

Abstract
Background and Purpose
The intermediate conductance calcium/calmodulin-regulated K+ channel KCa3.1 produces hyperpolarizing K+ currents that counteract depolarizing currents carried by transient receptor potential (TRP) channels, and provide the electrochemical driving force for Cl− and fluid movements. We investigated whether a deficiency in KCa3.1 (KCa3.1−/−) protects against fatal pulmonary circulatory collapse in mice after pharmacological activation of the calcium-permeable TRP subfamily vanilloid type 4 (TRPV4) channels.
Experimental Approach
An opener of TRPV4 channels, GSK1016790A, was infused in wild-type (wt) and KCa3.1−/− mice; haemodynamic parameters, histology and pulmonary vascular reactivity were measured; and patch clamp was performed on pulmonary arterial endothelial cells (PAEC).
Key Results
In wt mice, GSK1016790A decreased right ventricular and systemic pressure leading to a fatal circulatory collapse that was accompanied by increased protein permeability, lung haemorrhage and fluid extravasation. In contrast, KCa3.1−/− mice exhibited a significantly smaller drop in pressure to GSK1016790A infusion, no haemorrhage and fluid water extravasation, and the mice survived. Moreover, the GSK1016790A-induced relaxation of pulmonary arteries of KCa3.1−/− mice was significantly less than that of wt mice. GSK1016790A induced TRPV4 currents in PAEC from wt and KCa3.1−/− mice, which co-activated KCa3.1 and disrupted membrane resistance in wt PAEC, but not in KCa3.1−/− PAEC.
Conclusions and Implications
Our findings show that a genetic deficiency of KCa3.1 channels prevented fatal pulmonary circulatory collapse and reduced lung damage caused by pharmacological activation of calcium-permeable TRPV4 channels. Therefore, inhibition of KCa3.1channels may have therapeutic potential in conditions characterized by abnormal high endothelial calcium signalling, barrier disruption, lung oedema and pulmonary circulatory collapse.
Tables of Links
| LIGANDS | |
|---|---|
| ACh | Nitric oxide (NO) |
| GSK1016790A | Phenylephrine |
| HC067047 | TRAM-34 |
| Indomethacin | UCL1684 |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14
Alexander et al., 2013a,b,).
Introduction
The endothelial calcium/calmodulin-regulated K+ channel with intermediate conductance, KCa3.1 (Ishii et al., 1997; Alexander et al., 2013a) and Na+/Ca2+ permeable channels of the transient receptor potential (TRP) gene family regulate arterial tone by stimulating Ca2+ entry-triggered and hyperpolarization-mediated dilation to mechanical stress and stimulation of GPCRs (Kohler et al., 2006; Sonkusare et al., 2012). Besides these roles, some TRP channels control endothelial/epithelial barrier functions and vascular integrity (Alvarez et al., 2006; Mene et al., 2013; Nilius and Szallasi, 2014) while KCa3.1 provides the negative driving force required for Cl− and water transport in some cells and most secretory epithelia (Vandorpe et al., 1998; Devor et al., 2000; Grgic et al., 2009). Among the endothelial Ca2+-permeable TRPs, (vanilloid) type 4 (TRPV4) (Nilius and Szallasi, 2014) is best characterized (Watanabe et al., 2003). Its activation by the selective openers, GSK1016790A (Willette et al., 2008) and 4α-phorbol-12,13-didecanoate (4αPDD) (Watanabe et al., 2002), causes Ca2+ entry (Kohler et al., 2006) and produces arterial dilation by stimulating Ca2+-dependent NOS (eNOS) (Kohler et al., 2006) and endothelium-dependent hyperpolarization (EDH) in mice (Hartmannsgruber et al., 2007). Notably, stretch activation of endothelial TRPV4 channels followed by pulmonary vascular pressure-mediated Ca2+ uptake is thought to be involved in acute lung injury (Alvarez et al., 2006; Jian et al., 2008; Yin et al., 2008; 2011,). Moreover, systemic administration of GSK1016790A can cause fatal circulatory collapse, endothelial cell retraction and detachment, and ensuing loss of arterial tone and vascular leakage (Willette et al., 2008). This indicates that TRPV4 has a critical role in endothelial barrier integrity and circulatory homeostasis, which precludes the use of TRPV4 activators for blood pressure-lowering therapies but raises the possibility that TRPV4 and TRPV4-linked pathways can be exploited for the prevention of pathological vascular permeability, circulatory collapse and lung oedema to pulmonary venous pressure increases as suggested by recent studies in canine and rodent models of heart failure (Thorneloe et al., 2012; Hilfiker et al., 2013).
At present, the cellular signal transduction pathways used by TRPV4 to produce detrimental hypotension, endothelial damage and circulatory collapse are not well defined but are mostly related to calcium overload of the endothelium, as vascular responses to TRPV4 activation are abolished by clamping of the intracellular calcium concentration (Kohler et al., 2006). In particular, the functional coupling of TRPV4 with other determinants of endothelial ionic regulation to induce and sustain Ca2+ influx and overload is not yet clear. In the endothelium, KCa3.1 channels (Kohler and Ruth, 2010) are pivotal constituents of EDH-mediated vasodilatation to ACh receptor activation (for review see Weston et al., 2010; Feletou et al., 2012). Here, we hypothesize (i) that KCa3.1 channels serve as endogenous sensors of TRPV4-mediated Ca2+ entry and could amplify TRPV4-induced dilator responses by causing hyperpolarization (Qian et al., 2014) that exacerbates pulmonary vasorelaxation, hypotension or even circulatory collapse; (ii) that TRPV4 uses KCa3.1 to evoke cell swelling, blebbing and detachment. This could explain mechanistically the disruption of the endothelial barrier seen in the lung after pharmacological TRPV4 activation. (iii) That TRPV4-mediated activation may use endothelial/epithelial KCa3.1 channels (Li et al., 2011; Kroigaard et al., 2012) to produce K+ efflux and the concomitant fluid extravasation from the blood into the alveolar space. That the latter is a possible scenario is suggested by results from recent and former pharmacological studies in rats, which showed that pharmacological inhibition of KCa3.1 reduces fluid transport into the brain after ischaemic stroke (Chen et al., 2011; 2015,) and after trauma (Mauler et al., 2004).
In the present study, we tested our hypotheses of pathophysiological synergy of TRPV4 and KCa3.1 channels by measuring pulmonary pressure and arterial relaxation, vascular and epithelial lung damage, and fluid extravasation, as well as the interaction of TRPV4 and KCa3.1 membrane currents to induce pharmacological TRPV4 activation at the cellular level in wild-type (wt) and KCa3.1−/− mice (Si et al., 2006). Our main findings are that GSK1016790A through TRPV4 co-activates the calcium-sensing KCa3.1 in pulmonary endothelium and that a deficiency in KCa3.1 protects against TRPV4-induced pulmonary arterial relaxation, fluid extravasation, haemorrhage, pulmonary circulatory collapse and cardiac arrest in vivo. These data identify KCa3.1 channels as crucial molecular components in the downstream TRPV4 signal transduction pathway and as a potential target for the prevention of undesired fluid extravasation, vasorelaxation and circulatory collapse in situations of pharmacologically-induced endothelial Ca2+ overload.
Methods
Mice
KCa3.1−/− and wt mice were derived from our breeding colonies (Si et al., 2006; Lambertsen et al., 2012; Wandall-Frostholm et al., 2014). All animal protocols or in vitro studies were performed in agreement with the ARRIVE guidelines and approved by the Danish animal care committee (permission 2011/561–2011; Kilkenny et al., 2010; McGrath et al., 2010).
Right ventricular haemodynamic parameters
Mice were anaesthetized by s.c. injection of a cocktail of fentanyl (0.158 mg·mL−1) and midazolam (2.5 mg·mL−1). Anaesthesia was maintained by injection of 0.02 mL of the cocktail every 20 min. Mice were kept on a 37°C heated pad. The right jugular vein was isolated, occluded rostrally by a suture (Seralon 3/0, Serag Weissner, Germany), and a 1F microtip pressure catheter (SPC-1000, Millar Instruments Inc., Houston, TX, USA) was inserted and advanced into the right ventricle. To examine the effects of i.v. infusion of GSK1016790A on right ventricular pressure, heart rate and pressure were initially recorded for 3 min. When haemodynamic parameters were stable, cumulatively increasing amounts of GSK1016790A (0.01, 0.03, 0.1 and 0.2 mg·kg−1) were infused into the right jugular vein, and right ventricular haemodynamic parameters were measured continuously. GSK1016790A was dissolved in 50% PEG-400 (Sigma-Aldrich, Wacker Chemic AG, Bunghausen, Germany) and 50% NaCl, 0.9%. In a subset of experiments, NG-nitro-L-arginine (L-NOARG, 4 mg·kg−1) was infused before GSK1016790A. After measurements were completed, the mice were killed by cervical dislocation. All data were recorded and analysed using PowerLab and Chart7 software (AD Instruments, Oxfordshire, UK).
