

Abstract
p44 is a short isoform of the tumor suppressor protein p53 that is regulated in an age-dependent fashion. When overexpressed in the mouse it causes a progeroid phenotype that includes premature cognitive decline, synaptic defects and hyperphosphorylation of tau. The hyperphosphorylation of tau has recently been linked to the ability of p44 to regulate transcription of relevant tau kinases. Here, we report that the APP intracellular domain (AICD), which results from the processing of the amyloid precursor protein (APP), regulates translation of p44 through a cap-independent mechanism that requires direct binding to the second internal ribosome entry site (IRES) of the p53 mRNA. We also report that AICD associates with nucleolin, an already known IRES-specific trans-acting factor that binds to p53 IRES elements and regulates translation of p53 isoforms. The potential biological impact of our findings was assessed in a mouse model of Alzheimer’s disease. In conclusion, our study reveals a novel aspect of AICD and p53/p44 biology and provides a possible molecular link between, APP, p44 and tau.
Keywords: p53, p44, AICD, IRES, aging, Alzheimer’s disease
Graphical Abstract
1. Introduction
Due to a combination of alternative promoter usage and alternative initiation of translation, the TP53 gene generates at least 12 different proteins (Bourdon, 2007, Bourdon, et al., 2005). The full-length product (known as p53) has been extensively studied for its tumor-suppressor activity. In contrast, the shorter N-terminal truncated isoforms, Delta40p53, Delta133p53, and Delta160p53 are relatively new and their biological functions are largely unknown. Delta40p53 (referred to as p44 thereafter) lacks the first 39 amino acids of the full-length protein. Transgenic mice overexpressing p44 (p44+/+) develop a progeroid phenotype that includes premature cognitive decline, synaptic defects and hyperphosphorylation of the microtubule-binding protein tau (Maier, et al., 2004, Pehar, et al., 2010). The aberrant phosphorylation of tau is due to the ability of p44 to induce transcription of several tau kinases, including dual-specificity tyrosine-regulated kinase 1a (Dyrk1α), glycogen synthase kinase-3b (GSK3β), and the cyclin-dependent kinase 5 (Cdk5) regulatory partners, Cdkp35 and Cdkp39 (Pehar, et al., 2014). Importantly, haploinsufficiency of Mapt, the gene encoding tau, can rescue the synaptic impairments displayed by p44+/+ mice (Pehar, et al., 2010). Finally, p44 appears to be activated in an age-dependent fashion in the brain (Pehar, et al., 2014), suggesting a possible association with the cognitive decline associated with aging.
It is also worth stressing that hyperphosphorylation of tau has been linked to different forms of age-associated tauopathies as well as to the memory defects that characterize different mouse models of neurodegenerative diseases (Kurz and Perneczky, 2009, Lee, et al., 2001). Hyperphosphorylation and increased aggregation of tau in neurofibrillary tangles are also essential features of Alzheimer’s disease (AD) pathology, the most common form of dementia in the elderly (Pehar & Puglielli 2012).
The amyloid precursor protein (APP) is a type I membrane protein that is tightly linked to the pathogenesis of AD. More than 25 pathogenic mutations have been found in the APP gene (Loy, et al., 2014, Tanzi, 2012); they all cause an autosomal dominant form of AD. A sequence variant that confers strong protection against AD has also been identified (Jonsson, et al., 2012). The causative role of APP (and its proteolytic derivatives) in AD neuropathology is further supported by studies performed in mouse models of the disease (Duyckaerts, et al., 2008, Ghosal, et al., 2009, Gotz, et al., 2004). Finally, down-regulation of the Mapt gene in APP mouse models of AD can rescue the memory loss associated with the disease suggesting a possible role of tau down-stream of APP (Roberson, et al., 2007, Santacruz, et al., 2005).
2. Material and Methods
2.1. Animals and cells
APP695/swe, p53−/− and AICDTg mice were described before (Giliberto, et al., 2008, Maier, et al., 2004, Pehar, et al., 2010). APP−/− mice were from The Jackson Laboratory (stock number 004133). Intact frozen brains from young and old wild-type/non-transgenic mice (4, 18 and 32 months of age) were obtained from the National Institute on Aging (NIA) Rodent Tissue Bank. Animals were maintained in accordance to guidelines for the ethical care and treatment of animals from the Institutional Animal Care and Use Committee of the University of Wisconsin-Madison and the Madison Veterans Administration Hospital. The day of the experiment, mice were sacrificed and brains were immediately processed for analysis. Euthanasia was performed according to the NIH Guide for the Care and Use of Laboratory Animals.
Down syndrome (DS) fibroblasts were obtained from Coriell Cell Repository or from biopsies with approval from the University of Wisconsin Human Subjects Institutional Review Board. Induced pluripotent stem cells (iPSCs) were generated and characterized as previously described (Weick, et al., 2013). iPSCs were differentiated into neurons following established protocols (Johnson, et al., 2007, Li and Zhang, 2006, Zhang, et al., 2001). SH-SY5Y (human neuroblastoma) cells were cultured as described before (Costantini, et al., 2006).
2.2. Human brain tissue
Brain tissue from late-onset AD patients and age-matched controls was kindly provided by the Brain Bank of the Neuropathology Core of the Wisconsin Alzheimer’s Disease Research Center (established by grant P50-AG033514 from NIH/NIA). The use of human brain tissue was approved by the University of Wisconsin-Madison Institutional Review Board in accordance with US federal regulations (as defined under 45 CFR 46.102(f)).
2.3. Western blot
Protein extraction and Western blot procedures were described before (Costantini, et al., 2007, Costantini, et al., 2006, Jonas, et al., 2008, Jonas, et al., 2010, Pehar, et al., 2010, Peng, et al., 2014). The following antibodies were used: p53 (monoclonal-DO1; cat. n. ab1101; Abcam); p53 (monoclonal-PAb240; cat. n. 227-020; Ancell); H3 (polyclonal; cat. n. 07-690; Millipore); APP-C-terminal/AICD (polyclonal; cat. n. AB5352; Millipore); Fe65 (monoclonal; cat. n. 05-758; Millipore); GFP (monoclonal; cat. n. 32-160; Poway); Nucleolin (monoclonal; cat. n. sc-8031; Santa Cruz; monoclonal; cat. n. ab13541; Abcam); PTBP1 (polyclonal; cat. n. sc-16549; Santa Cruz).
2.4. Nuclear Fraction
Cells were suspended in 400 μl cold buffer A (10 mM Hepes pH 7.9, 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM DTT with complete protease and phosphatase inhibitors) and incubated on ice for 15 min. 25 μl NP-40 10% was added and centrifuged for 30 sec at 11,000g at 4°C. Supernatant was collected as cytosolic frac tion. Pellets were resuspended in 50 μl of cold buffer B (20 mM Hepes pH 7.9, 400 mM KCl, 1 mM EDTA, 1 mM EGTA, 1 mM DTT with complete protease and phosphatase inhibitors) and centrifuged at 11,000g for 5 min at 4°C. Supernatant was collected as nuclear fraction.
2.5. Real-Time PCR
Real-time PCR was performed as described (Pehar, et al., 2014). Primers for DYRK1A, GSK3β, CDK5, CDK5P35 and CDK5P39 are reported in (Pehar, et al., 2014). Primers for p44 “short” mRNA were: Forward-5′-GTGGATCCATTGGAAGGGCAG-3′; Reverse-5′-TTGGCAAAACATCTTGTTGAGGGC-3′.
