); HH-562, Feit et al. (1970) and Feit (1971); PF-1573, Feit (1971); PF-1574, Feit (1971); PF-1730, Feit and Nielsen (1976). Synthesis of BUM13 was first described by Nielsen and Feit (1978), but it was resynthesized by one of us (TE) both as free base and as oxalate (BUM13Ox) to enhance solubility. These compounds were analysed routinely for C, H, Cl, N and S contents. Furthermore, infrared and NMR spectroscopy were applied. The purity of the compounds was furthermore checked by thin-layer chromatography as detailed elsewhere (Feit et al., 1970; 1974; Feit, 1971; Feit and Nielsen, 1976). The characterization of BUM13 and BUM13Ox included 1H HMR, 13C NMR, MS and combustion analysis. All compounds were of high purity of ≥95%. Bumetanide (≥98% purity) was obtained from Sigma-Aldrich (Copenhagen, Denmark).
Lipophilicity (logP) and acidic dissociation constant (pKa) of the bumetanide derivatives were calculated by the Molecular Operating Environment (MOE 2012.10; Chemical Computing Group Inc., Montreal, QC, Canada). For comparison, logP and pKa of bumetanide were calculated by the same software and verified by published experimental approaches (Orita et al., 1976).
For testing the inhibitory effect of bumetanide and its derivatives on hNKCC2A, drugs had to be dissolved before addition to the test medium used for the transport experiments. However, some of the bumetanide derivatives, particularly BUM13 and BUM13Ox, HH-562, PF-1573 and PF-1730, were difficult to dissolve in vehicles that could subsequently be added to the medium used for the in vitro NKCC2 assay (see below), so that various preliminary experiments with different solvents were performed. The procedure finally used for the different compounds was as follows. Up to 10 mM stock solutions was prepared freshly with DMSO (≥98% purity) (for bumetanide, PF-2178, PF-1712, PF-1659, PF-1962 and PF-1574), absolute ethanol (for HH-562 and PF-1573), Tween®80 (polyoxyethylene 80 sorbitan monooleate; Merck-Schuchardt, München, Germany) and absolute ethanol (1:9; for BUM13 and BUM13Ox), or polyethylene glycol 400 (Rotipuran® Ph. Eur.; Sigma-Aldrich) and ethanol (1:1; for PF-1730). The stock solutions were added to the test medium used for the transport assays, resulting in final drug concentrations of 1, 10 and 100 μM, with respective vehicles included as required to obtain comparable solvent concentrations in all compared conditions (≤1%). The experiments were carried out no later than 3 h after preparation of the drug. Because BUM13 and its oxalate salt BUM13Ox did not differ in solubility or biological activity, only the term BUM13 will be used in this paper.
Oocyte preparation and NKCC2 protein expression
All animal care and experimental procedures complied with the European Community guidelines for the use of experimental animals. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). A total of 31 animals were used in the experiments described here.
Oocytes from Xenopus laevis were obtained from our own frogs (Nasco, Fort Atkinson, WI, USA, or National Centre for Scientific Research, Rennes, France) or purchased from Ecocyte Bioscience, Castrop-Rauxel, Germany. The surgical removal and preparation of defolliculated oocytes was performed essentially as previously described (Fenton et al., 2010). Human NKCC2A in the expression vector pTLN (obtained from Dr H. Castrop, NIH, Bethesda, MD, USA) was linearized downstream from the poly-A segment, and in vitro transcribed using SP6-mMessage mMachine according to manufacturer’s instructions (Ambion, Austin, TX, USA). cRNA was then extracted with MEGAclear (Ambion) and microinjected into defolliculated X. laevis oocytes (50 ng RNA/oocyte). The oocytes were kept in Kulori medium (in mM: 90 NaCl, 1 KCl, 1 CaCl2, 1 MgCl2, 5 HEPES, pH 7.4) for 5–6 days at 19°C before experiments.
NKCC2 activity assay
To activate NKCC2 before the uptake experiments, hNKCC2A-expressing oocytes or uninjected control oocytes were pre-incubated 30 min at room temperature in a K+-free solution containing (in mM: 5 choline chloride, 95 NaCl, 1 MgCl2, 1 CaCl2, 10 HEPES, pH 7.4, 207 mOsm) 5–15 oocytes per well. To measure K+ influx, oocytes were exposed to an isosmotic test solution in which KCl was substituted for choline chloride and 2–3 μCi·mL−1 86Rb+ (NEZ072; PerkinElmer, Germany) included as a tracer for K+. Osmolarities of the test media were verified by using an automatic osmometer type 15 (Löser; Berlin, Germany). Bumetanide (0.03–100 μM) (B3023, Sigma-Aldrich), its derivatives (1–100 μM), or control vehicle (≤1% relevant drug solvent) was added to the test solution. The uptake assay was performed at room temperature with mild agitation for 5 min, which we have demonstrated to be within the linear phase of K+ uptake (data not shown, and Zeuthen and MacAulay, 2012). The influx experiments were terminated by rapid washing three times in ice-cold 86Rb+-free assay solution after which the oocytes were individually dissolved in 200 μL 10% sodium dodecyl sulfate in scintillation vials. The radioactivity present was determined by liquid scintillation β-counting with Ultima Gold XR scintillation liquid (6013199; PerkinElmer, Waltham, MA, USA) using a Tri-Carb 2900TR Liquid Scintillation Analyzer (PerkinElmer). hNKCC2A-mediated K+ uptake was assessed as ([fluxNKCC2-expressing oocytes in the presence of x μM drug]-[fluxuninjected oocytes in the presence of x μM drug]) in order to correct for endogenous NKCC activity.
Data analysis
Sigmoidal curves were constructed for determination of the IC50 value for bumetanide and its derivatives on hNKCC2A using GraphPad Prism 6.0, assuming the curves would reach complete inhibition [going from 100% to 0 according to a dose–response inhibition curve with log(inhibitor) vs. response, variable slope; Y = 100/(1 + 10∧((LogIC50-X)*HillSlope))]. As we only had access to limited amounts of the bumetanide derivatives, these drugs were only tested in two to four concentrations, that is, in the range from 1 to 100 μM. With this limited number of data points, we only assigned an estimation of the IC50 values to these data. To distinguish these IC50 values from the ones obtained from a full data set, the estimated values are referred to as eIC50. eIC50 values (or IC50 in case of bumetanide) were obtained from each individual experiment and averaged across all experiments (at least three) with the given drug to obtain the average eIC50 ± SEM. Although different batches of oocytes express NKCC2 to different levels, we did not observe any trends towards batch-specific differences in the eIC50s and the difference in expression level should thus not affect the obtained values. All representative experiments are shown with mean ± SD whereas pooled experiments and IC50 values are presented as means ± SEM.
