

In this issue of the Journal, Jaccard et al. report the outcome of a retrospective multi-institutional experience with bortezomib, cyclophosphamide and dexamethasone (VCD, or CyBorD) used as up-front therapy in 60 Mayo stage III treatment naïve patients with systemic light chain amyloidosis (AL amyloidosis).1 Since cardiac damage rapidly progresses in this disease, the authors evaluated if this regimen, which is known to act rapidly inducing a substantial proportion of profound hematologic responses, was able to rescue cardiac function and extend survival in a population with advanced heart damage. In a multicenter setting, VCD produced an overall response rate of 68% with 42% of patients achieving a very good partial response or a complete response that translated into 32% cardiac responses. Cardiac response was predictive of survival with an estimated, remarkable, 1-year overall survival of 89% for responders. However, 24 patients (40%) died while on therapy and in patients with very advanced amyloid cardiac damage, identified uniquely by high values of the cardiac biomarker N-terminal pro-natriuretic peptide type-B (NT-proBNP) or BNP (NT-proBNP >9500 ng/L or BNP >1100 ng/L), the outcome was dismal with a median survival of only 4.4 months. The results open up a wide array of considerations and perspectives.
Cardiac damage is the determinant of survival in virtually all patients with AL amyloidosis.2 Patients who present with advanced heart involvement, defined by very high levels of NT-proBNP, survive only a few months, representing the major impediment to a further improvement in life expectancy in this disease.3 Clinical and experimental evidence indicate that cardiac damage in AL amyloidosis is mainly determined by a direct toxicity exerted by the circulating amyloidogenic free light chain (FLC). Thus, therapy for cardiac amyloidosis is aimed at obtaining a rapid and profound FLC reduction by targeting the amyloidogenic plasma cell clone with chemotherapy. Several groups have investigated this problem with studies evaluating the efficacy of front-line conventional and novel therapies in patients with amyloid cardiomyopathy.1,2 The outcome of these studies is disheartening, with 20–40% early mortality rates and median survival spanning from a few months to less than two years. The largest population has been reported by the European collaborative retrospective study of 337 patients with cardiac stage III AL amyloidosis, treated mainly by melphalan-dexamethasone (MDex) and cyclophosphamide-thalidomide-dexamethasone (CTD). Interestingly, the threshold of NT-proBNP identifying patients with good outcome has been elevated from 8500 ng/L, reported in the European study, to 9500 ng/L in the present study. The analysis of a joint database of the Pavia and London groups including all the patients (112 stage III) treated with VCD since 2004 confirms that most early deaths occur in subjects with NT-proBNP above 9500 ng/L (Figure 1) (G Palladini and A Wechalekar, unpublished data, 2014). All the outcomes so far available, mostly from retrospective studies, indicate that even the addition of novel agents used synergistically in combination with alkylators and dexamethasone is unable to alter the natural history of very high-risk patients. Two very recent companion papers reporting a matched comparison between two standards of care (MDex and CTD vs. CVD) showed that up-front therapy with CVD correlates with deeper response rates and improved clonal control. However, none of the regimens was able to overcome the expected early deaths seen in patients with severe cardiac damage.4,5 Heart transplantantaion may represent a viable solution, considering the powerful anti-clone regimens now available that can follow transplant, but the limited organ availability significantly narrows its clinical relevance. The treatment of patients with advanced cardiac damage in AL amyloidosis remains an unmet need, and innovative strategies are urgently needed to overcome the apparently insurmountable barrier of severe amyloid cardiomyopathy.
Figure 1.