2.6. Immunostaining
Immunostaining procedures were described before (Pehar, et al., 2010, Peng, et al., 2014). The following primary antibodies were used for tau phosphorylation: mouse anti-PHF-Tau (clone AT8; 10 lg/mL; Innogenetics-AutogenBioclear), rabbit anti-phospho-S356-Tau (polyclonal; 1:100; Abcam). Secondary antibodies and imaging were described before (Pehar, et al., 2010).
2.7. Polysome preparation
Cells were homogenized on ice in polysome lysis buffer (1 mM KCl, 2 mM MgCl2, 10 mM Tris-pH 7.6, 2 mM DTT, 100 units/ml RNasin and protease inhibitor cocktail). After centrifugation at 10,000g for 10 min, supernatants were layered over a cushion of polysome lysis buffer containing 30% sucrose and immediately centrifuged at 130,000g for 2.5 h. The polyribosome pellets were resuspended in NT2 buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.05% NP40, 200 units/ml RNasin, Protease inhibitor cocktail).
2.8. RNA-binding Protein Immunoprecipitation assay
RNA-binding Protein Immunoprecipitation (RIP) assay was used to isolate target mRNAs from polysomes. Polyribosome pellets, isolated as above, were used to immunoprecipitate AICD:mRNA complexes. Specifically, an anti-C-terminal APP antibody (Millipore) was incubated with protein-G Sepharose beads overnight; beads-antibody complexes were then incubated with polyribosome lysate at 4°C for 4 h. Target mRNAs we re purified by proteinase K digestion and then isolated with the RNasy Plus Mini Kit (Qiagen). 1 ug mRNAs were reverse-transcribed to cDNA and detected by PCR according to manufacturer’s protocol (Qiagen). The following primers were used: p53/p44 forward-5′-GAGCCGCAGTCAGATCCTA-3′; p53/p44 reverse-5′-TGCTTGGGACGGCAAGG-3′; LEF1 forward-5′-TATGATTCCCGGTCCTCCTGGTC-3′; LEF1 reverse-5′-TGG CTCCTGCTCCTTTCTCTGTTC-3′. LEF1 primers were from (Tsai, et al., 2014).
2.9. RNA-protein pull-down
RNA pull-down was performed with the Pierce Magnetic RNA-Protein Pull-Down Kit (cat. n. 20164; Pierce/Thermo Fisher Scientific). 1st and 2nd IRES of p53 fragments were cloned by PCR with primer sets as follows: 1st IRES: forward-5′-GACATTAATACGACTCACTATAGGGCGTCCAGGGAGCAGGTA-3′ (including T7 promoter); reverse-5′-CAGTGACCCGGAAGGCA-3′; 2nd IRES: forward-5′-GACATTAATACGACTCACTATAGGGGAGCCGCAGTCAGATCCTA-3′ (including T7 promoter); reverse-5′-TGCTTGGGACGGCAAGG-3′. RNA probes were in vitro synthesized with the T7 RNA polymerase (Promega). 50 pmol of RNA was labeled with biotin by using the RNA 3′ End Desthiobiotinylation kit (cat. n. 20163; Pierce). Labeled RNA was conjugated to streptavidin magnetic beads and incubated with cytosol at 4°C overnight. Unbound proteins were removed by washing; bound proteins were eluted by boiling for 10 min in SDS sample buffer (Invitrogen) and then analyzed by Western blotting. As negative control we used a fragment corresponding to the 3′ end of the p53 mRNA that was generated with the following primers: forward-5′-GACATTAATACGACTCACTATAGGGTTTACATTCTGCAAGCACATCT; reverse-5′-ACAGGTGGCAGCAAAGT.
2.10. Luciferase assay
An mRNA construct containing the green fluorescent protein (GFP), a hairpin structure, and the two IRES elements of p53 was generously obtained from Dr. Fahraeus (Bourougaa, et al., 2010). The GFP/hairpin component with the second IRES of the p53/p44 mRNA was inserted into a promoterless pGL4.18 plasmid (Promega) with the following primers: Forward-5′-CTAACTGGCCGGTACCTAATACGACTCACTATAGGGAGACC-3′, Reverse-5′-CCGGATTGCCAAGCTTTGCTTGGGACGGCAAGG-3′. SY5Y and SY5Y-APP cells were transfected with the above construct and luciferase activity was determined 36 h after transfection as described (Ko and Puglielli, 2007, Pehar, et al., 2014). Co-transfected Renilla luciferase was used for transfection efficiency.
2.11. Electrophysiology
Mice were rapidly decapitated and the brain was removed and submerged in ice cold cutting saline (CS [in mM]: 110 Sucrose, 60 NaCl, 3 KCl, 1.25 NaH2PO4, 28 NaHCO3, 0.5 CaCl2, 7 MgCl2, 5 Glucose, 0.6 Ascorbate). The hippocampi were sectioned transversely in a Vibratome (Vibratome) into 400μM slices immersed in ice-cold CS. After sorting, slices were allowed to recover for 45 min at room temperature (RT) in 50:50 CS:Artificial Cerebro Spinal Fluid (ACSF [in mM]: 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 2 CaCl2, 1MgCl2, 25 glucose), and a further 45 min in 100% ACSF at RT before being transferred into an interface chamber (Fine Science Tools) bathed in 100% ACSF (1–1.5 mL/min) at 32°C (Warnder Instrument Corporation, TC-324B) for 90 mins. All solutions were carb-oxygenated (95/5, O2/CO2). Enameled bipolar platinum-tungsten (92:8 Pt:Y) stimulating electrodes were placed at the border of Area CA3 and Area CA1 along the Schaffer-Collateral pathway. Field EPSP’s are recorded from the stratum radiatum, with ACSF filled recording electrodes (1–2 MOhm). Baseline synaptic transmission was assessed for each individual slice by applying gradually increasing stimuli (0.5V – 15V, 25nA – 1.5uA, A-M Systems model 2200 stimulus isolator) to determine the relationship between fiber volley and fEPSP slopes (input:output). All subsequent experimental stimuli were set to an intensity that invoked a fEPSP slope half that of the maximum recorded slope. Synaptic efficacy was continually monitored (0.05Hz). Every 2 min, 4 sweeps were averaged; the fEPSP’s were amplified (A-M Systems model 1800), digitized (Digidata 1322B, Molecular Devices) and then analyzed (pClamp, Molecular Devices).
2.12. Statistical analysis
Data analysis was performed using GraphPad InStat and GraphPad Prism statistical software (GraphPad Software). Data were expressed as mean ± sd and were analyzed using Student’s t-test or one-way analysis of variance followed by Tukey–Kramer multiple comparisons test. Differences were declared statistically significant if p ≤ 0.05.
3. Results
3.1. APP can induce the expression of p44
As mentioned above, p44+/+ transgenic mice display hyperphosphorylation of tau, synaptic deficits, and memory impairment (Pehar, et al., 2010). When engineered to overexpress APP, the animals develop a severe form of neurodegeneration that affects memory-forming and -retrieving areas of the brain (Pehar, et al., 2010). While analyzing the phenotype of p44+/+ and p44+/+; APP695/swe mice we noticed that the distribution pattern, as well as magnitude of changes, of tau hyperphosphorylation in these two different animal models was very similar (Pehar, et al., 2010). Recently, we reported that p44 regulates tau phosphorylation by controlling transcription of tau kinases Dyrk1A, GSK3β, Cdk5, p35, and p39 (Pehar, et al., 2014). To assess whether the similarities previously observed at the histological levels were caused by similar biochemical events, we determined mRNA levels of the above kinases in the hippocampus of p44+/+ and p44+/+; APP695/swe mice. Again, the results showed a strikingly similar outcome (Fig. 1A).
Fig. 1. APP can induce the expression of p44.