Results
Inhibitory potency of bumetanide in the hNKCC2A assay
To obtain an experimental system in which to determine the transport activity of NKCC2, and the inhibitory potency of a range of diuretics on hNKCC2A activity, with minor contamination of endogenous NKCC activity, we employed the X. laevis oocyte expression system. Heterologous expression of hNKCC2A in Xenopus oocytes increased the 86Rb+ uptake 6.3 ± 0.4-fold (n = 37) of that of the uninjected oocytes (a representative experiment is illustrated in Figure 2A). Bumetanide displayed dose-dependent inhibition of 86Rb+ uptake in NKCC2-expressing oocytes (Figure 2A); at 100 μM, the 86Rb+ uptake was reduced to background levels, as previously observed (Zeuthen and MacAulay, 2012). Bumetanide caused, in addition, a reduction of the 86Rb+ uptake in the uninjected oocytes (Figure 2A), which is compatible with the low levels of endogenous NKCC expressed by Xenopus oocytes (Suvitayavat et al., 1994). For determination of the IC50 for hNKCC2A (see Methods), the 86Rb+ uptake in uninjected oocytes was, for all bumetanide concentrations, established in parallel and subsequently subtracted from the 86Rb+ uptake obtained in the NKCC2-expressing oocytes (data summarized for illustrative purposes in Figure 2B, n = 6). The hNKCC2A displayed an IC50 for bumetanide of 4.0 ± 1.0 µM (calculated individually from n = 6).
Figure 2.
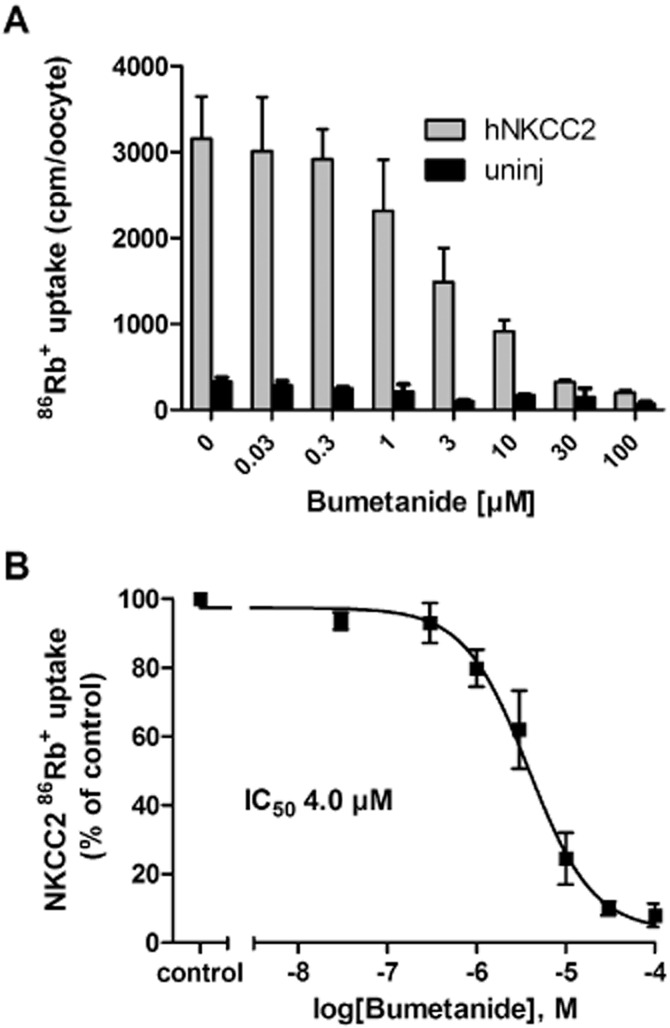
Effect of bumetanide on hNKCC2A-mediated 86Rb+ uptake in Xenopus oocytes. (A) A representative experiment demonstrating the inhibitory effect of 0.03–100 μM bumetanide on 86Rb+ uptake (in cpm, counted for 10 min) in hNKCC2A-expressing oocytes and on batch-matched uninjected oocytes, n = 5–10 oocytes per condition and error bars are SDs. (B) Dose–inhibition curve of bumetanide on hNKCC2A-mediated 86Rb+ uptake (corrected for endogenous NKCC contribution in uninjected oocytes) normalized to control (0 μM bumetanide) and averaged across six experiments, with the IC50 calculated from each individual experiment prior to averaging.
Inhibitory potency of bumetanide derivatives in the hNKCC2A assay
Only limited samples of bumetanide derivatives were available to us and since only freshly prepared solutions were used for each experiment, a restricted number of drug concentrations could be tested for each compound. Therefore, we decided to test each derivative at 1 and 10 μM. If no clear hNKCC2A inhibition was seen, the concentration was increased to 100 μM. Further increase in concentration was not possible because of the limited solubility of most of the bumetanide derivatives. If not otherwise indicated, all experiments were repeated at least three times, although for the two drugs (PF-1659 and BUM13), in which an initial experiment with 100 μM yielded no reduction in NKCC2-mediated 86Rb+ uptake, the experiment was not repeated (n = 1 for 100 μM). In addition to the three concentrations shown in Figure 3, BUM13 was also tested at 20 μM, indicating no inhibitory effect on hNKCC2A (not illustrated). Representative experiments are shown for the nine derivatives in Figure 3. Seven of the nine bumetanide derivatives inhibited NKCC2 at the concentrations applied, although at widely varying potencies. To allow comparison with bumetanide (Figure 2B) and to correlate to diuretic potency, we estimated IC50s from the concentration–response experiments of each derivative and assigned these eIC50 to distinguish these estimates from the IC50 obtained with a full data set for bumetanide (see Methods) (Table 2013).
Figure 3.

Inhibitory effect of bumetanide derivatives on hNKCC2A activity in the Xenopus oocyte assay. 86Rb+ uptake was measured in the absence or presence of different drug concentrations. Data are shown as a representative experiment (out of at least three identical experiments) with means ± SD from 5 to 15 oocytes per condition in each experiment. For PF-1659 and BUM13, the highest concentration of the compound (100 μM) was only tested once due to lack of inhibitory effect.
Table 1.
