

Abstract
Robust interpretation of experimental results measuring discreet biological domains remains a significant challenge in the face of complex biochemical regulation processes such as organismal versus tissue versus cellular metabolism, epigenetics, and protein post-translational modification. Integration of analyses carried out across multiple measurement or omic platforms is an emerging approach to help address these challenges. This review focuses on select methods and tools for the integration of metabolomic with genomic and proteomic data using a variety of approaches including biochemical pathway-, ontology-, network-, and empirical-correlation-based methods.
Keywords: data integration, omics, data analysis, networks, bioinformatics, metabolomics, proteomics, genomics
Introduction
Over the past decade, major advancements in omic technologies (eg, genomics, proteomics, and metabolomics) have enabled high-throughput monitoring of a variety of molecular and organismal processes. These techniques have been widely applied to identify biological variants (eg, biomarkers), to characterize complex biochemical systems and to study pathophysiological processes. While many omic platforms target comprehensive analysis of genes (genomics), mRNA (transcriptomics), proteins (proteomics), and metabolites (metabolomics),1 challenges remain for within and between omic-domain data integration.
Biological interpretation of changes in discreet omic domains is challenging in the face of complex biochemical regulation such as organismal versus tissue versus cellular- level processes, epigenetics,2 and mRNA or protein post- translational modification.3,4 Combining experimental results from multiple omic platforms is an emerging approach, which aims to help identify latent biological relationships that may become evident only through holistic analyses integrating measurements across multiple biochemical domains. This article focuses on select methods and tools for the integration of metabolomic with genomic and proteomic data.
Metabolomics, the analysis of small molecules (eg, <1200 Da) and biochemical intermediates (metabolites), has been widely used to study interactions between gene and protein downstream products and environmental stimuli. Over the past decade, metabolomics has been widely used to study various pathophysiological process including type 1 diabetes and cancer, with typical goals involving identification of biomarkers predictive of disease onset, prognosis, and treatment efficacy monitoring.5–8 The metabolome is highly responsive to both environmental and biological regulatory mechanisms (eg, epigenetics, transcription, post-translational modification), the analysis of which presents a unique approach to characterize the organismal phenotype. However, metabolomics by itself may not be sufficient to fully characterize complex biological systems or pathologies (eg, cancer). For example, many researchers focus on the analysis of circulating metabolites (eg, serum or plasma), but this pool is the integrated input and output of many biological systems, making it challenging to derive insights into tissue- and cellular-level mechanisms. Other challenges include effective integration of metabolomic-based analyses in cases of limited biochemical domain knowledge, which may result in sparse and disconnected biological interpretations.9
To date, a variety of software tools have been developed to help integrate multiple omic datasets based on biochemical pathway, ontology, network or empirical correlation (Table 1). A selection of approaches and tools for omic data integration are discussed below.
Table 1.
Key features of a selection of tools for omic data analysis and integration.
| NAME | KEY FEATURES | URL |
|---|---|---|
| Pathway enrichment analysis | ||
| IMPALA | - Integrated pathway-level analysis from gene or protein expression and metabolomics data - Identification of additional pathways from the combined datasets - Accepted inputs: gene or protein expression and metabolomics data - Web-based application - Difficulty: low |
http://impala.molgen.mpg.de/ |
| iPEAP | - Pathway enrichment analysis integrating multiple omic platforms - Identification of additional pathways from the combined datasets - Accepted inputs: transcriptomics, proteomics, metabolomics, and GWAS data - Java-based desktop software - Difficulty: moderate |
http://www.tongji.edu.cn/~qiliu/ipeap.html |
| MetaboAnalyst | - Comprehensive metabolomics including: metabolomics data processing, normalization, multivariate statistical analysis - Functional enrichment analysis for metabolites, including: SNPs, locations, pathways, and diseases - Integrated pathway analysis from gene expression and metabolomics data - Accepted inputs: transcriptomic and metabolomic data - Web-based application - Difficulty: low |
http://www.metaboanalyst.ca/faces/home.xhtml |
| Biological network analysis | ||
| SAMNetWeb | - Generate biological networks for genes, proteins, and transcription factors representing changes in protein and gene expression levels - Integrated network and pathway enrichment analysis - Accepted inputs: transcriptomics and proteomics - Web-based application - Difficulty: moderate |
http://fraenkel-nsf.csbi.mit.edu/samnetweb/ |
| pwOmics | - Compute consensus networks between signaling molecules (genes, proteins, and transcription factors) - Time-series data analysis for identification of co-regulation patterns across time - Accepted inputs: transcriptomic and proteomic data - R package - Difficulty: high |