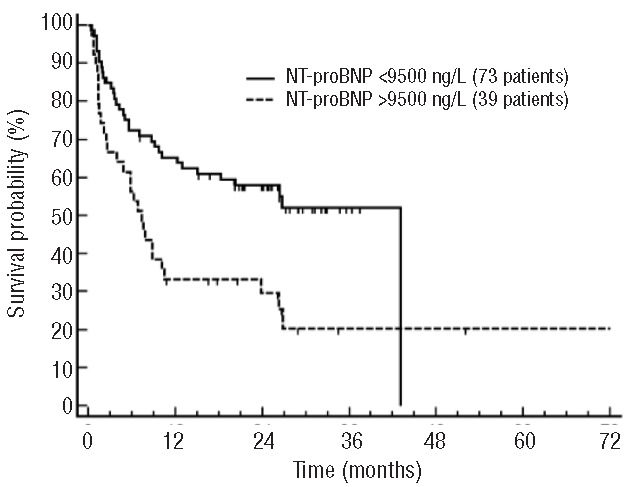
Survival of 112 patients with Mayo Clinic stage III AL amyloidosis treated frontline with CyBorD from a collaborative retrospective study of the Pavia Amyloidosis Research and Treatment Center and the London National Amyloidosis Centre. Median survival 43.0 vs. 7.3 months; P=0.003.
The most obvious intervention is to anticipate the advanced cardiac damage through early diagnosis. It is known that a NT-proBNP cut off of 332 ng/L has perfect sensitivity in detecting heart involvement in AL amyloidosis antedating echocardiographic abnormalities and preceding the onset of symptoms by several months.6 We have already proposed several times2,7 a feasible strategy that includes the addition of the measurement of NT-proBNP to the routine laboratory tests performed at yearly follow up of patients with MGUS and abnormal free light chain ratio (the patients who, according to current guidelines,8 are at higher risk of developing amyloidosis and should undergo lifelong monitoring for symptomatic myeloma). In the presence of abnormal values of NT-proBNP, the search for amyloid deposits in abdominal fat may rapidly confirm diagnosis and allow the prompt institution of rapidly acting regimens thus preventing progression of cardiac damage. Great advances have recently been made in cardiac amyloid imaging.9 In addition to echocardiography, which is the cornerstone for the diagnosis and management of patients with known or suspected cardiac amyloidosis, several techniques that have been recently developed, such as cardiac MRI with native T1 mapping, and molecular imaging methods using specific ligands for amyloid deposits such as aprotinin, Pittsburg compound B and florbetapir, and bone imaging agents have markedly improved the potential to detect cardiac amyloid deposits. The development of quantitative cardiac amyloid imaging, hopefully related to disease activity, as recently suggested by preliminary data obtained with 18F-florbetapir and MRI T1 mapping, offers a potential for detecting early cardiac amyloidosis and for monitoring changes after therapy.10 The combined use of sensitive cardiac biomarkers and cardiac amyloid imaging may greatly enhance the detection of amyloid cardiac involvement and may help widen our understanding of the mechanisms of cardiac amyloid disease.
Innovative therapies for amyloid cardiomyopathy may derive from a better understanding of the disease process. Clinical observations showed a lack of correlation between the amount of amyloid fibril deposition within the heart and the degree of cardiac dysfunction and survival.11 Furthermore, it has been reported that the decrease in serum FLCs translated into a decrease in NT-proBNP and improved survival independently of the amount of cardiac amyloid deposits.12 These clinical observations suggest a direct cardiotoxic effect of amyloid LC which is further supported by in vitro experiments on isolated cardiac muscle cells showing a direct and specific cardiotoxic response to LC purified from patients with amyloid cardiomyopathy.13 The LC cardiotoxic effect involves the activation of specific stress-responsive signaling cascades involving also the p38MAPK pathway resulting in increased oxidant stress.14 It is interesting that the p38MAPK pathway is also involved in the upregulation of the transcription of the BNP gene,15 thus providing an additional link between the secretion of NT-proBNP and the cardiotoxicity exerted by LC. The recent development of C. elegans and zebra fish model systems16,17 promises to accelerate the understanding of cardiotoxic response to amyloid light chain and the development of small-molecule targeted therapies. For instance, selective p38MAPK inhibitors significantly attenuated amyloid LC cardiotoxicity in vitro14 and rescued amyloid LC-induced cardiac dysfunction and attenuated mortality in zebrafish.16 In the C. elegans model, compounds such as epigallocatechin gallate and doxycycline, which are being tested in clinical trials for treating amyloid cardiomyopathy, reduced the toxicity of amyloid LC.17 These novel agents could alleviate the cardiomyocyte damage and improve function, and may act synergistically with chemotherapy to improve patient outcome. Finally, the presence of amyloid deposits might promote the oligomerization of the soluble LC, its amyloid conversion and, possibly, its cell toxicity. Approaches directed at accelerating the removal of amyloid deposits, through immunotherapy18,19 or small molecules20 may also contribute to improve cardiac function.