Diuretic effect of bumetanide derivatives with modifications in R1, R3, R4 or R5 position and inhibitory effect of these derivatives on hNKCC2A

| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Compound | R1 | R3 | R4 | R5 | Diuretic effect in dogs (mL·kg−1 or mEq·kg−1 per 3 h) | Relative diuretic potency (based on Na+ excretion) (bumetanide = 1) | eIC50 for hNKCC2A | |||||
| Dose (mg·kg−1 i.v.) | H2O (mL·kg−1) | Na+ (mEq·kg−1) | K+ (mEq·kg−1) | Cl− (mEq·kg−1) | eIC50 (μM) ± SEM | Relative to bumetanide (=1) | ||||||
| Control | – | 0.93 | 0.10 | 0.16 | 0.08 | |||||||
| Control | – | 2 | 0.19 | 0.13 | 0.13 | |||||||
| Bumetanide (PF-1593) | –COOH | –NH(CH2)3CH3 | –O–C6H5 | –SO2NH2 | 0.25 | 39 | 4.0 | 0.84 | 5.7 | 1 | 4.0 ± 1.0 | 1 |
| 0.1 | 26 | 2.4 | 0.44 | 3.5 | ||||||||
| 0.01 | 10.0 | 0.92 | 0.27 | 1.4 | ||||||||
| Modification in R1 position | ||||||||||||
| PF-2178 | –SO3H | –NH(CH2)3CH3 | –O–C6H5 | –SO2NH2 | 0.25 | 3.7 | 0.925 | 7.2 ± 2.1 | 0.56 | |||
| BUM13 | –CH2NHC6H5 | –NH(CH2)3CH3 | –O–C6H5 | –SO2NH2 | 1 | 33 (6 h) | 4.5 (6 h) | 1.15 (6 h) | 5.4 (6 h) | 0.14 | >100 | <0.04 |
| Modification in R3 position | ||||||||||||
| PF-1712 | –COOH | –NH(CH2)4CH3 | –O–C6H5 | –SO2NH2 | 0.25 | 29 | 2.9 | 0.6 | 4.5 | 0.725 | 3.7 ± 1.7 | 1.08 |
| PF-1659 | –COOH | –NHCH2C6H3, 3,4(–O–CH2–O–) | –O–C6H5 | –SO2NH2 | 0.25 | 2 | 0.1 | 0.3 | 0.3 | 0.025 | >100 | <0.04 |
| PF-1962 | –COOH | –S(CH2)3CH3 | –O–C6H5 | –SO2NH2 | 0.1 | 25.7 | 2.5 | 0.57 | 3.5 | 1.04 | 1.1 ± 0.3 | 3.8 |
| Modification in R3 and R4 positions | ||||||||||||
| HH-562 | –COOH | –NHCH2C6H5 | –CI | –SO2NH2 | 0.25 | 5 | 0.5 | 0.2 | 0.6 | 0.125 | 62.6 ± 18.6 | 0.06 |
| 10 | 21 | 2.0 | 0.53 | 2.6 | ||||||||
| PF-1573 | –COOH | –NHCH2C6H5 | NHC6H5, 4–CI | –SO2NH2 | 0.25 | 26 | 2.9 | 0.6 | 4 | 0.725 | 2.3 ± 1.1 | 1.7 |
| PF-1574 | –COOH | –NHCH2C6H5 | NHC6H5, 2–CH3, 4–CH3 | –SO2NH2 | 0.25 | 2 | 0.4 | 0.4 | 0.4 | 0.1 | 12.2 ± 2.8 | 0.33 |
| Modification in R3 and R5 positions | ||||||||||||
| PF-1730 | –COOH | –NHCH2C6H5 | –O–C6H5 | –SO2CH3 | 0.25 | 21 | 3.0 | 0.99 | 5.1 | 0.75 | 2.2 ± 1.1 | 1.8 |
| 0.1 | 9 | 1.1 | 0.20 | 1.4 | ||||||||
| 1 | 31 | 4.8 | 0.98 | 5.1 | ||||||||
For discussion of the inhibitory potency, the bumetanide derivatives were grouped according to the positions (R1, R3, R4, R5) within the bumetanide structure at which side-chain modifications were performed (Table 2013 and Figure 1, indicated on graphs in Figure 3). Two compounds were modified in position 1 of the molecule (R1 analogues; PF-2178 and BUM13), three had different substituents in position 3 only (R3 analogues; PF-1712, PF-1659 and PF-1962), three had different substituents in positions 3 and 4 (R3/R4 analogues; HH-562, PF-1573 and PF-1574), and one had different substituents in positions 3 and 5 (R3/R5 analogues; PF-1730). Figure 1 also indicates the lipophilicity (logP) of these compounds. As indicated in Figure 1 legend, the acidic dissociation constant, pKa, of bumetanide and its derivatives was similar (∼3.6) except for PF-2178 (0.7) and BUM13 (7.0).
R1 analogues
In R1 analogues (PF-2178 and BUM13), the carboxylic residue of bumetanide was substituted. PF-2178, in which the carboxylic group was replaced by sulfonic acid, was a less potent hNKCC2A inhibitor than bumetanide (eIC50 7.2 μM; relative potency vs. bumetanide 0.56; Table 2013), whereas its diuretic activity in dogs was similar to that of bumetanide (relative diuretic potency 0.925; Table 2013), indicating that the carboxylic group is not a prerequisite for high diuretic activity. However, BUM13, in which the carboxylic group was replaced by an anilinomethyl group, did not inhibit hNKCC2A in concentrations up to 100 μM (Figure 3). Its diuretic activity was only about one-tenth of that of bumetanide (Table 2013).
R3 analogues
R3 analogues (PF-1712, PF-1659, PF-1962) were substituted in the 3-butylamino side chain of bumetanide. PF-1712, in which this side chain was prolonged to a pentylamino group, did not differ in its hNKCC2A inhibitory potency (eIC50 3.7 μM) from bumetanide, and was only moderately less diuretic than bumetanide (Table 2013). In contrast, PF-1659, in which the butylamino side chain was replaced by a 1,3-benzodioxol-5-ylmethylamino group, was devoid of any hNKCC2A inhibitory potency (Figure 3) and almost inactive as a diuretic in dogs (Table 2013). PF-1962, in which the butylamino side chain was replaced by a butylthio side chain (Figure 1), displayed around four times higher potency towards NKCC2 inhibition than bumetanide (eIC50 1.1 μM), while the diuretic activity was comparable (Table 2013).
R3/R4 analogues
All analogues of this type (HH-562, PF-1574, PF-1573) contained a benzylamino residue instead of the butylamino side chain of bumetanide at position 3 (Figure 1). One compound, HH-562, lacked the phenoxy substituent at R position 4, which was replaced by a chloride atom. These modifications markedly reduced the potency to inhibit hNKCC2A (eIC50 62.6 μM) as well as the diuretic activity compared with bumetanide (Table 2013). When the phenoxy substituent of bumetanide was replaced by a 4-chloroanilino group (PF-1573), the inhibitory potency for hNKCC2A was almost doubled (eIC50 2.3 μM) compared with that of bumetanide, whereas the diuretic activity in the dog assay was slightly lower (Table 2013). When the phenoxy substituent of bumetanide was replaced by a 2,4-dimethylanilino group (PF-1574), both hNKCC2A inhibitory potency (eIC50 12.2 μM) and diuretic activity were considerably reduced (Table 2013).
