

Abstract
Chronic alcohol consumption causes multifaceted damage to the central nervous system (CNS), underlying mechanisms of which are gradually being unraveled. In our previous studies, activation of calpain, a calcium-activated neutral protease has been found to cause detrimental alterations in spinal motor neurons following ethanol (EtOH) exposure in vitro. However, it is not known whether calpain plays a pivotal role in chronic EtOH exposure-induced structural damage to CNS in vivo. To test the possible involvement of calpain in EtOH-associated neurodegenerative mechanisms the present investigation was conducted in a well-established mouse model of alcohol dependence - chronic intermittent EtOH (CIE) exposure and withdrawal. Our studies indicated significant loss of axonal proteins (neurofilament light and heavy, 50-60 %), myelin proteins (myelin basic protein, 20-40 % proteolipid protein, 25 %) and enzyme (2′, 3′-cyclic-nucleotide 3′-phosphodiesterase, 21-55 %) following CIE in multiple regions of brain including hippocampus, corpus callosum, cerebellum, and importantly in spinal cord. These CIE-induced deleterious effects escalated after withdrawal in each CNS region tested. Increased expression and activity of calpain along with enhanced ratio of active calpain to calpastatin (sole endogenous inhibitor) was observed after withdrawal compared to EtOH exposure. Pharmacological inhibition of calpain with calpeptin (25 μg/kg) prior to each EtOH vapor inhalation significantly attenuated damage to axons and myelin as demonstrated by immuno-profiles of axonal and myelin proteins, and Luxol Fast Blue staining. Calpain inhibition significantly protected the ultrastructural integrity of axons and myelin compared to control as confirmed by electron microscopy. Together, these findings confirm CIE exposure and withdrawal induced structural alterations in axons and myelin, predominantly after withdrawal and corroborate calpain inhibition as a potential protective strategy against EtOH associated CNS degeneration.
Keywords: axonal degeneration, calpain, calpastatin, ethanol withdrawal, myelin proteins and enzyme, neurofilament proteins
1. Introduction
Neuropathological hallmarks of heavy alcohol consumption include shrinkage of gray matter, enlargement of ventricles, and degeneration of the white matter leading to compromised brain structure and associated functional deficits (Harper, 2009; Harper and Kril, 1990; Laas and Hagel, 2000; Sullivan and Pfefferbaum, 2005). Evidence from postmortem human central nervous system (CNS) tissue from alcoholics indicated a loss in brain weight primarily due to the loss of white matter (de la Monte, 1988; Kril et al., 1997). In vivo imaging in human alcoholics further revealed perturbation in the microstructure of white matter in brain (Pfefferbaum et al., 2000; Pfefferbaum and Sullivan, 2002; Pfefferbaum and Sullivan, 2005; Pfefferbaum et al., 2006). Disruptions in circuitry identified in diverse brain regions were implicated as underlying causes for the cognitive and motor deficits seen in alcoholics (Chanraud et al., 2009; Colrain et al., 2011; Sullivan and Pfefferbaum, 2005). Importantly, these studies also revealed the protracted presence of altered microstructural profiles in recovering alcoholics (Rosenbloom et al., 2008), thus, warranting the need to address the mechanisms of white matter degeneration associated with chronic alcohol consumption and potential interventional strategies.
Postmortem studies in human alcoholics showed down regulation of several genes associated with axons and myelin as well as degeneration of myelin sheath (Lewohl et al., 2000; Pfefferbaum et al., 2009). Experimental studies in utero and in early postnatal days hypothesized that the degenerative effects of ethanol (EtOH) exposure on myelin lead to delayed myelination in cerebral cortex (Jacobson et al., 1979) and decreased number of myelinated axons in spinal cord (McNeill et al., 1991). Findings in an animal model of fetal alcohol syndrome have also confirmed decreased expression of mRNAs of the myelin components including 2′, 3′-cyclic nucleotide 3′- phosphodiesterase (CNPase), and myelin basic protein (MBP) associated with delayed myelination following in utero EtOH exposure (Kojima et al., 1994). Postnatal EtOH exposure permanently altered the expression of mRNAs encoding MBP and myelin-associated glycoprotein (MAG) and reduced the expression of selective isoforms of myelin proteins in the cerebellum of adult rodents (Zoeller et al., 1994). Thus, the EtOH exposure is known to cause abnormal effects during the early stages of brain development corresponding to the period of rapid myelination. Recently, the selective vulnerability of myelin to EtOH exposure in adolescent rodent brain was compared to adult and the involvement of TLR-4 was reported as a probable mechanism (Alfonso-Loeches et al., 2012; Pascual et al., 2014). However, less is known about the disruptive mechanisms of alcohol dependence on mature myelin in the adult brain. Disruption in myelin may eventually render the axons vulnerable. Axonal degeneration may also occur following damage to the neuronal cell bodies. Mechanisms by which EtOH trigger damage in brain is only partially understood, hence, this study was undertaken.
The effects of EtOH on the CNS are complex; in any rodent model these effects largely depend on the route of EtOH administration/exposure. Likewise, loss of axonal and myelin integrity in animal models of alcohol dependence, and the underlying mechanism of such degeneration are also subjective and may depend on the model being tested. The present study utilized a standardized chronic intermittent EtOH (CIE) vapor inhalation model that produces escalation in EtOH consumption in adult C57BL/6J mice (Becker and Lopez, 2004; Griffin et al., 2009a; Griffin et al., 2009b; Lopez and Becker, 2005). The model with alternating cycles of EtOH exposure and withdrawal has been extremely well investigated for behavioral cohorts. The model offers a strong platform for mechanistic studies. Further, the extent of EtOH-induced neurodegeneration can have site specificity in brain as reviewed recently (Szabo and Lippai, 2014); we chose to examine the EtOH effects in three regions in brain including hippocampus, corpus callosum, cerebellum and spinal cord - a novel CNS region to study the effects of EtOH.
While multiple factors have been implicated in the loss of axons and myelin in neurodegenerative diseases, and CNS injuries (Das et al., 2008; Geddes and Saatman, 2010; Podbielska et al., 2013; Ray et al., 2011; Samantaray et al., 2008) whether similar mechanisms such as protease activation, inflammatory factors and oxidative stress are also involved in degeneration of axons and myelin following chronic alcohol consumption is not clear. Over-activation of calpain is implicated in neurodegeneration in a wide range of neurological disorders (Bevers and Neumar, 2008; Saatman et al., 2010; Samantaray et al., 2008; Vosler et al., 2008). The challenge is to inhibit the pathological consequences of calpain over-activation while preserving the essential physiological aspects of calpain function. Since, calpain is present in the cytosol and myelin (Banik et al., 1985) and the substrates of two calpain isoforms are similar, and calpain inhibitors, e.g., calpeptin inhibits both the isoforms with similar potency (Geddes and Saatman, 2010; Goll et al., 2003); we tested the efficacy of calpeptin against CIE exposure and withdrawal-induced degeneration axons and myelin in vivo.
In a nutshell, the objective of the study was to determine, if CIE exposure and withdrawal perturbs morphological and molecular parameters of myelin, causes axonal degeneration and identify the mechanisms that may regulate the damaging effects of EtOH on CNS axons and myelin. Our findings suggest that EtOH withdrawal causes aggravated degenerative effects on axonal and myelin integrity. In addition, we demonstrated the protective efficacy of calpain inhibition against CIE exposure and against the specific neurodegenerative pathways induced by withdrawal.
2. Results
2.1. Blood EtOH concentration (BEC)
A stable level of EtOH was maintained in the chambers for the experimental groups (Fig. 1). Representative data from one of the three experiments indicated consistent levels of EtOH in the breathalyzer (0.166-0.238 g/L) over a 5-day period of CIE exposure. Pre-treatment with calpeptin did not significantly alter the levels of BEC in mice as shown in the calpeptin treated group (211.3 ± 10.75 mg/dL) compared to the saline group (214.5 ± 11.58 mg/dL), (p = 0.838; t = 0.2051; df = 72), (Fig. 1).
Fig. 1. Timeline of CIE exposure, withdrawal and calpeptin treatment.

(A) Mice (n = 4-6 in each group) were intermittently exposed to EtOH from day 1 through 5; 16 h of continuous EtOH vapor exposure in inhalation chambers (gray box) was followed by 8 h in home cages outside the chambers (striated gray-black box simulating declining BEC); the whole process was repeated 4 times. In parallel, control mice were exposed to air. All mice received pyrazole (1 mmol/kg, i.p.) prior to entry into the respective chambers. Mice were sacrificed at 2 time points: the exposure or CIE 0 h group was sacrificed immediately after the last (4th) exposure on day 5; the withdrawal or CIE 24 h group was given a prolonged withdrawal of a total 24 h (black box). Calpeptin (25 μg/kg, i.p.) was administered prior to each EtOH vapor inhalation (green arrows), whereas the drug control groups received saline correspondingly. (B) Plot of the average breathalyzer readings did not show any significant variation amongst chambers over the complete span of CIE model. (C) BEC levels showed no difference between saline (n = 39) and calpeptin treated (n = 35) groups (p = 0.838).