http://www.bioconductor.org/packages/release/bioc/html/pwOmics.html |
| MetaMapR | - Calculate biochemical reaction, structural similarity, mass spectral similarity, and correlation-based networks - Metabolite identifier translation for >200 common biological databases - Accepted inputs: metabolomic and mass spectral data - R package and user interface - Difficulty: low |
http://dgrapov.github.io/MetaMapR/ |
| MetScape | - Gene, enzyme, and metabolite networks analysis with emphasis on metabolic pathways - Pathway enrichment analysis based on gene expression data - Correlation networks - Accepted inputs: gene expression and metabolite data - Cytoscape plugin - Difficulty: moderate |
http://metscape.ncibi.org/ |
| Grinn | - Integrated neo4j graph-database supporting reconstruction metabolite-protein-gene-pathway - Includes correlation and differential correlation analysis methods - Graph-based integration of biological and empirical relationships - Pathway enrichment - Accepted inputs: genomic, proteomic, and metabolomic data - R package - Difficulty: high |
https://github.com/kwanjeeraw/grinn |
| Empirical correlation analysis | ||
| WGCNA | - Integrated analysis of correlation and network topology - Hierarchical clustering and graph-based module detection - Support for dimensional reduction - Accepted inputs: any - R package - Difficulty: high |
http://labs.genetics.ucla.edu/horvath/CoexpressionNetwork/Rpackages/WGCNA/ |
| MixOmic | - Variety of multivariate analysis and visualization methods - Comparison of two heterogeneous datasets - Multilevel analysis - Accepted inputs: biochemical domain independent - R package - Difficulty: high |
http://mixomics.qfab.org/ |
| DiffCorr | - Compare changes in correlation patterns between two experimental conditions - Accepted inputs: biochemical domain-independent - R package - Difficulty: high |
http://cran.r-project.org/web/packages/DiffCorr/index.html |
| qpgraph | - Estimation of partial correlation and q-order partial correlation - Optimized for small sample sizes and many variables - Accepted inputs: biochemical domain independent - R package - Difficulty: high |
http://www.bioconductor.org/packages/release/bioc/html/qpgraph.html |
| huge | - Fast computation for high-dimensional data using lasso estimate of the inverse covariance matrix - Integrated pipeline for data preprocessing, neighborhood screening, graph estimation, and model selection - Accepted inputs: biochemical domain-independent - R package - Difficulty: high |
https://cran.r-project.org/web/packages/huge/index.html |
Pathway- or Biochemical-Ontology-Based Integration
It is becoming increasingly evident that integrative analyses across multiple omic platforms are required to interrogate complex biological systems. Over the past several years, enrichment analyses methods such as gene set enrichment analysis (GSEA)10 have been widely used to help interpret gene expression data. These methods facilitate biological interpretation by integrating biological domain knowledge (eg, biochemical pathways, biological processes) with gene expression results. Even though these approaches are highly sensitive to the expert definitions of what constitutes a biochemical pathway or a set of related molecular functions, they remain key methods for omic data integration. Existing tools such as IMPALA,11 iPEAP,12 and the integrated pathway analysis in MetaboAnalyst 3.013 support integration of different omic platforms through pathway enrichment and overrepresentation analyses. However, pathway-based approaches rely on predefined pathways, which may not accurately represent the complexity of biological systems and could potentially bias the analysis results.
Biological-Network-Based Integration
Network-based analyses are another set of promising tools used to study a variety of organismal and cellular mechanisms.14 Biological networks represent complex connections among diverse types of cellular components such as genes, proteins, and metabolites. These networks can be used to integrate or map multiple omic experimental results and help identify altered graph neighborhoods, which do not depend on any predefined biochemical pathways. For example, SAMNetWeb15 and pwOmics16 support integration of transcriptomic, proteomic, and interactomic data for biological network computation, visualization and functional enrichment analysis. Metscape,17 a plug-in for the widely used network analysis software Cytoscape,18 supports calculation, analysis, and visualization of gene-to-metabolite networks in the context of metabolism.17 Another software, Meta-MapR,9 leverages the KEGG19 and PubChem20 databases to provide methods for integration and visualization of complex metabolomic results even in cases where biochemical domain knowledge or molecular annotations are unknown.9 For example, MetaMapR has been used to integrate both biochemical reaction information with molecular structural and mass spectral similarity to identify pathway-independent relationships, including, between molecules with unknown structure or biological function.7,8,21 However, biological-network-based methods alone may yield limited insight in cases of insufficient domain knowledge of gene, protein, and metabolite interactions, and are often extended through the incorporation of empirical relationships or correlations between measured species.
