

Abstract
Natural products that inhibit the proteasome have been fruitful starting points for the development of drug candidates. Those of the syringolin family have been under exploited in this context. Using the published model for substrate mimicry by the syringolins and knowledge about the substrate preferences of the proteolytic subunits of the human proteasome, we have designed, synthesized, and evaluated syringolin analogs. As some of our analogs inhibit the activity of the proteasome with second-order rate constants 5-fold greater than the methyl ester of syringolin B, we conclude that the substrate mimicry model for the syringolins is valid. The improvements in in vitro potency and the activities of particular analogs against leukemia cell lines are strong bases for further development of the syringolins as anticancer drugs.
Keywords: Syringolins, Proteasome, Leukemia, Inhibitors
Graphical Abstract
1. Introduction
The 26S proteasome is a multi-subunit complex that effects targeted protein degradation in eukaryotic organisms. It has emerged as one of the highest value targets in drug discovery and development programs focused on cancer treatments.1 Efforts to develop anti-cancer drugs that target the proteasome are motivated by the success of Bortezomib (Velcade), a frontline drug for the treatment of multiple myeloma and mantle cell lymphoma.2,3 This drug is a peptide boronate that reversibly inhibits the proteasome via substrate imitation and labile bonding between its boronic acid moiety and active site threonine residues of the proteasome's proteolytic β subunits. Interestingly, Bortezomib's substrate mimicry and its active site reactive warhead are features that it shares with virtually all naturally occurring and designed inhibitors of the proteasome that have been reported to date. Among these molecules are peptidyl aldehydes, peptidyl epoxyketones, and β–lactones.4,5 It is remarkable that, with the exception of the peptidyl aldehydes, molecules from each of these structural classes are currently in or have completed clinical trials. Notably, an analog of a peptidyl epoxyketone natural product, Carfilzomib (Kyprolis), has recently been approved for the treatment of multiple myeloma. Clearly, optimization of the reactive substrate mimics is a viable strategy for the development of proteasome inhibitors with potential in medicine.4–12
Although natural products in the β-lactone and peptidyl epoxyketone classes of proteasome inhibitors have been thoroughly optimized in medicinal chemistry programs, those in the syringolin family have received much less attention. Syringolins were first isolated in 1998 from Pseudomonas syringae pv. syringae (Figure 1)13 and are characterized by a 12-membered macrocyclic lactam and an exocyclic dipeptide urea.14 Irreversible proteasome inhibition by these molecules is a consequence of reaction of the α,β-unsaturated carbonyl moiety (i.e., the vinylogous amino acid) in their macrolactams with the catalytic threonine residues of the proteolytic subunits.13,15 Syringolin congeners mostly differ with respect to the dipeptide peptide urea moiety, but syringolins B and E are distinguished from the others by the absence of a unit of unsaturation in the macrolactam. The presence of the alkene likely strains the macrolactam such that it is more prone to form the inhibitory conjugate, as evidenced by the fact that syringolin A is the most potent congener. In the context of anticancer drug development, only a few syringolin analogs have been studied.14,16–19 For instance, analogs with esters rather than carboxylic acids in the dipeptide urea moiety are reportedly more active.16 The compounds with the most potent anti-cancer activities are the lipophilic variants of syringolin A (SylA-LIP)16 and a syringolin B (TIR-203)19 (Figure 1).
Figure 1.

Syringolin natural products (syringolins A-F) and synthetic analogs thereof (SylA-Lip and TIR-203).