R3/R5 analogues
As with the R3/R4 analogues, the butylamino group in position 3 was replaced by a benzylamino residue; furthermore, the sulfamoyl group in position 5, which is not a prerequisite for the loop diuretic activity of bumetanide (Nielsen and Feit, 1978), was replaced by a methylsulfonyl group, resulting in PF-1730 (Figure 1). This compound was almost twice as potent as bumetanide to inhibit hNKCC2A (eIC50 2.2 μM), but slightly less potent in the dog assay (Table 2013).
Diuretic activity in dogs
In order to determine whether the hNKCC2A inhibitory potencies of the various bumetanide derivatives correlated with their in vivo diuretic activities, we used previously reported data from the dog assay. The dog assay used to determine the diuretic activity of bumetanide and its derivatives has been described in detail by Ostergaard et al. (1972). Compounds were injected i.v. at a dose of 0.25 mg·kg−1 (dissolved by means of NaOH) and the urine volume and electrolyte excretion were determined over 3 h (in some experiments over 6 h). Depending on diuretic activity, some compounds were tested at lower or higher doses. Diuretic data shown in Table 2013 were taken from the following publications of P.W. Feit and H.-H. Frey’s group at Leo Pharma (Ballerup, Denmark): controls, Ostergaard et al. (1972), Feit et al. (1974) and Feit et al. (1970); bumetanide (PF-1593), Ostergaard et al. (1972); PF-2178, Palfrey et al. (1980); BUM13, Nielsen and Feit (1978); PF-1712, Feit (1971); PF-1659, Feit (1971); PF-1962, Feit et al. (1974); HH-562, Feit et al. (1970) and Feit (1971); PF-1573, Feit (1971); PF-1574, Feit (1971); PF-1730, Feit and Nielsen (1976).
For calculating the diuretic potency of the derivatives relative to bumetanide (Table 2013), the Na+ excretion measured after 0.25 mg·kg−1 was used. In case that a compound was not tested at 0.25 mg·kg−1 (BUM13, PF-1962), the diuretic potency of the derivative was compared with that of bumetanide at this dose (0.1 mg·kg−1; PF-1962) or the difference in dosing was taken into account (assuming that the dose–effect relationship is linear) when calculating relative diuretic potency (BUM13, only tested at 1 mg·kg−1).
As shown in Table 2013, most bumetanide derivatives exerted a lower diuretic activity than bumetanide in the dog assay. Only PF-1962 and PF-2178 exhibited potency comparable to bumetanide.
Correlation between inhibitory potency of bumetanide derivatives on hNKCC2A activity and their diuretic activity in dogs
To obtain a relation between the diuretic potency of the bumetanide and its nine tested derivatives and their ability to inhibit NKCC2 transport activity, their diuretic potency (relative to that of bumetanide) was plotted as a function of the eIC50s (Figure 4). Non-linear correlation analysis of these two parameters yielded a correlation coefficient (r2) of 0.817, P < 0.01, indicating a significant correlation between the inhibitory potency of these diuretics on the two experimental systems.
Figure 4.
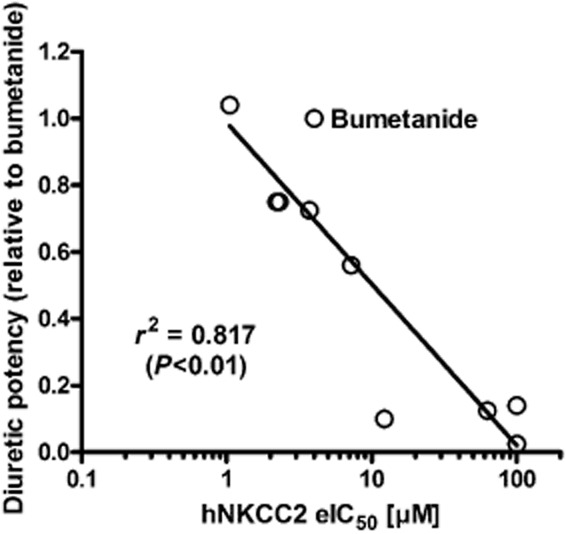
Correlation between log IC50 (or eIC50) for inhibition of hNKCC2A in the Xenopus oocyte assay and diuretic potency in the dog assay for the 10 compounds used in this study (see Figure 1 and Table 2013).
Differences in lipophilicity (logP; see Figure 1), which may affect the relative membrane permeability of the compounds, were not correlated with IC50 values for hNKCC2A (r2 = 0.02032). Furthermore, logP was not significantly correlated with diuretic activity in the dog assay (r2 = 0.06696). The same was true for the acidic dissociation constant, pKa. The most acidic compound (PF-2178) was only slightly less potent in the dog assay than bumetanide (Table 2013).
Discussion
The Na-K-2Cl cotransporter, isoform 2 (SLC12A1; NKCC2), is the major salt transport pathway in the apical membrane of the mammalian TAL and the clinical target for loop diuretics such as bumetanide and furosemide (Alvarez-Leefmans, 2012; Markadieu and Delpire, 2014). The sensitivity to bumetanide of the three full-length spliced variants of NKCC2 (A, B, F) has previously been studied in the Xenopus oocyte heterologous expression system and all variants expressed about the same sensitivity to this diuretic drug (Alvarez-Leefmans, 2012). In the present study, the human NKCC2A variant was inhibited by bumetanide with an IC50 of 4 μM, which is considerably higher than the 0.54 μM reported by Carota et al. (2010) for hNKCC2A expressed in Xenopus oocytes but similar to the 2 μM reported for the mouse NKCC2A by Plata et al. (2002). In order not to obscure the factual transport limit, and thus skew the IC50 obtained for the transport, we determined the NKCC2-mediated transport within the linear part of the time-dependent uptake curve (to prevent time-dependent saturation) and under conditions in which intracellular [Na+] is expected to be constant due to undisturbed Na+/K+-ATPase activity. Plata et al. (2002) and Carota et al. (2010) included the Na+/K+-ATPase inhibitor ouabain in the transport assay, which was carried out with prolonged uptake times (45–60 min), during which the accumulated [Na+]i could not be expelled by the blocked Na+/K+-ATPase. The NKCC1-mediated accumulation of intracellular [Na+] would alter the overall net free energy (kJ·mol−1), providing the driving force for NKCC2 activity, and thus decrease the NKCC2 cotransporter activity and possibly shift the IC50 of bumetanide to the lower value reported by Carota et al. (2010).