Evans blue distribution and histology
The experiments were performed in anaesthetized mice using isoflurane (5% for induction and 3% for maintenance). After a 3 min recording of right ventricular pressure, Evans blue (2 mL·kg−1) was infused into the left jugular vein, followed by infusion of GSK1016790A (0.1 mg·kg−1) or vehicle [NaCl/PEG (1:1)] and pressure was recorded over 15 min. Heart and lung were removed and photographed. Lungs were snap-frozen in liquid nitrogen after homogenization in PBS. Evans blue was extracted by incubation in two volumes of formamide at 60°C for 18 h. Then, samples were centrifuged at 1000× g for 30 min, supernatants were collected and absorbance was measured at 620 and 740 nm (Kaner et al., 2000). Absorbance values were calculated and corrected for contaminating haem pigments using the formula: AbsorbanceEvansblue = E620 − (1.426 × E740 + 0.030).
In another series, lungs were fixated by perfusion with 4% formaldehyde and kept in formaldehyde for another 2 days before being stored in 70% ethanol, until they were embedded in paraffin. Transverse sections (3 μm thick) were cut, mounted on slides and stained with haematoxylin and eosin (H&E). All histological measurements were performed on six slides from each animal.
Measurement of haemorrhage in histological lung sections was performed by measuring areas containing aggregation of red blood cells in alveolar space in a predefined area using the image analysis software (ImageJ, NIH, Bethesda, MD, USA).
The perivascular cuff area was measured using a superimposed grid and counting the line intersections occurring within the area between the outer layer of the smooth muscle and the edge of the vessel compared with the vessel size.
Isometric myography
Male and female mice of either genotype (n = 10) were killed by cervical dislocation and exsanguinated by decapitation. Lungs were removed and placed in a 4°C physiological saline solution (PSS). Segments of intrapulmonary first- and second-order branches of the main right pulmonary arteries (PA) were carefully dissected under a microscope by removing the surrounding tissue. Segments of approximately 2 mm were mounted on two 40 μm steel wires in microvascular myographs (Danish Myotechnology, model 310A, Aarhus, Denmark) for isometric tension recording. The baths were heated to 37°C and equilibrated with 5% CO2 in artificial air (21% O2, 74% N2) to keep pH = 7.4. The arterial segments were stretched to 2.4 kPa, corresponding to a transmural pressure of 18 mmHg (Elmedal et al., 2005), then the PA were stimulated with high potassium PSS (KPSS, 60 mM), washed and contracted with phenylephrine (0.1 μM). Only PAs that developed a tension of more than 2.7 kPa were used for the experiment. In other experiments, the endothelium was removed by gently rubbing the luminal surface with a human hair, a procedure confirmed by lack of relaxation to ACh (not shown). In other series, the segments were incubated for 20 min with L-NOARG (300 μM) or the TRPV4 channel blocker HC067047 (1 μM). At stable contraction, we determined relaxations to GSK1016790A (10 nM) or sodium nitroprusside (SNP, 0.1 nM–10 μM). Unless otherwise stated, experiments were performed in the presence of indomethacin (3 μM). Mean diameters of these PA segments did not differ between genotypes (not shown). Regarding endothelium-independent responses, PAs from either genotype behaved similarly: contractions to high potassium (KPSS, 60 mM) and to phenylephrine (PE; 0.1 μM) were similar in wt (0.71 ± 0.05 and 0.67 ± 0.50 N·m−1, respectively) and in KCa3.1−/− (0.64 ± 0.04 and 0.67 ± 0.06 N·m−1 respectively).
Isolation of pulmonary arterial endothelial cells (PAEC) and patch clamp electrophysiology
Isolation of endothelial cells from PAs was carried out using previously described protocols (Stankevicius et al., 2011). In brief, 0.4 cm segments of first-order branches of main right PA were carefully dissected from the excised lungs and cleaned from perivascular tissues. Segments were fixed on a glass capillary and filled with a trypsin (0.25%)/EDTA buffer (Biochrom KG, Berlin, Germany). After being filled with the buffer, PAs were sutured and incubated for 15 min at 37°C. Thereafter, PAs were cut open longitudinally and the luminal side was smoothly scraped with a 10 μL pipette tip. Detached single PAECs and small PAEC clusters (three to four cells) were soaked up and transferred to a culture dish containing coverslips and DMEM supplemented with 10% fetal calf serum and penicillin/streptomycin (Biochrom KG). Cells were allowed to attach to coverslips for 1–3 h and thereafter used for patch clamp experiments within the same day. Membrane currents were recorded in the whole-cell mode in PAEC from wt (cell capacitance: 8.2 ± 0.4 pF, n = 10) and KCa3.1−/− (9.2 ± 0.6 pF, n = 7) using an Axopatch patch clamp amplifier (Axon Instruments, Foster City, CA, USA). Data were analysed using Clampfit 9.2 software (Axon Instruments Inc, Foster City, CA, USA). The KCl pipette solution contained (in mM): 140 KCl, 1 Na2ATP, 1 MgCl2, 2 EGTA, 0.72 CaCl2, (0.1 μM [Ca2+]free) and 5 HEPES, pH 7.2. The NaCl bath solution contained (mM): 140 NaCl, 5 KCl, 1 MgCl2, 1 CaCl2, 10 glucose and 10 HEPES (pH 7.4). For activation of TRPV4-mediated currents, GSK1016790A was added to the bath solution (final concentration 200 nM). For blocking experiments, the selective TRPV4 blocker HC067047 (1 μM) was applied. Co-activated KCa3.1 and KCa2.3 channels were blocked by the selective KCa3.1 blocker TRAM-34 (1 μM) and the KCa2.X blocker UCL1684 (1 μM) respectively.
Morphological in vitro studies on HUVEC
HUVEC cells were a kind gift of Dr. Ángel-Luis García Otín (IACS/GIPASC, Hospital Universitario Miguel Servet, Zaragoza). The study on these cells was approved by the local ethics committee, (CEICA), permission nr. PI12/097. Cells (passages 4–5) were grown in EGM-2 medium (Lonza, Spain). HUVEC was grown to subconfluency (10–30%) in culture flask. The cells were stimulated with 200 nM GSK1016790A to ensure full activation of the TRPV4 channels, 200 nM GSK1016790A plus 1 μM TRAM-34, 200 nM GSK1016790A plus 1 μM HC067047 or vehicle (DMSO) was used. Photographs of cells were taken 45 min thereafter. The amount (%) of fully rounded cells and disrupted cells per high power field (200× magnification) was determined by counting. Cells were considered disrupted when exhibiting retracted cytoplasmic edges or loss of pseudopodia-like extensions (with or without visibly condensed nuclei or bleb formation).
Solutions and drugs
PSS was composed of (mM): 119 NaCl, 4.7 KCl, 1.17 MgSO4, 25 NaHCO3, 1.18 KH2PO4, 5.5 glucose, 1.6 CaCl2 and 0.026 EDTA. For KPSS, 60 mM Na+ was replaced by 60 mM K+. PE, GSK1016790A, TRAM-34, UCL1684, L-NOARG and indomethacin were purchased from Sigma Aldrich (St. Louis, MO, USA). HC067047 was from Tocris Bioscience (Bristol, UK). Stock solution of GSK1016790A, TRAM-34, UCL1684, indomethacin and HC067047 was prepared freshly with a mixture of DMSO and bath medium (1:10). The final DMSO concentration in the bath solution did not exceed 0.3% (with three compounds). Such DMSO concentrations have no unspecific effects on the channels investigated. L-NOARG was dissolved in Ca2+-free PSS. If not stated otherwise, all further dilutions were made in distilled water. All stock solutions were kept at −20°C until use.
Statistical analyses
In vivo haemodynamic data and Evans blue experiments were analysed using a two-way anova to test for genotype and drug effect followed by Bonferroni’s post hoc test. Relaxations are expressed as % of phenylephrine-induced contraction and analysed using one-way anova followed by Tukey’s post hoc test. All arteries were from different experimental animals, and n indicates the number of preparations (one preparation per animal) examined. All other data were tested using Student’s two-tailed unpaired t-test. The results are expressed as means ± SEM. In all cases, P < 0.05 was considered significant.