(A) Quantitative real-time PCR determination of indicated kinases in the hippocampal formation. Animals (males) were 2.5-month-old when analyzed. All values are mean (n=5) ± sd. *p < 0.05; **p < 0.005. (B) Schematic view showing APP upstream of p44. (C) Western blot analysis showing induction of p44 as a result of APP overexpression. A longer exposure of the membrane (high exp.) is included to show the down-regulation of Delta133p53α. The nuclear marker H3 was used as loading control. (D) Schematic view of p53 isoforms showing the domains targeted by D01 and PAb240 antibodies. (E) The nuclear fraction probed with PAb240 in (C) was probed again with D01. (F) Western blot showing migration profile of p53 and p44 following transfection in SH-SY5Y cells. Lane 1, empty vector; lane 2, p53 cDNA; lane 3, p44 cDNA.
The above results would suggest that the changes in tau metabolism, as well as tau kinases, in the single- and double-transgenics are caused by p44 alone and are not influenced by the APP transgene. Alternatively, they could also suggest that APP acts-at least in part-through p44 itself and that the overexpression of transgenic APP in p44+/+ mice is unable to further stimulate a pathway that is already saturated by the overexpression of its down-stream target (in this case p44; see Fig. 1B).
To discriminate between the above two possibilities, we analyzed the expression profile of p53 isoforms in human neuroblastoma (SH-SY5Y) cells following overexpression of human APP. It is already known that the different p53 isoforms distribute differently between the cytosol and the nucleus (Bourdon, et al., 2005); specifically, p53 is found in both compartments while p44 is preferentially found in the nucleus (Pehar, et al., 2014). Therefore, we separated nuclear from cytosolic proteins and analyzed them separately.
Figure 1C shows that over-expression of APP caused a marked increase in the levels of both full-length p53 and p44. The up-regulation of p44 was only evident in the nuclear fraction while the upregulation of p53 was evident in both the nuclear and cytosolic fractions. Interestingly, APP overexpression also affected the levels of Delta133p53α (Fig. 1C), another short and naturally-occurring p53 isoform that is preferentially found in the nucleus (Bourdon, et al., 2005). In contrast, no effect was observed with Delta133p53γ (Fig. 1C), which is known to distribute to the cytosol (Bourdon, et al., 2005).
When analyzed on SDS-PAGE, human p44 migrates with a molecular mass of ~47-kDa, which is similar to p53β, a C-terminal truncated isoform of p53 (Bourdon, et al., 2005). Since the antibody (PAb240) used in Figure 1C cannot differentiate between the two isoforms, we decided to use antibody D01, which binds to the N-terminal 20–25 aa. region that is missing in p44 (Fig. 1D) and recognizes p53β but not p44 (Bourdon, 2007). Figure 1E shows that the band labeled as p44 in Figure 1C was not observed when we used antibody D01. A similar behavior was observed with Delta133p53α. Finally, we confirmed the migration profile of both p53 and p44 in SH-SY5Y cells following transient overexpression (Fig. 1F).
To further confirm the results obtained with APP over-expressing cell lines, we analyzed levels of p44 in brain tissue of late-onset AD patients. Tissue was processed to isolate nuclei and then analyzed by Western blot. Figure 2A shows a very modest (not significant) or no increase in the levels of p44 in AD patients. The analysis of post-mortem brain tissue is hampered by several confounding factors, which include the long duration of the disease, the degeneration that affects the brain tissue, and, finally, the intrinsic subject variability (discussed later). To compensate for these potential confounding factors we decided to analyze the nuclear levels of p44 in human neurons developed from induced pluripotent stem cells (iPSCs) established from skin fibroblasts mosaic for Down syndrome (DS; trisomy 21) that yielded isogenic control and trisomy 21 iPSCs. It is worth remembering that DS patients have higher propensity of developing a clinical and pathological phenotype that resembles AD (Lott and Dierssen, 2010, Nelson, et al., 2011, Zigman, 2013). This increased risk for AD has been linked to the extra copy of the APP gene, which is located on chromosome 21. Therefore, DS neurons were used in this study because-in light of the trisomy-they naturally overexpress APP. Analysis of differentiated neurons from DS (trisomy 21) and control iPSCs revealed a marked increase in the levels of p44 in the nucleus of DS neurons (Fig. 2B and 2C), supporting the results obtained with APP over-expressing cell lines (Fig. 1C). It is worth stressing that all DS iPSCs lines used here had a third copy of APP. Finally, control and DS neurons appeared morphologically similar (Fig. 2B; see also (Weick, et al., 2013)). Importantly, p44 levels were unchanged in non-differentiated iPSCs and only slightly increased in skin fibroblasts (data not shown) suggesting that the APP-mediated up-regulation of p44 is neuron-specific.
Fig. 2. p44 levels in the brain of late-onset AD patients, human trisomy 21 neurons, and brain of APP695/swe and AICDTg mice.

(A) Western blot showing levels of p53 and p44 in the brain of AD patients (mean age: 71 yr; range: 67–91 yr; sex: males) and age-matched controls (mean age: 70 yr; range: 66–78 yr; sex: males). Only the nuclear fraction was analyzed. Representative images are shown in the left panel while quantification is shown in the right panel. (B) Neurons differentiated from DS and control iPSCs. (C) Western blot showing increased levels of p44 in neurons differentiated from DS and control iPSCs. Representative images are shown in the left panel while quantification is shown in the right panel. Two independent lines are shown. (D) Western blot showing increased levels of p44 in the brain of APP695/swe mice. Representative images are shown in the left panel while quantification is shown in the right panel. NTG, non-transgenic/wild-type. (E) Western blot showing increased levels of p44 in the brain of AICDTg mice. Representative images are shown in the left panel while quantification is shown in the right panel. NTG, non-transgenic/wild-type. All values are mean ± sd. **p < 0.005.
Next, we analyzed levels of p44 in mice overexpressing (APP695/swe) and lacking (APP−/−) APP. Analysis of the nuclear fraction of APP695/swe mouse brains showed a consistent and statistically significant increase in the levels of p44 (Fig. 2D). However, no change in p44 levels was observed in APP−/− animals (Fig. 2D) suggesting that although APP can induce the expression of p44, it is not essential for its baseline levels. Finally, we determined the nuclear levels of p44 in transgenic mice overexpressing AICD alone (AICDTg; (Giliberto, et al., 2008)). The result revealed a significant up-regulation (Fig. 2E).
In conclusion, when taken together, the results displayed in Figure 1 and Figure 2 indicate that APP can induce the expression of p44. These results were obtained in a variety of experimental systems, which included cell lines overexpressing APP, mice overexpressing APP and AICD, and human trisomy 21 iPSCs-induced neurons that express a third copy of APP. Therefore, the similar behavior of p44+/+ and p44+/+; APP695/swe mice (see Fig. 1A and (Pehar, et al., 2010)) could be explained by the fact that the overexpression of transgenic APP in p44+/+ mice is unable to further stimulate a pathway that is already saturated by the overexpression of its down-stream target. Under these conditions (mice overexpressing transgenic p44), further expression of APP (and induction of endogenous p44) is ineffective (see Fig. 1B).