The aim (and hope) is that the combination of these approaches could rescue a significant proportion of patients with severe heart involvement, expressed by cardiac failure (NYHA class III and IV) and very high concentrations of NT-proBNP, who are now beyond the reach of available remedies. While this therapeutic armamentarium is under development, we should focus on improving our diagnostic skills in order to prevent cardiac damage. The alarm raised by the presentation of symptoms themselves can be too late, and cardiac biomarkers and new imaging technologies are powerful, sensitive, diagnostic tools that should be deployed earlier and more extensively in clinical practice.
Acknowledgments
The authors would like to thank the Associazione Italiana per la Ricerca sul Cancro, special program “5 per mille” (grant n. 9965), and Cariplo Foundation (grant n. 2013-0964) for their support.
Footnotes
Giampaolo Merlini is a professor of clinical biochemistry at the Department of Molecular Medicine of the University of Pavia, and the director of the Amyloidosis Research and Treatment Center of the Fondazione IRCCS Policlinico San Matteo, Pavia, Italy. Giovanni Palladini is a lecturer in clinical biochemistry at the Department of Molecular Medicine of the University of Pavia, and an attending physician at the Amyloidosis Research and Treatment Center, Fondazione IRCCS Policlinico San Matteo, Pavia, Italy. Their main research interests are the molecular mechanisms, diagnosis and treatment of systemic amyloidoses and monoclonal gammopathies, with a particular focus on amyloid cardiomyopathy and related biomarkers.
Financial and other disclosures provided by the author using the ICMJE (www.icmje.org) Uniform Format for Disclosure of Competing Interests are available with the full text of this paper at www.haematologica.org.
References
- 1.Jaccard A, Comenzo RL, Hari P, Hawkins PN, Roussel M, Morel P, et al. Efficacy of Bortezomib, Cyclophosphamide and Dexamethasone in treatment of naive patients with high risk cardiac AL amyloidosis (Mayo Clinic stage III). Haematologica. 2014. May 23 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Merlini G, Palladini G. Light chain amyloidosis: the heart of the problem. Haematologica. 2013;98(10):1492–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wechalekar AD, Schonland SO, Kastritis E, Gillmore JD, Dimopoulos MA, Lane T, et al. A European collaborative study of treatment outcomes in 346 patients with cardiac stage III AL amyloidosis. Blood. 2013;121(17):3420–7. [DOI] [PubMed] [Google Scholar]
- 4.Venner CP, Gillmore JD, Sachchithanantham S, Mahmood S, Lane T, Foard D, et al. A Matched Comparison of Cyclophosphamide, Bortezomib and Dexamethasone (CVD) versus risk adapted Cyclophosphamide, Thalidomide and Dexamethasone (CTD) in AL Amyloidosis. Leukemia. 2014. July 16 [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 5.Palladini G, Milani P, Foli A, Rosin MV, Basset M, Lavatelli F, et al. Melphalan and dexamethasone with or without bortezomib in newly-diagnosed AL amyloidosis: a matched case control study on 174 patients. Leukemia. 2014. July 25 [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 6.Palladini G, Campana C, Klersy C, Balduini A, Vadacca G, Perfetti V, et al. Serum N-terminal pro-brain natriuretic peptide is a sensitive marker of myocardial dysfunction in AL amyloidosis. Circulation. 2003;107(19):2440–5. [DOI] [PubMed] [Google Scholar]