Results
Alterations of right ventricular pressure profiles by pharmacological TRPV4 activation in wt and KCa3.1−/− mice
To study the deleterious consequences of TRPV4 activation in the pulmonary circulation of wt and KCa3.1−/− mice, we infused GSK1016790A via the left jugular vein at cumulatively increasing dosages of 0.01, 0.03, 0.1 and 0.2 mg·kg−1, and measured the right ventricular pressure profiles and heart rate (Figure 1A, see Supporting Information Table S1). Right ventricular mean pressure (RVMP) prior to infusion of GSK1016790A was similar in wt (10.1 ± 0.1 mmHg, n = 7) and KCa3.1−/− (10.7 ± 1.2 mmHg, n = 7) mice (Figure 1A). Infusion of GSK1016790A at dosages of 0.03 mg·kg−1 and above severely decreased RVMP to zero in wt mice resulting in circulatory collapse and death of all mice (Figure 1A and Supporting Information Fig. S1). This drop in RVMP was caused by the severe drop in right ventricular systolic pressure (Figure 1B) with no or minor change in the very low right ventricular diastolic pressure (Figure 1C). The individual responses to the different dosages of GSK1016790A varied: at 0.03 mg·kg−1, RVMP dropped to zero in two mice, in three mice at 0.1 mg·kg−1 and in two mice at 0.2 mg·kg−1. Infusion of L-NOARG (4 mg·kg−1) increased basal RVMP by about 3 mmHg but failed to prevent the depressor response to any GSK1016790A dosage (Figure 1A). In KCa3.1−/−, GSK1016790A at all dosages produced weak depressor responses, which were statistically not different from vehicle, and all seven KCa3.1−/− mice survived 30 min after 0.2 mg·kg−1 GSK1016790A infusion (Figure 1A–C, Supporting Information Fig. S1) before being killed. These data suggest that KCa3.1−/− mice are protected against pulmonary circulatory collapse caused by pharmacological TRPV4 activation.
Figure 1.
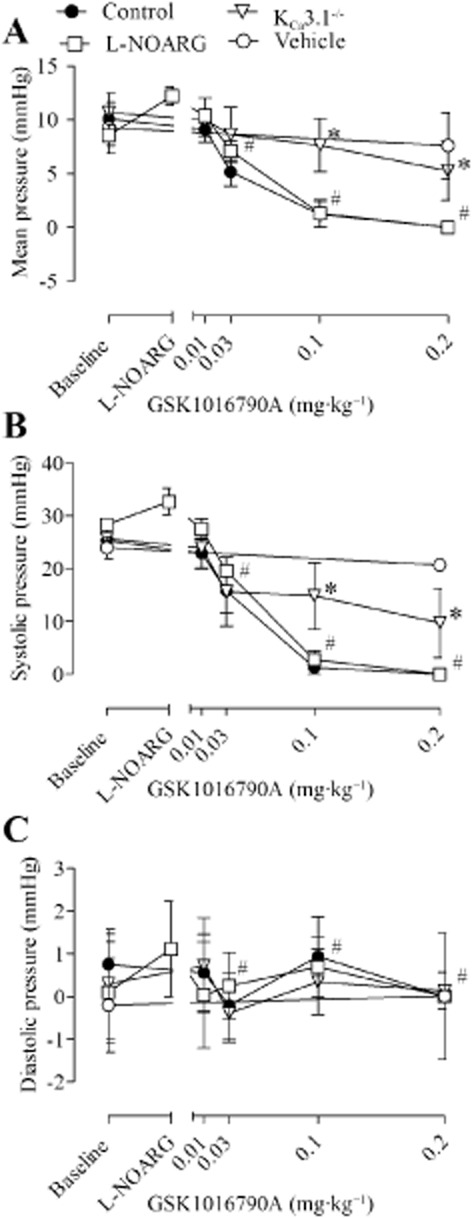
Right ventricular mean (A), systolic (B) and diastolic (C) pressure after i.v. infusion of GSK1016790A (0.01, 0.03, 0.1 and 0.2 mg·kg−1) in wt mice in the absence and presence of L-NOARG (4 mg·kg−1) and in KCa3.1−/− mice. Data are means ± SEM (n = 4–7). Two-way anova. *P < 0.05 from wt. #Indicates dosages of GSK1016790A at which wt mice were experiencing circulatory collapse.
Lung damage to GSK1016790A infusion in wt and KCa3.1−/− mice
To measure differences in protein extravasation caused by GSK1016790A stimulation, we infused Evans blue either together with vehicle or GSK1016790A. We found that combined Evans blue and GSK1016790A infusion led to substantial stain of lung tissue while the infusion of the vehicle did not produce stain (Figure 2). Yet, there was no difference between wt and KCa3.1−/− mice (Figure 2B), suggesting that protein extravasation was unaffected by KCa3.1 deficiency.
Figure 2.
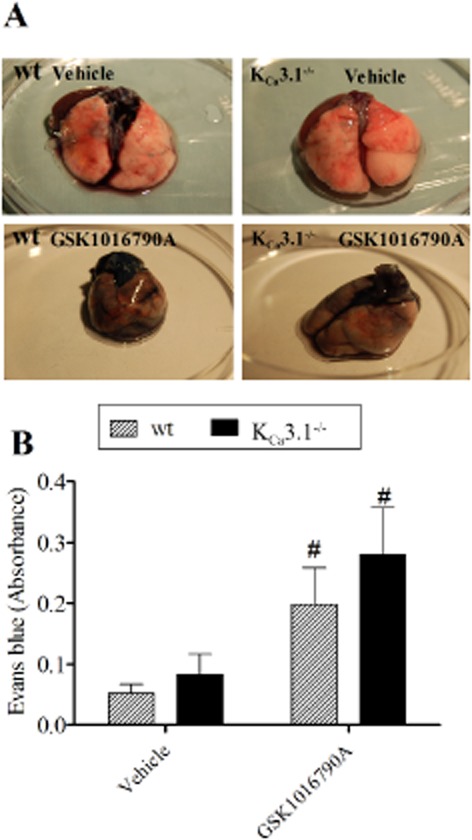
GSK1016790A induced lung barrier disruption in wt mice and KCa3.1−/− mice. (A) Evans blue stain showing barrier disruption in lungs from wt (left) versus KCa3.1−/− (right) mice following vehicle (top) or GSK1016790A (bottom) infusion. (B) Summary data. Data points are means ± SEM (n = 4–6). Two-way anova. #P < 0.05 from vehicle.
Histological examination of lung sections revealed that GSK1016790A infusion induced cuffing around the arteries in both genotypes (Figure 3A–D). Morphometric measurements revealed that the cuffing area was significantly larger in arteries with internal lumen diameters below 50 μm in GSK1016790A-infused wt mice versus KCa3.1−/− mice (Figure 3E–F). Moreover, GSK1016790A increased extracellular space (being indicative of fluid extravasation) in the wt mice but not in the KCa3.1−/− (Supporting Information Fig. S2). GSK1016790A also produced localized haemorrhage in lungs of wt mice, which was virtually absent in the lungs of GSK1016790A-treated KCa3.1−/− mice and in vehicle-treated mice (Figure 4A–E).
Figure 3.
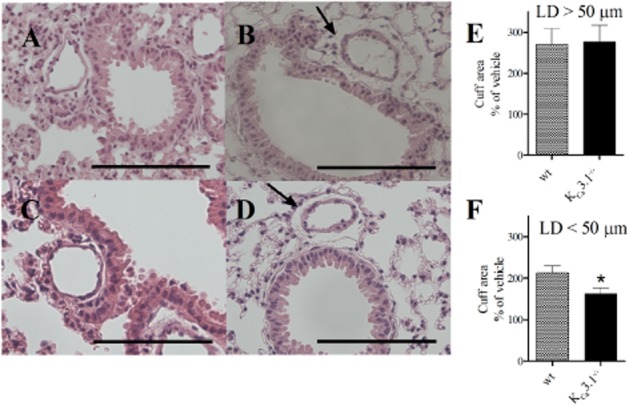
GSK1016790A induced perivascular fluid cuffs in wt mice and KCa3.1−/− mice. H&E staining in sections of lungs infused with vehicle (A, C) or GSK1016790A (B, D) in wt (A, B) and KCa3.1−/− mice. Perivascular fluid cuffs shown with arrow. (E, F) Average changes in cuff areas in vessels with lumen diameter larger than 50 μm (E) and smaller than 50 μm (F). Scale bar = 100 μm in panels. Data points are means ± SEM (n = 4–6). Student’s t-test: *P < 0.05 from wt.
Figure 4.