3.2. AICD regulates translation of p53/p44 through an IRES-dependent mechanism
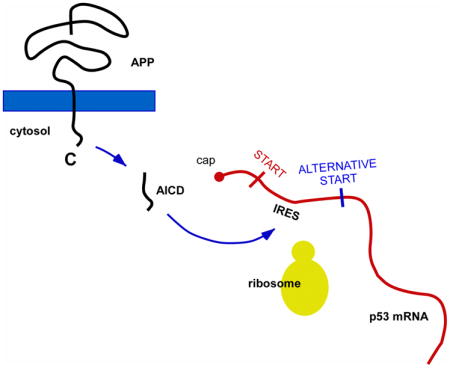
Next, we decided to determine the molecular mechanism responsible for the APP-mediated upregulation of p44. The different p53 isoforms are generated by alternative splicing, alternative promoter usage or alternative initiation of translation (Bourdon, 2007). Specifically, p44 preferentially originates from alternative initiation of translation of the full-length p53 mRNA (reviewed in (Scrable, et al., 2005)). The alternative translation of the p53 mRNA is achieved through two internal ribosome entry sites (IRESs) that regulate cap-independent translation of full-length p53 and p44 (see Fig. 3A). The first IRES, mediating translation at codon 1, is in the 5′-untranslated region of the mRNA and leads to translation of full-length p53; in contrast, the second IRES, mediating translation at codon 40 (p44), extends into the protein coding region and leads to translation of p44 (Ray, et al., 2006). In addition to the above IRES-dependent alternative initiation of translation, p44 can also originate from a shorter mRNA generated by alternative splicing of intron 2 (Ghosh, et al., 2004).
Fig. 3. AICD binds to IRES elements of the p53 mRNA and can induce cap-independent translation of p44.

(A) Schematic view of the IRES elements in the p53 mRNA. (B) Quantitative real-time PCR showing that the levels of p44-specific mRNA are not affected by APP overexpression. Results are mean (n=6) ± sd. (C) Western blot showing the presence of AICD in the polysome fraction. N, nucleus; P, polysome; C, cytosol; Ctrl, control (non-transfected) cells; Empty, cells transfected with empty plasmid; APP, cells transfected with APP. (D) RNA-binding protein immunoprecipitation showing that AICD binds to the p53 mRNA in vivo. Input (lane 5) and negative controls (lanes 1–2) are also shown. (E–F) mRNA-protein pull-down showing that AICD binds to the IRES elements of the p53 mRNA in vitro. The pull-down was done with DS fibroblasts (E) and SH-SY5Y cells overexpressing APP (SH-SY5YAPP; F). A fragment corresponding to the 3′ end of the p53 mRNA was used as negative control. (G) Schematic view of the bicistronic mRNA construct used in (H-J). The GFP was placed upstream of a hairpin structure (shown as a black circle) that impedes ribosomal read-through. The p44-specific IRES element without the first AUG site was placed after the hairpin structure and immediately upstream of the luciferase reporter system. For comparison, see a scheme of the normal p53 mRNA in (A). H-J, Overexpression of APP can induce cap-independent translation of p44. Expression of GFP (H and I) served as control. The luciferase reporter assay (J) was normalized to Renilla and expressed as mean (n=4) + sd. **p < 0.005.
To discriminate between the two possibilities, we initially determined the levels of the short, p44-specific mRNA in SH-SY5Y cells overexpressing APP. However, no changes were observed (Fig. 3B) suggesting an IRES-dependent mechanism. This was somehow expected because the above mRNA appears to be limited to certain cellular situations and does not seem to be the preferential form of regulation of p44 levels (Bourdon, 2007, Scrable, et al., 2005). Therefore, we decided to assess whether APP, or its proteolytic derivatives, could regulate IRES-dependent translation of the p53 mRNA.
APP undergoes proteolytic processing, which results in at least three major fragments: the N-terminal ectodomain, which is secreted in the extracellular milieu, the Aβ peptide, which tends to aggregate in extracellular amyloid plaques, and the C-terminal tail (also known as AICD), which is found in the cytosol and nucleus. AICD is known to act as a transcriptional activator/regulator ((Cao and Sudhof, 2001, Pardossi-Piquard, et al., 2005); reviewed in (Beckett, et al., 2012, Pardossi-Piquard and Checler, 2012)). Therefore, we decided to focus our attention to AICD.
We initially determined the distribution profile of AICD. In fact, we reasoned that if AICD regulates translation of the p53 mRNA it must be found in messenger-ribonucleoprotein (mRNP) complexes as well as in the cytosol. Consistent with its biological functions, AICD was found in both the cytosol and nucleus (Fig. 3C). Importantly, AICD was also found in polysomes containing highly purified mRNP particles that were isolated from the cytosolic fraction (Fig. 3C). Next, we assessed whether the AICD found in the cytosol and polysome fractions was tightly bound to the p53 mRNA in the mRNP complexes. For this purpose, we performed a RNA-binding protein immunoprecipitation assay. Specifically, we purified polysomes from the cytosol of APP overexpressing SH-SY5Y cells and used them to immunoprecipitate AICD:mRNA complexes. The immunoprecipitated complexes were then digested with proteinase K to eliminate the protein component; the free mRNA was retrotranscribed and amplified by PCR. Primers used for the assay covered the second IRES located between the +1 and the +120 codons of the p53 mRNA (see Fig. 3A). The results indicate that AICD is bound to the p53 mRNA in native mRNP complexes (Fig. 3D). The band corresponding to the p53 mRNA was not resolved when the polysomes were immunoprecipitated with an anti-myc antibody (Fig. 3D), thus providing specificity to our results. As additional control we used the same AICD:mRNA complexes and tried to expand the IRES element of the lymphoid enhancer factor-1 (LEF-1) mRNA (Jimenez, et al., 2005, Tsai, et al., 2014). The results show that AICD does not bind to the IRES element of LEF-1 and, therefore, does not appear to behave as a promiscuous IRES-binding protein.
To further confirm the above results, we assessed whether AICD was able to bind to the p53 mRNA in vitro. Specifically, we generated a mRNA construct corresponding to either the IRES1 or IRES2 area and then cross-linked it to biotin at the 3′ end. The biotinylated mRNA probe was used to pull-down AICD from the cytosol. Following purification with streptavidin, the mRNA:protein complex was digested with RNAses prior to SDS-PAGE and immunoblotting with an anti-AICD antibody. The AICD fragment was successfully pulled down from the cytosol of both DS fibroblasts and SH-SY5YAPP cells (Fig. 3E and 3F). Interestingly, Fe65, which is known to associate to AICD and potentiate its transcription-regulatory activity (Pardossi-Piquard and Checler, 2012), was not found in the pull-down (Fig. 3E). These findings might indicate that Fe65 is not required for AICD translation-regulatory activities. As internal control for the pull-down experiment, we also used a probe corresponding to the 3′end of the p53 mRNA. The results indicate that the AICD does not simply bind any mRNA probe (Fig. 3E and 3F; see lanes labeled as 3′end); binding is specific for IRES structures (Fig. 3E and 3F).
Finally, we used a bicistronic mRNA construct with a hairpin structure inserted at the 3′ of the green fluorescent protein (GFP) and immediately before the second IRES (IRES2) of the p53 mRNA, which controls translation of p44. IRES2, was-in turn-placed upstream of a luciferase-reporter system (see Fig. 3G). The hairpin element was used to minimize cap-dependent ribosomal read-through. Therefore, in this system translation of GFP is cap-dependent while translation of the luciferase system is IRES-dependent. When the bicistronic mRNA construct was transfected in SH-SY5Y cells, we detected successful translation of GFP (Fig. 3H and 3I) as well as luciferase activity (Fig. 3J). However, only the translation of the luciferase-reporter system was significantly affected by the overexpression of APP (Fig. 3H–3J). The above findings indicate that AICD can bind to the second IRES of the p53 mRNA and regulate translation of p44 through a cap-independent mechanism.