To our knowledge, the sensitivity of NKCC2 to bumetanide derivatives has not been reported previously. Overall, we found a significant correlation between inhibition of hNKCC2A in the Xenopus oocyte heterologous expression system and diuretic potency in the in vivo dog assay. Only three of the nine derivatives, PF-1962, PF-1573 and PF-1730, were more potent as inhibitor of hNKCC2A than bumetanide. However, this higher NKCC2 inhibitory potency did not translate into a higher diuretic activity in dogs. For instance, PF-1730 was almost twice as potent as bumetanide against hNKCC2A, but its relative diuretic potency was only 0.75. One possible explanation is a difference in bumetanide sensitivity between human and dog NKCC2. Another possibility is that diuretic activity of bumetanide and its derivatives in dogs depends not only on inhibition of NKCC2 but also on carrier-mediated transport from blood into the kidney (Burckhardt, 2012). Bumetanide is 99% negatively charged at pH 7.4, as the acidic dissociation constant (pKa) of the carboxylic group is 3.6, whereas the pKa of the amino group is 7.7 (Orita et al., 1976). As a consequence of its high ionization rate at pH 7.4 and extensive plasma protein binding (∼97–98%), which restrict membrane penetration from blood into tissues by passive diffusion, bumetanide penetrates only poorly into most tissues, including the brain (Löscher et al., 2013; Puskarjov et al., 2014). In contrast, bumetanide rapidly accumulates in kidney and liver (Cohen et al., 1976) as a result of carrier-mediated transport by organic anion transporters (OATs) in the kidney and organic anion-transporting polypeptides (OATPs) in the liver (Petzinger et al., 1996; VanWert et al., 2010; Burckhardt, 2012). The affinity of bumetanide derivatives to OATs responsible for bumetanide uptake into the kidney is not known, but Petzinger et al. (1993) studied the interaction of various bumetanide derivatives, including most of the compounds used in the present study, with a transport system for bile acids in isolated rat hepatocytes. No correlation was found between the diuretic potency of bumetanide derivatives and their affinity for the hepatic bile salt transport system (Petzinger et al., 1993).
With respect to the present correlation between the natriuretic effect of bumetanide and its derivatives, as measured in vivo in the dog assay system, and their eIC50 values in the in vitro hNKCC2A expression system, it is possible that an even better correlation would have been obtained if the IC50 values had been compared with the renal concentration of each compound that caused a fixed natriuretic response. However, this would require a complete dose–response curve and information on the metabolic fate of each compound. Nevertheless, compounds eliciting weak diuretic responses proved to be poor inhibitors of NKCC2, while compounds promoting active diuresis inhibited NKCC2 at low concentrations (Figure 4 and Table 2013). It must be kept in mind that the eIC50s were obtained from only two to four drug concentrations due to our limited access to these derivatives, and that their value could shift slightly if based on a full inhibitory curve.
The nature of the specific substituents at the R3 and R4 positions had a marked effect on the potency of the molecule. Based on these results, it could be stated that a butylthio substituent in R3 is most favourable. Exchange of the phenoxy group for a 4-chloroanilino group yielded better NKCC2 inhibition results, compared with bumetanide. Compounds like PF-1730 with a methylsulfonyl group instead of sulfamoyl group in position 5 should be considered most interesting drug candidates. Furthermore, an anionic function at R1 appears to be essential for activity. Both the active carboxylic acid and the sulfonic acid derivatives are ionized at physiological pH, but when this position is substituted with a non-ionic residue, for example, an anilinomethyl group as in BUM13, inhibitory activity against hNKCC2A was totally lost. Related substituted aryl- or alkyl-amino-methyl derivatives (see Nielsen and Feit, 1978) produce considerable diuresis but may require metabolism to their corresponding benzoic acids before showing diuretic activity. Thus, the finding that BUM13, which lacks an acidic group at R1, still exerts a relatively potent diuretic effect is most likely a consequence of metabolism to bumetanide, which is substantiated by experiments in mice (C. Brandt, K. Töllner and W. Löscher, unpubl. data). For a more extensive discussion of the structure–activity relationships of these compounds as diuretics, the reader is referred to a previous review by Nielsen and Feit (1978).
In addition to structural requirements for hNKCC2A inhibition, it may be possible to explain the differences between the potencies of the analogues in terms of differences in their relative membrane permeability. This impinges on the issue of sidedness of the bumetanide binding site. If as in NKCC1, the high affinity binding sites for bumetanide are on the cytoplasmic side of the translocation cavity (Somasekharan et al., 2012), differences in relative permeability should be considered in addition to differences in chemical structure. Drug lipophilicity is a very important descriptor governing permeation across a biological membrane (Mälkiä et al., 2004). Lipophilicity is generally expressed quantitatively as the log10 of the partitioning of a neutral drug species between n-octanol and water (logP) and is the most widely used predictor for drug permeation. For the present compounds, logP was neither correlated with their hNKCC2A inhibitory potency in the oocyte expression system nor their diuretic potency in the dog assay. In addition to lipophilicity, passive membrane transport is affected by ionization of the compound in that the passive membrane transport of ions is usually much slower than that of neutral compounds (Mälkiä et al., 2004). Except BUM13, all of the bumetanide derivatives evaluated in this study are highly ionized at plasma pH because of their carboxylic or, in case of PF-2178, sulfonic acid group (Figure 1). Hence, the most acidic compound (PF-2178) was only moderately less potent than bumetanide, which, together with the data from logP, indicates that differences in passive membrane permeability play no major role in the differences between the potencies of the analogues observed in this study. Instead, as discussed above, carrier-mediated transport of bumetanide and its derivatives in the kidney is a prerequisite of any diuretic activity, so that we cannot exclude the possibility that differences in such OAT-mediated transport among the derivatives are involved in the differences in their diuretic potency in the dog assay, nor whether putative endogenous expression of OATs in the plasma membrane of Xenopus oocytes might affect our data. These oocytes possess a large variety of endogenous membrane transporters (Sobczak et al., 2010) and active organic anion transport has been suggested from taurocholate efflux experiments (Shneider and Moyer, 1993), but we are not aware of any studies to the effect of mapping endogenous expression of OATs or OATPs in Xenopus oocyte plasma membranes.