2.2. CIE-induced loss of myelin proteins
To test the damaging effects of CIE in adult mice, three CNS regions (hippocampus, cerebellum and spinal cord) from mice under four experimental groups: naïve, control, CIE 0 h and CIE 24 h (n = 4, naïve and n = 6 mice in remaining three groups) were examined. Immunoblots for myelin basic protein MBP revealed all three isoforms (21.5, 18.5 and 14 kDa) in hippocampus, cerebellum and spinal cord (Fig. 2A). Corresponding densitometric analysis in these regions when subjected to ANOVA followed by Bonferroni's multiple comparison suggested that the levels of the three MBP isoforms were not statistically different in naïve, control or CIE 0 h (exposure) groups of mice. Multiple comparison showed statistical significant difference between CIE 24 h (withdrawal) group when compared with naïve or control or CIE 0 h (Fig. 2B). A plot of the comparison between naïve and CIE 24 h groups are represented in Fig. 2B. In hippocampus, changes in MBP isoforms were as follows, MBP (21.5 kDa): t = 19.75, F = 24.01; MBP (18.5 kDa): t = 17.61, F = 2.101; MBP (14 kDa): t = 11.38, F = 2.099; all these comparisons of MBP isoforms in hippocampus were significant at p < 0.0001, (DFn = 3, DFd = 16). In cerebellum, MBP (21.5 kDa): t = 3.643, F = 11.75, p = 0.0066; MBP (18.5 kDa): t = 8.766, F = 6.78; p < 0.0001; MBP (14 kDa): t = 4.77, F = 20.24; p = 0.0021; all these comparisons of MBP isoforms in cerebellum were significant at p < 0.001, (DFn = 3, DFd = 16). In spinal cord, MBP (21.5 kDa): t = 12.90, F = 14.99; MBP (18.5 kDa): t = 7.15, F = 11.4; MBP (14 kDa): t = 8.476, F = 9.457; all these comparisons of MBP isoforms in spinal cord were significant at p < 0.0001, (DFn = 3, DFd = 16). Likewise, immunoprofiles of PLP and CNPase-2 and -1 (Fig. 2B, C) respectively showed selective and statistically significant reduction in CIE 24 h mice compared to naïve (p < 0.0001, DFn = 3, DFd = 16); data shown in hippocampus as comparison of naïve and CIE 24 h; PLP: t = 8.592, F = 23.05; CNPase-2: t = 13.36, F = 2.157; CNPase-1: t = 12.89, F = 36.67 (Fig. 2B, C). Thus, the significant reduction of myelin proteins MBP (20-40 %) and PLP (45 %) and the enzyme CNPase-1, 2 (21 %, 55 %, respectively) occurred in each of these CNS regions tested, only following the final withdrawal stage.
Fig. 2. CIE-induced loss of myelin proteins and enzyme in brain and spinal cord.
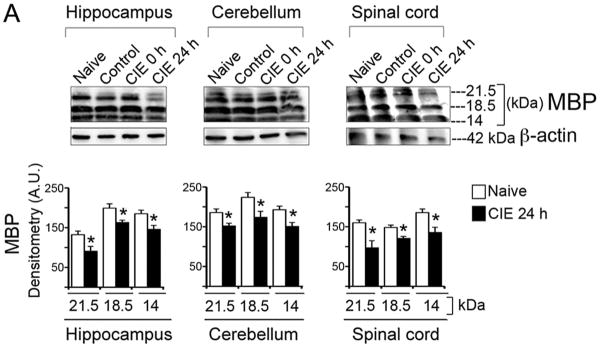
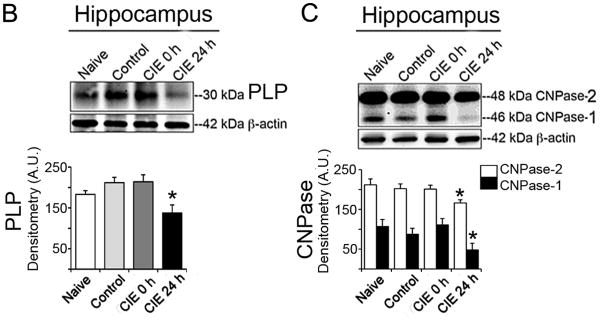
(A) Immunoblots and corresponding densitometric analysis (A.U., mean ± S.E.M.) of MBP showed significantly (*p < 0.001) reduced MBP isoforms (21.5, 18.5 and 14 kDa) in hippocampus, cerebellum and spinal cord in CIE 24 h (withdrawal) compared with the respective MBP isoforms in naïve mice (n = 4-6). Likewise significant reduction was seen in (B) PLP (30 kDa) and (C) CNPase-1 and-2 (46 and 48 kDa) in hippocampus in CIE 24 h (withdrawal) compared with naïve group of mice (*p < 0.0001; n = 4-6). Each immunoblot was re-probed for β-actin (42 kDa), which showed tendency towards marginal loss in CIE 24 h group compared to rest of the groups.
2.3. CIE-induced loss of axonal proteins and axonal degeneration
To test whether disruption in myelin was associated with axonal degeneration, the effect on axonal intermediary filaments or NFPs (NFH and NFL) was examined in the four groups of mice: naïve, control, CIE 0 h and CIE 24 h. Tissue samples were analyzed with immunoblotting (in hippocampus) and immunofluorescent staining (in cerebellum, corpus callosum, and spinal cord); representative data are shown in Fig. 3. In hippocampus, densitometric analysis of the levels of NFL (68 kDa) when subjected to ANOVA followed by Bonferroni's multiple comparison suggested that the levels of NFL were not statistically different in naïve, control or CIE 0 h (exposure) groups of mice, but significantly reduced in CIE 24 h group (59 %) compared with naïve [t = 35.74, F = 6.46 (DFn = 3, DFd = 16), p < 0.0001; Fig. 3A]. Immunofluorescent images in spinal cord indicated intense NFL and NFH immunoreactivity (IR) in control spinal cord, which showed tendency towards reduction in CIE 0 h, and were significantly reduced in CIE 24 h compared with control (Fig. 3A, lower panel). In confirmation, the deNFP immunofluorescent staining, an indicator of axonal degeneration as it immunostains the NFPs that are undergoing dephosphorylation, was marginally increased in CIE 0 h, but significantly increased in CIE 24 h. The trend of axonal degeneration was seen in all three CNS regions (spinal cord, corpus callosum, and cerebellum) relative to the respective controls (Fig. 3B). Reductions in NFL and NFH, and a concomitant increase of deNFP (an index of axonal degeneration) were escalated following withdrawal.
Fig. 3. CIE-induced loss of axonal NFPs and elevation of deNFP in brain and spinal cord.
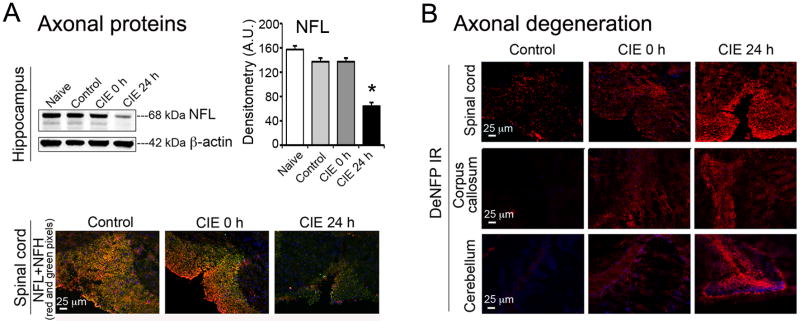
Double or single immunofluorescent staining was performed in coronal spinal cord slices and longitudinal brain slices to illustrate corpus callosum. Images were captured at 200 × magnification; n = 3-4. (A) Immunoblot and corresponding bar graphs (% change, naïve taken as 100%) showed reduction in NFL (68 kDa) in hippocampus in CIE 24 h compared with naive (*p < 0.0001; n = 4-6). Blots were re-probed for β-actin (42 kDa). Immunofluorescent images (lower panel) indicated substantially reduced levels of NFL (red) and NFH (green) IR in spinal cord in CIE 24 h (withdrawal) compared with control. (B) Immunofluorescent images showed elevated levels of deNFP in spinal cord, corpus callosum and cerebellum, indicating significant axonal degeneration in CIE 24 h compared to control.
2.4. CIE-induced up-regulation of calpain in CNS
To investigate the underlying mechanism of CIE-induced myelin disruption and axonal degeneration, profile of calpain expression and activity, and possible elevation of calpain: calpastatin ratio was examined. In hippocampus, the level of active calpain expression was upregulated in CIE 0 h (12%) and CIE 24 h (26%) mice compared to control. A parallel decrease in calpastatin CIE 0 h (15%) and CIE 24 h (36%) was observed. Ratio of active calpain to calpastatin was significantly higher in CIE 24 h (*p < 0.01) compared to control (Fig 4A). These findings were further confirmed by detection of spectrin breakdown products (SBDP) – a measure of calpain activity. Results showed increased levels of degradation of 280 kDa α-spectrin to the calpain-specific 145 kDa SBDP and downstream caspase-3-specific 120 kDa SBDP. Although increased SBDP levels were seen in CIE 0 h, but, statistically significant increases (86% and 98% for 145 kDa SBDP and 120 kDa SBDP, respectively at (*p < 0.01) were seen only in CIE 24 h compared to control (Fig. 4A). Co-localization studies with active calpain and neuronal marker NeuN (as shown in coronal sections of spinal cord) represented overlap of active calpain with NeuN in CIE 0 h and CIE 24 h compared to control, but more pronounced following withdrawal (Fig. 4B).