When taken together, the above results indicate that AICD is able to regulate translation of p53/p44 through a cap-independent mechanism that involves direct binding of the IRES structures on the p53 mRNA. Importantly, our results were obtained with different strategies, all of which included internal controls.
When the DNA binding properties of AICD were first described, it was shown that AICD acts as part of a complex that includes the adaptor protein Fe65 and the histone acetyltransferase Tip60 activity (Cao and Sudhof, 2001). Therefore, we decided to investigate whether the translational properties of AICD also required additional co-factors. Specifically, we used the IRES1 and IRES2 mRNA probes described in Figure 3E and 3F to pull-down potential IRES-specific trans-acting factors (ITAFs) that bind to p53 IRES elements. Mass-spectrometry (MS) of the pull-down identified two proteins: nucleolin (UniProt/Swiss-Prot: P19338) and polypyrimidine tract-binding protein 1 (PTBP1; UniProt/Swiss-Prot: P26599). Both proteins had previously been shown to bind to the IRES elements of the p53 mRNA and regulate translation of p53 isoforms (Grover, et al., 2009, Grover, et al., 2008, Takagi, et al., 2005). To confirm the MS results, we repeated the mRNA:protein pull-down and assessed the presence of the proteins by immunoblotting. The results show successful pull-down for both nucleolin and PTBP1 (Fig. 4A and 4B). Again, a mRNA probe corresponding the 3′end of the p53 mRNA was used to confirm specificity (Fig. 4A and 4B; see lanes labeled as 3′end). Next, to explore functional association with AICD, we performed co-immunoprecipitation experiments in DS fibroblasts. Immunoprecipitation of AICD successfully pulled-down nucleolin but not PTBP1 (Fig. 4C). Nucleolin is known to acts as a “negative” regulator of IRES-dependent translation (Takagi, et al., 2005) whereas the AICD appears to act as a “positive” regulator (see Fig. 3). To assess whether age-associated changes in the expression profile of nucleolin contribute to the age-associated upregulation of p44 that we previously described (Pehar, et al., 2014), we assessed levels of nucleolin in the frontal cortex of wild-type/non-transgenic mice. The results show a significant downregulation of nucleolin as a result of normal aging (Fig. 4D).
Fig. 4. AICD interacts with nucleolin.

(A–B) mRNA-protein pull-down showing that nucleolin (A) and PTBP1 (B) bind to the IRES elements of the p53 mRNA in vitro. The pull-down was done with DS fibroblasts. A fragment corresponding to the 3′ end of the p53 mRNA was used as negative control. (C) Immunoprecipitation of AICD from the cytosolic fraction of DS fibroblasts was able to pull-down nucleolin but not PTBP1. Immunoprecipitation was done with an antibody against the AICD peptide and Protein A (negative control). (D) Western blot showing levels of NCL in the brain of wild-type/non-transgenic mice. Representative Western blots are shown in the upper panel while quantification is shown in lower panel. Values are mean (n=3) ± sd. *p < 0.05.
When taken together, the above results suggest that functional association between AICD and nucleolin may affect translation of the p53 mRNA in an age-dependent fashion (discussed later).
3.3. Down-regulation of p53/p44 rescues the APP phenotype
To assess biological significance of our findings, we analyzed mice overexpressing APP (APP695/swe). The animals displayed increased mRNA levels of Dyrk1A, GSK3β, p35, and p39 (Fig. 5A) as well as hyperphosphorylation of tau (Fig. 5B). The upregulation of the above kinases was already evident at 2.5-months of age. These changes were all rescued by the haploinsufficiency of Tp53, the gene that encodes all p53 isoforms (Fig. 5A and 5B). APP695/swe mice display defects in the post-synaptic component of long-term potentiation (LTP) (reviewed in (Duyckaerts, et al., 2008)). These defects were also rescued by the haploinsufficiency of Tp53 (Fig. 5C).
Fig. 5. Haploinsufficiency of p53/p44 can rescue some of the phenotypic features caused by the overexpression of APP in the mouse.

(A) Quantitative real-time PCR determination of indicated kinases in the hippocampal formation. Animals (males) were 2.5-month-old when analyzed. All values are mean (n=12) ± sd. *p < 0.05; **p < 0.005. (B) Immunostaining with anti-phospho-tau antibodies shows reduced labeling in APP695/swe;p53+/− mice. Two different antibodies were used: AT8 (against pSer202 and pThr205) and S356 (against pSer356). Mice (males) were ~1-year-old when analyzed. (C) Long-term potentiation (LTP) induction in hippocampal slices. Mice (males) were 2.5-month-old when analyzed. APP695/swe mice display deficits in the late component of LTP. These deficits are rescued by p53/p44 haploinsufficiency. **p < 0.005.
When taken together, the above studies suggest that some of the changes induced by the overexpression of APP, specifically those impinging on tau, might depend on the induction of p44. Tp53 encodes both full-length p53 and p44; therefore, p53- and p44-specific events cannot be truly differentiated in the above p53+/− model. However, overexpression of p44 alone (p44+/+ mice) caused altered metabolism of tau as well as synaptic deficits (Pehar, et al., 2010), while over-expression of full-length p53 (Super-p53 mice) did not (Garcia-Cao, et al., 2002). Therefore, it is likely that the changes induced by the genetic disruption of Tp53 are directly linked to p44 itself or to the imbalance in p44:p53 expression caused by AICD.
4. Discussion
In the present study we show that AICD, the cytosolic tail of APP, can bind to the IRES elements of the p53 mRNA and regulate translation of p44, the longevity-assurance isoform of p53. Specificity of binding as well as translational-regulatory activity was established with a combination of in vitro and in vivo strategies. We also show that AICD functionally associates with NCL, an already known ITAF that binds to the IRES elements of the p53 mRNA (Grover, et al., 2009, Grover, et al., 2008). Finally, the possible biological implications of our findings were assessed in a mouse model of AD. Combined with already published data (Pehar, et al., 2014, Pehar, et al., 2010), our results provide a possible molecular link between APP, p44, and tau (discussed below).
4.1. IRES elements and protein translation
The initiation of protein translation in eukaryotes is typically mediated by a cap- and 5′end-dependent mechanism. Specifically, the “ribosomal scanning machinery”, which includes the 40S ribosomal subunit and the initiation factor complex, recognizes and binds to the 5′end (methylated) cap structure of the mRNA. Translation starts when the “scanning machinery” reaches the intiation codon (Voet & Voet 2011). IRES elements are highly structured internal segments of the mRNA that allow cap- and 5′end-independent binding of the “ribosomal scanning machinery”. They were initially identified on virus mRNAs. In eukaryotes they ensure protein translation under conditions that are characterized by a partial block of cap-dependent translation (i.e., ER stress, mitosis, etc.) (reviewed in (Baird, et al., 2006)). IRES elements across mRNAs tend to have limited sequence homology and are most typically characterized by their secondary and tertiary structure. Comparative sequence homology and minimum free energy structure modeling have been employed to search for IRES elements across the mRNA database; however, they have not been able to correctly predict a common structural signature (Wu, et al., 2009). As a result, the identification of IRES structures has mostly been driven by direct testing of individual mRNAs through bicistronic constructs (Baird, et al., 2006). IRES-dependent translation is regulated and enhanced by mRNA binding proteins known as ITAFs. In general ITAFs are found associated with mRNP complexes and “free” in the cytosol. Some ITAFs also participate in processing and export of mRNAs from the nucleus and, as a result, can also be found in nuclear fractions (Baird, et al., 2006). Finally, ITAFs with both transcription- and translation-regulatory functions have also been identified (Baird, et al., 2006).
