

Abstract
A pathological hallmark of Parkinson disease (PD) is progressive degeneration of nigrostriatal dopamine (NSDA) neurons, which underlies the motor symptoms of PD. While there is severe loss of midbrain NSDA neurons, tuberoinfundibular (TI) DA neurons in the mediobasal hypothalamus (MBH) remain intact. In the present study, confocal microscopic analysis revealed that mitochondrial content and numbers of mitophagosomes were lower in NSDA neuronal cell bodies in the substantia nigra pars compacta (SNpc) compared to TIDA neuronal cell bodies in the arcuate nucleus (ARC) of C57BL/6J male mice. Mitochondrial respiration, mass, membrane potential and morphology were determined using bioenergetic, flow cytometric and transmission electron microscopic analyses of synaptosomes isolated from discrete brain regions containing axon terminals of NSDA and TIDA neurons. Maximum and spare respiratory capacities, and mitochondrial mass were lower in synaptosomal mitochondria derived from the striatum (ST) as compared with the MBH, which correlated with lower numbers of mitochondria per synaptosome in these brain regions. In contrast, there was no regional difference in mitochondrial basal, maximum or spare respirations following inhibition of Complex I activity with rotenone. These results reveal that higher numbers of viable mitochondria are correlated with more extensive autophagic mitochondrial quality maintenance in TIDA neurons as compared with NSDA neurons.
Keywords: Neurodegeneration, Parkinson disease, mitochondria, respiration, autophagy, striatum, mediobasal hypothalamus, nigrostriatal DA neurons, tuberoinfundibular DA neurons, substantia nigra, transmission electron microscopy, flow cytometry, confocal microscopy
1. Introduction
Nigrostriatal dopamine (NSDA) neurons located in the substantia nigra pars compacta (SNpc) of the ventral midbrain have axons that project rostrally via the median forebrain bundle and terminate in the striatum (ST). NSDA neurons modulate the function of the basal ganglia voluntary motor control circuits (Albin, Young et al. 1989) and degeneration of these neurons is associated with resting tremor, rigidity, and bradykinesia, i.e., the classic motor features of Parkinson disease (PD). The motor symptoms are a major source of disability in PD and effective treatment of these symptoms markedly reduces morbidity and mortality in PD (Ahlskog 2001, Connolly and Lang 2014). As such, understanding the mechanisms underlying the degeneration of NSDA neurons is of significant importance (Sulzer and Surmeier 2013).
Tuberoinfundibular (TI) DA neurons located in the arcuate nucleus (ARC) project axons that course ventrally and terminate in the median eminence of the mediobasal hypothalamus (MBH). DA released from these neurons regulates anterior pituitary hormone secretion (Moore, et al., 1987). Disruption of the function of TIDA neurons results in hyperprolactinemia, and associated gynecomastia and infertility (Cookson et al., 2012). In contrast to NSDA neurons, TIDA neurons are relatively unaffected in Parkinson disease (Matzuk and Saper, 1985; Langston and Forno, 1978; Jellinger and Kurt, 1991; Braak and Braak, 2000).
There are notable differences between NSDA and TIDA neurons with respect to the location of axon terminals in relation to the blood-brain barrier, regulation of DA synthesis and release from axon terminals, and the susceptibility and response of these neurons to neurotoxicant exposure. NSDA axons terminate in classic synapses with target neurons within the blood-brain barrier and contain abundant DA transporters for re-uptake of released DA (Vaughan and Foster, 2013). NSDA neurons are regulated by pre-synaptic inhibitory D2 autoreceptors that couple the synthesis and release of DA in axon terminals (Ford, 2014). NSDA neurons are susceptible to both acute and chronic exposure to the mitochondrial toxicant 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) resulting in sustained loss of axon terminal DA stores, and compensatory activation of DA synthesis and metabolism in the surviving axon terminals (Behrouz et al., 2007; Benskey et al., 2012; 2013).
In contrast, TIDA neurons terminate outside the blood-brain barrier in the median eminence of the MBH and release DA in close proximity to the hypophysial portal system, which transports DA to the anterior pituitary to act via D2 receptors to inhibit prolactin release (Lookingland and Moore, 2005). TIDA neurons lack high affinity DA re-uptake transporters and pre-synaptic D2 autoreceptors, and are regulated instead by the stimulatory feedback effects of elevated prolactin in the circulation (Moore et al., 1987). TIDA neurons are injured by acute MPTP, but recovery of axon terminal DA stores occurs within hours following exposure (Behrouz et al., 2007; Benskey et al., 2012). Recovery of TIDA neurons is protein synthesis dependent and correlates with up-regulation of synthesis of the E3 ligase parkin (Benskey et al., 2012), an enzyme involved in protein homeostasis (Heo and Rutter, 2011; Cook et al., 2012) and mitochondrial maintenance (Davison et al., 2009; Guo, 2010; Tanaka, 2010; Taylor and Rutter, 2011; Youle and Narendra, 2011; Koh and Chung, 2012).
Deficient mitochondrial Complex I activity is present in the midbrain of Parkinson disease patients (Schapira, 1989; Mizuno et al., 1989) suggesting that mitochondrial dysfunction may play a role in determining NSDA neuronal susceptibility in PD, similar to the differential susceptibility of NSDA and TIDA neurons to MPTP exposure (Behrouz et al., 2007; Benskey et al., 2012; 2013). In the present study, regional differences in maintenance of mitochondrial homeostasis were evaluated using bioenergetic, flow cytometric, transmission electron and confocal microscopic analyses in C57BL/6J male mice. The results reveal that diminished mitochondrial bioenergetics, and mass in synaptosomes containing axon terminals of NSDA neurons were correlated with fewer mitochondria and mitophagosomes in cell bodies in the SNpc as compared with those of TIDA neurons. The region-dependent disparity in mitochondrial mass and function associated with corresponding changes in mitophagosome formation suggests the possibility that differences in mitochondrial autophagic maintenance could play a role in differential susceptibility of central DA neurons to degeneration.
2. Results
2.1 Mitochondrial content and mitophagosome numbers in tyrosine hydroxylase (TH) neurons in the SNpc and ARC
In situ analysis of mitochondrial numbers localized more specifically within TH immunoreactive NSDA and TIDA neurons was performed using confocal microscopy. Coronal sections through the midbrain and MBH were immunohistochemically stained for TH using brain sections selected approximately -3.16 and -1.46 mm relative to Bregma for SNpc (Figures 1A & 1C) and ARC (Figures 1B & 1C), respectively. TH, a phenotypic marker of DA neurons, is labeled with secondary antibody linked to Alexa Fluor 405 and visualized as blue. The mitochondrial marker cytochrome c oxidase (COX IV) is labeled with secondary antibody linked to Alexa Fluor 594 and is visualized as red. The mitochondrial distribution in TH immunoreactive neurons was quantified and expressed as a percentage of the total pixels of COX IV localized within TH immunoreactive neurons divided by the total number of pixels representing TH. This percentage represents the area within the TH immunoreactive neurons that is occupied by mitochondria. The co-localization of COX IV staining within TH immunoreactive neurons in the ARC was higher than that of SNpc, consistent with a greater density of mitochondria in cell bodies of TIDA versus NSDA neurons (Figure 1E).