Curiously, the medicinal chemistry efforts on the syringolins described in the literature were not fully driven by considerations of the means by which the molecules bind to the proteasome. From the published crystal structure of syringolin A bound to the yeast proteasome,15 it is evident that the side chains of the macrolactam's vinylogous amino acid and the amino acid residue appended to the macrocycle mimic those of amino acids at the P1 and P3 positions of a proteasome substrate, respectively (Figure 2).14 (A substrate's residue at the P1 position has the scissile bond and is separated from the one at the upstream P3 position by a single residue.) This model for substrate mimicry by the syringolins is consistent with their preferential (but not exclusive) reactivity with the β5 subunit of eukaryotic proteasomes.16 This subunit has a substrate specificity reminiscent of chymotrypsin, a protease that prefers substrates with aromatic amino acid residues (e.g., phenylalanine, tyrosine, and tryptophan) at the P1 position. In contrast, the β1 and β2 subunits have substrate preferences similar to caspase and trypsin, respectively. The former protease prefers acidic residues at the scissile bond, while the latter prefers those that are basic. Accordingly, the biased reactivity of the syringolins towards the β5 subunit is likely a consequence of their macrolactams having a hydrophobic, vinylogous amino acid (derived from valine) rather than one that is polar. In principle, the potency and selectivity of β5 subunit inhibition by the syringolins could be enhanced by replacing this mimic of valine with aromatic moieties, like those preferred at the P1 position of chymotrypsin substrates. Such design considerations could be coupled with recently reported findings of Chiba and co-workers that a syringolin A analog with a phenylalanine in the dipeptide urea (mimicking a substrate's P3 residue) was much more potent than the parent compound.20 Herein, we report the synthesis and evaluation of syringolin analogs designed to closely mimic the preferred substrates of the proteasome subunit having specificity like chymotrypsin.
Figure 2.

Model for Substrate Mimicry by the Syringolins. R and R′ mimic the side chains of P1 and P3 residues of the proteasome substrate, respectively.
2. Results and Discussion
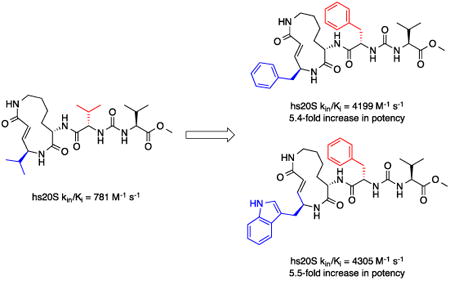
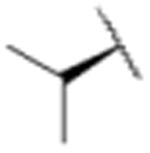
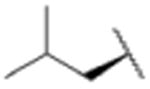
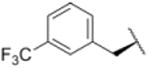
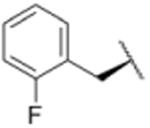
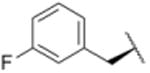
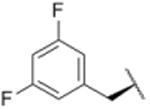
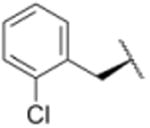
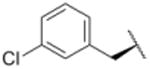
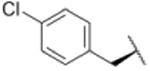
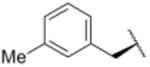
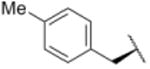
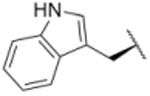
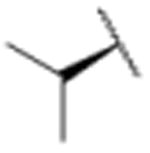
We sought to test the prediction that the capacity of the syringolins to inhibit the proteasome could be improved by rendering their structures more like those of the preferred substrates of the β and β5 subunits of the proteasome. Our attention was focused on the vinylogous amino acid of the macrolactam (mimic of P1 residue) and the amino acid appended to the macrocycle (mimic of P3 residue). Although several syntheses have been presented in the literature,16,20–24, we used a convergent synthetic approach developed by Pirrung and coworkers for the syntheses of syringolin B and analogs thereof (Schemes 1 and 2).24 It is modular and thus amenable to diversity-oriented synthesis. For example, the linear precursor of the macrolactam is prepared from a Cbz-protected lysine residue having an acetyl phosphonate moiety on the ε-amino group and commercially available or easily synthesized 1,2-amino alcohols (Scheme 1).25 Our design strategy dictated the selection of phenylalaninol, tryptophanol, or close analogs of these 1,2-amino alcohols as building blocks because their substituents mimic the aromatic side chains of the P1 residues of the preferred substrates of chymotrypsin substrates (see R groups in Scheme 1, Table 1). For the purposes of comparing analogs with aromatic substituents to those having aliphatic substituents at R, we used valinol and leucinol to prepare linear precursors of syringolin B macrolactam and the closely related compound having an isobutyl group at R, respectively (Table 1, entries 1 and 2). After coupling of the amino alcohols to the protected lysine and oxidation of products' primary alcohols, the reactive functionality mimicking the P1 residue of proteasome substrates was formed via an intramolecular Horner-Wadsworth-Emmons reaction. A modular route was also used for syntheses of the dipeptide urea side chain fragments whose constituents mimic the P3 residue of a proteasome substrate. Indeed, the selection of phenylalanine was informed by the report that a syringolin B analog with this amino acid at the same position was much more potent than those with glycine, alanine, leucine, or isoleucine.20 In total, we synthesized 16 syringolin analogs having esterified side chains and varying degrees of similarity to the substrates preferred by the β5-subunit of the human proteasome (Table 1).