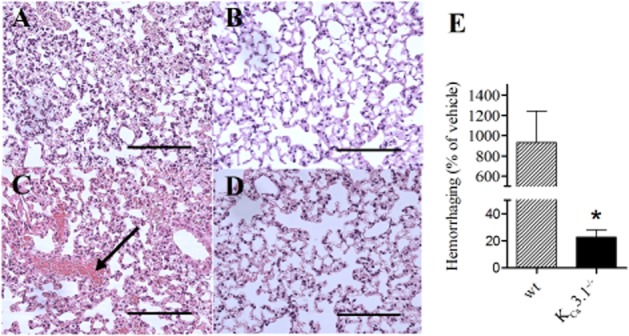
GSK1016790A induced lung damage in wt mice and KCa3.1−/− mice. H&E staining in sections of lungs infused with vehicle (A, B) or GSK1016790A (C, D) from wt (A, C) and KCa3.1−/− mice (B, D). Note the haemorrhage in wt lungs that was not seen in the KCa3.1−/− lungs. (E) Summary data. Scale bar = 100 μm in panels. Data points are means ± SEM (n = 4–6). Student’s t-test: *P < 0.05 versus wt mice.
In both genotypes, GSK1016790A produced detachment of the epithelium in bronchioles, whereas detachment of the endothelium was not obvious but cell rounding was evident and the degree of endothelial cell rounding was alike in wt mice and KCa3.1−/− (results not shown).
KCa3.1 contributes to GSK1016790A-induced relaxation of PA
To elucidate arterial mechanisms, by which a KCa3.1 deficiency protected agonist circulatory collapse provoked by GSK1016790A-mediated TRPV4 activation, we performed isometric myography on PAs from wt and KCa3.1−/− mice. A single high dose of GSK1016790A (10 nM) induced relaxation in intrapulmonary arteries from wt while, remarkably, the response was markedly reduced in arteries from KCa3.1−/− mice (Figure 5A), suggesting loss of endothelial function, downstream of TRPV4 signalling in these small resistance-sized PAs from the KCa3.1−/− mice. As expected, relaxations were abolished by inhibition of eNOS with L-NOARG and by removal of the endothelium (Figure 5B), indicating that endothelial TRPV4 and KCa3.1 functions converge on NO-mediated vascular relaxation. Both blockers of KCa2 and KCa3.1 channels, UCL1684 and TRAM-34, respectively, inhibited GSK1016790A relaxations in wt PA, while only UCL1684 blocked relaxations in PA from KCa3.1−/− mice (Figure 5C). This is in line with the results of previous studies, showing the TRPV4-mediated relaxation involves mainly NO and KCa3.1 deficiency reduces NO-mediated dilatations in carotid arteries, presumably by impairing the electrochemical driving force for Ca2+ entry (Si et al., 2006). Relaxations induced by the NO donor, SNP were enhanced in PAs from KCa3.1−/− mice versus wt mice (Figure 5D), suggesting that there was some compensatory changes in the NO system in arteries from the KCa3.1−/− mice, a difference that disappeared in vessels without endothelium. In conclusion, these functional studies on PAs suggested that disruption of the TRPV4/KCa3.1 interaction in the KCa3.1−/− mice impairs arterial relaxation in the pulmonary circulation after pharmacological TRPV4 activation.
Figure 5.
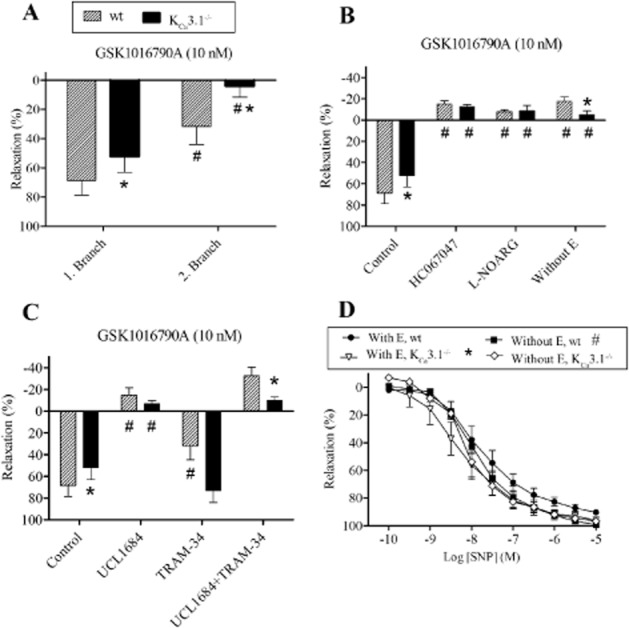
TRPV4 and relaxation of pulmonary arteries from wt and KCa3.1−/− mice. (A) Relaxations induced by GSK1016790A in the first- and second-order intrapulmonary branches of the right main pulmonary artery from wt and KCa3.1−/− mice. (B) Relaxations induced by GSK1016790A in the absence and the presence of a blocker of TRPV4 channels, HC067047, an inhibitor of NOS, L-NOARG, and after removal of the endothelium (without E) in first-order intrapulmonary arteries of the right main pulmonary artery from either genotype. (C) Relaxations induced by GSK1016790A in first-order intrapulmonary arteries of the right main pulmonary artery in the absence and the presence of blockers of KCa2, UCL1684 and KCa3.1 channels, TRAM-34. (D) Relaxations induced by SNP in first-order intrapulmonary arteries of the right main pulmonary artery from wt and KCa3.1−/− mice. The experiments were performed in the presence of indomethacin. Data are means ± SEM (n = 7). One-way anova followed by Tukey’s post hoc test: *P < 0.05 versus wt, #P < 0.05 versus control pulmonary artery with endothelium (E).
TRPV4 and KCa currents in PAEC from wt and KCa3.1−/− mice
To further explore the direct functional interactions of KCa3.1 and TRPV4, we characterized and compared GSK1016790A-induced TRPV4- and KCa3.1-mediated currents in freshly isolated PAEC from both genotypes using the whole-cell patch clamp electrophysiology (Figure 6). Initial currents recorded after establishing the fast whole-cell configuration were similar in both genotypes (Figure 6A). In PAEC of either genotype, GSK1016790A (200 nM) activated small inward and outward cation currents that were clearly distinguishable from background currents and similar to previously described TRPV4 currents (Figure 6B) (Hartmannsgruber et al., 2007). Current amplitudes were similar in both genotypes (Figure 6B). These instantaneous GSK1016790A-induced currents were sensitive to the selective TRPV4 blocker, HC067047 (1 μM) (Figure 6B), demonstrating that the currents were carried by endothelial TRPV4 channels. In a subset of experiments, GSK1016790A produced further activation of K+ outward currents shifting the membrane potential towards the K+ equilibrium potential (Figure 6C). These K+ currents were largely sensitive to the KCa3.1 blocker TRAM-34 (1 μM) and to a smaller extent to the KCa2 blocker UCL1684 (1 μM) in wt PAEC (Figure 6C), indicating that these K+ currents were carried by mainly endothelial KCa3.1 and to some extent by KCa2.3 channels (Brahler et al., 2009). In KCa3.1−/− PAEC, the KCa current was of smaller amplitude and UCL1684 (1 μM) was able to induce complete blockade, indicating that GSK1016790A activated residual KCa2.3 channels in these PAEC. In another subset of experiments, the relatively small currents produced by GSK1016790A were followed by massive and HC067047-insensitive leak currents (>10 nS) in 4 out of 9 wt cells (Figure 7A,B) that reflected breakdown of membrane resistance as this is an electrical sign of deleterious alterations of the cell membrane and perhaps of bleb formation as described previously (Alvarez et al., 2006). GSK1016790A produced no leak currents in the KCa3.1−/− cells, as we did not observe appreciable HC067047-insensitive currents (0 out of 7 cells; Figure 7C).
Figure 6.

Activation of TRPV4 currents and co-activation of KCa channels in pulmonary artery endothelial cells from wt and KCa3.1−/− mice. (A) Representative whole-cell currents as recorded immediately after establishing the fast whole-cell configuration. (B) TRPV4 currents activated by the GSK1016790A (200 nM) and blockade by HC067047 (1 μM). (C) Co-activation of TRAM-34 (1 μM)-sensitive KCa3.1 currents and UCL1684 (1 μM)-sensitive KCa2.3 currents in wt and of UCL1684-sensitive KCa2.3 currents in KCa3.1−/−. Panels on right: currents normalized to cell capacitance. Numbers refer to the n of cells studied per genotype. Data represent means ± SEM.
Figure 7.