Figure 1. Comparison of mitochondrial content in cell bodies of TH immunoreactive neurons in the SNpc and ARC.
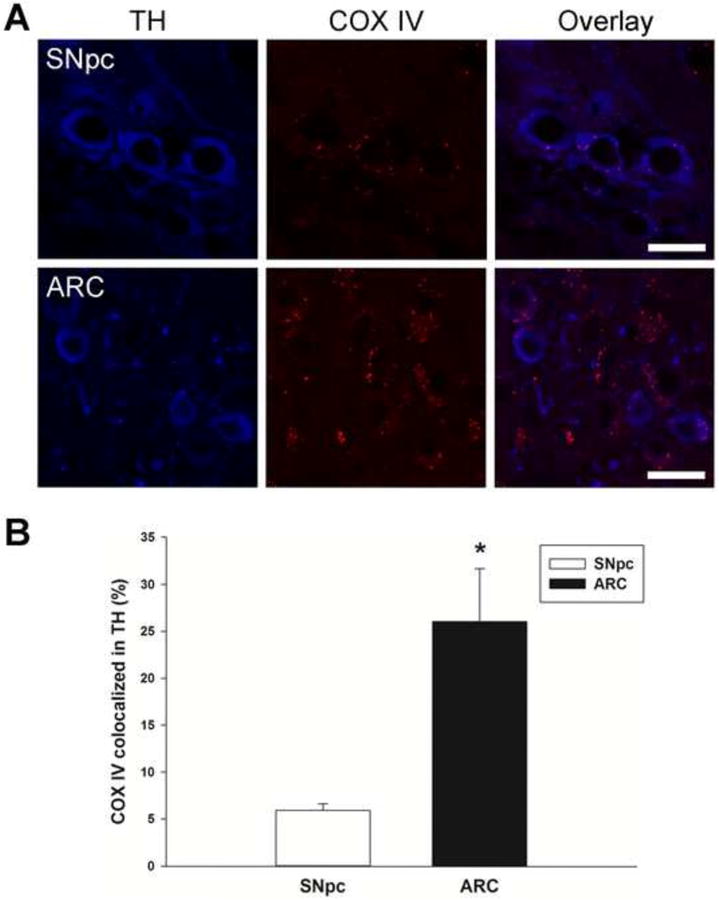
A, Representative high power confocal microscopic single plane focal images are depicted for the SNpc and ARC. GFP-LC3 mice were perfused with fixative and brains were removed and cut into 20 μm sections for immunohistochemical analyses. Mitochondrial distribution in TH immunoreactive neurons was quantitated in SNpc at approximately Bregma -3.16 and in ARC at Bregma -1.46. Blue represents staining for TH and red represents COX IV staining in mitochondria. (Scale bar = 20 μm). B, Representative bar graphs of the percentage of COX IV co-localized with TH in SNpc and ARC. Pixels were quantified by upper right quadrant scatter plot analyses, and the percent distribution of COX IV within TH neurons was calculated. Percent values were converted to parametric values by ASIN(SQRT(% value/100)). Columns represent the means and error bars +1 standard error of the mean. * indicates percentage COX IV co-localization in TH immunoreactive neurons in the ARC (filled column) that were significantly different (p<0.05) from those in the SNpc (open column).
Mitochondrial quality control is mediated, in part, by dynamic recycling of mitochondria. Functional mitochondria are maintained through fission of existing mitochondria or fusion with other mitochondria while irreparably damaged mitochondria are removed by autophagy, i.e., mitophagy (Lemasters, 2005; Green and Houten, 2011; Youle and Narendra, 2011; Youle and van der Bleik, 2012). In the present study, the extent of mitophagy was assessed by immunohistochemical co-localization of mitochondria with autophagosomes in SNpc and ARC using GFP-microtubule-associated protein 1A/1B-light chain 3 (LC3) mice and dual immunohistochemical staining for the mitochondrial protein COX IV and TH. TH immunoreactive neurons were visualized as blue (Alexa Fluor 405), mitochondria as red (Alexa Fluor 594), and autophagosomes as green punctate structures containing GFP-LC3.
LC3 exists as a cytosolic protein, but becomes membrane-bound once autophagosomes are formed. As shown in Figure 2A for the ARC, autophagosomal GFP-LC3 appears as a green punctate signal associated with mitochondrial COX IV within TH immunoreactive neurons. The co-localization of COX IV immunoreactivity (mitochondria) and GFP punctate structures (autophagosomes) within TH immunoreactive neurons located in the SNpc and ARC were compared using the Pearson Coefficient value. Pearson Coefficient values range from 0 to 1, with 1 as highest co-localization of the two fluorescence signals representing mitochondria located within autophagosomes. The Pearson Coefficient was 3-4 times higher in the ARC compared to the SNpc (Figure 2C), which suggests that mitochondria were more frequently found to co-localize with autophagosomes in TIDA neurons in the ARC as compared with NSDA neurons in the SNpc.
Figure 2. Comparison of the co-localization of mitochondria with autophagosomes in TH immunoreactive neurons in the SNpc and ARC.
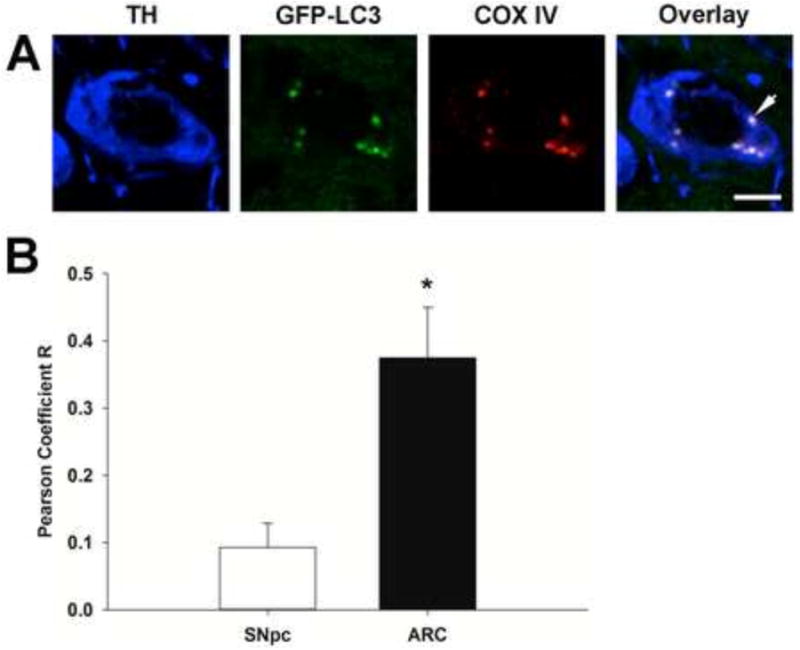
Representative confocal microscopic single plane focal image of a single cell in the ARC (A). Blue represents staining for TH, GFP-LC3 labeled autophagosomes were visualized as green puncta, and red represents COX IV staining in mitochondria. Arrow denotes co-localization of GFP-LC3 and COX IV in a TH immunoreactive neuron. Scale bar is 5 μm. (B); Pearson Coefficient of mitochondrial co-localization with autophagosomes in TH immunoreactive cells in the SNpc and ARC. Pearson coefficients were measured using Fiji with Image J software. TH immunoreactive neurons were masked and pixels with TH immunoreactive neurons were analyzed for co-localization using Fiji Coloc_2. Columns represent the means and error bars +1 standard error of the mean. * indicates Pearson coefficients in TH immunoreactive neurons in the ARC (filled column) that were significantly different (p<0.05) from those in the SNpc (open column).