Fig. 4. CIE-induced upregulation of calpain expression and activity in hippocampus and spinal cord.

(A) Elevated ratio of active calpain: calpastatin following exposure compared to control, which intensified during withdrawal. Increased levels of calpain activity was indicated by increased levels of 145 kDa calpain specific as well as 120 kDa caspase-3 SBDP following CIE specifically in CIE 24 h (withdrawal) compared to control; (n = 4, *p < 0.01). (B) Double immunofluorescent staining in cross section of cervical spinal cord, with active calpain and NeuN, showed co-localization following CIE exposure compared to control; greater intensity was seen in CIE 24 h than CIE 0 h. Images in the panel of active calpain staining are also suggestive of other neural cells that might be positively stained with active calpain in both CIE 0 h and CIE 24 h.
2.5. Calpain inhibition attenuated loss of myelin and axonal proteins
The protective efficacy of calpain inhibition was tested with calpeptin pre-treatment in six groups (n = 6 /group). Mice received calpeptin (25 μg/kg, i.p.) before entering into the EtOH vapor chamber; a cumulative dose of 100-125 μg/kg over 4-5 days is well tolerated in adult mice (Samantaray S., 2013). Immunoblotting for myelin and axonal proteins showed significant protection with calpeptin pre-treatment; data are presented in hippocampus (Fig. 5). One-way ANOVA with post-hoc Bonferroni's multiple comparison test of the densitometric A.U. revealed that the levels of MBP isoforms in hippocampus were significantly reduced (20 % and 23 %) in CIE 24 h group compared with those in control, [MBP (21.5 kDa), t = 8.351 and MBP (18.5 kDa) t = 11.2], p < 0.0001 (DFn = 5, DFd = 30). MBP levels were markedly retained in calpeptin pre-treated CIE 24 h group compared with CIE 24 h [MBP (21.5 kDa), t = 30.45 and MBP (18.5 kDa), t = 3.309], p < 0.0001, (DFn = 5, DFd = 30). Likewise, the reduced levels of PLP (49 %) in CIE 24 h compared with controls (t = 26.67, p < 0.0001, DFn = 5, DFd = 30) were retained in calpeptin pre-treated CIE 24 h group (t = 24.46, p < 0.0001). For CNPase-2 there was a 28 % reduction in CIE 24 h compared to control (t = 15.68, p < 0.0001, DFn = 5, DFd = 30), which was significantly protected with calpeptin pre-treatment (t = 23.59, p < 0.0001). For CNPase-1, there was 43% and 72% reduction in CIE 0 h (t = 12.53, p < 0.0001, DFn = 5, DFd = 30), and CIE 24 h (t = 12.53, p < 0.0001, DFn = 5, DFd = 30), respectively; calpeptin pre-treatment significantly protected in CIE 0 h (t = 8.34, p < 0.0001) and CIE 24 h (t = 12.98, p < 0.0001) respectively.
Fig. 5. Calpeptin prevented loss of myelin and axonal proteins following CIE exposure and withdrawal.

Mice received calpeptin (25 μg/kg, i.p.) prior to each EtOH vapor inhalation. A parallel group of control mice received calpeptin prior to the entry into air chambers, which served as the drug control. (A) Immunoblot of MBP and corresponding densitometric analysis (A.U.) showed significantly reduced levels of MBP isoforms (21.5 and 18.5 kDa) in hippocampus in CIE 24 h (withdrawal) compared with those in control (*p < 0.0001; n = 3), and markedly retained levels of MBP isoforms in the group, pre-treated with calpeptin (@p < 0.0001, compared with CIE 24 h). Likewise, immunoblots of PLP and CNPase -2 and -1 and corresponding densitometric analysis also showed significantly reduced levels of PLP (30 kDa) and CNPase-2 and -1 (48 and 46 kDa, respectively) in CIE 24 h group compared with control or calpeptin (*p < 0.0001; n = 3), and significantly retained levels of PLP and CNPase by calpeptin in CIE 24 h groups compared with CIE 24 h (@p < 0.0001). Myelin proteins or enzyme were unaffected in CIE 0 h except 46 kDa CNPase, which could be significantly protected by calpeptin. (B) Both axonal NFPs (NFH and NFL) were selectively reduced in hippocampus of CIE 24 h group of mice compared to controls (*p < 0.0001; n = 3) whereas only NFH was affected by CIE 0 h; calpeptin pre-treatment significantly protected against such degeneration (@p < 0.0001). Re-probed β-actin served as the loading control.
The significant loss of NFH in CIE 0 h (40 %) compared to control (t = 11.09, p < 0.0001, DFn = 5, DFd = 30) was protected with calpeptin pre-treatment (45 %) relative to CIE 0 h (t = 15.7, p < 0.0001). Loss in NFH escalated in CIE 24 h (53 %) compared to control (t = 15.12, p < 0.0001, DFn = 5, DFd = 30), and the loss was significantly protected with calpeptin (61 %) relative to CIE 24 h (t = 21.33, p < 0.0001). Axonal protein NFL was found diminished in hippocampus in CIE 24 h (67 %) compared to control (t = 9.12, p < 0.0001, DFn = 5, DFd = 30), which was substantially preserved (74 %) in calpeptin pre-treated group relative to CIE 24 h (t = 14.5, p < 0.0001); no significant changes were found in CIE 0 h.
2.6. Calpain inhibition preserved myelin and reduced axonal degeneration
The protective effect of calpeptin in preservation of CNS axons and myelin was further tested with LFB followed by H & E staining. In cervical spinal cord, LFB staining in coronal sections demonstrated healthy neurons in both dorsal horn and ventral horn, and axons and myelin in calpeptin controls (Fig. 6, upper panel as 40× and magnified at 200× in the dorsal and ventral horns). In CIE 24 h group, magnified images at 200× clearly demonstrated that motoneurons with chromatolysis-like changes which were not as prominent in dorsal horn (Fig. 6, middle panel). Compared to CIE 24 h, these alterations were not as prominent in CIE 0 h (data not presented). Calpeptin pre-treatment significantly attenuated the chromatolysis-like changes seen in motoneurons in the ventral horn following CIE 24 h (Fig. 6, lower panel). LFB staining also showed healthy neuropil (stained pink, as shown in Fig. 6, upper panel); CIE 24 h spinal cord demonstrated dislodging of myelin lipoproteins in neuropil (Fig. 6, middle panel), which was partially protected in calpeptin pre-treated group (Fig. 6, lower panel).
Fig. 6.

Neuroprotective efficacies of calpeptin following CIE exposure and withdrawal. Coronal slices (8 μm) from cervical spinal cord were stained with LFB and counterstained with H&E; myelin was stained as blue-green color and gray matter as pink. Images represent LFB staining in spinal cord slices from 3 experimental groups (n = 3): calpeptin (as drug control), CIE 24 h (withdrawal), and calpeptin + CIE 24 h (mice received calpeptin before each EtOH vapor inhalation). In 1st column, low magnification images (40 ×) are presented, which are magnified (200 ×) in the next two columns at dorsal horn and ventral horn respectively. Calpeptin control group showed normal sensory neurons at dorsal horn and motoneurons at ventral horn respectively (upper panel). In CIE 24h spinal cord sections prominent chromatolysis-like changes were seen in the motoneuron (middle panel), which were significantly attenuated with calpeptin-pre-treatment (lower panel).
Next degeneration of axons and myelin was confirmed with EM studies. Photomicrographs of EM demonstrated (Fig. 7) ultrastructural changes in corpus callosum following CIE exposure and protective effects of calpeptin pre-treatment. As seen in corpus callosum of control or calpeptin pre-treated mice, axons were surrounded with compact healthy myelin forming spherical shape; neurofilaments and microtubules were evenly distributed within the myelinated axons containing intact mitochondria (Fig. 7A). The model showed progressive degeneration of white matter with increasing degree of disruption in myelin including disarrangement of the sheath and focal damage accompanied with degeneration of axons. Damaging effects were enhanced during CIE 24 h (Fig. 7CI-CIII) than CIE 0 h (Fig. 7 BI-BIII) when compared to control or calpeptin alone group. Myelin disruptions were followed by axonal degeneration; axonal swelling was predominantly seen in all sections of CIE 0 h and CIE 24 h mice examined compared to control. Disintegration of axonal filaments and tubules within the axons were found in association with hydropic mitochondria. EM studies on the calpeptin pre-treated group clearly demonstrated the attenuation of degenerative changes in myelin (reduced levels of disarrangements in myelin sheath, absence of focal damage or vacuolation or granulation in the myelin sheath, and significantly compact myelin) and in axonal spheres (well organized NFP and tubules with intact mitochondria), (Fig. 7D, E). Axonal swelling was also markedly absent in this group. A significant calpeptin mediated protection was seen in CIE 0 h group than CIE 24 h group (Fig. 7D, E). Similar findings were also observed in other CNS regions (data not shown).
Fig. 7. Calpeptin preserved ultrastructural integrity of axons and myelin following CIE exposure and withdrawal.