Scheme 1.

Synthetic Route for Analogs of the Syringolin Macrolactam.24
Scheme 2.

Synthetic Route for Analogs of the Syringolin Dipeptide Urea and Their Coupling to Macrolactams.24
Table 1. In Vitro Evaluation of Human Proteasome Inhibition by Syringolin Analogs.
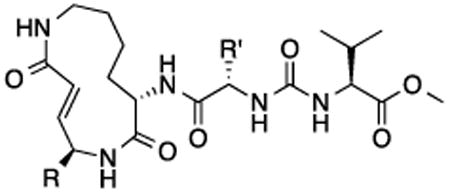
| |||
|---|---|---|---|
|
| |||
| Compound | R | R′ | hs20S kin/Ki (M-1 s-1) |
| 1 |
|

|
781 |
| 2 |
|
|
571 |
| 3 |
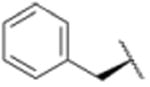
|
|
1912 |
| 4 |
|
|
187 |
| 5 |
|
|
1591 |
| 6 |
|
|
2471 |
| 7 |
|
|
1579 |
| 8 |
|
|
735 |
| 9 |
|
|
1527 |
| 10 |
|
|
1214 |
| 11 |
|
|
635 |
| 12 |
|
|
1321 |
| 13 |
|
|
904 |
| 14 |
|
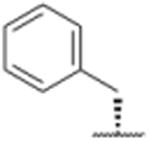
|
1897 |
| 15 |
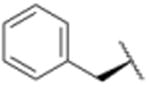
|
|
4199 |
| 16 |
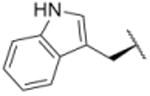
|
|
4305 |
Using purified human 20S proteasome (hs20S) and a fluorogenic substrate (Suc-LLVY-AMC), we performed in vitro assays to systematically assess proteasome inhibition by the syringolin analogs. Our choice of substrate was based on the fact that it is preferentially acted upon by the chymotrypsin-like β5-subunits of the proteasome due to the aromatic tyrosine residue at the scissile bond (i.e., P1 position).26 From measurements of the rates of hydrolysis of the fluorogenic substrate by the proteasome in the presence of increasing concentrations of each inhibitor, we determined second-order rate constants, kin/Ki (M-1 s-1) (Table 1), which reflect both the affinity of the non-covalent binding (Ki) and the rate of the chemical reaction with the enzyme (kin).27 All of the syringolin analogs were capable of inhibiting the chymotrypsin-like activity of hs20S. As is consistent with our design predictions, the most significant contributor to the apparent second-order rate constants of the compounds was their binding affinity (Ki) rather than the rates of inhibition (see supporting information). Accordingly, we found that an analog of syringolin B with an isobutyl substituent at R rather than the isopropyl substituent of the parent exhibited a diminished capacity to inhibit the proteasome. In contrast, compounds with aromatic substituents (Table 1, entries 3-13) at the same position were markedly more potent. Of these macrocyclic analogs, compounds 5 and 6, both containing fluorine-substituted benzyl groups, were the most effective in inhibiting the hs20S proteasome. We were surprised that a syringolin analog with a methylindole substituent was only slightly more active than syringolin B (Table 1, entries 1 and 13). Importantly, the enhanced inhibitory activities of syringolin B analogs having aromatic substituents at the R position on the macrolactam are consistent with the substrate preference of the β5 subunits of the proteasome and with the model for substrate mimicry.