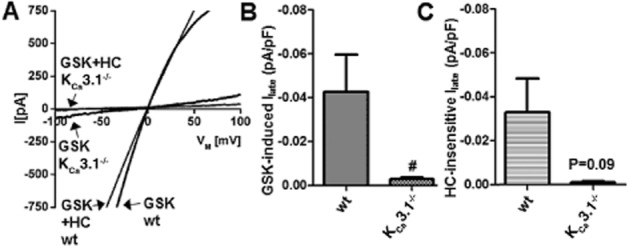
GSK1016790A-induced leak currents in pulmonary artery endothelial cells. (A) Whole-cell patch clamp recordings showing that GSK1016790A (GSK, 200 nM)-induced TRPV4 activation was followed by massive leak currents in some PAEC (4 out of 9) from wt mice. These leak currents were largely insensitive to the blocker of TRPV4 channels, HC067047 (HC, 1 μM). KCa3.1−/− PAEC did not show leak currents (0 out of 7 cells) and currents were blocked by HC067047. (B) Average GSK1016790A-induced currents at −80 mV in wt (n = 9) and KCa3.1−/− PAEC (n = 7). (C) Average HC067047-insensitive currents in PAEC from both genotypes (wt, n = 6; KCa3.1−/−, n = 3). Data represent means ± SEM. Student’s t-test. #P < 0.05.
These findings from our electrophysiological experiments suggest that GSK1016790A effectively activated TRPV4 channels in PAEC and that the Ca2+ influx through TRPV4 produced hyperpolarization and K+ efflux mainly through co-activation of KCa3.1 channels and to a minor extent KCa2.3 channels. Moreover, TRPV4 activation and KCa3.1 co-activation together have deleterious effects on cell membrane stability in PAEC and KCa3.1 deficiency protects against this breakdown of the cell membrane.
GSK1016790A-induced endothelial cell damage in vitro
To study GSK1016790A-induced endothelial cell damage, we switched for technical reasons and too low amounts of murine material to an in vitro model of human endothelium, that is, confluent HUVECs, which are known to express TRPV4 (Willette et al., 2008) and KCa3.1 channels (Brakemeier et al., 2003) and allow us to characterize the structural damage in a similar way as reported previously (Willette et al., 2008). As shown in Figure 8, 45 min of exposure to 200 nM GSK1016790A produced rounding of ≈30% of the cells and ≈70% of the cells showed apparent disarrangements as indicated by retracted cytoplasmic edges, condensed nuclei and bleb formation (for examples see scans in Figure 8). TRAM-34 at 1 μM prevented the cell disarrangements and rounding by ≈25% (P < 0.01) and ≈70% (P < 0.01; Figure 8). As expected, the TRPV4 blocker, HC067047 (1 μM), almost completely prevented cell disarrangements and rounding (P < 0.01 vs. GSK1016790A). The vehicle (DMSO) itself did not produce significant cell rounding or disarrangement (P < 0.01 vs. GSK1016790A). These data suggest that co-activation of KCa3.1 was required for significant TRPV4-induced cell disarrangement and detachment in this in vitro model of human endothelium.
Figure 8.
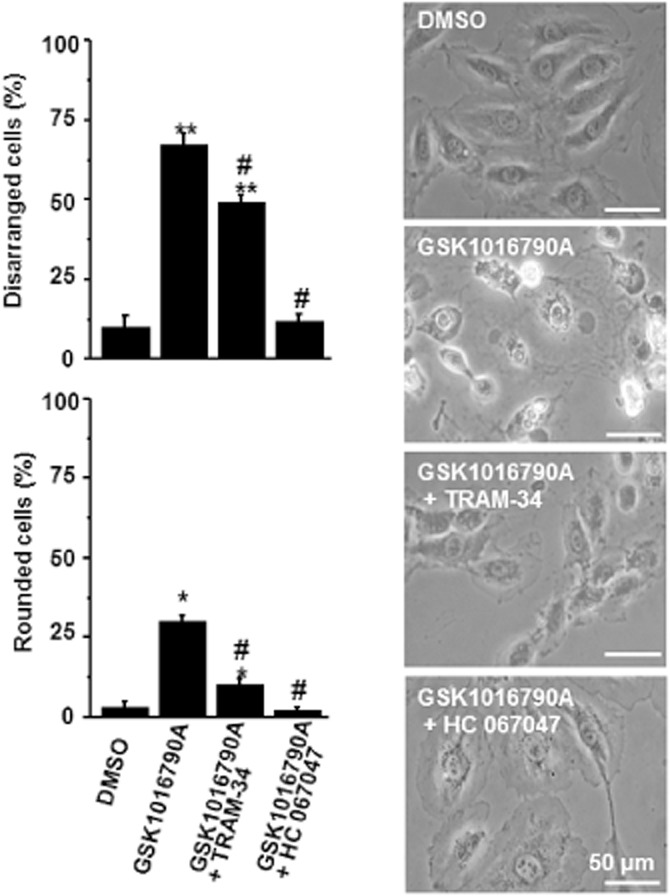
KCa3.1 inhibition antagonizes cell disarrangement and rounding of HUVEC caused by TRPV4 activation. Panels on right: representative scans (200×) of HUVEC treated for 45 min with either vehicle (DMSO), GSK1016790A (200 nM) or the combinations of TRAM-34(1 μM) and GSK1016790A or of HC067047 (1 μM) and GSK1016790A. Panels on left: summary data. Data represent means ± SEM (n = 4, independent experiments). *P < 0.05, **P < 0.01 versus vehicle, #P < 0.01 versus GSK1016790A; one-way anova followed by Tukey’s post hoc test.
Discussion
The main purpose of this study using KCa3.1−/− mice was to investigate whether the Ca2+-activated KCa3.1 channel acts as endogenous sensor of calcium entry through TRPV4 channels in lung endothelium and whether interruption of this interaction in KCa3.1−/− mice protects against undesired endothelial damage and barrier disruption, lung oedema, pulmonary arterial relaxation, deleterious pulmonary circulatory collapse and death caused by pharmacological TRPV4 activation. The experimental evidence that we provide here to support this pathophysiological role of KCa3.1 is (i) at the endothelial cell level, TRPV4 activation by GSK1016790A produced strong co-activation of KCa3.1 and led to breakdown of membrane resistance, while KCa3.1-deficient endothelial cells supposedly lack this co-activation and were resistant to breakdown of membrane resistance. (ii) Regarding GSK1016790A-induced lung damage, lungs of KCa3.1−/− showed a lower degree of fluid extravasation, virtually no haemorrhage. (iii) PA from KCa3.1−/− mice showed a substantially reduced relaxation to TRPV4 activation. (iv) GSK1016790A infusion caused pulmonary circulatory collapse, cardiac arrest and death in wt mice while KCa3.1−/− mice survived the experiment.
Electrophysiological findings
The biophysical and pharmacological properties of TRPV4 and KCa channels in the murine PAEC (Figure 6) were similar to those reported earlier for TRPV4 and KCa in other endothelial preparations from mice and rat arteries (Kohler et al., 2006) (Hartmannsgruber et al., 2007). Still, it is worth mentioning that in the wt PAEC the activation of Ca2+- and Na+-permeable TRPV4 channels was able to co-activate calcium/calmodulin-gated KCa3.1 and KCa2.3 channels. Moreover, we showed that TRPV4 primarily activated KCa3.1 channels because a large fraction of the KCa current was sensitive to the KCa3.1 blocker, TRAM-34, while a smaller fraction was sensitive to the KCa2 blocker, UCL1684, in wt and KCa3.1−/− PAEC. Interestingly, a functional interaction of TRPV4 with KCa1.1 (Maxi K) has been reported in rat and human bronchial epithelial cell lines (Alvarez et al., 2006; Fernandez-Fernandez et al., 2008). Although activation of KCa1.1 was not found to contribute to the TRPV4-induced disruption of the alveolar septal barrier (Alvarez et al., 2006), these findings support the view that TRPV4 channels indeed co-operate with KCa channels despite the fact that the role of the individual KCa channel subtype can vary. Yet in pulmonary endothelium, co-operation of TRPV4 and KCa3.1 can be interpreted as a physiological mechanism to provide countercurrents to depolarizing currents and thus to regulate electrical neutrality and cellular water movements.