Only one of the nine bumetanide derivatives evaluated in the present study (PF-2178) has previously been tested for inhibition of a NKCC isoform (Palfrey et al., 1980). In this study, a series of diuretically active substituted 3-aminobenzoic acid derivatives and related compounds was investigated on a cyclic AMP-activated Na+-K+ cotransport system in avian erythrocytes (Palfrey et al., 1980), most likely representing NKCC1 that is known to be expressed by erythrocytes of different species, including humans (Flatman and Creanor, 1999; Hannaert et al., 2002; Matskevich et al., 2005; Garay and Alda, 2007). Palfrey et al. (1980) reported a good correlation between the diuretic potency of loop diuretics in the dog and their inhibition of cation cotransport in turkey erythrocytes. However, bumetanide was about 20 times more potent than PF-2178 as an inhibitor of cation transport in the avian erythrocytes, whereas the diuretic potency of the two compounds in dogs is almost the same (Palfrey et al., 1980). Therefore, the correlation between diuretic activity and inhibition of avian NKCC1 was poor for some of the tested compounds. In the present study, PF-2178 was only slightly less potent than bumetanide in inhibiting hNKCC2A, indicating that the hNKCC2A expression system used is a useful predictor of diuretic potency of such compounds. Indeed, although bumetanide inhibits NKCC1 and NKCC2 at about the same potency (Alvarez-Leefmans, 2012), putative isoform-specific inhibitory potencies are not known for bumetanide derivatives. Any correlation with diuretic activity should thus relate to the actual renal target of such compounds, the transporter NKCC2.
From a clinical perspective, it would be interesting to search for bumetanide derivatives with high diuretic potency but low or absent inhibitory effects on NKCC1. Bumetanide, particularly when administered at high doses, is associated with a potential risk of ototoxicity, resulting in a temporary or, in some cases, a permanent loss of hearing in patients, including infants (Ward and Heel, 1984; Rybak, 1993; Rybak and Ramkumar, 2007; Pressler et al., 2015). The ototoxicity of bumetanide is markedly increased by concomitant use of aminoglycoside antibiotics such as gentamycin (Rybak and Ramkumar, 2007; Pressler et al., 2015). Inhibition of NKCC1 by bumetanide has been proposed as a cause of deafness by blocking the generation of the endocochlear potential which is necessary for cochlear amplification (Delpire et al., 1999). Thus, compounds that potently inhibit NKCC2, but not NKCC1, would be interesting candidates in the search of loop diuretics without ototoxicity. It remains to be studied whether PF-1712, PF-1962 and PF-1730, which were the most potent hNKCC2A inhibitors found in this study, are less potent inhibitors of hNKCC1 than bumetanide.
On the other hand, bumetanide derivatives with selectivity for NKCC1 versus NKCC2 would be of high clinical interest because of the recent clinical use of bumetanide in treatment of brain disorders, such as neonatal seizures, epilepsy or autism, in which abnormal function of NKCC1 has been implicated (Kahle and Staley, 2008; Kahle et al., 2008; Ben Ari, 2012; Miles et al., 2012; Löscher et al., 2013; Pressler and Mangum, 2013; Kaila et al., 2014; Puskarjov et al., 2014). Use of bumetanide for treatment of brain disorders is associated with problems including poor brain penetration and systemic adverse effects such as diuresis, hypokalemic alkalosis and hearing loss (Löscher et al., 2013; Puskarjov et al., 2014). Thus, a bumetanide derivative with better brain penetration, high affinity for neuronal NKCC1, but lower diuretic activity than bumetanide would be an interesting option (Löscher et al., 2013; Töllner et al., 2014). It remains to be studied whether the structural requirements for inhibition of NKCC1 differ from those of NKCC2.
In conclusion, as shown by the present structure–activity relationships of bumetanide derivatives in the X. laevis oocyte expression system, an acidic group at R1 is a requirement for inhibition of NKCC2. We also show that modifications at the R3 or R5 position of bumetanide can lead to compounds which are more potent hNKCC2A inhibitors than the parent drug, which is associated with high-ceiling diuretic activity. Our investigation on bumetanide derivatives has established a close correlation between the inhibition of hNKCC2A in Xenopus oocytes and the diuretic potency in vivo and allows analysis of the structural requirements for potent inhibition of NKCC2.
Acknowledgments
We thank the late Professor Ernst Petzinger for extremely helpful discussions during this project and providing samples of several bumetanide derivatives that were previously synthesized by P. W. F. Furthermore, we thank Charlotte G. Iversen and Catia C. G. Andersen for technical assistance, and Dr H. Castrop, NIH, Bethesda (MD, USA), for the hNKCC2A construct.
Glossary
- NKCC
Na-K-Cl cotransporter
- OAT
organic anion transporter
- OATP
organic anion-transporting polypeptide
- TAL
thick ascending limb
Author contributions
K. L. performed the in vitro experiments, analysed and interpreted the data, and critically revised the manuscript. K. T. compiled the in vivo experimental data, interpreted the data and critically revised the manuscript. K. R. performed the drug solution experiments, interpreted the data and critically revised the manuscript. P. W. F. substantially contributed to the conception of the work, synthesized most of the bumetanide derivatives, interpreted the data and critically revised the manuscript. T. E. synthesized part of the bumetanide derivatives, calculated and interpreted part of the data, and critically revised the manuscript. N. M. designed the study, interpreted the data and critically revised the manuscript. W. L. designed the study, interpreted the data and wrote the manuscript.