4.2. AICD binds to IRES elements on the p53 mRNA
The first evidence that AICD is released in the cytosol following γ cleavage of APP was reported by Passer et al. (Passer, et al., 2000). Soon after, Cao et al. showed that a functional complex of AICD, Fe65 and Tip60 had DNA binding activity (Cao and Sudhof, 2001). The transcriptional properties of AICD have now been confirmed by several groups (reviewed in (Beckett, et al., 2012, Pardossi-Piquard and Checler, 2012)). Targeted genes include APP, BACE1, and NEP, among others (reviewed in (Beckett, et al., 2012, Pardossi-Piquard and Checler, 2012)). Previous work from da Costa et al. showed that AICD can induce transcription of the p53 mRNA (Alves da Costa, et al., 2006). Here, we report yet another surprising feature of AICD biology, specifically, its ability to bind to IRES structures on the translating mRNA of p53. This activity seems to be Fe65 independent and results in increased translation of p44, a short isoform of p53 with “longevity-assurance” activity. Whether or not AICD can bind to other IRES structures is currently unknown and it remains to be assessed. Direct analysis of known IRES structures (http://www.iresite.org/IRESite_web.php) did not reveal specific features that would make p53 IRES elements unique. Therefore, it is possible that AICD might regulate translation of other mRNAs. However, as stressed above, no prediction model has been able to find a common IRES or ITAF signature (Baird, et al., 2006). As such, identification of other putative targets will require direct testing.
The two IRES elements on the p53 mRNA have been shown to regulate translation of p53 and p44 under different cellular conditions (Grover, et al., 2009). Specific ITAFs have also been identified. They include, p53 itself, PTBP1, ribosomal protein L26, and nucleolin (Grover, et al., 2009, Grover, et al., 2008, Mosner, et al., 1995, Takagi, et al., 2005). Our work provided evidence that AICD is a novel p53 ITAF. Interestingly, immunoprecipitation of AICD was also able to pull-down nucleolin suggesting functional interaction between the two proteins. The interaction appears to be specific since PTBP1 was not pulled-down with the AICD. The biological significance of the AICD-nucleolin interaction remains to be fully dissected. However, as mentioned above, nucleolin seems to be a “negative” ITAF (Takagi, et al., 2005) while, like PTB, AICD appears to be a “positive” factor (present study). In our study we also show that nucleolin levels in the brain decrease in an age-dependent fashion. Unfortunately, the AICD peptide displays a very short half-life (reviewed in (Nalivaeva and Turner, 2013)) impeding direct and definitive assessment of its endogenous levels throughout age. However, both expression and β processing of APP appear to be regulated in an age-dependent fashion (reviewed in (Nalivaeva and Turner, 2013)). Since AICD is stoichiometrically released with the Aβ peptide as a result of the β/γ processing of APP we could expect a parallel increase in intracellular levels of AICD. Obviously, as mentioned above, due to the intrinsic difficulties in assessing endogenous levels of AICD in the brain, the above scenario remains speculative. Of interest is the fact that the expression of p44 is also regulated (at least in the brain) in an age-dependent fashion (Pehar, et al., 2014). Perhaps AICD and nucleolin interact in order to regulate p53 translation in a coordinated manner. It is likely that molecular dissection of their interaction will yield important information.
4.3. AICD-p44 and possible impact for aging, AD, and age-associated tauopathies
Although mostly known for its tumor-suppressor activities, TP53 encodes a large family of proteins retaining different domains/motifs of the full-length protein (Bourdon, 2007, Bourdon, et al., 2005). The different isoforms result from a combination of alternative promoter usage and alternative initiation of translation (Bourdon, 2007, Bourdon, et al., 2005). The alternative translation of p53 and p44 is tightly regulated by several ITAFs. As mentioned above, p53 itself can regulate its own translation by binding to the first IRES element (reviewed in (Scrable, et al., 2005)). In fact, full-length p53 binds to the stem-loop element of its own mRNA and, by doing so, prevents translation of codon 1 while allowing translation of codon 40. This blockage can be released by the Mdm2 E3 ligase, which regulates the degradation of p53 itself (Murray-Zmijewski, et al., 2008), thereby reactivating translation of codon 1. The blockage is reinstated when p53 levels increase again, either because of increased translation or reduced degradation. Importantly, Mdm2 also regulates degradation of p53 by binding to the same N-terminal region of p53 that is missing in the Δ40p53 isoform (Yin, et al., 2002). In conclusion, the expression of the p53 isoforms appears to be tightly regulated by a complex array of molecular events.
The p53 isoforms are likely to retain some of the functions elicited by the full-length protein. However, they are also likely to elicit biological events that are either unique or different. Specifically, p44 appears to possess “longevity-assurance” activity. When overexpressed in the mouse (p44+/+), it causes a progeroid phenotype and reduced lifespan (Maier, et al., 2004). Importantly, the phenotype requires co-expression of the full-length protein (Maier, et al., 2004) suggesting that the ratio (or functional interaction) of the different p53 isoforms, rather than the individual proteins, determine the biological outcomes. When engineered to overexpress APP, p44+/+ mice display an accelerated form of AD-like neuropathology (Pehar, et al., 2010) offering a possible link between aging and AD.
Although highly suggestive, the impact of our findings for AD neuropathology remains to be fully determined. Since p44 can regulate transcription of several tau kinases (Pehar, et al., 2014) and, therefore, affect the phosphorylation status of tau, we could envision a molecular pathway that connects AICD, p44 and tau (present study and (Pehar, et al., 2014, Pehar, et al., 2010)). This pathway could be implicated in AD as well as in other forms of tauopathies associated with aging. Assessment of p44 levels in the brain of late-onset AD patients did not yield striking results. However, AD is a heterogeneous disease and not all AD patients are identical. For example, AD patients with a duplication of the APP gene have been identified; they express levels of APP mRNA that are in the same range of DS (trisomy 21) patients (Israel, et al., 2012). Since differentiated neurons of DS patients display increased levels of p44, we could conceive that AD patients that display either duplication of APP or clear upregulation of APP β processing will share similar features. Interestingly, studies have shown that late-onset AD patients (who do not have a duplication of the APP gene) express increased levels of a p53 isoform that migrates with the molecular mass of p44 on polyacrylamide gels (Uberti, et al., 2006, Uberti, et al., 2008). It is also worth stressing that transgenic mice overexpressing AICD (alone or with Fe65) develop some of the features that characterize AD (Ghosal, et al., 2009, Giliberto, et al., 2008). These include abnormal activation of tau kinases and abnormal phosphorylation of mouse tau as well as synaptic deficits and increased neuronal susceptibility to exogenous stress (Ghosal, et al., 2009, Giliberto, et al., 2008).
In conclusion, because of the impact of the role that p44 plays in longevity and cognitive-related events, our findings might offer novel insights on an important topic of biomedical research.
Highlights.
p44 is a short isoform of the tumor suppressor protein p53 with longevity-assurance activity.
AICD is a cytosolic and nuclear fragment of APP.
AICD binds to the second IRES of the p53 mRNA and regulates translation of p44.
Haploinsufficiency of p53/p44 can rescue the phenotype of APP695/swe mice.
Acknowledgments
The authors wish to remember Dr. Heidi Scrable, who died on February 13, 2013 after a prolonged battle with a disease. Although she participated in all intellectual aspects of this work, she never saw the final version of this manuscript. The authors thank Dr. Robin Fahraeus for the bicistronic construct. This work was supported by: VA Merit Award (BX001638), NIH (P50 AG033514 and GM103542), and Thome Memorial Foundation.