In a separate part of our structure-activity relationship study, we examined the moiety of the syringolins thought to imitate the P3 residue of proteasome substrates. Consistent with a recent report about the preferred amino acid at this position,20 we found that a molecule with the syringolin B macrolactam and a phenylalanine in the dipeptide urea had a second order rate constant for inhibition 2.4-fold greater than the methyl ester of syringolin B (Table 1, entries 1 and 14). Strikingly, the same compound reacts with hs20S at only 45% the rate of a molecule with syringolin analog with a benzyl group at the R position on the macrolactam and a phenylalanine in the dipeptide urea (Table 1, entries 14 and 15). We were particularly surprised to find that our most potent inhibitor had a methylindole substituent on the macrolactam and a phenylalanine residue in the dipeptide urea side chain (Table 1, entry 16) because syringolins whose macrolactam substituents were reminiscent of phenylalanine side chain were more potent (Table 1, entries 3-12) than one with having the tryptophan side chain (13).
The data in Table 1 reflect inhibition of the β5-subunits of the proteasome, which have substrate specificities like chymotrypsin. We performed experiments to assess whether the improvements in potency also translated to greater selectivity. Specifically, we carried out experiments to assess the capacities of syringolin B methyl ester (1) and the most potent inhibitor of the chymotrypsin-like activity (16) to inhibit the trypsin-like activity of the hs20S that is mediated by the β2 subunit. Using a fluorogenic peptide substrate (Boc-LRR-AMC), we measured the second-order rate constants of proteasome inhibition by both compounds (see supplementary material). As expected,12 compound 1 preferentially inhibited the chymotrypsin-like activity of the proteasome by 2.4-fold (Table 2).12 Disappointingly, the degree of selectivity of compound 16 (2.5-fold greater in favor of the chymotrypsin-like activity) was similar to that of compound 1; despite the fact that the former is a 5.5-fold greater inhibitor than the latter in assays of chymotrypsin-like activity (Table 1).
Table 2. Evaluation of Subunit-Specific Inhibition of Syringolin B Methyl Ester and Analogs Thereof.
| Compound | Chymotrypsin-Like Activity kin/Ki (M-1 s-1) | Trypsin-Like Activity kin/Ki (M-1 s-1) |
|---|---|---|
| 1 | 781 | 326 |
| 16 | 4305 | 1701 |
To assess their growth inhibitory activities against cancer cells, we submitted syringolin B methyl ester (1) and analogs 13, 14, 15, and 16 to the National Cancer Institute (NCI) where they were initially evaluated in single-dose assays at a 10 micromolar concentration with 60 different cancer cell lines (see supporting information). Though it was not the most potent inhibitor of the human proteasome (Table 1), analog 15 exhibited the broadest spectrum of growth inhibitory activity against the panel of cell lines. Specifically, cell lines incubated in media with 10 micromolar of compound 15 grew at an average of 30% of those incubated in media without compound (see supporting information). In contrast, the mean growth of the cell lines in media with the same concentration of compound 16 was approximately 71% of those in negative control experiments. As a follow up to the single-dose experiments, the dose-dependent growth inhibition by syringolin B methyl ester (1) and the most potent inhibitors (15 and 16) was assessed at the NCI (see supporting information). We found that the three compounds were particularly active against leukemia cell lines. Though compound 16 is slightly more potent than compound 15 as a proteasome inhibitor (Table 3), it is markedly more potent in cell culture, which could reflect differences in the cell permeabilities or stabilities of the two compounds. Notably, compound 16 is as much as 11.5-fold more potent than syringolin B methyl ester (1) against the leukemia cell lines.
Table 3. Growth Inhibitory Activities of Syringolin B Methyl Ester and Analogs Thereof Against Various Leukemia Cell Lines.
| GI50 (μM) | |||
|---|---|---|---|
|
|
|||
| Cell Line | Compound 1 | Compound 15 | Compound 16 |
| HL-60 | 5.41 | 4.34 | 1.29 |
| K-562 | 6.12 | 14.6 | 2.94 |
| MOLT-4 | 4.73 | 3.32 | 0.599 |
| RPMI-8226 | 3.97 | 2.72 | 0.343 |
| SR | 5.13 | 2.78 | 0.453 |
Data provided by the National Cancer Institute (NCI): “GI50 is the concentration of test drug where 100 × (T - T0)/(C - T0) = 50. The optical density of the test well after a 48-h period of exposure to test drug is T, the optical density at time zero is T0, and the control optical density is C. The “50” is called the GI50PRCNT, a T/C-like parameter that can have values from +100 to -100.”