Empirical Correlation Analysis
Correlation-based analyses are useful for omic data integration when there is a lack of biochemical domain knowledge and to integrate biological and other meta data (eg, clinical outcomes). The R package22 mixOmics supports correlation analysis between two high-dimensional datasets through methods such as regularized sparse principal component analysis (sPCA), canonical correlation analysis (rCCA), and sparse PLS discriminant analysis (sPLS-DA).23 Weighted gene correlation network analysis (WGCNA) R package extends the concept of correlations to also include measures of graph topology and has been widely used to analyze gene coexpression networks.24 WGCNA can be used to relate clusters of highly connected genes to additional information such as single-nucleotide polymorphisms (SNPs) as well as proteomic and clinical data. Other correlation-based approaches, such as the R package DiffCorr, can be used to focus on differences in patterns of relationships between two physiological conditions.25 Other tools such as MetaMapR incorporate correlation analysis with other relationships such as biochemical reactions and molecular structural and mass spectral similarity.9 The recently developed R package Grinn26 implements a Neo4j27 graph database27 to provide a dynamic interface to rapidly integrate gene, protein, and metabolite data using both biological-network-based and correlation-based approaches.
While correlation-based analyses are relatively simple to implement and widely used for multi-omic data integration, these approaches may provide limited insight in cases of highly multicollinear systems (eg, hairball graphs). Gaussian graphical models, partial correlation and Bayesian networks are more sophisticated approaches that are gaining favor over simple correlations due to their ability to decouple direct from indirect variable associations. For example, the R packages glasso,28 qpgraph,29 and huge30 have been used to identify conditionally independent pairwise relationships (ie, adjusting for all other possible relationships), which can greatly simplify network interpretation. However, these methods may be computationally challenging to implement on typical omic data, which contains far many more measured variables than samples. Bayesian-network-based analyses have been used to robustly integrate multiple high-dimensional datasets even in cases of low sample sizes.31,32 However, one potential limitation of this approach is the need to use prior knowledge to estimate probabilistic interactions between31 modeled variables,31 which may lead to biased conclusions.
Future Directions
Development of methods that can deal with both large, complex, high-dimensional data and sparse biological domain knowledge are required to effectively integrate the massive amounts of biochemical information produced from current and next-generation omic platforms. Developers of future tools need to consider the variety of steps required to effectively integrate multi-omic experiments. Incorporation of scalable and quickly searchable databases, machine-learning methods, and scientific application programming interfaces (APIs) are promising approaches to meet the rapidly growing needs to support current and future omic data analysis and integration pipelines. For example, current state-of-the-field metabolomic experiments may require integration of multiple analytical instruments, data processing methods, robust statistical analyses, machine-learning-based predictive modeling, pathway enrichment and network-based analyses to fully interrogate and interpret the biological systems in question (Fig. 1).7 Development of comprehensive omic analysis tools combining statistical and multivariate analysis with biochemical domain knowledge, such as MetaboAna-lyst13 or DeviumWeb,33 are required to enable efficient omic data analysis and integration. Moreover, it is important that advanced statistical methods, computational packages, and tools are easily accessible and well documented, in order to gain wide adoption by the scientific community. As omic technologies proceed to become higher throughput and grow in coverage and complexity, the bottleneck for omic data analysis will become increasingly shifted to effective integration and interpretation. To meet this need, it will become increasingly necessary to expand currently used data integration approaches including pathway analysis, biochemical and empirical networks to include scalable databases, intuitive user interface, interactive visualizations, machine-learning tools and scientific APIs.
Figure 1.