Footnotes
Disclosure Statement
None of the authors report any potential or actual conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alves da Costa C, Sunyach C, Pardossi-Piquard R, Sevalle J, Vincent B, Boyer N, Kawarai T, Girardot N, St George-Hyslop P, Checler F. Presenilin-dependent gamma-secretase-mediated control of p53-associated cell death in Alzheimer’s disease. J Neurosci. 2006;26:6377–85. doi: 10.1523/JNEUROSCI.0651-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baird SD, Turcotte M, Korneluk RG, Holcik M. Searching for IRES. Rna. 2006;12(10):1755–85. doi: 10.1261/rna.157806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckett C, Nalivaeva NN, Belyaev ND, Turner AJ. Nuclear signalling by membrane protein intracellular domains: the AICD enigma. Cellular signalling. 2012;24(2):402–9. doi: 10.1016/j.cellsig.2011.10.007. [DOI] [PubMed] [Google Scholar]
- Bourdon JC. p53 and its isoforms in cancer. Br J Cancer. 2007;97:277–82. doi: 10.1038/sj.bjc.6603886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourdon JC, Fernandes K, Murray-Zmijewski F, Liu G, Diot A, Xirodimas DP, Saville MK, Lane DP. p53 isoforms can regulate p53 transcriptional activity. Genes Dev. 2005;19:2122–37. doi: 10.1101/gad.1339905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourougaa K, Naski N, Boularan C, Mlynarczyk C, Candeias MM, Marullo S, Fahraeus R. Endoplasmic reticulum stress induces G2 cell-cycle arrest via mRNA translation of the p53 isoform p53/47. Mol Cell. 2010;38:78–88. doi: 10.1016/j.molcel.2010.01.041. [DOI] [PubMed] [Google Scholar]
- Cao X, Sudhof TC. A transcriptionally [correction of transcriptively] active complex of APP with Fe65 and histone acetyltransferase Tip60. Science. 2001;293:115–20. doi: 10.1126/science.1058783. [DOI] [PubMed] [Google Scholar]
- Costantini C, Ko MH, Jonas MC, Puglielli L. A reversible form of lysine acetylation in the ER and Golgi lumen controls the molecular stabilization of BACE1. Biochem J. 2007;407:383–95. doi: 10.1042/BJ20070040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantini C, Scrable H, Puglielli L. An aging pathway controls the TrkA to p75(NTR) receptor switch and amyloid beta-peptide generation. EMBO J. 2006;25:1997–2006. doi: 10.1038/sj.emboj.7601062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duyckaerts C, Potier MC, Delatour B. Alzheimer disease models and human neuropathology: similarities and differences. Acta Neuropathol. 2008;115:5–38. doi: 10.1007/s00401-007-0312-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Cao I, Garcia-Cao M, Martin-Caballero J, Criado LM, Klatt P, Flores JM, Weill JC, Blasco MA, Serrano M. “Super p53” mice exhibit enhanced DNA damage response, are tumor resistant and age normally. EMBO J. 2002;21:6225–35. doi: 10.1093/emboj/cdf595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosal K, Vogt DL, Liang M, Shen Y, Lamb BT, Pimplikar SW. Alzheimer’s disease-like pathological features in transgenic mice expressing the APP intracellular domain. Proc Natl Acad Sci USA. 2009;106:18367–72. doi: 10.1073/pnas.0907652106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A, Stewart D, Matlashewski G. Regulation of human p53 activity and cell localization by alternative splicing. Mol Cell Biol. 2004;24:7987–97. doi: 10.1128/MCB.24.18.7987-7997.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giliberto L, Zhou D, Weldon R, Tamagno E, De Luca P, Tabaton M, D’Adamio L. Evidence that the Amyloid beta Precursor Protein-intracellular domain lowers the stress threshold of neurons and has a “regulated” transcriptional role. Mol Neurodegener. 2008;3:12. doi: 10.1186/1750-1326-3-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotz J, Streffer JR, David D, Schild A, Hoerndli F, Pennanen L, Kurosinski P, Chen F. Transgenic animal models of Alzheimer’s disease and related disorders: histopathology, behavior and therapy. Mol Psychiatry. 2004;9:664–83. doi: 10.1038/sj.mp.4001508. [DOI] [PubMed] [Google Scholar]
- Grover R, Candeias MM, Fahraeus R, Das S. p53 and little brother p53/47: linking IRES activities with protein functions. Oncogene. 2009;28:2766–72. doi: 10.1038/onc.2009.138. [DOI] [PubMed] [Google Scholar]
- Grover R, Ray PS, Das S. Polypyrimidine tract binding protein regulates IRES-mediated translation of p53 isoforms. Cell Cycle. 2008;7:2189–98. doi: 10.4161/cc.7.14.6271. [DOI] [PubMed] [Google Scholar]
- Israel MA, Yuan SH, Bardy C, Reyna SM, Mu Y, Herrera C, Hefferan MP, Van Gorp S, Nazor KL, Boscolo FS, Carson CT, Laurent LC, Marsala M, Gage FH, Remes AM, Koo EH, Goldstein LS. Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature. 2012;482:216–20. doi: 10.1038/nature10821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez J, Jang GM, Semler BL, Waterman ML. An internal ribosome entry site mediates translation of lymphoid enhancer factor-1. Rna. 2005;1:1385–99. doi: 10.1261/rna.7226105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson MA, Weick JP, Pearce RA, Zhang SC. Functional neural development from human embryonic stem cells: accelerated synaptic activity via astrocyte coculture. J Neurosci. 2007;27:3069–77. doi: 10.1523/JNEUROSCI.4562-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas MC, Costantini C, Puglielli L. PCSK9 is required for the disposal of non-acetylated intermediates of the nascent membrane protein BACE1. EMBO Rep. 2008;9:916–22. doi: 10.1038/embor.2008.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas MC, Pehar M, Puglielli L. AT-1 is the ER membrane acetyl-CoA transporter and is essential for cell viability. J Cell Sci. 2010;123:3378–88. doi: 10.1242/jcs.068841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson T, Atwal JK, Steinberg S, Snaedal J, Jonsson PV, Bjornsson S, Stefansson H, Sulem P, Gudbjartsson D, Maloney J, Hoyte K, Gustafson A, Liu Y, Lu Y, Bhangale T, Graham RR, Huttenlocher J, Bjornsdottir G, Andreassen OA, Jonsson EG, Palotie A, Behrens TW, Magnusson OT, Kong A, Thorsteinsdottir U, Watts RJ, Stefansson K. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature. 2012;488:96–9. doi: 10.1038/nature11283. [DOI] [PubMed] [Google Scholar]
- Ko MH, Puglielli L. The sterol carrier protein SCP-x/pro-SCP-2 gene has transcriptional activity and regulates the Alzheimer disease gamma-secretase. J Biol Chem. 2007;282:19742–52. doi: 10.1074/jbc.M611426200. [DOI] [PubMed] [Google Scholar]
- Kurz A, Perneczky R. Neurobiology of cognitive disorders. Curr Opin Psychiatry. 2009;22:546–51. doi: 10.1097/YCO.0b013e328330588b. [DOI] [PubMed] [Google Scholar]
- Lee VM, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annu Rev Neurosci. 2001;24:1121–59. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- Li XJ, Zhang SC. In vitro differentiation of neural precursors from human embryonic stem cells. Methods in Mol Biol. 2006;331:169–77. doi: 10.1385/1-59745-046-4:168. [DOI] [PubMed] [Google Scholar]