3. Conclusions
At present, the syringolins and analogs thereof are poorly represented in cancer drug development pipelines. The experiments reported herein were motivated by an effort to fill in some gaps on the structure-activity relationships of the syringolins and to test the idea that proteasome inhibitors can be rationally optimized based on knowledge of the substrate preferences of the proteolytic subunits of the proteasome. We found that syringolin analogs that are reminiscent of chymotrypsin substrates are more potent inhibitors of the β5-subunits of the proteasome than the parent compound, yet their subunit-selectivities for inhibition are comparable. The fact that potency, and not selectivity, has been improved could be an asset in that inhibition of multiple proteolytic subunits by a compound is highly correlated with significant suppression of protein degradation.28,29 Indeed, we found that the syringolin analogs are potent suppressors of the growth of cancer lines in vitro, especially those derived from leukemias. These observations provide a foundation for further rational optimization of the syringolins as lead compounds for anti-leukemic drugs.
Supplementary Material
Acknowledgments
The work was supported in part by funding from Brown University to JKS and NIH grant AI-16892 to RTS. DB was supported by Deutsche Forschungsgemeinschaft Grants BA 4890/1-1 and BA 4890/3-1. PS was supported by a undergraduate teaching and research assistantship from Brown University. Expert technical support on NMR and mass spectrometry were provided by R. Hopson and T. Shen in the Brown University Department of Chemistry.
Footnotes
Supplementary Material: Supplementary data associated with this article can be found, in the online version, at
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Rajkumar SV, Richardson PG, Hideshima T, Anderson KC. J Clin Oncol. 2005;23:630–639. doi: 10.1200/JCO.2005.11.030. [DOI] [PubMed] [Google Scholar]
- 2.Adams J, Kauffman M. Cancer Invest. 2004;22:304–311. doi: 10.1081/cnv-120030218. [DOI] [PubMed] [Google Scholar]
- 3.Bonvini P, Zorzi E, Basso G, Rosolen A. Leukemia. 2007;21:838–842. doi: 10.1038/sj.leu.2404528. [DOI] [PubMed] [Google Scholar]
- 4.Gräwert MA, Groll M. Chem Commun. 2012;48:1364–137. doi: 10.1039/c1cc15273d. [DOI] [PubMed] [Google Scholar]
- 5.Kisselev AF, Van Der Linden Wa, Overkleeft HS. Chem Biol. 2012;19:99–115. doi: 10.1016/j.chembiol.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Metcalf R, Scott LM, Daniel KG, Dou QP. Expert Opin Ther Pat. 2014:1–14. doi: 10.1517/13543776.2014.877444. [DOI] [PubMed] [Google Scholar]
- 7.Hasegawa M, Yasuda Y, Tanaka M, Nakata K, Umeda E, Wang Y, Watanabe C, Uetake S, Kunoh T, Shionyu M, Sasaki R, Shiina I, Mizukami T. Eur J Med Chem. 2014;71:290–305. doi: 10.1016/j.ejmech.2013.11.009. [DOI] [PubMed] [Google Scholar]
- 8.Steele JM. J Oncol Pharm Pract. 2013;19:348–354. doi: 10.1177/1078155212470388. [DOI] [PubMed] [Google Scholar]