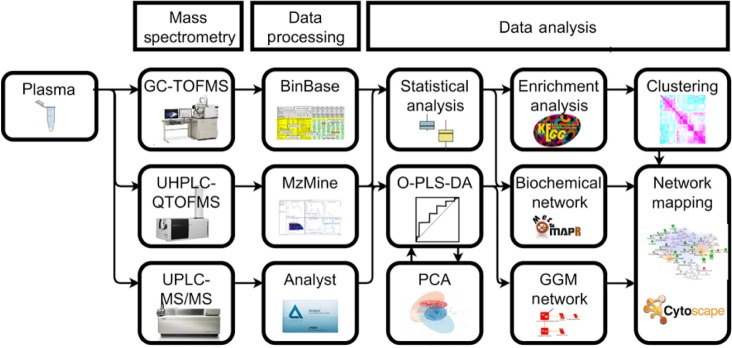
Example of a modern metabolomic data analysis workflow integrating three discreet mass spectral analysis platforms.7 Data from three independent analytical platforms were merged and evaluated using statistical and machine-learning methods to identify significant metabolomic differences and top 10% discriminants between experimental treatments. Partial correlation networks, biochemical enrichment analysis, hierarchical clustering, and biochemical network integration were used to visualize and integrate the high-dimensional omic data within a biological context.
Acknowledgments
The authors thank Prof Oliver Fiehn for his support.
Footnotes
ACADEMIC EDITOR: Dr Karen Pulford, Editor in Chief
PEER REVIEW: Five peer reviewers contributed to the peer review report. Reviewers’ reports totaled 632 words, excluding any confidential comments to the academic editor.
FUNDING: We acknowledge National Institutes of Health grant NIH 1 U24 DK097154 for the West Coast Metabolomics Center. The authors confirm that the funder had no influence over the study design, content of the article, or selection of this journal.
COMPETING INTERESTS: Authors disclose no potential conflicts of interest.
Paper subject to independent expert blind peer review. All editorial decisions made by independent academic editor. Upon submission manuscript was subject to anti-plagiarism scanning. Prior to publication all authors have given signed confirmation of agreement to article publication and compliance with all applicable ethical and legal requirements, including the accuracy of author and contributor information, disclosure of competing interests and funding sources, compliance with ethical requirements relating to human and animal study participants, and compliance with any copyright requirements of third parties. This journal is a member of the Committee on Publication Ethics (COPE).
Author Contributions
Conceived and designed experiments: DG. Analyzed the data: KW, JF, DG. Wrote the first draft of the manuscript: KW, JF, DG. Contributed to the writing of the manuscript: KW, JF, DG. Agree with the manuscripts results and conclusions: KW, JF, DG. Jointly developed the structure and arguments for the paper: KW, JF, DG. Made critical revisions and approved final version: DG. All authors reviewed and approved of the final manuscript.
REFERENCES
- 1.Gracie S, Pennell C, Ekman-Ordeberg G, et al. PREBIC “-Omics” Research Group An integrated systems biology approach to the study of preterm birth using “-omic” technology – a guideline for research. BMC Pregnancy Childbirth. 2011;11:71. doi: 10.1186/1471-2393-11-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33(suppl):245–54. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- 3.Fabian MR, Sonenberg N, Filipowicz W. Regulation of mRNA translation and stability by microRNAs. Annu Rev Biochem. 2010;79:351–79. doi: 10.1146/annurev-biochem-060308-103103. [DOI] [PubMed] [Google Scholar]
- 4.Wang YC, Peterson SE, Loring JF. Protein post-translational modifications and regulation of pluripotency in human stem cells. Cell Res. 2014;24(2):143–60. doi: 10.1038/cr.2013.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Friedrich N. Metabolomics in diabetes research. J Endocrinol. 2012;215(1):29–42. doi: 10.1530/JOE-12-0120. [DOI] [PubMed] [Google Scholar]
- 6.Spratlin JL, Serkova NJ, Eckhardt SG. Clinical applications of metabolomics in oncology: a review. Clin Cancer Res. 2009;15(2):431–40. doi: 10.1158/1078-0432.CCR-08-1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fahrmann J, Grapov D, Yang J, et al. Systemic alterations in the metabolome of diabetic nod mice delineate increased oxidative stress accompanied by reduced inflammation and hypertriglyceridemia. Am J Physiol Endocrinol Metab. 2015;308(11):E978–89. doi: 10.1152/ajpendo.00019.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wikoff WR, Grapov D, Fahrmann JF, et al. Metabolomic markers of altered nucleotide metabolism in early stage adenocarcinoma. Cancer Prev Res (Phila) 2015;8(5):410–8. doi: 10.1158/1940-6207.CAPR-14-0329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grapov D, Wanichthanarak K, Fiehn O. MetaMapR: pathway independent metabolomic network analysis incorporating unknowns. Bioinformatics. 2015;31(16):2757–60. doi: 10.1093/bioinformatics/btv194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–50. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kamburov A, Cavill R, Ebbels TM, Herwig R, Keun HC. Integrated pathway-level analysis of transcriptomics and metabolomics data with IMPaLA. Bioinformatics. 2011;27(20):2917–8. doi: 10.1093/bioinformatics/btr499. [DOI] [PubMed] [Google Scholar]