Conflict of interest
The authors have no conflicts to report.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Transporters. Br J Pharmacol. 2013;170:1706–1796. doi: 10.1111/bph.12450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Leefmans FJ. Intracellular chloride regulation. In: Sperelakis N, editor. Cell physiology Sourcebook. Fourth Edition. Essentials of Membrane Biophysics. London: Academic Press; 2012. pp. 221–259. [Google Scholar]
- Ben Ari Y. Blocking seizures with the diuretic bumetanide: promises and pitfalls. Epilepsia. 2012;53:394–396. doi: 10.1111/j.1528-1167.2011.03378.x. [DOI] [PubMed] [Google Scholar]
- Burckhardt G. Drug transport by Organic Anion Transporters (OATs) Pharmacol Ther. 2012;136:106–130. doi: 10.1016/j.pharmthera.2012.07.010. [DOI] [PubMed] [Google Scholar]
- Carota I, Theilig F, Oppermann M, Kongsuphol P, Rosenauer A, Schreiber R, et al. Localization and functional characterization of the human NKCC2 isoforms. Acta Physiol (Oxf) 2010;199:327–338. doi: 10.1111/j.1748-1716.2010.02099.x. [DOI] [PubMed] [Google Scholar]
- Castrop H, Schnermann J. Isoforms of renal Na-K-2Cl cotransporter NKCC2: expression and functional significance. Am J Physiol Renal Physiol. 2008;295:F859–F866. doi: 10.1152/ajprenal.00106.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen M. Pharmacology of bumetanide. J Clin Pharmacol. 1981;21:537–542. doi: 10.1002/j.1552-4604.1981.tb05662.x. [DOI] [PubMed] [Google Scholar]
- Cohen MR, Hinsch E, Vergona R, Ryan J, Kolis SJ, Schwartz MA. A comparative diuretic and tissue distribution study of bumetanide and furosemide in the dog. J Pharmacol Exp Ther. 1976;197:697–702. [PubMed] [Google Scholar]
- Delpire E, Rauchman MI, Beier DR, Hebert SC, Gullans SR. Molecular cloning and chromosome localization of a putative basolateral Na(+)-K(+)-2Cl- cotransporter from mouse inner medullary collecting duct (mIMCD-3) cells. J Biol Chem. 1994;269:25677–25683. [PubMed] [Google Scholar]
- Delpire E, Lu J, England R, Dull C, Thorne T. Deafness and imbalance associated with inactivation of the secretory Na-K-2Cl co-transporter. Nat Genet. 1999;22:192–195. doi: 10.1038/9713. [DOI] [PubMed] [Google Scholar]
- Feit PW. Aminobenzoic acid diuretics. 2. 4-Substituted-3-amino-5-sulfamylbenzoic acid derivatives. J Med Chem. 1971;14:432–439. doi: 10.1021/jm00287a014. [DOI] [PubMed] [Google Scholar]
- Feit PW. Bumetanide – the way to its chemical structure. J Clin Pharmacol. 1981;21:531–536. doi: 10.1002/j.1552-4604.1981.tb05661.x. [DOI] [PubMed] [Google Scholar]
- Feit PW. Bumetanide: historical background, taxonomy and chemistry. In: Lant AF, editor. Bumetanide. Camforth, UK: Marius Press; 1990. pp. 1–13. [Google Scholar]
- Feit PW, Nielsen OB. Aminobenzoic acid diuretics. 8.(2) 3, 4-Disubstituted 5-methylsulfonylbenzioc acids and related compounds. J Med Chem. 1976;19:402–406. doi: 10.1021/jm00225a013. [DOI] [PubMed] [Google Scholar]
- Feit PW, Bruun H, Nielsen CK. Aminobenzoic acid diuretics. 1. 4-Halogeno-5-sulfamylmetanilic acid derivatives. J Med Chem. 1970;13:1071–1075. doi: 10.1021/jm00300a012. [DOI] [PubMed] [Google Scholar]
- Feit PW, Nielsen OB, Brunn H. Aminobenzoic acid diuretics. 6. 4-Substituted 3-alkylthio-5-sulfamoylbenzoic and 5-sulfamoylthiosalicylic acids. J Med Chem. 1974;17:572–578. doi: 10.1021/jm00252a002. [DOI] [PubMed] [Google Scholar]
- Fenton RA, Moeller HB, Zelenina M, Snaebjornsson MT, Holen T, Macaulay N. Differential water permeability and regulation of three aquaporin 4 isoforms. Cell Mol Life Sci. 2010;67:829–840. doi: 10.1007/s00018-009-0218-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flatman PW, Creanor J. Regulation of Na+-K+-2Cl- cotransport by protein phosphorylation in ferret erythrocytes. J Physiol. 1999;517(Pt 3):699–708. doi: 10.1111/j.1469-7793.1999.0699s.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey HH. Pharmacology of bumetanide. Postgrad Med J. 1975;51(Suppl. 6):14–18. [PubMed] [Google Scholar]
- Frizzell RA, Field M, Schultz SG. Sodium-coupled chloride transport by epithelial tissues. Am J Physiol. 1979;236:F1–F8. doi: 10.1152/ajprenal.1979.236.1.F1. [DOI] [PubMed] [Google Scholar]
- Gamba G, Miyanoshita A, Lombardi M, Lytton J, Lee WS, Hediger MA, et al. Molecular cloning, primary structure, and characterization of two members of the mammalian electroneutral sodium-(potassium)-chloride cotransporter family expressed in kidney. J Biol Chem. 1994;269:17713–17722. [PubMed] [Google Scholar]
- Garay RP, Alda O. What can we learn from erythrocyte Na-K-Cl cotransporter NKCC1 in human hypertension? Pathophysiology. 2007;14:167–170. doi: 10.1016/j.pathophys.2007.09.006. [DOI] [PubMed] [Google Scholar]
- Haas M, Forbush B., III The Na-K-Cl cotransporters. J Bioenerg Biomembr. 1998;30:161–172. doi: 10.1023/a:1020521308985. [DOI] [PubMed] [Google Scholar]
- Hannaert P, Alvarez-Guerra M, Pirot D, Nazaret C, Garay RP. Rat NKCC2/NKCC1 cotransporter selectivity for loop diuretic drugs. Naunyn Schmiedebergs Arch Pharmacol. 2002;365:193–199. doi: 10.1007/s00210-001-0521-y. [DOI] [PubMed] [Google Scholar]
- Kahle KT, Staley KJ. The bumetanide-sensitive Na-K-2Cl cotransporter NKCC1 as a potential target of a novel mechanism-based treatment strategy for neonatal seizures. Neurosurg Focus. 2008;25:1–8. doi: 10.3171/FOC/2008/25/9/E22. [DOI] [PubMed] [Google Scholar]
- Kahle KT, Staley KJ, Nahed BV, Gamba G, Hebert SC, Lifton RP, et al. Roles of the cation-chloride cotransporters in neurological disease. Nat Clin Pract Neurol. 2008;4:490–503. doi: 10.1038/ncpneuro0883. [DOI] [PubMed] [Google Scholar]
- Kaila K, Price TJ, Payne JA, Puskarjov M, Voipio J. Cation-chloride cotransporters in neuronal development, plasticity and disease. Nat Rev Neurosci. 2014;15:637–654. doi: 10.1038/nrn3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: Reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löscher W, Puskarjov M, Kaila K. Cation-chloride cotransporters NKCC1 and KCC2 as potential targets for novel antiepileptic and antiepileptogenic treatments. Neuropharmacology. 2013;69:62–74. doi: 10.1016/j.neuropharm.2012.05.045. [DOI] [PubMed] [Google Scholar]