Ultrathin sections of corpus callosum from 5 groups: control, CIE 0 h with or without calpeptin, and CIE 24 h with or without calpeptin were examined. EM microphotographs depicting ultrastructure of axons and myelin are represented at 60,000× magnification. In control group, axons were surrounded with compact myelin (green arrowheads) forming spherical shape (green arrows); contained evenly distributed neurofilaments, microtubules and intact mitochondria (green asterisk) within the intact axonal sphere. Progressive degeneration of myelin and axons were seen following CIE; less damage in CIE 0 h (represented in BI through BIII) and greater damage in CIE 24 h (represented in CI through CIII). In CIE 0 h, myelin was disrupted (BI), hydropic mitochondria were seen (BI), granules were formed (BII), and axonal filaments disintegration had begun (BIII). In CIE 24 h, disarrangements within myelin sheaths were intense with focal damge or valuolation (CI-CIII), progressive loss of axonal filament arrangements and ruptured mitochondria were seen (CIII). Axonal swelling was also seen through out in CIE 0 h and 24 h (BI-BIII and CI-CIII compared to A). Calpeptin pre-treatment significantly attenuated each of these parameters of myelin disruption and axonal degeneration; preserved myelin sheath, intact mitochondria and healthy axons were seen in calpeptin + CIE 0 h (D), and calpeptin + CIE 24 h (E) respectively. A semi-quantitative analysis of damage to myelin and axons is presented via % scores of the frequencies of disarrangement in myelin sheath, focal damage in myelin sheath or vacuolation, granular axonal degeneration, and presence of hydropic mitochondria within axons. Results expressed as percent change of control. (*p < 0.05 or **p < 0.01 compared to control and @p < 0.05 or @@p < 0.01 compared to CIE exposed groups).
Semi-quantitative analysis of the total damage in myelin sheath and axons were assessed. All the parameters that were investigated (disarrangement in myelin sheath, focal damage in myelin sheath or vacuolation, granular axonal degeneration, and presence of hydropic mitochondria), showed that there were no significant differences between the control and the calpeptin alone treated groups of mice; however, all CIE exposed groups presented statistically significant differences compared to control or calpeptin alone groups (*p < 0.05 or **p < 0.01). Percent scores of the frequencies of disarrangement in myelin sheath (Fig. 7F) and focal damage in myelin sheath or vacuolation (Fig. 7G), revealed graded damage in CIE 0 h and CIE 24 h compared to control (*p < 0.05 or **p < 0.01 respectively, Fig. 7F-G); calpeptin treatment prior to CIE-exposure significantly attenuated these effects (@p < 0.05 or @@p < 0.01), Fig 7F-G. Concomitant axonal damage as assessed by scoring the frequencies of granular axonal degeneration (Fig. 7H), and presence of hydropic mitochondria (Fig. 7I) within the axons was significantly higher in CIE 0 h and CIE 24 h (*p < 0.05 or **p < 0.01) compared to control, Fig. 7H-I; calpeptin treatment prior to exposure significantly attenuated these effects (@p < 0.05 or @@p < 0.01), Fig. 7H-I.
3. Discussion
Present investigation is a hypothesis driven study that attempts to unravel the underlying mechanisms of the damaging effects of chronic alcohol consumption in CNS. It demonstrates calpain-mediated degenerative effects on axons and myelin caused by EtOH exposure in adult mice convincingly. Significant losses of axonal and myelin proteins in discrete CNS regions including hippocampus, corpus callosum, cerebellum, spinal cord, and corresponding alterations in ultra-structural integrity of axons and myelin was conclusively established in a well characterized mouse model of alcohol dependence. While the alcohol withdrawal substantially escalated the degenerative changes, a causal link was found between the calpain-calpastatin dysregulation and CIE exposure and withdrawal. Importantly, significant protection was rendered by the pharmacological inhibition of calpain against the CIE-induced axons and myelin degeneration.
3.1. CIE perturbed mature myelin in CNS of adult mice
The study is important, as it demonstrates the compelling evidence of damaging effects of chronic EtOH exposure in adult mice, thus corroborating the existing literature in utero (Jacobson et al., 1979; Kojima et al., 1994; McNeill et al., 1991), during early stages of brain development (Zoeller et al., 1994), and the active period of intense synaptogenesis in adolescence (Alfonso-Loeches et al., 2012; Pascual et al., 2014). In this context, the formation of myelin and its maturation is an age-dependent process (Branson, 2013; Hartman et al., 1982). Therefore, EtOH exposure to brain, depending upon the age, may have different degree of detrimental consequences. The current study is unique as it investigated the EtOH effects on axon and myelin in CIE model in adult mice and the underlying mechanisms. Previously, it was reported that mature myelin may be resistant to EtOH-induced damage in a intermittent binge model of alcohol dependence in adult rats (Pascual et al., 2014). In contrast, in the present study the ultrastructural changes found in myelin, including splitting of the myelin sheath, focal damage including vesiculation, and granular degeneration of axons, were demonstrated with EM in CIE model in adult mice. These discrepancies may have been due to different alcohol exposure models used. Also, the repeating cycles of withdrawal, as studied and reported in the current manuscript may have significant detrimental effects on the CNS as suggested previously by escalating behavioral discrepancies (Becker and Lopez, 2004). Such changes in myelin rendered the axons vulnerable, and the current study on CIE-induced axon-myelin degeneration needs attention as damage to the axon-myelin unit may affect coordination and movement.
Recently a gender bias has been reported in several aspects of alcohol dependence (Forbes et al., 2013; Rosenwasser et al., 2014), in our studies adult male mice were used. However, it would be of interest to examine whether axon-myelin degeneration in CIE model also reflects a gender bias.
3.2. Repeated cycles of withdrawal escalated the CNS damage
Importantly, the alcohol-mediated changes may greatly depend on the animal model being used. In order to simulate human-like repeat binge drinking, BEC level in rodents has to be experimentally elevated. The CIE model is uniquely designed for behavioral and mechanism studies, as the repeated cycles of withdrawal escalated the alcohol dependence (Griffin et al., 2009a; Griffin et al., 2009b; Lopez and Becker, 2005). High level of BEC is attainable in mice in this model, which is ideal for investigating the EtOH effects comparable to chronic alcoholism. This model is specifically relevant as it clearly discerns between abusive alcohol consumption and social drinking. It is based on experimentally induced escalated alcohol dependence. It also gives the latitude to distinguish between events following EtOH exposure and withdrawal; and provides a platform where pharmacological agents can be administered either pre-exposure (prior to each EtOH vapor inhalation) or pre-withdrawal (prior to each withdrawal stage) and to examine various effects following EtOH exposure or withdrawal. The present study tested the efficacy of calpeptin pre-treatment regimen prior to each EtOH exposure. However, the findings of more detrimental responses in axon-myelin following withdrawal suggest that it may also be beneficial to test the efficacy of calpeptin in another regimen - prior to withdrawal. Nonetheless, current study on axonal and myelin degeneration clearly establishes a possible mechanism of damage induced during the withdrawal phase of CIE model.
3.3. Spinal cord was detected as a novel CNS target in addition to hippocampus, corpus calloasum and cerebellum in CIE
While, alcohol does not have any preferred access to any CNS region, yet there are region specific vulnerability in different parts of CNS depending upon the neural cyto-architecture and the propensity of EtOH-induced damaging pathways. In this study different CNS areas were examined with relation to axonal and myelin degeneration following CIE exposure and withdrawal. First, hippocampus is a site of major emphasis as it has been previously reported to be a predominant region of impairment leading to alcohol-associated dysfunctions (Vilpoux et al., 2009). Loss of myelin proteins has been previously found in hippocampus (Alfonso-Loeches et al., 2012; Lee et al., 2010; Okamoto et al., 2006). Alterations in myelin and axonal integrity in hippocampus may underlie cognitive deficits as previously reported (Dutta et al., 2011; Dutta et al., 2013). Whether such damage is associated with chronic alcoholism and mediated by inflammatory mechanisms is not known. Nonetheless, inflammatory mediators TLR-4, Cox-2, NFκ-B have been previously suggested to be involved in myelin disruption in hippocampus of adolescent rats (Alfonso-Loeches et al., 2012; Pascual et al., 2014). Thus, it is possible that the morphological changes found in the hippocampal axons and myelin as confirmed in the present study may have been due to the inflammatory factors. In the current study, we conclusively demonstrate calpain-calpastatin dysregulation and over-activation of calpain in hippocampus; more prominently during withdrawal. Second, white matter degeneration in corpus callosum has been extensively studied with in vivo imaging in human alcoholics (Kashem et al., 2008; Muller-Oehring et al., 2009; Pfefferbaum et al., 2006; Pitel et al., 2010; Schulte et al., 2005; Schulte et al., 2008), but the mechanism of this degenerative process is not clearly known. Nonetheless, recent studies in adolescent rat (Alfonso-Loeches et al., 2012; Pascual et al., 2014) and current study in adult mice further elucidated the mechanisms of such degeneration. Third, while EtOH effects in cerebellum have been extensively studied in vitro and in vivo; such studies have not been thoroughly investigated in EtOH-induced axon-myelin degeneration in spinal cord. The morphological alterations in axons and myelin seen in different regions of the brain due to EtOH exposure have been very well correlated with the loss of their structural components (e.g. axonal and myelin protein), destabilizing the membrane integrity. Understanding the mechanism involved may help develop strategies to preserve the axon-myelin structural unit. Finally, the normal physiological function of spinal cord is to control movement, coordination, and motor function. Examining axon-myelin degeneration in spinal cord in CIE model is critical because significant spinal cord neurodegeneration can impair movement and coordination, and cause dysfunction. Thus, the present investigation on axon-myelin degeneration in multiple CNS regions may explain the impairment in movement and coordination and cognitive deficits associated with EtOH dependence.