2.2 DA concentrations in synaptosomes derived from ST and MBH
In order to determine if there are differences in mitochondrial structure and function in regions of the brain containing axon terminals of NSDA and TIDA neurons, synaptosomal preparations were isolated from the ST and MBH for transmission electron microscopic, bioenergetic, and flow cytometric analyses. HPLC-ED analysis revealed appreciable concentrations of DA in synaptosomes derived from both the ST (46 ±8 ng/mg) and MBH (7.1 ±1.1 ng/mg), whereas the DA metabolite homovanillic acid (HVA) was not detected. Since generation of HVA in the brain is dependent on the DA catabolic enzyme catechol-O-methyl transferase present in glia, the lack of detectable HVA in the context of robust DA levels indicates little, if any, glial contamination in the synaptosomal preparations analyzed in these experiments.
2.3 Differential mitochondrial respiration in synaptosomes derived from ST and MBH
Mitochondrial respiration was determined by measuring the oxygen consumption rate (OCR) of synaptosomes derived from the ST and MBH. Non-mitochondrial respiration was determined following pharmacological inhibition of the electron transport chain. Preliminary studies found that there were no differences when using the Complex I inhibitor rotenone or the Complex II inhibitor antimycin A to measure non-mitochondrial respiration in our synaptosome samples. Under basal conditions, ST- and MBH-derived synaptosomal mitochondria showed no difference in OCR (Figure 3A). Basal respiration of ST-derived synaptosomal mitochondria was 8.9 ± 1.7 (pmoles/min/μg) versus 7.4 ± 1.6 (pmoles/min/μg) for MBH-derived synaptosomal mitochondria. However, maximum respiration measured by changes in OCR following treatment with FCCP in MBH-derived synaptosomal mitochondria (45.8 ± 1.3 pmoles/min/μg) was higher than in ST-derived synaptosomal mitochondria (36.0 ± 1.5 pmoles/min/μg). Moreover, synaptosomal mitochondria from MBH had a higher spare respiratory capacity (37.5 ± 2.8 pmoles/min/μg) than synaptosomal mitochondria from ST (27.0 ± 2.5 pmoles/min/μg).
Figure 3. Comparison of oxygen consumption rates (OCR) and extracellular acidification rates (ECAR) in synaptosomal mitochondria derived from the striatum (ST) and mediobasal hypothalamus (MBH).
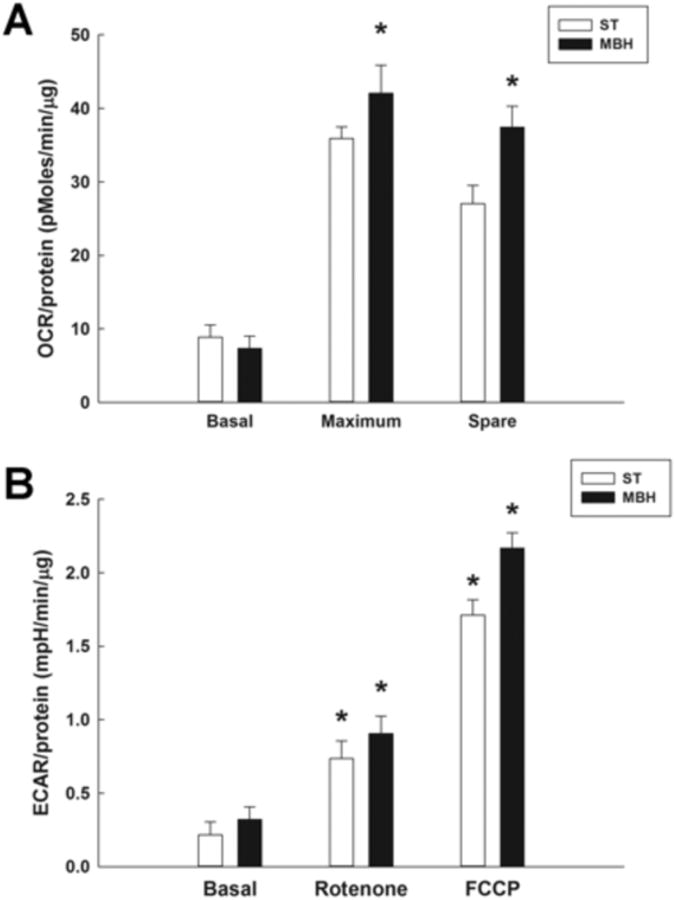
OCR and ECAR measurements were performed using a Seahorse XF24 analyzer, with 20 μg of synaptosomal protein derived from each region yielding a respiratory control ratio' (RCR') of greater than 3. A; OCR (pmoles/min/μg) of ST- and MBH-derived synaptosomal mitochondria during basal, maximum and spare aerobic respiration measured using a Seahorse XF analyzer. Columns represent mean OCR of synaptosomal mitochondria and vertical lines + 1 SEM of determinations in 5 mice. * indicates OCR of maximum and spare respiration in MBH that are significantly different (p < 0.05) from those in ST. B; ECAR (mpH/min/μg) of ST- and MBH-derived synaptosomal mitochondria under basal conditions, and following inhibition (rotenone) or uncoupling (FCCP) of the mitochondrial electron transport chain. * indicates ECAR in FCCP- and rotenone-treated groups that are significantly different (p < 0.05) from basal ECAR values for each region.
In order to investigate possible non-aerobic metabolic profile differences between brain regions, extracellular acidification rate (ECAR) was determined and used as an index of glycolysis (Gohil et al., 2010). Basal ECAR was similar in ST-derived synaptosomal mitochondria (0.26 ± 0.03 mpH/min/μg) and MBH-derived synaptosomal mitochondria (0.35 ± 0.05 mpH/min/μg) (Figure 3B). When mitochondrial Complex I activity was inhibited with rotenone, ECAR increased, but there was no difference in the response between synaptosomal mitochondria derived from these two brain regions. When the mitochondrial proton gradient was dissipated by FCCP, ECAR was further increased to a similar extent in both ST- and MBH-derived synaptosomal mitochondria.