- 9.Trivella DBB, Pereira AR, Stein ML, Kasai Y, Byrum T, Valeriote Fa, Tantillo DJ, Groll M, Gerwick WH, Moore BS. Chem Biol. 2014;21:782–791. doi: 10.1016/j.chembiol.2014.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Britton M, Lucas MM, Downey SL, Screen M, Pletnev AA, Verdoes M, Tokhunts RA, Amir O, Goddard AL, Pelphrey PM, Wright DL, Overkleeft HS, Kisselev AF. Chem Biol. 2009;16:1278–1289. doi: 10.1016/j.chembiol.2009.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Elofsson M, Splittgerber U, Myung J, Mohan R, Crews CM. Chem Biol. 1999;6:811–822. doi: 10.1016/s1074-5521(99)80128-8. [DOI] [PubMed] [Google Scholar]
- 12.Momose I, Umezawa Y, Hirosawa S, Iinuma H, Ikeda D. Bioorganic Med Chem Lett. 2005;15:1867–1871. doi: 10.1016/j.bmcl.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 13.Wäspi U, Misteli B, Hasslacher M, Jandrositz A, Kohlwein SD, Schwab H, Dudler R. Eur J Biochem. 1998;254:32–37. doi: 10.1046/j.1432-1327.1998.2540032.x. [DOI] [PubMed] [Google Scholar]
- 14.Krahn D, Ottmann C, Kaiser M. Nat Prod Rep. 2011;28:1854–1867. doi: 10.1039/c1np00048a. [DOI] [PubMed] [Google Scholar]
- 15.Groll M, Schellenberg B, Bachmann AS, Archer CR, Huber R, Powell TK, Lindow S, Kaiser M, Dudler R. Nature. 2008;452:755–758. doi: 10.1038/nature06782. [DOI] [PubMed] [Google Scholar]
- 16.Clerc J, Groll M, Illich DJ, Bachmann AS, Huber R, Schellenberg B, Dudler R, Kaiser M. Proc Natl Acad Sci U S A. 2009;106:6507–6512. doi: 10.1073/pnas.0901982106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ibarra-Rivera TR, Opoku-Ansah J, Ambadi S, Bachmann AS, Pirrung MC. Tetrahedron. 2011;67:9950–9956. [Google Scholar]
- 18.Clerc J, Li N, Krahn D, Groll M, Bachmann AS, Florea BI, Overkleeft HS, Kaiser M. Chem Commun (Camb) 2011;47:385–387. doi: 10.1039/c0cc02238a. [DOI] [PubMed] [Google Scholar]
- 19.Opoku-Ansah J, Ibarra-Rivera TR, Pirrung MC, Bachmann AS. Pharm Biol. 2012;50:25–29. doi: 10.3109/13880209.2011.626784. [DOI] [PubMed] [Google Scholar]
- 20.Chiba T, Hosono H, Nakagawa K, Asaka M, Takeda H, Matsuda A, Ichikawa S. Angew Chemie - Int Ed. 2014;53:4836–4839. doi: 10.1002/anie.201402428. [DOI] [PubMed] [Google Scholar]
- 21.Clerc J, Schellenberg B, Groll M, Bachmann AS, Huber R, Dudler R, Kaiser M. European J Org Chem. 2010:3991–4003. [Google Scholar]
- 22.Schmidt U, Kleefeldt A, Mangold RJ. Chem Soc Chem Commun. 1992:1687–1689. [Google Scholar]
- 23.Dai C, Stephenson CRJ. Org Lett. 2010;12:3453–3455. doi: 10.1021/ol101252y. [DOI] [PubMed] [Google Scholar]
- 24.Pirrung MC, Biswas G, Ibarra-Rivera TR. Org Lett. 2010;12:2402–2405. doi: 10.1021/ol100761z. [DOI] [PubMed] [Google Scholar]
- 25.McKenon MJ, Meyers AI. J Org Chem. 1993;58:3568–3571. [Google Scholar]
- 26.Harris JL, Alper PB, Li J, Rechsteiner M, Backes BJ. Chem Biol. 2001;8:1131–1141. doi: 10.1016/s1074-5521(01)00080-1. [DOI] [PubMed] [Google Scholar]
- 27.Singh J, Petter RC, Baillie TA, Whitty A. Nat Rev Drug Discov. 2011;10:307–317. doi: 10.1038/nrd3410. [DOI] [PubMed] [Google Scholar]
- 28.Kisselev AF, Callard A, Goldberg AL. J Biol Chem. 2006;281:8582–8590. doi: 10.1074/jbc.M509043200. [DOI] [PubMed] [Google Scholar]
- 29.Oberdorf J, Carlson EJ, Skach WR. Biochemistry. 2001;40:13397–13405. doi: 10.1021/bi011322y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.