3.4. Calpain-calpastatin dysregulation were established as underlying mechanisms in CIE
Although EtOH mediated mechanisms remain unclear, the loss of axonal and myelin proteins (e.g. MBP/NFP) indicates a pivotal role for calpain; furthermore, these proteins are substrates of calpain. Pathological calpain has been implicated in many neurodegenerative diseases (Bevers and Neumar, 2008; Saatman et al., 2010; Samantaray et al., 2008; Vosler et al., 2008). In order to understand the degenerative mechanisms in CIE, a possible role of calpain was investigated. Calpain may play regulatory role in alcohol-associated damage because calpain substrates Cox-2, NFκ-B and TLR4 have been implicated in alcohol dependence and associated cognitive and motor behavioral deficits (Alfonso-Loeches et al., 2012; Dhir et al., 2005; Kelley and Dantzer, 2011; Mayfield et al., 2013; Wu et al., 2012; Yakovleva et al., 2011). Alcohol-induced neuroinflammation may also lead to neurodegeneration (Sullivan and Zahr, 2008), and if intervened adequately by therapeutic agents, they may attenuate alcohol-associated pathologies. It is known that withdrawal aggravates alcohol dependence and impairs behavior. Likwise, alcohol-associated neuroinflammation may also be intensified with withdrawal (Freeman et al., 2012; Zahr et al., 2010). Up-regulation of calpain expression and activity in multiple CNS regions following CIE causes significant axon-myelin as indicated by LFB staining and loss of its proteins. Degradation of these proteins will not only destabilize myelin sheath and promote vesiculation, but also, lead to granular changes in axons by destroying neurofilaments and microtubules, losing axonal transport; calpain inhibitor may provide protection and preservation of these structures. Importantly, LFB staining clearly showed the dislodging of myelin lipoproteins in the neuropil, which was partly reduced by calpeptin pre-treatment. The significance of such alterations in myelin cannot be fully accounted for in the current study. We aim to objectively study these alterations in myelin in future via comparison of single versus multiple cycle of CIE, wherein the withdrawal driven CNS pathology will be more pronounced due to the repeated cycles of CIE. Thus, these various findings suggest that calpain is a potential therapeutic target and calpain inhibition may attenuate the structural degeneration induced by EtOH exposure and withdrawal. Calpeptin pre-treatment partially prevented the detrimental process suggesting a regulatory role of calpain in the degenerative mechanisms. Present study being our pilot attempt, was kept limited to pre-exposure administration of calpeptin as an intervening agent. Current findings are also suggestive of a possibility of better protection if calpeptin was administered at the pre-withdrawal stage of CIE. It was demonstrated previously that a calpain inhibitor has an effect on brain injury, it may be possible that calpain inhibitors may have similar effects also during exposure to alcohol and the associated CNS dysfunction. Studies are underway with water soluble calpain inhibitors like SNJ-1945 (Knaryan et al., 2013), for translational initiatives against alcohol-associated CNS dysfunctions.
4. Experimental procedures
4.1. Animals
Adult male C57BL/6 mice, (10 weeks old) from the Jackson Laboratory (Bar Harbor) were divided into groups (n = 6 in each group) as per the experimental design. Mice were individually housed in polycarbonate cages and maintained in standard temperature and humidity conditions (23 °C, 55 % relative humidity), and 12 h light/dark cycle with access to food and water. All procedures with mice followed the National Institutes of Health (NIH, Bethesda) Guide for the Care and Use of Laboratory Animals (NIH publication 80-23, revised 1996) and approved by the Institutional Animal Care and Use Committee (IACUC) of the Medical University of South Carolina (MUSC), Charleston, SC.
A well-established mouse model of CIE exposure was used to study axon-myelin degeneration, and to test the neuroprotective efficacy of calpeptin. All standard operating procedures of EtOH exposure were performed in the core facility of the Charleston Center of Alcohol Research at MUSC with technical support from highly trained lab personnel. EtOH (or air) was supplied in Plexiglas inhalation chambers (60 × 36 × 60 cm), following the described protocols (Becker and Lopez, 2004; Becker and Baros, 2006; Lopez and Becker, 2005). The EtOH vapor mixed with fresh air was delivered at a rate of 10 L/min, which maintained the EtOH concentration in the range of 15 to 20 mg/L of air in chambers and 150 to 200 mg/dL in blood. Before each entry to the vapor chamber, all mice were injected with alcohol dehydrogenase inhibitor, pyrazole (1 mmol/kg, i.p.), to stabilize blood EtOH concentration (BEC). The BEC was monitored daily in blood samples obtained from retro-orbital sinus, as described previously (Becker and Lopez, 2004). The results are shown in Fig. 1C. A set of naïve mice (n = 4) was also included in the study that did not receive any treatment.
4.2. Chronic Intermittent EtOH and calpeptin treatment
Experimental mice were intermittently exposed to EtOH vapor in inhalation chambers (16 h) followed by withdrawal intervals (8 h) from day 1 through day 5; the timeline of CIE exposure is illustrated in Fig. 1A. In parallel, control mice were placed in air chambers, at similar “in and out” schedule for experimental groups. Naïve mice were placed in individual cages and kept in a separate area. The EtOH vapor exposure was started in chambers at 4:00 PM (day 1) which ended at 8:00 AM of the next day (day 2). Mice undergoing withdrawal were maintained in cages placed outside the chambers during the day.
Mice were distributed into either 4 or 6 groups (n = 6 mice in each group), according to the experimental design: (1) naïve, control, CIE 0 h (exposure) or CIE 24 h (withdrawal); or (2) control, calpeptin, CIE 0 h, calpeptin + CIE 0 h, CIE 24 h, calpeptin + CIE 24 h. Calpeptin (25 μg/kg, i.p.) was injected before each entry to the EtOH vapor or air chamber. CIE mice were sacrificed at 2 time points: immediately after the last (4th 16 h EtOH vapor) exposure on day 5, which served as the immediate end point of exposure or CIE 0 h group; another group was given a prolonged 24 h of withdrawal which served as the final withdrawal point or CIE 24 h group. Experiments were repeated twice.
4.3. Tissue processing
Upon completion, mice were sacrificed at designated time points; whole brain and spinal cord tissues were dissected and freshly frozen at -80 °C for further analysis. Tissue samples were analyzed using Western blotting or immunofluorescent staining.
A separate set of mice were processed for histological staining, Luxol Fast Blue (LFB) followed by H&E and ultrastructural electron microscopic (EM) studies for integrity of axons and myelin, which included the six groups according to the experimental design as mentioned above (n = 6, mice in each group). Mice were deeply anesthetized with isoflurane (VEDCO, Inc.), and transcardially perfused; first with 5 mL of saline (0.9% NaCl), followed by 10 mL of 2.5 % glutaraldehyde in 0.1 M sodium cacodylate buffer, pH 7.4 (sodium cacodylate trihydrate: #12300 Electron Microscopy Sciences). Mice were decapitated; whole brain and spinal cord within chorda were dissected, stored in glutaraldehyde containing cacodylate buffer at 4 °C over night for thorough fixation. Corpus callosum and spinal cord were dissected on the next day and processed for EM, and LFB with H&E respectively.
4.4. Western blot analysis
Immunoblotting was performed following the described protocol (Samantaray et al., 2007; Samantaray et al., 2013a; Samantaray et al., 2013b). Briefly, dissected tissues (hippocampus, cerebellum, spinal cord) were homogenized in ice-cold homogenizing buffer (50 mM Tris-HCl, pH 7.4; 5 mM EGTA) with freshly added phenylmethylsulfonyl fluoride (1 mM), and protein was estimated using Coomassie plus Protein Assay Reagent (Pierce). Samples were equilibrated (1:1 v/v) with sample buffer (62.5 mM Tris-HCl, pH 6.8, 2 % SDS, 5 mM β-mercaptoethanol, 10 % glycerol), boiled and briefly centrifuged. Samples were diluted with homogenizing and sample buffers (1:1 v/v), containing bromophenol blue (0.01%), to attain a final concentration of protein 1.5 mg/mL. Protein samples were resolved in 4–20 or 7.5 % (for SBDP) precast sodium dodecyl sulfate–polyacrylamide gel (Bio-Rad Laboratories, Hercules, CA) at 100 V for 1 h or 1 and 1/2 h respectively; transferred to the Immobilon™-polyvinylidene fluoride microporous membranes (Millipore). Membranes were incubated with 5 % non-fat milk in Tris-HCl buffer (20 mM Tris-HCl, pH 7.6, 0.1 % Tween-20), then incubated overnight at 4 °C with following primary IgG antibodies: rabbit polyclonal MBP and PLP, (1:500; raised and characterized in the lab; (Li and Banik, 1995), mouse monoclonal CNPase (1:500, Sigma), neurofilaments light and heavy (NFL and NFH; both 1:1000; Sigma), active calpain (1:500; raised and characterized in the lab; (Banik et al., 1983), calpastatin (1:250, Santa Cruz Biotechnology), α Fodrin (1:10,000, Enzo, Biomol). Next day, the membranes were washed 3 × 5 min in Tris-HCl buffer, and then incubated with horseradish peroxidase-conjugated corresponding secondary IgG antibodies (1:2000; MP Biomedicals) for 1 h. Immunoblots were developed with chemiluminescent reagent (ECL, Amersham) and imaged on Alpha-Innotech, equipped with FluorChem FC2 Imaging System (Cell Biosciences). All blots were re-probed for β-actin except for the spectrin blots. The immunoreactive bands of interest were quantified by densitometric analysis using Image J 1.45 software.