2.4 Effects of rotenone on mitochondrial respiration in synaptosomes derived from ST and MBH
Regional differences in maximum and spare aerobic respiratory responses could be associated with differences in the degree of mitochondrial response to Complex I inhibition. To determine if this was the case, ex vivo rotenone concentration responses of mitochondria in synaptosomes derived from the ST and MBH were compared (Figure 4), and the estimated half maximal inhibitor concentrations (IC50) and 95% confidence intervals for rotenone were calculated (Table 1). Synaptosomes were isolated from the ST and MBH, and OCR was determined beginning 5 min after ex vivo treatment with either vehicle (incubation buffer) or rotenone (0.0125, 0.25 or 2 uM). Rotenone 0.0125, 0.25 and 2 μM inhibited basal respiration by 23%, 67%, and 89%, respectively, in ST-derived synaptosomes, and likewise by 21%, 64%, and 73% in MBH-derived synaptosomes (Figure 4A). Estimated IC50 for inhibition of basal respiration in ST-derived synaptosomes was 0.22 μM versus 0.91 μM for MBH-derived synaptosomes (Table 1).
Figure 4. Comparison of the effects of rotenone on mitochondrial aerobic respiration in synaptosomes derived from the ST and MBH.
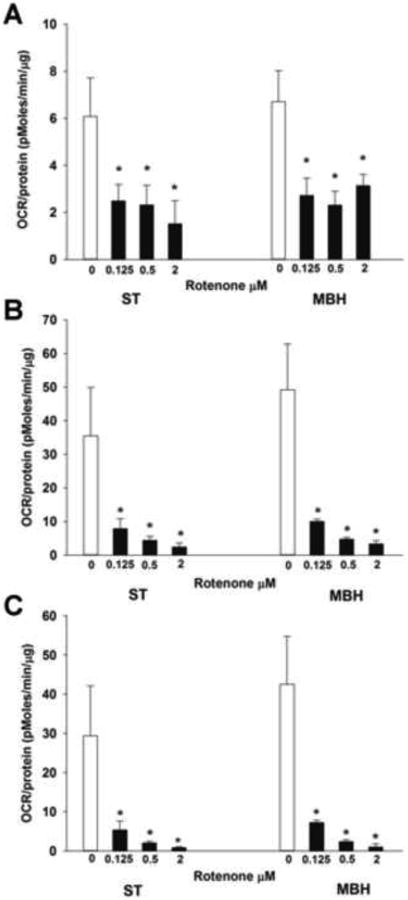
OCR measurements were performed using a Seahorse XF24 analyzer, with twenty μg of synaptosomal protein derived from each region yeilding a respiratory control ratio' (RCR') of greater than 3. OCR (pmoles/min/μg) was measured beginning 5 min after treatment with either vehicle (incubation buffer) or rotenone (0.0125, 0.25, or 2 μM). Columns represent mean OCR of synaptosomal mitochondria basal (A), maximum (B) and spare (C) respirations, and vertical lines + 1 SEM of 3 determinations. * indicates values in rotenone-treated synaptosomes that were significantly different from vehicle-treated (0 μM) controls, p < 0.05.
Table 1. Comparison of half maximal inhibitory concentrations (IC50) of rotenone on synaptosomal mitochondria derived from ST and MBH.
ST and MBH were dissected in male C57B1/6J mice (n=4) and synaptosomes were isolated from the ST and MBH using Percoll gradients and centrifugation. Twenty μg synaptosomes were examined. Spare respiration of synaptosomal mitochondria from ST and MBH were measured beginning 5 min after vehicle (synaptosome ionic media) or rotenone (0.0125, 0.25, or 2 μM) treatment and OCR was measured using a Seahorse XF24 analyzer. The RCR' was greater than 3. IC50 was calculated using ED50 plus (v1.0) software. Values in parenthesis are IC50 95% confidence intervals.
| IC50 | ST (μM) | MBH (μM) |
|---|---|---|
| Basal | 0.22 (0.16 – 0.30) | 0.31 (0.12 – 0.78) |
| Maximum | 0.11 (0.10 – 0.11) | 0.11 (0.11 – 0.12) |
| Spare | 0.09 (0.08 – 0.10) | 0.10 (0.09 – 0.10) |
Similar to basal respiration, both maximum (Figure 4B) and spare (Figure 4C) respirations were inhibited by rotenone in a concentration-dependent manner. Rotenone at 0.25 and 2 μM reduced maximum respiration by 85% and 96%, respectively, in ST-derived synaptosomes versus 87% and 96% for MBH-derived synaptosomes. Rotenone (2 μM) reduced spare respiration by 98% in ST-derived synaptosomes and by 99.7% in MBH-derived synaptosomes. The mean estimated IC50 values for maximum (0.11 μM rotenone) and spare (0.09 μM rotenone) respirations were similar (Table 1). Taken together, these data demonstrate that there is no difference in the ability of rotenone to inhibit respiration in ST and MBH synaptosomal preparations.
2.5 Mitochondrial mass and membrane potential of ST and MBH synaptosomal mitochondria
Flow cytometric assessment of ST- and MBH-derived synaptosomal mitochondria was performed to investigate the differential mitochondrial maximum and spare respiratory capacities observed in the ST and MBH. The mitochondrial mass within the synaptosomes, representing mitochondrial density in axon terminals, was measured in synaptosomes (20 μg) from each brain region using MitoTracker Green uptake. The amount of MitoTracker Green detected in synaptosomes is directly related to both the number and volume of healthy mitochondria (Kaasik, 2006). In the present study, synaptosomal populations from the ST and MBH were found using flow cytometry to have similar size/volume and inner complexity/granularity. MBH- derived synaptosomal mitochondria showed a shift of the green fluorescence toward a higher mode of MitoTracker Green with an average MitoTracker Green fluorescence intensity 35% higher in synaptosomes derived from MBH as compared with ST (Figure 5A). These results are consistent with a greater overall mitochondrial mass due to either higher numbers or larger volume of individual mitochondrion in synaptosomes derived from the MBH versus ST.
Figure 5. Flow cytometric analysis of mitochondrial mass and membrane potential in synaptosomes derived from the ST and MBH.
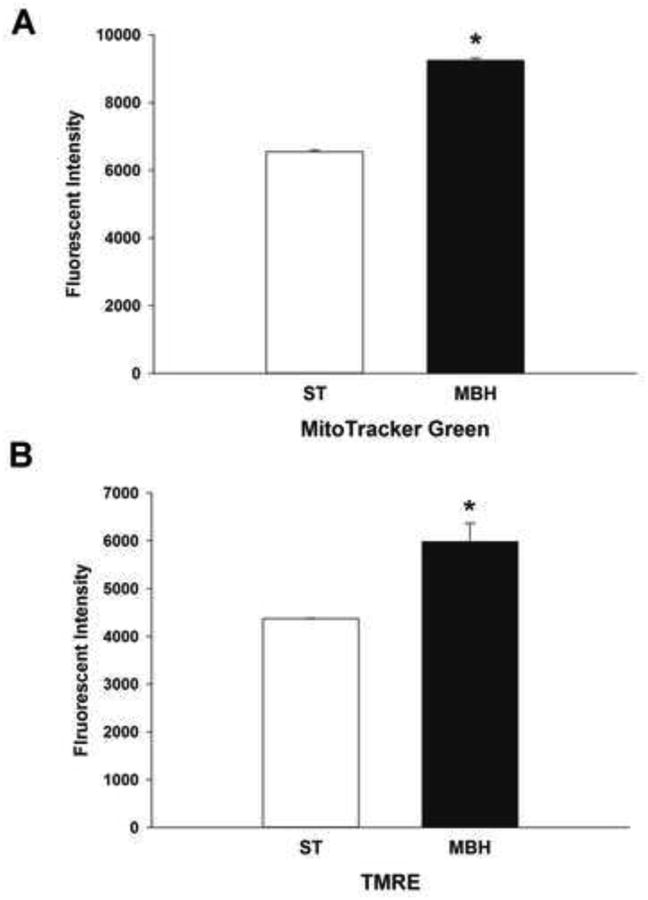
Flow cytometric assessment of mitochondrial mass and membrane potential were performed using the fluorescent dyes MitoTracker Green and TMRE, respectively. Twenty μg of synaptosomal protein was examined for mitochondrial mass and membrane potential by flow cytometry. Columns represent the mean fluorescent intensity of MitoTracker Green (A) and TMRE (B) in ST- and MBH-derived synaptosomal mitochondria and vertical lines + 1 SEM of determinations in 3 mice. * indicates values in MBH-derived synaptosomes that are significantly different (p < 0.05) from those derived from the ST.