- Lott IT, Dierssen M. Cognitive deficits and associated neurological complications in individuals with Down’s syndrome. Lancet Neurol. 2010;9:623–33. doi: 10.1016/S1474-4422(10)70112-5. [DOI] [PubMed] [Google Scholar]
- Loy CT, Schofield PR, Turner AM, Kwok JB. Genetics of dementia. Lancet. 2014;383:828–40. doi: 10.1016/S0140-6736(13)60630-3. [DOI] [PubMed] [Google Scholar]
- Maier B, Gluba W, Bernier B, Turner T, Mohammad K, Guise T, Sutherland A, Thorner M, Scrable H. Modulation of mammalian life span by the short isoform of p53. Genes Dev. 2004;18:306–19. doi: 10.1101/gad.1162404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosner J, Mummenbrauer T, Bauer C, Sczakiel G, Grosse F, Deppert W. Negative feedback regulation of wild-type p53 biosynthesis. EMBO J. 1995;14:4442–9. doi: 10.1002/j.1460-2075.1995.tb00123.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray-Zmijewski F, Slee EA, Lu X. A complex barcode underlies the heterogeneous response of p53 to stress. Nat Rev Mol Cell Biol. 2008;9:702–12. doi: 10.1038/nrm2451. [DOI] [PubMed] [Google Scholar]
- Nalivaeva NN, Turner AJ. The amyloid precursor protein: a biochemical enigma in brain development, function and disease. FEBS Lett. 2013;587:2046–54. doi: 10.1016/j.febslet.2013.05.010. [DOI] [PubMed] [Google Scholar]
- Nelson LD, Siddarth P, Kepe V, Scheibel KE, Huang SC, Barrio JR, Small GW. Positron emission tomography of brain beta-amyloid and tau levels in adults with Down syndrome. Arch Neurol. 2011;68:768–74. doi: 10.1001/archneurol.2011.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardossi-Piquard R, Checler F. The physiology of the beta-amyloid precursor protein intracellular domain AICD. J Neurochem. 2012;120(Suppl 1):109–24. doi: 10.1111/j.1471-4159.2011.07475.x. [DOI] [PubMed] [Google Scholar]
- Pardossi-Piquard R, Petit A, Kawarai T, Sunyach C, Alves da Costa C, Vincent B, Ring S, D’Adamio L, Shen J, Muller U, St George Hyslop P, Checler F. Presenilin-dependent transcriptional control of the Abeta-degrading enzyme neprilysin by intracellular domains of betaAPP and APLP. Neuron. 2005;46:541–54. doi: 10.1016/j.neuron.2005.04.008. [DOI] [PubMed] [Google Scholar]
- Passer B, Pellegrini L, Russo C, Siegel RM, Lenardo MJ, Schettini G, Bachmann M, Tabaton M, D’Adamio L. Generation of an apoptotic intracellular peptide by gamma-secretase cleavage of Alzheimer’s amyloid beta protein precursor. JAD. 2000;2:289–301. doi: 10.3233/jad-2000-23-408. [DOI] [PubMed] [Google Scholar]
- Pehar M, Ko MH, Li M, Scrable H, Puglielli L. P44, the ‘longevity-assurance’ isoform of P53, regulates tau phosphorylation and is activated in an age-dependent fashion. Aging Cell. 2014;13:449–56. doi: 10.1111/acel.12192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pehar M, O’Riordan KJ, Burns-Cusato M, Andrzejewski ME, del Alcazar CG, Burger C, Scrable H, Puglielli L. Altered longevity-assurance activity of p53:p44 in the mouse causes memory loss, neurodegeneration and premature death. Aging Cell. 2010;9:174–90. doi: 10.1111/j.1474-9726.2010.00547.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y, Li M, Clarkson BD, Pehar M, Lao PJ, Hillmer AT, Barnhart TE, Christian BT, Mitchell HA, Bendlin BB, Sandor M, Puglielli L. Deficient Import of Acetyl-CoA into the ER Lumen Causes Neurodegeneration and Propensity to Infections, Inflammation, and Cancer. J Neurosci. 2014;34:6772–89. doi: 10.1523/JNEUROSCI.0077-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray PS, Grover R, Das S. Two internal ribosome entry sites mediate the translation of p53 isoforms. EMBO Rep. 2006;7:404–10. doi: 10.1038/sj.embor.7400623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ, Mucke L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science. 2007;316:750–4. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, Forster C, Yue M, Orne J, Janus C, Mariash A, Kuskowski M, Hyman B, Hutton M, Ashe KH. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309:476–81. doi: 10.1126/science.1113694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scrable H, Sasaki T, Maier B. DeltaNp53 or p44: priming the p53 pump. Int J Biochem Cell Biol. 2005;37:913–9. doi: 10.1016/j.biocel.2004.11.014. [DOI] [PubMed] [Google Scholar]
- Takagi M, Absalon MJ, McLure KG, Kastan MB. Regulation of p53 translation and induction after DNA damage by ribosomal protein L26 and nucleolin. Cell. 2005;123:49–63. doi: 10.1016/j.cell.2005.07.034. [DOI] [PubMed] [Google Scholar]
- Tanzi RE. The genetics of Alzheimer disease. Cold Spring Harbor perspectives in medicine. 2012;2(10) doi: 10.1101/cshperspect.a006296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai BP, Jimenez J, Lim S, Fitzgerald KD, Zhang M, Chuah CT, Axelrod H, Wilson L, Ong ST, Semler BL, Waterman ML. A novel Bcr-Abl-mTOR-eIF4A axis regulates IRES-mediated translation of LEF-1. Open Biology. 2014;4(11) doi: 10.1098/rsob.140180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uberti D, Lanni C, Carsana T, Francisconi S, Missale C, Racchi M, Govoni S, Memo M. Identification of a mutant-like conformation of p53 in fibroblasts from sporadic Alzheimer’s disease patients. Neurobiol Aging. 2006;27:1193–201. doi: 10.1016/j.neurobiolaging.2005.06.013. [DOI] [PubMed] [Google Scholar]
- Uberti D, Lanni C, Racchi M, Govoni S, Memo M. Conformationally altered p53: a putative peripheral marker for Alzheimer’s disease. Neurodegener Dis. 2008;5:209–11. doi: 10.1159/000113704. [DOI] [PubMed] [Google Scholar]
- Weick JP, Held DL, Bonadurer GF, 3rd, Doers ME, Liu Y, Maguire C, Clark A, Knackert JA, Molinarolo K, Musser M, Yao L, Yin Y, Lu J, Zhang X, Zhang SC, Bhattacharyya A. Deficits in human trisomy 21 iPSCs and neurons. Proc Natl Acad Sci USA. 2013;110:9962–7. doi: 10.1073/pnas.1216575110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu TY, Hsieh CC, Hong JJ, Chen CY, Tsai YS. IRSS: a web-based tool for automatic layout and analysis of IRES secondary structure prediction and searching system in silico. BMC bioinformatics. 2009;10:160. doi: 10.1186/1471-2105-10-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Y, Stephen CW, Luciani MG, Fahraeus R. p53 Stability and activity is regulated by Mdm2-mediated induction of alternative p53 translation products. Nat Cell Biol. 2002;4:462–7. doi: 10.1038/ncb801. [DOI] [PubMed] [Google Scholar]
- Zhang SC, Wernig M, Duncan ID, Brustle O, Thomson JA. In vitro differentiation of transplantable neural precursors from human embryonic stem cells. Nat Biotechnol. 2001;19:1129–33. doi: 10.1038/nbt1201-1129. [DOI] [PubMed] [Google Scholar]
- Zigman WB. Atypical aging in Down syndrome. Developmental disabilities research reviews. 2013;18:51–67. doi: 10.1002/ddrr.1128. [DOI] [PubMed] [Google Scholar]