4.5. Immunofluorescent staining
Double or single immunofluorescent staining was performed in the brain and spinal cord slices, following previously described procedures (Samantaray et al., 2007; Samantaray et al., 2013a). Tissue sections (5-10 μm) were fixed in 95% EtOH, rinsed 3 × 5 min in phosphate-buffered saline (PBS, containing 137 mM NaCl, 2.7 mM KCl, 11.9 mM phosphates, pH 7.4). For double staining, sections were blocked in PBS containing 2 % horse and goat serum for 1 h, next incubated with respective mouse monoclonal NFL and rabbit polyclonal NFH primary IgG antibodies (1:1000; Sigma) or mouse monoclonal NeuN and rabbit polyclonal active calpain (Banik et al., 1983; Samantaray et al., 2007) overnight at 4 °C. Next day, slices were rinsed in PBS and incubated with secondary horse anti-mouse IgG, DyLight™ 594 (red) or goat anti-rabbit, DyLight™ 488 (green) conjugated antibodies (Thermo Scientific) for 1 h. For single staining, mouse monoclonal SMI 311 (1:1000; Covance) antibody was used for detection of dephosphorylated neurofilament protein (deNFP). Prior to deFNP staining, antigen retrieval was done by autoclaving the samples in 10 mM citrate buffer solution (pH 6.0) for 5 minutes, cooled to room temperature (23 °C), and next washed in PBS. Sections were blocked in 2% horse serum containing PBS for 1 h, then incubated with deNFP overnight at 4 °C, rinsed with PBS 3 × 5 min, and incubated with secondary horse anti-mouse IgG, DyLight 594. Slices were mounted with antifade Vectashield™ (Vector Laboratories). Fluorescent photomicrographs were viewed and captured in Olympus BH-2 microscope (Olympus) equipped with MagnaFire SP image acquisition software (Media Cybernetics). Images captured at 200x magnification were formatted with Adobe Photoshop software, CS4 extended, Version 11.0.2 (Adobe).
4.6. Luxol Fast Blue and H&E staining
Myelin was studied with LFB staining, which binds to the bases of the lipoproteins in the myelin sheath. The staining was performed according to Hans B. Snyder's write up in Armed Forces Institute of Pathology manual (Luna, 1968). Briefly, after fixation, spinal cord specimens were embedded in paraffin, cut into 8-10 μm sections, dried at 37°C overnight, de-paraffinized, cleared, then hydrated in 95% EtOH, rinsed in distilled water, and stained in 0.1% LFB (Solvent Blue 38; Sigma). Slides were rinsed in 95% EtOH, to remove excess stain, then in water, differentiated in 0.05% lithium carbonate and 70% EtOH, and counterstained with H&E. Finally, sections were dehydrated, cleaned in xylene and mounted. Upon LFB staining, myelin fibers appeared blue, neuropils appeared pink, and neural cell bodies appeared purple. Slices were viewed and images were captured in Olympus microscope equipped with MagnaFire SP image acquisition software. Images were formatted with Adobe Photoshop software.
4.7. Electron microscopy
Ultrastructural integrity of axons and myelin were studied with EM. Sample preparation was performed following fixation protocol (Hayat, 1981) and staining on grids procedure (Mollenhauer, 1964). Briefly, small blocks of corpus callosum tissue were fixed in 2.5% glutaraldehyde in cacodylate-buffered, post-fixed in 1% osmium tetroxide (OsO4), dehydrated, infiltrated with epoxy resin (EMbed 812; Electron Microscopy Sciences), and sliced as 70 nm sections in ultramicrotome. Ultrathin sections were contrast-stained on grids with aqueous uranyl acetate followed by lead citrate, and dehydrated in EtOH. Specimens from each experimental group were observed in EM (Jeol 1010) in three different grids. Locations for myelinated axons were randomly selected. EM images were photographed at lower (2.5; 10; 20 K) to higher (60 and 150 K) magnifications. About 200 photomicrographs were studied to find out characteristic changes in axonal and myelin ultrastructures; selected representative photomicrographs were formatted by Adobe Photoshop software. The ultrastructural features of the myelin sheaths and axons were semi-quantitatively analyzed after the observation of approximately 100 randomly selected myelinated axons per group in three different grids and the percentage was established in each of them. Normal characteristics were shown in the control mice. The results are expressed as percentages compared to control mice.
4.8. Statistical analysis
The data were obtained from three independent experiments, n = 4-6 mice in each group. The immunoreactive bands of Western blotting were quantified by densitometric analysis; the mean ± S.E.M. of arbitrary units (A.U.) was plotted. Statistical significance of differences between groups was analyzed with one–way ANOVA with Bonferroni's post hoc test at > 95% confidence interval. Changes between groups were considered significant at *p < 0.05 or **p < 0.01 compared to naïve or control or @p < 0.05 compared to calpeptin pre-treatment.
Supplementary Material
Highlights.
Withdrawal escalates myelin damage following chronic intermittent ethanol (CIE)
Concomitant axonal degeneration is associated with CIE-induced alterations in myelin
CIE-induces degeneration of axons and myelin in spinal cord in addition to brain
Calpain activation and dysregulation of calpain-calpastatin causes axon-myelin degeneration
Calpain inhibition protects against CIE-induced axon-myelin degeneration
Acknowledgments
This work was funded in part by the R01 Grants from National Institute of Neurological Disorders and Stroke of the National Institutes of Health (NINDS-NIH; NS-62327-01A2), Ralph H. Johnson Veterans Administration Medical Center (1I01BX001262), Alcohol Research Center (P50AA10761) and the State of South Carolina Spinal Cord Injury Foundation.
Authors thank Ms. Nancy M. Smythe and Ms. Margaret H. Romano for excellent technical assistance; Mr. Guilherme Porto and Mr. Harold Haun for invaluable assistance in the animal model; Drs. E.L. Hogan and Catrina Sims-Robinson for discourse and critical revision of the manuscript.
Abbreviations
- A.U.
arbitrary units
- CIE
chronic intermittent ethanol
- CNPase
2′, 3′-cyclic nucleotide 3′-phosphodiesterase
- deNFP
dephosphorylated neurofilament protein
- EM
electron microscopy
- EtOH
ethanol
- IR
immunoreactivity
- LFB
Luxol Fast Blue
- MBP
myelin basic protein
- PBS
phosphate-buffered saline
- PLP
proteolipid protein
- NFP
neurofilament protein
- NFH
neurofilament heavy
- NFL
neurofilament light
Footnotes
Conflict of interest statement: The authors declare that they have no competing interests.
Author contributions: SS: Participated in design, research, data analysis and interpretation, and manuscript preparation
VHK: Participated in research, data analysis and interpretation, and manuscript writing
KSP: Carried out molecular techniques and data analysis
PJM: Participated in the research, interpretation of results and revision of manuscript
HCB: Participated in the research, interpretation of results and revision of manuscript
NLB: Participated in the research, interpretation of results and preparation of manuscript
Contributor Information
Varduhi H. Knaryan, Email: vknaryan@yahoo.com.
Kaushal S. Patel, Email: kspatel1990@gmail.com.
Patrick J. Mulholland, Email: mulholl@musc.edu.
Howard C. Becker, Email: beckerh@musc.edu.
Naren L. Banik, Email: baniknl@musc.edu.