While MitoTracker Green enters and labels mitochondria independent of membrane potential, TMRE entry into active mitochondria is dependent on membrane polarization (Pendergrass et al., 2004). Flow cytometric assessment of TMRE fluorescence revealed a shift toward a higher TMRE signaling in synaptosomes derived from MBH compared to those from the ST with approximately 30% higher TMRE fluorescence in synaptosomes derived from MBH versus those from the ST (Figure 5B). These results are consistent with a greater overall mitochondrial membrane potential in synaptosomes derived from the MBH as compared with the ST, corresponding with an overall higher numbers and/or larger volume of individual mitochondrion.
2.6 Transmission electron microscopy analyses of ST- and MBH-derived synaptosomal mitochondrial morphology and integrity
Bioenergetics, mass and membrane potential analyses represent the characteristics of overall mitochondria function and density in the ST- and MBH-derived synaptosomes. In order to investigate the ultrastructural integrity of individual mitochondrion, morphology was examined in synaptosomes using transmission electron microscopy. Visualized mitochondrial morphology was semi-quantified to evaluate mitochondrial quality using Flameng grading (Sun et al., 2012; Flameng et al., 1980). The Flameng mitochondrial score evaluates mitochondrial function based on the mitochondrial morphology using an ordinal scale of 1 through 5. As depicted by representative images of synatosomal mitochondria in Figure 6A, a score of 1 indicates that the mitochondria have disrupted mitochondrial membranes and broken cristae; a score of 2 indicates that the mitochondria have broken cristae, but maintain intact membranes; a score of 3 indicates swollen mitochondria with matrix clearance while cristae and membrane are intact; a score of 4 indicates that the mitochondria are intact, but devoid of granules; and a score of 5 indicates that the mitochondria are intact with granules.
Figure 6. Transmission electron microscopic analyses of ST- and MBH-derived synaptosomal mitochondria.
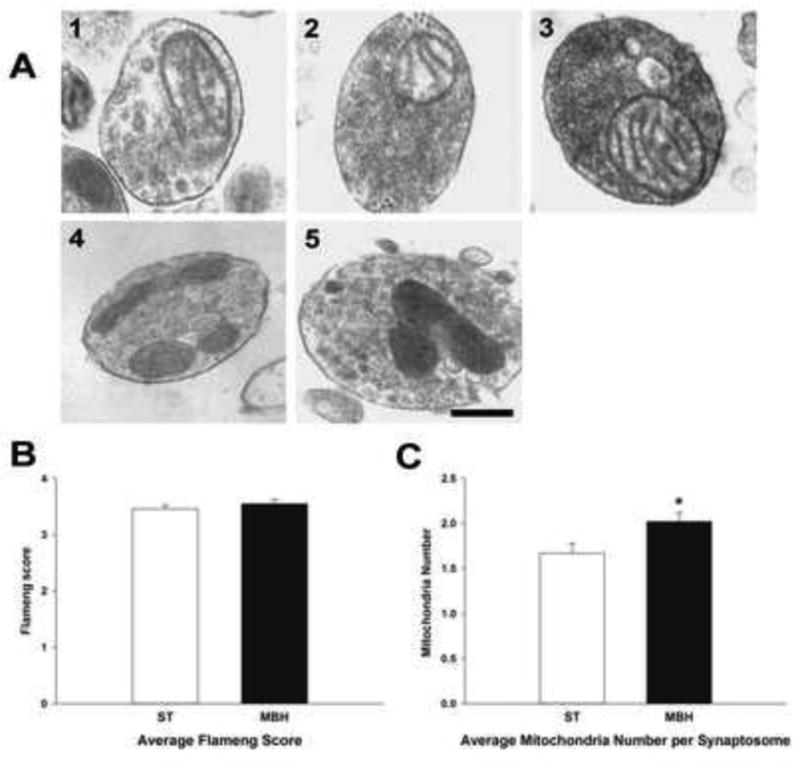
A; Photomicrographs depicting representative Flameng mitochondrial scores of ST-derived synaptosomes. Score 1: broken cristae with ruptured mitochondrial membrane, Score 2: broken cristae with matrix clearing but membrane intact, Score 3: swelled mitochondria with cleared matrix but intact cristae and membrane, Score 4: intact mitochondria, Score 5: intact mitochondria with mitochondrial granules. (Scale bar = 50 μm). Average Flameng mitochondrial scores (B) and average mitochondria number per synaptosome (C) of ST-derived (open columns) and MBH-derived (closed columns) synaptosomes. Synaptosomes were fixed, sectioned and at least 200 mitochondria from randomly selected images were analyzed. Mean mitochondrial functional score was calculated by averaging Flameng mitochondrial functional scores from at least 200 mitochondria from each experimental group. Mean number of mitochondria per synaptosome was derived by dividing total number of mitochondria by total number of synaptosomes in each experimental group. The experiment was repeated four times with + 1 standard error of the mean. * indicates mean number of mitochondria per synaptosomes in the MBH that were significantly different (p<0.05) from those in the ST.
There was no difference in the average Flameng score of mitochondria from the ST and MBH (Figure 6B), but the average numbers of mitochondria per synaptosome were higher in the MBH as compared with the ST (Figure 6C). These results directly correlate with observations of increased mass and membrane potentials of mitochondria in synaptosomes derived from the MBH, and suggest that greater maximum and spare respiratory capacities may be due to increased numbers of viable mitochondria in axons terminating in this MBH versus the ST.
3. Discussion
The results from the present study provide correlative evidence that mitochondrial quality control may underlie differences in mitochondrial function amongst distinct central DA neurons. Reduced aerobic mitochondrial capacity is correlated with diminished mitochondrial mass in synaptosomes derived from brain regions containing axon terminals of NSDA neurons, and mitophagy in perikarya of these neurons as compared with TIDA neurons. The lower numbers of viable mitochondria in NSDA neurons is correlated with limited mitochondrial autophagic maintenance capacity.