An important observation that we made during the course of the whole-cell patch clamp experiments was that TRPV4 activation by GSK1016790A ended with breakdown of membrane resistance, which was not seen in KCa3.1−/−-deficient PAEC. This showed that pharmacologically activated and Ca2+-conducting TRPV4 used KCa3.1 channels and K+ efflux to produce deleterious breakdown of cell architecture. However, in the present study we did not reveal the molecular details of the structural damage. Yet, in an in vitro model of human endothelium we found that GSK1016790A caused disarrangement and rounding of HUVEC that could be blocked by a TRPV4 blocker but importantly also to a significant extent by a KCa3.1 blocker. This endothelial damage caused by pharmacological TRPV4 activation described here was similar to the endothelial blebbing and cellular disarrangement caused by another TRPV4 activator, 4αPDD, or to GSK1016790A (Alvarez et al., 2006; Willette et al., 2008; Balakrishna et al., 2014). In summary, the present data stress the mechanistic importance of KCa3.1 as a downstream sensor of TRPV4-mediated calcium signalling and ensuing deleterious alterations of endothelial morphology.
Findings from myography
TRPV4 channel activation has previously been suggested to mainly involve NO and also EDH-type relaxation in rat PA (Sukumaran et al., 2013). In the present study of vasorelaxation to GSK1016790A in mouse PA, our results confirm the ability of pharmacologically-activated TRPV4 to produce vasorelaxation that required NO signalling, but also involved KCa2 and KCa3.1 channels sensitive to, respectively, UCL1684 and TRAM-34. This is in line with earlier reports in other vascular beds, such as the carotid and mesenteric arteries of the mice and rats (Hartmannsgruber et al., 2007; Sonkusare et al., 2012; 2014,). A significant contribution of EDH-type relaxation was not evident in both genotypes identifying NO as the major endothelium-derived vasodilator in the murine PA. Importantly, GSK1016790A-mediated relaxation of small-sized PAs was virtually absent in the arteries from KCa3.1−/− mice fostering the view of substantial endothelial vasodilator dysfunction to this type of pharmacological calcium influx-dependent signalling (Si et al., 2006; Wolfle et al., 2009; Hasenau et al., 2011). This is in line with earlier results showing defective ACh-induced and NO-mediated as well as EDH-type mediated dilations of carotid arteries and in the cremaster microcirculation during a genetic KCa3.1 deficiency (Si et al., 2006; Wolfle et al., 2009), which is mainly caused by defective hyperpolarization and the loss of a positive feedback on calcium dynamics in the endothelium (Qian et al., 2014), thus explaining the lower calcium-dependent NO-mediated relaxation in small-calibre PAs of KCa3.1−/− mice. Previous studies suggested that a hydrostatic pressure above 28 cmH2O (∼20.5 mmHg) was required for increased permeability through TRPV4-dependent activation in mouse isolated lung (Jian et al., 2008). The initial mean pulmonary pressure when GSK1016790A was infused in the present study was at this level (Figure 1), and the sudden vasodilatation induced as well as inhibition of myogenic tone by TRPV4 activation (Bagher et al., 2012) may allow transmission of high pressure to the capillary endothelium followed by increased permeability. Therefore, with regard to the pulmonary circulatory collapse caused by TRPV4 activation, the reduced endothelial vasodilator function in the KCa3.1−/− mice may provide a mechanistic explanation for the resistance of these animals to GSK1016790A-induced pulmonary circulatory collapse and death. However, at present we do not wish to exclude that other, for example, neurological alterations found in KCa3.1−/− may have an additional effect (Lambertsen et al., 2012).
Histological findings
Besides the overt vascular collapse, infusion of GSK1016790A was associated with the formation of cuffs around the small-sized PAs, fluid extravasation, haemorrhage, endothelial cell blebbing and epithelial cell detachment in larger airways. These findings agree with previous findings in isolated lungs showing blebs and breaks in the endothelial and epithelial layers, and hence disruption of the alveolar septal barrier to GSK1016790A (Alvarez et al., 2006; Jian et al., 2008; Wu et al., 2009; Villalta et al., 2014). Moreover, we found that TRPV4 activation markedly increased the extravasation of albumin-binding Evans blue. Recent studies suggest that part of the lung injury after infusion of GSK1016790A involves metalloproteinase (MMP) 2 and 9 in the TRPV4-induced lung injury as a blocker of these enzymes, SB3CT, markedly reduced total protein in broncho-alveolar lavage fluid, while the effect on lung wet-to-dry ratio was less pronounced (Villalta et al., 2014). In the present study, the KCa3.1−/− protected against haemorrhage and reduced the formation of cuffs in smaller PAs indicative of fluid extravasation, while there was no effect on extravasation of Evans blue and epithelial detachment. Taken together these studies suggest that different downstream signal pathways are involved in the TPRV4-induced lung injury and vascular collapse, where, intriguingly, the KCa3.1 channel appears to be mechanistically important as its loss protects the mice from injury.
Concerning the protective mechanism against haemorrhage, we speculate that this is a consequence of the higher capability of KCa3.1−/− lungs to maintain resistance and thereby to avoid deleterious hyperaemia and rupture of the downstream capillary system, while other mechanisms appear to underlie the effect on the permeability reflected by reduced cuff formation and hence fluid extravasation in the lung. Indeed, a recent report on the beneficial effects of endothelial KCa3.1 blockade on brain oedema formation after ischaemic stroke (Chen et al., 2011; 2015,) and after trauma (Mauler et al., 2004) have suggested therapeutic utility of KCa3.1 blockers to prevent K+ and concomitant Cl− and fluid movement. Although our studies were performed in HUVECs and may not directly reflect pulmonary microvascular endothelial cells, the protective mechanism against TRPV4-induced cell swelling and blebs, induced by blocking the KCa3.1 channel using TRAM-34 in cultures, suggests that the K efflux plays a role in the pathogenesis of increased permeability and breakdown of the cell resistance.
In conclusion, we suggest that KCa3.1 channels are part of a novel pathophysiological mechanism in the lung as the channel forms a functional unit with TRPV4 to maintain abnormally high calcium levels to drive fluid extravasation and endothelial retraction, as well as to produce abnormal relaxation of pulmonary resistance arteries leading to haemorrhage and pulmonary circulatory collapse. Finally, inhibitors of KCa3.1 by TRAM-34 or ICA-17043 (Senicapoc) (Ataga et al., 2008) and the pan-KCa2/KCa3.1 inhibitor (13b) (Olivan-Viguera et al., 2013) may have therapeutic utility in situations of oedema formation, hypotension and circulatory collapse associated with abnormally high endothelial calcium signalling, barrier disruption and pulmonary circulatory collapse.
Acknowledgments
The present study was supported by the Deutsche Forschungsgemeinschaft (KO1899/11-1 to R. K.), European Community (FP7-PEOPLE-CIG-321721 to R. K.), Department of Industry and Innovation, Government of Aragon (GIPASC-B105), the Fondo de Investigación Sanitaria (Red HERACLES RD12/0042/0014), The Danish Heart Foundation (R. K. and U. S.) and the NovoNordisk Foundation (U. S. and R. K.).
Glossary
- L-NOARG
NG-nitro-L-arginine
- PA
pulmonary arteries
- PAEC
pulmonary arterial endothelial cells
- PSS
physiological saline solution
- TRP
transient receptor potential channels
- TRPV4
transient receptor potential vanilloid type 4 channels
- wt
wild-type mice
Author contributions
U. S., R. K., C. W.-F., A. O.-V. and T. D. conceived and designed the experiments. C. W. F., T. D., V. B., A. O.-V., V. S., L. B., S. M. and E. S. collected, analysed and interpreted the data. T. D., A. O.-V., R. K., C. W.-F. and U. S. drafted and C. W.-F., R. K. and U. S. revised the article. All authors approved the final version of the manuscript. C. W.-F., T. D., V. B. and A. O.-V. contributed equally to this paper.
Conflict of interest
None.
Supporting Information
Figure S1 Systemic aortic mean (A), systolic (B) and diastolic (C) pressure after i.v. infusion of GSK1016790A (0.01, 0.03, 0.1 and 0.2 mg·kg−1) in wild-type (wt) mice (n = 7) and KCa3.1−/− mice (n = 3). Results are means ± SEM. #Indicates concentration of GSK1016790A, where mice are experiencing circulatory collapse. *P < 0.05 versus wt.
Figure S2 GSK1016790A induced lung damage in wt mice and KCa3.1−/− mice. H&E staining in sections of lungs infused with vehicle (A, B) or GSK1016790A (C, D) from wt (A, C) and KCa3.1−/− mice (B, D). The changes in extracellular space in the lungs were measured by superimposing a grid on randomized histological sections of the lungs. The line intersections in the extracellular space of each sections was counted and expressed relative to the number of intersections counted in vehicle-infused mice lungs. Alteration in extracellular space in wt but not KCa3.1−/− mice caused by infusion of GSK1016790A (C, D). (E) Summary data. Scale bar = 100 μm in panels. Data points are means ± SEM (n = 4–6). Student’s t-test: *P < 0.05 versus wt mice.