- 7.Merlini G, Wechalekar AD, Palladini G. Systemic light chain amyloidosis: an update for treating physicians. Blood. 2013;121(26):5124–30. [DOI] [PubMed] [Google Scholar]
- 8.Kyle RA, Durie BG, Rajkumar SV, Landgren O, Blade J, Merlini G, et al. Monoclonal gammopathy of undetermined significance (MGUS) and smoldering (asymptomatic) multiple myeloma: IMWG consensus perspectives risk factors for progression and guidelines for monitoring and management. Leukemia. 2010;24(6):1121–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Falk RH, Quarta CC, Dorbala S. How to image cardiac amyloidosis. Circ Cardiovasc Imaging. 2014;7(3):552–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Merlini G, Narula J, Arbustini E. Molecular imaging of misfolded protein pathology for early clues to involvement of the heart. Eur J Nucl Med Mol Imaging. 2014;41(9):1649–51. [DOI] [PubMed] [Google Scholar]
- 11.Rapezzi C, Merlini G, Quarta CC, Riva L, Longhi S, Leone O, et al. Systemic cardiac amyloidoses: disease profiles and clinical courses of the 3 main types. Circulation. 2009;120(13):1203–12. [DOI] [PubMed] [Google Scholar]
- 12.Palladini G, Lavatelli F, Russo P, Perlini S, Perfetti V, Bosoni T, et al. Circulating amyloidogenic free light chains and serum N-terminal natriuretic peptide type B decrease simultaneously in association with improvement of survival in AL. Blood. 2006;107(10):3854–8. [DOI] [PubMed] [Google Scholar]
- 13.Brenner DA, Jain M, Pimentel DR, Wang B, Connors LH, Skinner M, et al. Human amyloidogenic light chains directly impair cardiomyocyte function through an increase in cellular oxidant stress. Circ Res. 2004;94(8):1008–10. [DOI] [PubMed] [Google Scholar]
- 14.Shi J, Guan J, Jiang B, Brenner DA, Del Monte F, Ward JE, et al. Amyloidogenic light chains induce cardiomyocyte contractile dysfunction and apoptosis via a non-canonical p38alpha MAPK pathway. Proc Natl Acad Sci USA. 2010;107(9):4188–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liang F, Gardner DG. Mechanical strain activates BNP gene transcription through a p38/NF-kappaB-dependent mechanism. J Clin Invest. 1999;104(11):1603–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mishra S, Guan J, Plovie E, Seldin DC, Connors LH, Merlini G, et al. Human amyloidogenic light chain proteins result in cardiac dysfunction, cell death, and early mortality in zebrafish. Am J Physiol Heart Circ Physiol. 2013;305(1):H95–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Diomede L, Rognoni P, Lavatelli F, Romeo M, del Favero E, Cantu L, et al. A Caenorhabditis elegans-based assay recognizes immunoglobulin light chains causing heart amyloidosis. Blood. 2014;123(23):3543–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bodin K, Ellmerich S, Kahan MC, Tennent GA, Loesch A, Gilbertson JA, et al. Antibodies to human serum amyloid P component eliminate visceral amyloid deposits. Nature. 2010;468(7320):93–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wall JS, Kennel SJ, Williams A, Richey T, Stuckey A, Huang Y, et al. AL amyloid imaging and therapy with a monoclonal antibody to a cryptic epitope on amyloid fibrils. PLoS One. 2012;7(12):e52686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ward JE, Ren R, Toraldo G, Soohoo P, Guan J, O’Hara C, et al. Doxycycline reduces fibril formation in a transgenic mouse model of AL amyloidosis. Blood. 2011;118(25):6610–7. [DOI] [PMC free article] [PubMed] [Google Scholar]