3.1 Characteristics of synaptosomal mitochondria
In vivo studies offer an opportunity to assess mitochondrial function in the intact nervous system. The heterogeneity of cells within brain parenchyma, however, constrains the interpretation of mitochondrial bioenergetic respiratory end points. Brain tissue samples contain a variety of non-neuronal components including glial, immune, and endothelial cells, as well as phenotypically distinct neuronal cell bodies and axon terminals. As an alternative to whole brain tissue samples, synaptosomes derived from brain tissue homogenates offer a platform that is enriched with neuronal axon components. During homogenization, axon terminal membranes pinch off forming synaptosomes that contain cytosolic organelles including mitochondria, pre-synaptic machinery for neurotransmitter storage, release, re-uptake and metabolism (Nicholls, 2003; Dunkley et el., 2008). In the present study, synaptosome preparations from both the ST and MBH were found to contain DA, but not its metabolite HVA. Since HVA in brain is generated by catechol-o-methyl transferase located in glial cells (Huotari et al., 2002), absence of HVA is consistent with the conclusion that synaptosomes isolated in these experiments are unlikely to have appreciable glia contamination (Barrett et al, 2012). These results are congruent with those of a previous report showing that 84% of synaptosomes are of neuronal origin, while only 5-10% are derived from glia (Wolf and Kapatos, 1989).
NSDA neurons originating in the SNpc provide major afferent neuronal innervation to the ST and represent major contributors to the mitochondria derived from the ST synaptosomes (Smith et al., 1994; Moss and Bolam, 2008; Lehéricy et al., 2012). ST synaptosomes have been reported to be predominately comprised of pre-synaptic NSDA axon terminals (Barrett et al, 2012). Accordingly, measurements of mitochondrial respiration in the present study reflect, in large part, bioenergetics, mass, and membrane potential of mitochondria contained within NSDA axon terminals. Synaptosomes derived from the MBH contain axon terminals of TIDA neurons and release DA in response to depolarizing stimuli (Gregerson and Selmanoff, 1987). Nevertheless, differences in mitochondrial indices could be due to the presence of non-DA axon terminals. To complement axon terminal measurements, mitochondrial indices were also obtained specifically in TH immunoreactive neuronal perikarya in the SNpc and ARC using in situ confocal microscopy analysis. The results of these latter experiments are in agreement with data derived from synaptosomes and provide corroborative evidence that changes observed in synaptosomes reflect changes in NSDA and TIDA axon terminals.
3.2 Distinct characteristics of ST- and MBH-derived synaptosomal mitochondria
Under basal conditions, indices of mitochondrial respiration (OCR) and glycolysis (ECAR) were similar in synaptosomes derived from the ST and MBH, despite regional differences in mitochondrial mass (MitoTracker Green), membrane potential (TMRE) and numbers of mitochondria (ultrastructural analysis). Although there is a significant difference in mitochondrial membrane potentials between the ST- and MBH-derived synaptosomes, this could be explained by similar differences in mitochondrial mass. There was also no difference in the overall quality of individual mitochondrion contained within synaptosomes (ultrastructural analysis) or the responsiveness of these mitochondria to Complex I inhibition (estimated IC50 for rotenone). These results indicate that under basal conditions, i.e., in the absence of metabolic or oxidative stress, the respiratory performance of the existing viable mitochondria in ST synaptosomes is comparable to that of MBH synaptosomes.
On the other hand, when mitochondrial respiratory performance was challenged following dissipation of the proton gradient with FCCP, synaptosomes derived from the ST had lower maximum OCR as compared with the MBH. Accordingly, the maximum respiratory capacity and the calculated (maximum minus basal) spare respiratory capacity were both lower in ST versus MBH. Lower mitochondrial maximum respiratory capacity in ST synaptosomes is likely due to lower numbers or size of mitochondrion. Flow cytometric results showing both lower mitochondrial mass and membrane potential in ST versus MBH synaptosomes are consistent with this conclusion and suggest that axon terminals of NSDA neurons contain fewer active polarized mitochondria than TIDA neurons.
3.3 Mitochondrial density and autophagic maintenance in NSDA and TIDA neurons
To determine if perikarya of NSDA and TIDA neurons show similar differences in mitochondrial density as demonstrated for synaptosomes containing axon terminals of these neurons, confocal microscopic analysis of the extent of co-localization of the mitochondrial marker COX IV in TH immunoreactive neurons was performed. The results reveal a corresponding higher mitochondrial density in TIDA neurons in the ARC compared with NSDA neurons in the SNpc, which is consistent with the increased mitochondrial mass observed in MBH versus ST synaptosomes.
Numbers of viable mitochondria are maintained in cells by continuous fusion, fission, and mitophagy (Green and Houten, 2011; Youle and Narendra, 2011; Youle and van der Bleik, 2012). In neurons, an unhealthy mitochondrion undergoes fission or fusion with a healthy mitochondrion, which distributes nutrients and facilitates repair of damaged mitochondrial DNA (Chen and Chan, 2009). However, if a mitochondrion is irreparably damaged, either the entire organelle or the damaged portion of the mitochondrion may be segregated and targeted for mitophagy, which occurs normally as a maintenance and oxidative stress-induced compensatory process (Sugiura, McLelland et al. 2014) (Goldman et al., 2010).
In the present study, confocal microscopic analyses of the extent of co-localization of the autophagosome marker LC3 and mitochondrial marker COX IV (Pearson Coefficient) was 3-fold lower in TH immunoreactive neurons in the SNpc as compared with those in the ARC. These results correlate with a lower density of COX IV labeled mitochondria in TH immunoreactive neurons in the SNpc versus the ARC, and suggest that NSDA neurons have less autophagic elimination of damage mitochondria than TIDA neurons.
4. Conclusions
The results presented here represent novel findings that illustrate differences between NSDA and TIDA neurons with respect to the numbers of viable mitochondria and extent of ongoing mitophagy under basal conditions. Lower numbers of mitochondria in NSDA neurons, in turn, likely account for the impaired evoked maximum respiratory responses of mitochondria in ST synaptosomes following perturbation of the electron transport chain proton gradient. Due to the location of axon terminals outside the blood-brain barrier and increased exposure to substances that cause oxidative stress, TIDA neurons may have developed a robust mitophagic capacity to maintain numbers of viable mitochondria in the context of frequent toxicant and metabolic stress. Further investigation into the mechanisms underlying the unique ability of TIDA neurons to recover from injury may reveal novel targets for neuroprotective therapy development for diseases like PD.
5. Experimental Procedures
5.1 Animals
C57BL/6J male WT mice were obtained from Jackson Laboratories. GFP-LC3 transgenic mice (RIKEN Bio-Resource Center, #BRC00806) were bred in house and used between 20 – 24 weeks of age. Mice were housed in a room with a 12-h light/dark cycle, with food and water provided ad libitum. Michigan State University Institutional Animal Care & Use Committee approved all the experiments using live animals (AUF 01/14-001-00).