Table S1 Heart rate after GSK1016790A infusion.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Catterall WA, et al. The concise guide to pharmacology 2013/14: ion channels. Br J Pharmacol. 2013a;170:1607–1651. doi: 10.1111/bph.12447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The concise guide to pharmacology 2013/14: enzymes. Br J Pharmacol. 2013b;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez DF, King JA, Weber D, Addison E, Liedtke W, Townsley MI. Transient receptor potential vanilloid 4-mediated disruption of the alveolar septal barrier: a novel mechanism of acute lung injury. Circ Res. 2006;99:988–995. doi: 10.1161/01.RES.0000247065.11756.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ataga KI, Smith WR, De Castro LM, Swerdlow P, Saunthararajah Y, Castro O, et al. Efficacy and safety of the Gardos channel blocker, senicapoc (ICA-17043), in patients with sickle cell anemia. Blood. 2008;111:3991–3997. doi: 10.1182/blood-2007-08-110098. [DOI] [PubMed] [Google Scholar]
- Bagher P, Beleznai T, Kansui Y, Mitchell R, Garland CJ, Dora KA. Low intravascular pressure activates endothelial cell TRPV4 channels, local Ca2+ events, and IKCa channels, reducing arteriolar tone. Proc Natl Acad Sci USA. 2012;109:18174–18179. doi: 10.1073/pnas.1211946109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balakrishna S, Song W, Achanta S, Doran SF, Liu B, Kaelberer MM, et al. TRPV4 inhibition counteracts edema and inflammation and improves pulmonary function and oxygen saturation in chemically induced acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2014;307:L158–L172. doi: 10.1152/ajplung.00065.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brahler S, Kaistha A, Schmidt VJ, Wolfle SE, Busch C, Kaistha BP, et al. Genetic deficit of SK3 and IK1 channels disrupts the endothelium-derived hyperpolarizing factor vasodilator pathway and causes hypertension. Circulation. 2009;119:2323–2332. doi: 10.1161/CIRCULATIONAHA.108.846634. [DOI] [PubMed] [Google Scholar]
- Brakemeier S, Kersten A, Eichler I, Grgic I, Zakrzewicz A, Hopp H, et al. Shear stress-induced up-regulation of the intermediate-conductance Ca(2+)-activated K(+) channel in human endothelium. Cardiovasc Res. 2003;60:488–496. doi: 10.1016/j.cardiores.2003.09.010. [DOI] [PubMed] [Google Scholar]
- Chen YJ, Raman G, Bodendiek S, O’Donnell ME, Wulff H. The KCa3.1 blocker TRAM-34 reduces infarction and neurological deficit in a rat model of ischemia/reperfusion stroke. J Cereb Blood Flow Metab. 2011;31:2363–2374. doi: 10.1038/jcbfm.2011.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YJ, Wallace BK, Yuen N, Jenkins DP, Wulff H, O’Donnell ME. Blood-brain barrier KCa3.1 channels: evidence for a role in brain Na uptake and edema in ischemic stroke. Stroke. 2015;46:237–244. doi: 10.1161/STROKEAHA.114.007445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devor DC, Bridges RJ, Pilewski JM. Pharmacological modulation of ion transport across wild-type and DeltaF508 CFTR-expressing human bronchial epithelia. Am J Physiol Cell Physiol. 2000;279:C461–C479. doi: 10.1152/ajpcell.2000.279.2.C461. [DOI] [PubMed] [Google Scholar]
- Elmedal B, Mulvany MJ, Simonsen U. Dual impact of a nitric oxide donor, GEA 3175, in human pulmonary smooth muscle. Eur J Pharmacol. 2005;516:78–84. doi: 10.1016/j.ejphar.2005.04.017. [DOI] [PubMed] [Google Scholar]
- Feletou M, Kohler R, Vanhoutte PM. Nitric oxide: orchestrator of endothelium-dependent responses. Ann Med. 2012;44:694–716. doi: 10.3109/07853890.2011.585658. [DOI] [PubMed] [Google Scholar]
- Fernandez-Fernandez JM, Andrade YN, Arniges M, Fernandes J, Plata C, Rubio-Moscardo F, et al. Functional coupling of TRPV4 cationic channel and large conductance, calcium-dependent potassium channel in human bronchial epithelial cell lines. Pflugers Arch. 2008;457:149–159. doi: 10.1007/s00424-008-0516-3. [DOI] [PubMed] [Google Scholar]
- Grgic I, Kaistha BP, Paschen S, Kaistha A, Busch C, Si H, et al. Disruption of the Gardos channel (KCa3.1) in mice causes subtle erythrocyte macrocytosis and progressive splenomegaly. Pflugers Arch. 2009;458:291–302. doi: 10.1007/s00424-008-0619-x. [DOI] [PubMed] [Google Scholar]
- Hartmannsgruber V, Heyken WT, Kacik M, Kaistha A, Grgic I, Harteneck C, et al. Arterial response to shear stress critically depends on endothelial TRPV4 expression. PLoS ONE. 2007;2:e827. doi: 10.1371/journal.pone.0000827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasenau AL, Nielsen G, Morisseau C, Hammock BD, Wulff H, Kohler R. Improvement of endothelium-dependent vasodilations by SKA-31 and SKA-20, activators of small- and intermediate-conductance Ca2+ -activated K+ -channels. Acta Physiol (Oxf) 2011;203:117–126. doi: 10.1111/j.1748-1716.2010.02240.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilfiker MA, Hoang TH, Cornil J, Eidam HS, Matasic DS, Roethke TJ, et al. Optimization of a novel series of TRPV4 antagonists with in vivo activity in a model of pulmonary edema. ACS Med Chem Lett. 2013;4:293–296. doi: 10.1021/ml300449k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii TM, Silvia C, Hirschberg B, Bond CT, Adelman JP, Maylie J. A human intermediate conductance calcium-activated potassium channel. Proc Natl Acad Sci USA. 1997;94:11651–11656. doi: 10.1073/pnas.94.21.11651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jian MY, King JA, Al-Mehdi AB, Liedtke W, Townsley MI. High vascular pressure-induced lung injury requires P450 epoxygenase-dependent activation of TRPV4. Am J Respir Cell Mol Biol. 2008;38:386–392. doi: 10.1165/rcmb.2007-0192OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaner RJ, Ladetto JV, Singh R, Fukuda N, Matthay MA, Crystal RG. Lung overexpression of the vascular endothelial growth factor gene induces pulmonary edema. Am J Respir Cell Mol Biol. 2000;22:657–664. doi: 10.1165/ajrcmb.22.6.3779. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohler R, Ruth P. Endothelial dysfunction and blood pressure alterations in K+-channel transgenic mice. Pflugers Arch. 2010;459:969–976. doi: 10.1007/s00424-010-0819-z. [DOI] [PubMed] [Google Scholar]
- Kohler R, Heyken WT, Heinau P, Schubert R, Si H, Kacik M, et al. Evidence for a functional role of endothelial transient receptor potential V4 in shear stress-induced vasodilatation. Arterioscler Thromb Vasc Biol. 2006;26:1495–1502. doi: 10.1161/01.ATV.0000225698.36212.6a. [DOI] [PubMed] [Google Scholar]
- Kroigaard C, Dalsgaard T, Nielsen G, Laursen BE, Pilegaard H, Kohler R, et al. Activation of endothelial and epithelial K(Ca) 2.3 calcium-activated potassium channels by NS309 relaxes human small pulmonary arteries and bronchioles. Br J Pharmacol. 2012;167:37–47. doi: 10.1111/j.1476-5381.2012.01986.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambertsen KL, Gramsbergen JB, Sivasaravanaparan M, Ditzel N, Sevelsted-Moller LM, Olivan-Viguera A, et al. Genetic KCa3.1-deficiency produces locomotor hyperactivity and alterations in cerebral monoamine levels. PLoS ONE. 2012;7:e47744. doi: 10.1371/journal.pone.0047744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Kanju P, Patterson M, Chew WL, Cho SH, Gilmour I, et al. TRPV4-mediated calcium influx into human bronchial epithelia upon exposure to diesel exhaust particles. Environ Health Perspect. 2011;119:784–793. doi: 10.1289/ehp.1002807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauler F, Hinz V, Horvath E, Schuhmacher J, Hofmann HA, Wirtz S, et al. Selective intermediate-/small-conductance calcium-activated potassium channel (KCNN4) blockers are potent and effective therapeutics in experimental brain oedema and traumatic brain injury caused by acute subdural haematoma. Eur J Neurosci. 2004;20:1761–1768. doi: 10.1111/j.1460-9568.2004.03615.x. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mene P, Punzo G, Pirozzi N. TRP channels as therapeutic targets in kidney disease and hypertension. Curr Top Med Chem. 2013;13:386–397. doi: 10.2174/1568026611313030013. [DOI] [PubMed] [Google Scholar]