- 12.Sun H, Wang H, Zhu R, et al. iPEAP: integrating multiple omics and genetic data for pathway enrichment analysis. Bioinformatics. 2014;30(5):737–9. doi: 10.1093/bioinformatics/btt576. [DOI] [PubMed] [Google Scholar]
- 13.Xia J, Sinelnikov IV, Han B, Wishart DS. MetaboAnalyst 3.0-making metabolomics more meaningful. Nucleic Acids Res. 2015;43(W1):W251–7. doi: 10.1093/nar/gkv380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sung J, Wang Y, Chandrasekaran S, Witten DM, Price ND. Molecular signatures from omics data: from chaos to consensus. Biotechnol J. 2012;7(8):946–57. doi: 10.1002/biot.201100305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gosline SJ, Oh C, Fraenkel E. SAMNetWeb: identifying condition-specific networks linking signaling and transcription. Bioinformatics. 2015;31(7):1124–6. doi: 10.1093/bioinformatics/btu748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wachter A, Beissbarth T. pwOmics: an R package for pathway-based integration of time-series omics data using public database knowledge. Bioinformatics. 2015 doi: 10.1093/bioinformatics/btv323. [DOI] [PubMed] [Google Scholar]
- 17.Karnovsky A, Weymouth T, Hull T, et al. Metscape 2 bioinformatics tool for the analysis and visualization of metabolomics and gene expression data. Bioinformatics. 2012;28(3):373–80. doi: 10.1093/bioinformatics/btr661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012;40(Database issue):D109–114. doi: 10.1093/nar/gkr988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bolton EE, Chen J, Kim S, et al. PubChem3D: a new resource for scientists. J Cheminform. 2011;3(1):32. doi: 10.1186/1758-2946-3-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grapov D, Fahrmann J, Hwang J, et al. Diabetes associated metabolomic perturbations in NOD mice. Metabolomics. 2015;11(2):425–37. doi: 10.1007/s11306-014-0706-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.R Development Core Team . R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing; 2011. [Google Scholar]
- 23.González I, Lé Cao K, Déjean S. mixOmics: Omics Data Integration Project. 2011. http://www.mixomics.org.
- 24.Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fukushima A. DiffCorr: an R package to analyze and visualize differential correlations in biological networks. Gene. 2013;518(1):209–14. doi: 10.1016/j.gene.2012.11.028. [DOI] [PubMed] [Google Scholar]
- 26.Wanichthanarak K. Grinn: Graph Database and R Package for Omics Integration. Available at http://github.com/kwanjeera/grinn.
- 27.Neo Technology Inc Neo4j: The World’s Leading Graph Database. 2014. http://neo4j.com/
- 28.Friedman J, Hastie T, Tibshirani R. Sparse inverse covariance estimation with the graphical lasso. Biostatistics. 2008;9(3):432–41. doi: 10.1093/biostatistics/kxm045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Castelo R, Roverato A. Reverse engineering molecular regulatory networks from microarray data with qpgraphs. J Comput Biol. 2009;16(2):213–27. doi: 10.1089/cmb.2008.08TT. [DOI] [PubMed] [Google Scholar]
- 30.Zhao T, Liu H, Roeder K, Lafferty J, Wasserman L. The huge Package for High-dimensional Undirected Graph Estimation in R. J Mach Learn Res. 2012;13:1059–62. [PMC free article] [PubMed] [Google Scholar]
- 31.Mukherjee S, Speed TP. Network inference using informative priors. Proc Natl Acad Sci U S A. 2008;105(38):14313–8. doi: 10.1073/pnas.0802272105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang J, Zuo Y, Man YG, et al. Pathway and network approaches for identification of cancer signature markers from omics data. J Cancer. 2015;6(1):54–65. doi: 10.7150/jca.10631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.DeviumWeb: Dynamic Multivariate Data Analysis and Visualization Platform [Computer Program] 2014. http://github.com/dgrapov/DeviumWeb.