5.2 Perfusion, fixation and sectioning of mouse brains
Mice were anesthetized with a lethal dose of ketamine:xylazine (26.6 mg/kg : 4 mg/kg; i.p.) and perfused with 0.09% saline followed by 4% paraformaldehyde via a transcardial approach. After perfusion, mice were decapitated and the brains were removed. The whole brains were stored in 4% paraformaldehyde solution overnight and transferred into 20% sucrose in 0.1 M phosphate buffered saline for an additional day. Twenty μm sections were prepared using a cryostat at -20°C. Sections containing the SNpc and MBH were collected and immunohistochemistry was performed.
5.3 TH/COX IV immunohistochemistry
The sections were washed with phosphate buffer (PB) followed by 0.05M tris-buffered saline containing 1% Triton-X (TBS-TX, pH 7.4). The sections were blocked with 5% bovine serum albumin for 1 h and incubated with 1:500 COX IV (Santa Cruz, sc-69360, goat monoclonal) and 1:2000 TH (Millipore, Ab152, rabbit monoclonal) in TBS-TX overnight. Sections were washed with PB-TX and incubated with secondary antibodies 1:500 Alexa Fluor 594 donkey anti-goat IgG (Invitrogen, A11058) and Alexa Fluor 405 donkey anti-rabbit IgG (Abcam, ab175649 or ab175651) for 1 h at room temperature. After rinsing with PB, sections were mounted on glass slides with Prolong Gold antifade reagent (Invitrogen).
5.4 Fluorescent confocal microscopic analysis
COX IV was measured using an Alexa Fluor 594 filter set at 575 to 675 nm, GFP-LC3 using a EGFP filter set at 500 to 545 nm, and TH using a Alexa Fluor 405 filter set at 425 to 475nm on a Olympus Fluoview FV1000 confocal microscope. Hi/Lo settings were used to make certain that the fluorescence intensities recorded were within the detectable range of the photomultiplier tube detector, thus assuring accurate quantitative measurements. The detector voltage and offset were adjusted so that the fluorescence intensities recorded were below the detector saturation and above the detector lower limit (zero). The Hi/Low settings were used to set background levels for TH (threshold of 1209) and COX IV (threshold of 1000). Percent mitochondrial distribution in TH immunoreactive cells was calculated by dividing the number of pixels co-localized with TH and COX IV by TH immunoreactive pixels. The co-localization of mitochondria and TH immunoreactive neurons are analyzed using FV1000 ASW software. Mitochondria co-localized with autophagosomes in TH immunoreactive neurons were analyzed by Fiji coloc_2 with Image J (Rasband, W.S., ImageJ, U. S. National Institutes of Health, Bethesda, Maryland, USA, http://imagej.nih.gov/ij/, 1997-2015). TH immunoreactive neurons were masked by converting TH immunoreactive signal as 225 and background as 0 and only TH immunoreactive pixels were selected for co-localization analysis.
Co-localization was analyzed by comparison of Pearson Coefficients measured by Fiji Coloc_2 (Pompey et al., 2013). Point spread function (PSF) was estimated using equation; resolution = 0.61 λ/NA (Cole et al., 2011), where λ was 450 for Alexa Fluor 405, 522.5 for EGFP, and 625 for Alexa Fluor 594. NA is the numerical aperture of the objective (NA of 10× objective = 0.40; NA of oil-immersion 40× objective = 1.30; NA of oil-immersion 60× = 1.42). A blinded experimenter collected and analyzed the images.
5.5 Synaptosome isolation
After decapitation, ST and MBH were dissected on ice and synaptosomes were isolated from ST and MBH by the method of Dunkley with some modifications (Dunkley et al., 2008; Anderson and Sims, 2008). Dissected brain regions were collected in mitochondrial isolation buffer (300 mM sucrose, 10 mM HEPES, 1 mM EGTA, pH 7.4 at 4°C) and homogenized using 10 strokes of a pre-chilled Teflon/glass homogenizer on ice (Gray and Whittaker, 1962). The homogenates were transferred to 15-ml polycarbonate tubes and centrifuged at 21,000 g at 4 °C for 10 min in a fixed-angle rotor. The hard pellets were re-suspended in 15% Percoll and layered onto a gradient consisting of 2 ml of 23% and 2 ml of 40% Percoll in mitochondrial isolation buffer. The Percoll gradients were centrifuged at 26,000 g at 4 °C for 10 min. Synaptosomes were collected from the second band and washed with mitochondrial isolation buffer and sucrose buffer (300 mM sucrose and 10 mM HEPES, pH 7.4 at 4°C) by centrifugation and stored in sucrose buffer. The protein content of individual synaptosome samples was quantified using a bicinchoninic acid (BCA) assay (Noble and Bailey, 2009).
5.6 Synaptosome plating
Aliquots (10-15 μl) of sucrose buffer containing 20 μg of synaptosomal protein were added to wells of a poly-D-lysine coated Seahorse XF 24 plate (Seahorse Bioscience, North Bilerica, MA) containing a final volume of 600 μl ionic medium (20 mM HEPES, 10 mM D-Glucose, 1.2mM Na2HPO4, 1 mM MgCl2, 5 mM NaHCO3, 5 mM KCl, 140 mM NaCl, pH 7.4 at 4°C), and centrifuged at 2,500 g for 1 h at 4°C in swinging bucket rotor in a Fisher Scientific 3000R centrifuge. Synaptosomes attached to the Seahorse plates were incubated at 37°C in 600 μl incubation medium (3.5 mM KCl, 120 mM NaCl, 1.3 mM CaCl2, 0.4 mM KH2PO4, 1.2 mM Na2SO4, 2 mM MgSO4, 4 mg/ml BSA, 15 mM D-glucose, 5 mM pyruvate, 2.5 mM malate, pH 7.4 at 37°C).
5.7 Preparation of the Seahorse XF 24 cartridges
XF 24 cartridges (Seahorse Bioscience) were hydrated by incubation with 1 ml calibrant (Seahorse Bioscience) per well at 37°C overnight. Before loading a cartridge onto the XF 24 analyzer, 72 μl of 30 μM oligomycin, 80 μl of 30 μM FCCP, 89 μl of 25 μM rotenone (XF Cell Mito Stress Kit, Seahorse Bioscience, 101706-100) were added onto the cartridges (final concentrations of oligomycin, FCCP, and rotenone injected during the assay were 3μM, 3μM and 2.5 μM, respectively) and incubated for 10 min at 37°C. The cartridges were loaded onto the XF 24 analyzer for calibration.
5.8 Mitochondrial respiratory assay by Seahorse XF analyzer
Mitochondrial OCR was measured in micro-chambers created by immersing the fluorescent probes onto the sensor in the XF 24 well plate. The micro-chambers were re-mixed after each measurement cycle by emersion and immersion of the probes to reinstate initial oxygen concentrations for each cycle. OCR and ECAR reflect the mean rates of the measurement cycle. Appropriate mixing and measurement time were pre-set and operated following manufacturer's instructions.