- Nilius B, Szallasi A. Transient receptor potential channels as drug targets: from the science of basic research to the art of medicine. Pharmacol Rev. 2014;66:676–814. doi: 10.1124/pr.113.008268. [DOI] [PubMed] [Google Scholar]
- Olivan-Viguera A, Valero MS, Murillo MD, Wulff H, Garcia-Otin AL, Arbones-Mainar JM, et al. Novel phenolic inhibitors of small/intermediate-conductance Ca(2)(+)-activated K(+) channels, KCa3.1 and KCa2.3. PLoS ONE. 2013;8:e58614. doi: 10.1371/journal.pone.0058614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledgebase of drug targets and their ligands. Nucl Acids Res. 2014;42(Database Issue):D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian X, Francis M, Kohler R, Solodushko V, Lin M, Taylor MS. Positive feedback regulation of agonist-stimulated endothelial Ca2+ dynamics by KCa3.1 channels in mouse mesenteric arteries. Arterioscler Thromb Vasc Biol. 2014;34:127–135. doi: 10.1161/ATVBAHA.113.302506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Si H, Heyken WT, Wolfle SE, Tysiac M, Schubert R, Grgic I, et al. Impaired endothelium-derived hyperpolarizing factor-mediated dilations and increased blood pressure in mice deficient of the intermediate-conductance Ca2+-activated K+ channel. Circ Res. 2006;99:537–544. doi: 10.1161/01.RES.0000238377.08219.0c. [DOI] [PubMed] [Google Scholar]
- Sonkusare SK, Bonev AD, Ledoux J, Liedtke W, Kotlikoff MI, Heppner TJ, et al. Elementary Ca2+ signals through endothelial TRPV4 channels regulate vascular function. Science. 2012;336:597–601. doi: 10.1126/science.1216283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonkusare SK, Dalsgaard T, Bonev AD, Hill-Eubanks DC, Kotlikoff MI, Scott JD, et al. AKAP150-dependent cooperative TRPV4 channel gating is central to endothelium-dependent vasodilation and is disrupted in hypertension. Sci Signal. 2014;7:ra66. doi: 10.1126/scisignal.2005052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankevicius E, Dalsgaard T, Kroigaard C, Beck L, Boedtkjer E, Misfeldt MW, et al. Opening of small and intermediate calcium-activated potassium channels induces relaxation mainly mediated by nitric-oxide release in large arteries and endothelium-derived hyperpolarizing factor in small arteries from rat. J Pharmacol Exp Ther. 2011;339:842–850. doi: 10.1124/jpet.111.179242. [DOI] [PubMed] [Google Scholar]
- Sukumaran SV, Singh TU, Parida S, Narasimha RC, Thangamalai R, Kandasamy K, et al. TRPV4 channel activation leads to endothelium-dependent relaxation mediated by nitric oxide and endothelium-derived hyperpolarizing factor in rat pulmonary artery. Pharmacol Res. 2013;78:18–27. doi: 10.1016/j.phrs.2013.09.005. [DOI] [PubMed] [Google Scholar]
- Thorneloe KS, Cheung M, Bao W, Alsaid H, Lenhard S, Jian MY, et al. An orally active TRPV4 channel blocker prevents and resolves pulmonary edema induced by heart failure. Sci Transl Med. 2012;4:159ra148. doi: 10.1126/scitranslmed.3004276. [DOI] [PubMed] [Google Scholar]
- Vandorpe DH, Shmukler BE, Jiang L, Lim B, Maylie J, Adelman JP, et al. cDNA cloning and functional characterization of the mouse Ca2+-gated K+ channel, mIK1. Roles in regulatory volume decrease and erythroid differentiation. J Biol Chem. 1998;273:21542–21553. doi: 10.1074/jbc.273.34.21542. [DOI] [PubMed] [Google Scholar]
- Villalta PC, Rocic P, Townsley MI. Role of MMP2 and MMP9 in TRPV4-induced lung injury. Am J Physiol Lung Cell Mol Physiol. 2014;307:L652–L659. doi: 10.1152/ajplung.00113.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wandall-Frostholm C, Skaarup LM, Sadda V, Nielsen G, Hedegaard ER, Mogensen S, et al. Pulmonary hypertension in wild type mice and animals with genetic deficit in KCa2.3 and KCa3.1 channels. PLoS ONE. 2014;9:e97687. doi: 10.1371/journal.pone.0097687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe H, Davis JB, Smart D, Jerman JC, Smith GD, Hayes P, et al. Activation of TRPV4 channels (hVRL-2/mTRP12) by phorbol derivatives. J Biol Chem. 2002;277:13569–13577. doi: 10.1074/jbc.M200062200. [DOI] [PubMed] [Google Scholar]
- Watanabe H, Vriens J, Prenen J, Droogmans G, Voets T, Nilius B. Anandamide and arachidonic acid use epoxyeicosatrienoic acids to activate TRPV4 channels. Nature. 2003;424:434–438. doi: 10.1038/nature01807. [DOI] [PubMed] [Google Scholar]
- Weston AH, Porter EL, Harno E, Edwards G. Impairment of endothelial SK(Ca) channels and of downstream hyperpolarizing pathways in mesenteric arteries from spontaneously hypertensive rats. Br J Pharmacol. 2010;160:836–843. doi: 10.1111/j.1476-5381.2010.00657.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willette RN, Bao W, Nerurkar S, Yue TL, Doe CP, Stankus G, et al. Systemic activation of the transient receptor potential vanilloid subtype 4 channel causes endothelial failure and circulatory collapse: part 2. J Pharmacol Exp Ther. 2008;326:443–452. doi: 10.1124/jpet.107.134551. [DOI] [PubMed] [Google Scholar]
- Wolfle SE, Schmidt VJ, Hoyer J, Kohler R, de Wit C. Prominent role of KCa3.1 in endothelium-derived hyperpolarizing factor-type dilations and conducted responses in the microcirculation in vivo. Cardiovasc Res. 2009;82:476–483. doi: 10.1093/cvr/cvp060. [DOI] [PubMed] [Google Scholar]
- Wu S, Jian MY, Xu YC, Zhou C, Al-Mehdi AB, Liedtke W, et al. Ca2+ entry via alpha1G and TRPV4 channels differentially regulates surface expression of P-selectin and barrier integrity in pulmonary capillary endothelium. Am J Physiol Lung Cell Mol Physiol. 2009;297:L650–L657. doi: 10.1152/ajplung.00015.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin J, Hoffmann J, Kaestle SM, Neye N, Wang L, Baeurle J, et al. Negative-feedback loop attenuates hydrostatic lung edema via a cGMP-dependent regulation of transient receptor potential vanilloid 4. Circ Res. 2008;102:966–974. doi: 10.1161/CIRCRESAHA.107.168724. [DOI] [PubMed] [Google Scholar]
- Yin J, Kukucka M, Hoffmann J, Sterner-Kock A, Burhenne J, Haefeli WE, et al. Sildenafil preserves lung endothelial function and prevents pulmonary vascular remodeling in a rat model of diastolic heart failure. Circ Heart Fail. 2011;4:198–206. doi: 10.1161/CIRCHEARTFAILURE.110.957050. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Systemic aortic mean (A), systolic (B) and diastolic (C) pressure after i.v. infusion of GSK1016790A (0.01, 0.03, 0.1 and 0.2 mg·kg−1) in wild-type (wt) mice (n = 7) and KCa3.1−/− mice (n = 3). Results are means ± SEM. #Indicates concentration of GSK1016790A, where mice are experiencing circulatory collapse. *P < 0.05 versus wt.
Figure S2 GSK1016790A induced lung damage in wt mice and KCa3.1−/− mice. H&E staining in sections of lungs infused with vehicle (A, B) or GSK1016790A (C, D) from wt (A, C) and KCa3.1−/− mice (B, D). The changes in extracellular space in the lungs were measured by superimposing a grid on randomized histological sections of the lungs. The line intersections in the extracellular space of each sections was counted and expressed relative to the number of intersections counted in vehicle-infused mice lungs. Alteration in extracellular space in wt but not KCa3.1−/− mice caused by infusion of GSK1016790A (C, D). (E) Summary data. Scale bar = 100 μm in panels. Data points are means ± SEM (n = 4–6). Student’s t-test: *P < 0.05 versus wt mice.
Table S1 Heart rate after GSK1016790A infusion.