- Markadieu N, Delpire E. Physiology and pathophysiology of SLC12A1/2 transporters. Pflugers Arch. 2014;466:91–105. doi: 10.1007/s00424-013-1370-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matskevich I, Hegney KL, Flatman PW. Regulation of erythrocyte Na-K-2Cl cotransport by threonine phosphorylation. Biochim Biophys Acta. 2005;1714:25–34. doi: 10.1016/j.bbamem.2005.06.001. [DOI] [PubMed] [Google Scholar]
- Mälkiä A, Murtomaki L, Urtti A, Kontturi K. Drug permeation in biomembranes: in vitro and in silico prediction and influence of physicochemical properties. Eur J Pharm Sci. 2004;23:13–47. doi: 10.1016/j.ejps.2004.05.009. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles R, Blaesse P, Huberfeld G, Wittner L, Kaila K. Chloride homeostasis and GABA signaling in temporal lobe epilepsy. In: Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV, editors. Jasper’s Basic Mechanisms of the Epilepsies. 4th edn. New York: Oxford University Press; 2012. pp. 581–590. [PubMed] [Google Scholar]
- Nielsen OBT, Feit PW. Structure-activity relationships of aminobenzoic acid diuretics and related compounds. Am Chem Soc Symp Ser, Diuretic Agents. 1978;83:12–23. [Google Scholar]
- Olsen UB. The pharmacology of bumetanide. Acta Pharmacol Toxicol (Copenh) 1977;41:1–29. doi: 10.1111/j.1600-0773.1977.tb03209.x. [DOI] [PubMed] [Google Scholar]
- Orita Y, Ando A, Urakabe S, Abe H. A metal complexing property of furosemide and bumetanide: determination of pK and stability constant. Arzneimittelforschung. 1976;26:11–13. [PubMed] [Google Scholar]
- Ostergaard EH, Magnussen MP, Nielsen CK, Eilertsen E, Frey HH. Pharmacological properties of 3-n-butylamino-4-phenoxy-5-sulfamylbenzoic acid (Bumetanide), a new potent diuretic. Arzneimittelforschung. 1972;22:66–72. [PubMed] [Google Scholar]
- Palfrey HC, Feit PW, Greengard P. cAMP-stimulated cation cotransport in avian erythrocytes: inhibition by ‘loop’ diuretics. Am J Physiol. 1980;238:C139–C148. doi: 10.1152/ajpcell.1980.238.3.C139. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledge base of drug targets and their ligands. Nucl Acids Res. 2014;42(Database Issue):D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petzinger E, Follmann W, Blumrich M, Schermuly R, Schulz S, Hahnen J, et al. Interaction of bumetanide derivatives with hepatocellular bile acid uptake. Am J Physiol. 1993;265:G942–G954. doi: 10.1152/ajpgi.1993.265.5.G942. [DOI] [PubMed] [Google Scholar]
- Petzinger E, Blumrich M, Bruhl B, Eckhardt U, Follmann W, Honscha W, et al. What we have learned about bumetanide and the concept of multispecific bile acid/drug transporters from the liver. J Hepatol. 1996;24(Suppl. 1):42–46. [PubMed] [Google Scholar]
- Plata C, Meade P, Vazquez N, Hebert SC, Gamba G. Functional properties of the apical Na+-K+-2Cl- cotransporter isoforms. J Biol Chem. 2002;277:11004–11012. doi: 10.1074/jbc.M110442200. [DOI] [PubMed] [Google Scholar]
- Pressler RM, Mangum B. Newly emerging therapies for neonatal seizures. Semin Fetal Neonatal Med. 2013;18:216–223. doi: 10.1016/j.siny.2013.04.005. [DOI] [PubMed] [Google Scholar]
- Pressler RM, Boylan GB, Marlow N, Blennow M, Chiron C, Cross JH, et al. Bumetanide for the treatment of seizures in newborn babies with hypoxic ischaemic encephalopathy (NEMO): an open-label, dose finding, and feasibility phase 1/2 trial. Lancet Neurol. 2015;14:469–477. doi: 10.1016/S1474-4422(14)70303-5. [DOI] [PubMed] [Google Scholar]
- Puskarjov M, Kahle KT, Ruusuvuori E, Kaila K. Pharmacotherapeutic targeting of cation-chloride cotransporters in neonatal seizures. Epilepsia. 2014;55:806–818. doi: 10.1111/epi.12620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rybak LP. Ototoxicity of loop diuretics. Otolaryngol Clin North Am. 1993;26:829–844. [PubMed] [Google Scholar]
- Rybak LP, Ramkumar V. Ototoxicity. Kidney Int. 2007;72:931–935. doi: 10.1038/sj.ki.5002434. [DOI] [PubMed] [Google Scholar]
- Shneider BL, Moyer MS. Characterization of endogenous carrier-mediated taurocholate efflux from Xenopus laevis oocytes. J Biol Chem. 1993;268:6985–6988. [PubMed] [Google Scholar]
- Sobczak K, Bangel-Ruland N, Leier G, Weber WM. Endogenous transport systems in the Xenopus laevis oocyte plasma membrane. Methods. 2010;51:183–189. doi: 10.1016/j.ymeth.2009.12.001. [DOI] [PubMed] [Google Scholar]
- Somasekharan S, Tanis J, Forbush B. Loop diuretic and ion-binding residues revealed by scanning mutagenesis of transmembrane helix 3 (TM3) of Na-K-Cl cotransporter (NKCC1) J Biol Chem. 2012;287:17308–17317. doi: 10.1074/jbc.M112.356014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suvitayavat W, Palfrey HC, Haas M, Dunham PB, Kalmar F, Rao MC. Characterization of the endogenous Na(+)-K(+)-2Cl- cotransporter in Xenopus oocytes. Am J Physiol. 1994;266:C284–C292. doi: 10.1152/ajpcell.1994.266.1.C284. [DOI] [PubMed] [Google Scholar]
- Töllner K, Brandt C, Töpfer M, Brunhofer G, Erker T, Gabriel M, et al. A novel prodrug-based strategy to increase effects of bumetanide in epilepsy. Ann Neurol. 2014;75:550–562. doi: 10.1002/ana.24124. [DOI] [PubMed] [Google Scholar]
- Töpfer M, Töllner K, Brandt C, Twele F, Bröer S, Löscher W. Consequences of inhibition of bumetanide metabolism in rodents on brain penetration and effects of bumetanide in chronic models of epilepsy. Eur J Neurosci. 2014;39:673–687. doi: 10.1111/ejn.12424. [DOI] [PubMed] [Google Scholar]
- VanWert AL, Gionfriddo MR, Sweet DH. Organic anion transporters: discovery, pharmacology, regulation and roles in pathophysiology. Biopharm Drug Dispos. 2010;31:1–71. doi: 10.1002/bdd.693. [DOI] [PubMed] [Google Scholar]
- Ward A, Heel RC. Bumetanide. A review of its pharmacodynamic and pharmacokinetic properties and therapeutic use. Drugs. 1984;28:426–464. doi: 10.2165/00003495-198428050-00003. [DOI] [PubMed] [Google Scholar]
- Xu JC, Lytle C, Zhu TT, Payne JA, Benz E, Jr, Forbush B., III Molecular cloning and functional expression of the bumetanide-sensitive Na-K-Cl cotransporter. Proc Natl Acad Sci U S A. 1994;91:2201–2205. doi: 10.1073/pnas.91.6.2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeuthen T, MacAulay N. Cotransport of water by Na(+)-K(+)-2Cl(-) cotransporters expressed in Xenopus oocytes: NKCC1 versus NKCC2. J Physiol. 2012;590:1139–1154. doi: 10.1113/jphysiol.2011.226316. [DOI] [PMC free article] [PubMed] [Google Scholar]