References
- Alfonso-Loeches S, Pascual M, Gomez-Pinedo U, Pascual-Lucas M, Renau-Piqueras J, Guerri C. Toll-like receptor 4 participates in the myelin disruptions associated with chronic alcohol abuse. Glia. 2012;60:948–64. doi: 10.1002/glia.22327. [DOI] [PubMed] [Google Scholar]
- Banik NL, Hogan EL, Jenkins MG, McDonald JK, McAlhaney WW, Sostek MB. Purification of a calcium-activated neutral proteinase from bovine brain. Neurochem Res. 1983;8:1389–405. doi: 10.1007/BF00964996. [DOI] [PubMed] [Google Scholar]
- Banik NL, McAlhaney WW, Hogan EL. Calcium-stimulated proteolysis in myelin: evidence for a Ca2+-activated neutral proteinase associated with purified myelin of rat. CNS J Neurochem. 1985;45:581–8. doi: 10.1111/j.1471-4159.1985.tb04026.x. [DOI] [PubMed] [Google Scholar]
- Becker HC, Lopez MF. Increased ethanol drinking after repeated chronic ethanol exposure and withdrawal experience in C57BL/6 mice. Alcohol Clin Exp Res. 2004;28:1829–38. doi: 10.1097/01.alc.0000149977.95306.3a. [DOI] [PubMed] [Google Scholar]
- Becker HC, Baros AM. Effect of duration and pattern of chronic ethanol exposure on tolerance to the discriminative stimulus effects of ethanol in C57BL/6J mice. J Pharmacol Exp Ther. 2006;319:871–8. doi: 10.1124/jpet.106.108795. [DOI] [PubMed] [Google Scholar]
- Bevers MB, Neumar RW. Mechanistic role of calpains in postischemic neurodegeneration. J Cereb Blood Flow Metab. 2008;28:655–73. doi: 10.1038/sj.jcbfm.9600595. [DOI] [PubMed] [Google Scholar]
- Branson HM. Normal myelination: a practical pictorial review. Neuroimaging Clin N Am. 2013;23:183–95. doi: 10.1016/j.nic.2012.12.001. [DOI] [PubMed] [Google Scholar]
- Chanraud S, Leroy C, Martelli C, Kostogianni N, Delain F, Aubin HJ, Reynaud M, Martinot JL. Episodic memory in detoxified alcoholics: contribution of grey matter microstructure alteration. PLoS One. 2009;4:e6786. doi: 10.1371/journal.pone.0006786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colrain IM, Sullivan EV, Ford JM, Mathalon DH, McPherson SL, Roach BJ, Crowley KE, Pfefferbaum A. Frontally mediated inhibitory processing and white matter microstructure: age and alcoholism effects. Psychopharmacology (Berl) 2011;213:669–79. doi: 10.1007/s00213-010-2073-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das A, Guyton MK, Butler JT, Ray SK, Banik NL. Activation of calpain and caspase pathways in demyelination and neurodegeneration in animal model of multiple sclerosis. CNS Neurol Disord Drug Targets. 2008;7:313–20. doi: 10.2174/187152708784936699. [DOI] [PubMed] [Google Scholar]
- de la Monte SM. Disproportionate atrophy of cerebral white matter in chronic alcoholics. Arch Neurol. 1988;45:990–2. doi: 10.1001/archneur.1988.00520330076013. [DOI] [PubMed] [Google Scholar]
- Dhir A, Naidu PS, Kulkarni SK. Protective effect of cyclooxygenase-2 (COX-2) inhibitors but not non-selective cyclooxygenase (COX)-inhibitors on ethanol withdrawal-induced behavioural changes. Addict Biol. 2005;10:329–35. doi: 10.1080/13556210500352964. [DOI] [PubMed] [Google Scholar]
- Dutta R, Chomyk AM, Chang A, Ribaudo MV, Deckard SA, Doud MK, Edberg DD, Bai B, Li M, Baranzini SE, Fox RJ, Staugaitis SM, Macklin WB, Trapp BD. Hippocampal demyelination and memory dysfunction are associated with increased levels of the neuronal microRNA miR-124 and reduced AMPA receptors. Ann Neurol. 2011;73:637–45. doi: 10.1002/ana.23860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta R, Chang A, Doud MK, Kidd GJ, Ribaudo MV, Young EA, Fox RJ, Staugaitis SM, Trapp BD. Demyelination causes synaptic alterations in hippocampi from multiple sclerosis patients. Ann Neurol. 2013;69:445–54. doi: 10.1002/ana.22337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes A, Cooze J, Malone C, French V, Weber JT. Effects of intermittent binge alcohol exposure on long-term motor function in young rats. Alcohol. 2013;47:95–102. doi: 10.1016/j.alcohol.2012.12.007. [DOI] [PubMed] [Google Scholar]
- Freeman K, Brureau A, Vadigepalli R, Staehle MM, Brureau MM, Gonye GE, Hoek JB, Hooper DC, Schwaber JS. Temporal changes in innate immune signals in a rat model of alcohol withdrawal in emotional and cardiorespiratory homeostatic nuclei. J Neuroinflammation. 2012;9:97. doi: 10.1186/1742-2094-9-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geddes JW, Saatman KE. Targeting individual calpain isoforms for neuroprotection. Exp Neurol. 2010;226:6–7. doi: 10.1016/j.expneurol.2010.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goll DE, Thompson VF, Li H, Wei W, Cong J. The calpain system. Physiol Rev. 2003;83:731–801. doi: 10.1152/physrev.00029.2002. [DOI] [PubMed] [Google Scholar]
- Griffin WC, 3rd, Lopez MF, Becker HC. Intensity and duration of chronic ethanol exposure is critical for subsequent escalation of voluntary ethanol drinking in mice. Alcohol Clin Exp Res. 2009a;33:1893–900. doi: 10.1111/j.1530-0277.2009.01027.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin WC, 3rd, Lopez MF, Yanke AB, Middaugh LD, Becker HC. Repeated cycles of chronic intermittent ethanol exposure in mice increases voluntary ethanol drinking and ethanol concentrations in the nucleus accumbens. Psychopharmacology (Berl) 2009b;201:569–80. doi: 10.1007/s00213-008-1324-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper C. The neuropathology of alcohol-related brain damage. Alcohol Alcohol. 2009;44:136–40. doi: 10.1093/alcalc/agn102. [DOI] [PubMed] [Google Scholar]
- Harper CG, Kril JJ. Neuropathology of alcoholism. Alcohol Alcohol. 1990;25:207–16. doi: 10.1093/oxfordjournals.alcalc.a044994. [DOI] [PubMed] [Google Scholar]
- Hartman BK, Agrawal HC, Agrawal D, Kalmbach S. Development and maturation of central nervous system myelin: comparison of immunohistochemical localization of proteolipid protein and basic protein in myelin and oligodendrocytes. Proc Natl Acad Sci U S A. 1982;79:4217–20. doi: 10.1073/pnas.79.13.4217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayat MA. Fixation for Electron Microscopy. Academic Press Inc.; 1981. Vol. [Google Scholar]
- Jacobson S, Rich J, Tovsky NJ. Delayed myelination and lamination in the cerebral cortex of the albino rat as a result of the fetal alcohol syndrome. Curr Alcohol. 1979;5:123–33. [PubMed] [Google Scholar]
- Kashem MA, Harper C, Matsumoto I. Differential protein expression in the corpus callosum (genu) of human alcoholics. Neurochem Int. 2008;53:1–11. doi: 10.1016/j.neuint.2008.04.003. [DOI] [PubMed] [Google Scholar]
- Kelley KW, Dantzer R. Alcoholism and inflammation: neuroimmunology of behavioral and mood disorders. Brain Behav Immun. 2011;25(Suppl 1):S13–20. doi: 10.1016/j.bbi.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knaryan VH, Samantaray S, Park S, Azuma M, Inoue J, Banik NL. SNJ-1945, a calpain inhibitor, protects SH-SY5Y cells against MPP and rotenone. J Neurochem. 2013 doi: 10.1111/jnc.12629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima H, Mineta-Kitajima R, Saitoh-Harada N, Kurihara T, Takahashi Y, Furudate S, Shirataka M, Nakamura K, Tamai Y. Prenatal ethanol exposure affects the activity and mRNA expression of neuronal membrane enzymes in rat offspring. Life Sci. 1994;55:1433–42. doi: 10.1016/0024-3205(94)00758-6. [DOI] [PubMed] [Google Scholar]
- Kril JJ, Halliday GM, Svoboda MD, Cartwright H. The cerebral cortex is damaged in chronic alcoholics. Neuroscience. 1997;79:983–98. doi: 10.1016/s0306-4522(97)00083-3. [DOI] [PubMed] [Google Scholar]
- Laas R, Hagel C. Neuropathology of chronic alcoholism. Clin Neuropathol. 2000;19:252–3. [PubMed] [Google Scholar]
- Lee DH, Jeong JY, Kim YS, Kim JS, Cho YW, Roh GS, Kim HJ, Kang SS, Cho GJ, Choi WS. Ethanol down regulates the expression of myelin proteolipid protein in the rat hippocampus. Anat Cell Biol. 2010;43:194–200. doi: 10.5115/acb.2010.43.3.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewohl JM, Wang L, Miles MF, Zhang L, Dodd PR, Harris RA. Gene expression in human alcoholism: microarray analysis of frontal cortex. Alcohol Clin Exp Res. 2000;24:1873–82. [PubMed] [Google Scholar]
- Li Z, Banik NL. The localization of mcalpain in myelin: immunocytochemical evidence in different areas of rat brain and nerves. Brain Res. 1995;697:112–21. doi: 10.1016/0006-8993(95)00949-q. [DOI] [PubMed] [Google Scholar]
- Lopez MF, Becker HC. Effect of pattern and number of chronic ethanol exposures on subsequent voluntary ethanol intake in C57BL/6J mice. Psychopharmacology (Berl) 2005;181:688–96. doi: 10.1007/s00213-005-0026-3. [DOI] [PubMed] [Google Scholar]
- Luna LG. Manual of Histologic Staining Methods of the Armed Forces Institute of Pathology. MacGRAW-HILL Book Company; New York: 1968. Vol. [Google Scholar]
- Mayfield J, Ferguson L, Harris RA. Neuroimmune signaling: a key component of alcohol abuse. Curr Opin Neurobiol. 2013;23:513–20. doi: 10.1016/j.conb.2013.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeill DL, Shew RL, Papka RE. Effects of ethanol exposure on myelination and axon numbers in the L2 dorsal root of the neonatal rat. Dev Neurosci. 1991;13:171–5. doi: 10.1159/000112154. [DOI] [PubMed] [Google Scholar]
- Mollenhauer HH. Plastic embedding mixtures for use in electron microscopy. Stain Technol. 1964;39:111–114. [PubMed] [Google Scholar]
- Muller-Oehring EM, Schulte T, Fama R, Pfefferbaum A, Sullivan EV. Global-local interference is related to callosal compromise in alcoholism: a behavior-DTI association study. Alcohol Clin Exp Res. 2009;33:477–89. doi: 10.1111/j.1530-0277.2008.00858.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto H, Miki T, Lee KY, Yokoyama T, Kuma H, Wang ZY, Gu H, Li HP, Matsumoto Y, Irawan S, Bedi KS, Nakamura Y, Takeuchi Y. Oligodendrocyte myelin glycoprotein (OMgp) in rat hippocampus is depleted by chronic ethanol consumption. Neurosci Lett. 2006;406:76–80. doi: 10.1016/j.neulet.2006.07.023. [DOI] [PubMed] [Google Scholar]
- Pascual M, Pla A, Minarro J, Guerri C. Neuroimmune Activation and Myelin Changes in Adolescent Rats Exposed to High-Dose Alcohol and Associated Cognitive Dysfunction: A Review with Reference to Human Adolescent Drinking. Alcohol Alcohol. 2014 doi: 10.1093/alcalc/agt164. [DOI] [PubMed] [Google Scholar]
- Pfefferbaum A, Sullivan EV, Hedehus M, Adalsteinsson E, Lim KO, Moseley M. In vivo detection and functional correlates of white matter microstructural disruption in chronic alcoholism. Alcohol Clin Exp Res. 2000;24:1214–21. [PubMed] [Google Scholar]
- Pfefferbaum A, Sullivan EV. Microstructural but not macrostructural disruption of white matter in women with chronic alcoholism. Neuroimage. 2002;15:708–18. doi: 10.1006/nimg.2001.1018. [DOI] [PubMed] [Google Scholar]
- Pfefferbaum A, Sullivan EV. Disruption of brain white matter microstructure by excessive intracellular and extracellular fluid in alcoholism: evidence from diffusion tensor imaging. Neuropsychopharmacology. 2005;30:423–32. doi: 10.1038/sj.npp.1300623. [DOI] [PubMed] [Google Scholar]
- Pfefferbaum A, Adalsteinsson E, Sullivan EV. Dysmorphology and microstructural degradation of the corpus callosum: Interaction of age and alcoholism. Neurobiol Aging. 2006;27:994–1009. doi: 10.1016/j.neurobiolaging.2005.05.007. [DOI] [PubMed] [Google Scholar]
- Pfefferbaum A, Rosenbloom M, Rohlfing T, Sullivan EV. Degradation of association and projection white matter systems in alcoholism detected with quantitative fiber tracking. Biol Psychiatry. 2009;65:680–90. doi: 10.1016/j.biopsych.2008.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitel AL, Chanraud S, Sullivan EV, Pfefferbaum A. Callosal microstructural abnormalities in Alzheimer's disease and alcoholism: same phenotype, different mechanisms. Psychiatry Res. 2010;184:49–56. doi: 10.1016/j.pscychresns.2010.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podbielska M, Banik NL, Kurowska E, Hogan EL. Myelin recovery in multiple sclerosis: the challenge of remyelination. Brain Sci. 2013;3:1282–324. doi: 10.3390/brainsci3031282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray SK, Samantaray S, Smith JA, Matzelle DD, Das A, Banik NL. Inhibition of cysteine proteases in acute and chronic spinal cord injury. Neurotherapeutics. 2011;8:180–6. doi: 10.1007/s13311-011-0037-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbloom MJ, Sassoon SA, Fama R, Sullivan EV, Pfefferbaum A. Frontal Callosal Fiber Integrity Selectively Predicts Coordinated Psychomotor Performance in Chronic Alcoholism. Brain Imaging Behav. 2008;2:74–83. doi: 10.1007/s11682-007-9017-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenwasser AM, McCulley WD, 3rd, Fecteau M. Circadian activity rhythms and voluntary ethanol intake in male and female ethanol-preferring rats: effects of long-term ethanol access. Alcohol. 2014;48:647–55. doi: 10.1016/j.alcohol.2014.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saatman KE, Creed J, Raghupathi R. Calpain as a therapeutic target in traumatic brain injury. Neurotherapeutics. 2010;7:31–42. doi: 10.1016/j.nurt.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samantaray S, Knaryan VH, Guyton MK, Matzelle DD, Ray SK, Banik NL. The parkinsonian neurotoxin rotenone activates calpain and caspase-3 leading to motoneuron degeneration in spinal cord of Lewis rats. Neuroscience. 2007;146:741–55. doi: 10.1016/j.neuroscience.2007.01.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samantaray S, Ray SK, Banik NL. Calpain as a potential therapeutic target in Parkinson's disease. CNS Neurol Disord Drug Targets. 2008;7:305–12. doi: 10.2174/187152708784936680. [DOI] [PubMed] [Google Scholar]
- Samantaray S, Knaryan VH, Shields DC, Banik NL. Critical role of calpain in spinal cord degeneration in Parkinson's disease. J Neurochem. 2013a;127:880–90. doi: 10.1111/jnc.12374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samantaray S, Patel KS, Knaryan VH, Thakore NP, Roudabush S, Heissenbuttle JH, Becker HC, Banik NL. Calpain inhibition prevents ethanol-induced alterations in spinal motoneurons. Neurochem Res. 2013b;38:1734–41. doi: 10.1007/s11064-013-1077-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samantaray S, K VH, Shields DC, Banik NL. Possible calpain-mediated spinal cord degeneration of Parkinson's disease. J Neurochem. 2013;125(Suppl.1) doi: 10.1111/jnc.12374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte T, Sullivan EV, Muller-Oehring EM, Adalsteinsson E, Pfefferbaum A. Corpus callosal microstructural integrity influences interhemispheric processing: a diffusion tensor imaging study. Cereb Cortex. 2005;15:1384–92. doi: 10.1093/cercor/bhi020. [DOI] [PubMed] [Google Scholar]
- Schulte T, Muller-Oehring EM, Javitz H, Pfefferbaum A, Sullivan EV. Callosal Compromise Differentially Affects Conflict Processing and Attentional Allocation in Alcoholism, HIV, and Their Comorbidity. Brain Imaging Behav. 2008;2:27–38. doi: 10.1007/s11682-007-9014-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan EV, Pfefferbaum A. Neurocircuitry in alcoholism: a substrate of disruption and repair. Psychopharmacology (Berl) 2005;180:583–94. doi: 10.1007/s00213-005-2267-6. [DOI] [PubMed] [Google Scholar]
- Sullivan EV, Zahr NM. Neuroinflammation as a neurotoxic mechanism in alcoholism: commentary on “Increased MCP-1 and microglia in various regions of human alcoholic brain’. Exp Neurol. 2008;213:10–7. doi: 10.1016/j.expneurol.2008.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo G, Lippai D. Converging actions of alcohol on liver and brain immune signaling. Int Rev Neurobiol. 2014;118:359–80. doi: 10.1016/B978-0-12-801284-0.00011-7. [DOI] [PubMed] [Google Scholar]
- Vilpoux C, Warnault V, Pierrefiche O, Daoust M, Naassila M. Ethanol-sensitive brain regions in rat and mouse: a cartographic review, using immediate early gene expression. Alcohol Clin Exp Res. 2009;33:945–69. doi: 10.1111/j.1530-0277.2009.00916.x. [DOI] [PubMed] [Google Scholar]
- Vosler PS, Brennan CS, Chen J. Calpain-mediated signaling mechanisms in neuronal injury and neurodegeneration. Mol Neurobiol. 2008;38:78–100. doi: 10.1007/s12035-008-8036-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Lousberg EL, Moldenhauer LM, Hayball JD, Coller JK, Rice KC, Watkins LR, Somogyi AA, Hutchinson MR. Inhibiting the TLR4-MyD88 signalling cascade by genetic or pharmacological strategies reduces acute alcohol-induced sedation and motor impairment in mice. Br J Pharmacol. 2012;165:1319–29. doi: 10.1111/j.1476-5381.2011.01572.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakovleva T, Bazov I, Watanabe H, Hauser KF, Bakalkin G. Transcriptional control of maladaptive and protective responses in alcoholics: a role of the NF-kappaB system. Brain Behav Immun. 2011;25(Suppl 1):S29–38. doi: 10.1016/j.bbi.2010.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahr NM, Luong R, Sullivan EV, Pfefferbaum A. Measurement of serum, liver, and brain cytokine induction, thiamine levels, and hepatopathology in rats exposed to a 4-day alcohol binge protocol. Alcohol Clin Exp Res. 2010;34:1858–70. doi: 10.1111/j.1530-0277.2010.01274.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoeller RT, Butnariu OV, Fletcher DL, Riley EP. Limited postnatal ethanol exposure permanently alters the expression of mRNAS encoding myelin basic protein and myelin-associated glycoprotein in cerebellum. Alcohol Clin Exp Res. 1994;18:909–16. doi: 10.1111/j.1530-0277.1994.tb00059.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.