5.9 Ex vivo rotenone treatment of synaptosomal mitochondria
Prior to OCR measurements, 75 μl of 1 mM rotenone (Sigma, Cat. No R8875-1G, MW 394.42) was added into 3 ml incubation buffer for a final concentration of 25 μM rotenone. OCR were measured beginning 5 min after treatment with either vehicle (incubation buffer) or rotenone (0.0125, 0.25, or 2 μM). OCR measurements were performed using a Seahorse XF24 analyzer, with 20 μg of synaptosomal protein derived from each region yielding a respiratory control ratio' (RCR') of greater than 3.
5.10 Mitochondrial mass and membrane potential quantification by flow cytometry
Twenty μg of isolated synaptosomes were added to 1 mL of staining buffer containing 200 nM MitoTracker™ Green (Invitrogen, cat. no. M-7514) or 500 nM tetramethylrhodamine ethyl ester perchlorate (TMRE, Sigma-Aldrich Cat. No. 87917) in KCl assay buffer (125 mM KCl, 20 mM HEPES, 4 mM MgCl2 and 2 mM KH2PO4, pH 7.4) supplemented with 5 mM pyruvate and 2.5 mM malate, and immediately incubated for 5 min at room temperature. Two μM FCCP was used as a positive control for mitochondrial depolarization. The membrane potential of isolated synaptosomal mitochondria was analyzed with FACS analyzer (BD Biosciences). The MitoTracker™ Green positive mitochondria were measured using the fluorescein or FITC channel (excitation at 488 nm and emission measured at 520 nm) and TMRE positive mitochondria were measured using the R-phycoerythrin or PE channel (excitation at 488 nm and emission measured at 575 nm). The FACS data were analyzed using FlowJo software (Tree Star, Ashland, OR).
5.11 Transmission electron microscopy of synaptosomal mitochondria
Isolated synaptosomes were fixed in 2% glutaraldehyde and 4% paraformaldehyde in KCl mitochondria buffer (125 mM KCl, 20 mM HEPES, 4 mM MgCl2, 2 mM KH2PO4, pH 7.4) for 1 h at room temperature. Samples were washed with 0.1 M sodium cacodylate buffer (pH 7.4) and post fixed for 30 min with 1% OsO4 in ddH2O. Samples were treated with 0.2% tannic acid in ddH2O for 10 min and washed in ddH2O. After the last wash, samples were dehydrated in a graded acetone series, infiltrated and embedded in Spurr's resin, and cured at 65°C for approximately 48 h (Spurr, 1969). Samples were trimmed and sectioned into approximately 85 nm thin sections (or to a silver-gold color), collected on copper grids, stained with 0.2% Reynold's lead citrate and 2% uranyl acetate, and imaged with a JEOL100 CXII transmission electron microscope (Reynolds, 1963). A rastering pattern was used to image synaptosomes within a random grid opening and analyses of the images using magnification ranging from 40,000 to 72,000 × were completed by an investigator blinded to the experimental groups.
5.12 High performance liquid chromatography - electrochemical detection (HPLC-ED)analysis
Frozen synaptosomes were thawed and sonicated with 3 one-sec bursts (Sonicator Cell Disruptor, Heat Systems-Ultrasonic, Plainview, NY, USA). The samples were centrifuged at 18,000 rpm for 5 min and 50 μl of the supernatants were collected with addition of tissue buffer to a final volume of 65 ul. Content of DA in the supernatants was measured by HPLC-ED (Lindley et al., 1990), the detector potential set at -0.40 V and the mobile phase was composed of 20% methanol, pH of 2.64, containing 0.020-0.035% sodium octyl sulfate for optimal resolution. DA and HVA contents in the ST- and MBH-derived synaptosomes were determined by comparison of peak height values (Hewlett Packard Integrator, Model 3395) with those of external standards. Quantification of the protein content of the pellet samples was attained using a BCA protein assay. DA and HVA were normalized to protein content and expressed as a concentration of ng per mg protein.
5.13 Statistical analyses and experimental rigor
One-way analysis of variance (ANOVA) tests were used to make statistical comparison among three or more groups with single independent variable using SigmaPlot 12.0. Two way ANOVA equal variance test was performed to compare the samples with three or more independent variables and Tukey's or Holm-Sidak test were utilized for all pairwise multiple comparison tests. Multiple non-parametric groups were statistically analyzed by Kruskal Wallis multiple comparison test or converted to parametric value by equation ASIN(SQRT(% value/100)). For comparing the two parametric groups with equal variance, a student's t-test was performed using a two-tailed distribution. The statistical difference with probability of error of 0.05 or less was considered statistically significant.
Power analyses were performed a priori using expected inter-group difference of 23% for ANOVA and an α of 0.05. Sample size of 5 yields a power of 0.98 for the mitochondrial bioenergetics end points. A sample size of 3 for flow cytometric analysis yields a power of 1.0 with 20% group difference, while a sample size of 4 for transmission electron microscopy analysis yields power of 0.81 with 18% group difference. Investigators performing endpoint assays for flow cytometry, transmission electron microscopy and confocal microscopy were blinded to the experimental treatment groups.
Highlights.
There are more mitophagosomes in tuberoinfundibular than nigrostriatal DA neurons.
Lower maximum respiration in striatal versus mediobasal hypothalamic synaptosomes.
Lower mitochondrial mass in striatal synaptosomes.
Acknowledgments
This work was supported by the National Institute of Health grant R01 NS065338-01A1. The authors would like to thank Dr. Norbert Kaminski and Robert Crawford for providing access to the flow cytometry facility, Dr. Melinda Frame for providing access to the confocal microscope facility, and Theresa Lansdell for her editorial help in preparation of the manuscript.
Footnotes
Conflict of Interest: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahlskog JE. Parkinson's disease: medical and surgical treatment. Neurol Clin. 2001;19(3):579–605. doi: 10.1016/s0733-8619(05)70036-0. [DOI] [PubMed] [Google Scholar]
- Albin RL, Young AB, Penney JB. The functional anatomy of basal ganglia disorders. Trends in Neurosciences. 1989;12:366–75. doi: 10.1016/0166-2236(89)90074-x. [DOI] [PubMed] [Google Scholar]
- Anderson MF, Sims NR. Isolation of mitochondria from rat brain using Percoll density gradient centrifugation. Nature Protocols. 2008;3:1228–39. doi: 10.1038/nprot.2008.105. [DOI] [PubMed] [Google Scholar]
- Barrett KE, Barman SM, Boitano S, Brooks HL. Ganon's review of medical physiology. 24th. McGraw-Hill; 2012. p. 146. [Google Scholar]
- Behrouz B, Drolet RE, Sayed ZA, Lookingland KJ, Goudreau JL. Unique responses to mitochondrial complex I inhibition in tuberoinfundibular dopamine neurons may impart resistance to toxic insult. Neuroscience. 2007;147:592–98. doi: 10.1016/j.neuroscience.2007.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benskey M, Behrouz B, Sunryd J, Pappas SS, Baek SH, Huebner M, Lookingland KJ, Goudreau JL. Recovery of hypothalamic tuberoinfundibular dopamine neurons from acute toxicant exposure is dependent upon protein synthesis and associated with an increase in parkin and ubiquitin carboxy-terminal hydrolase-L1 expression. Neurotoxicology. 2012;33:321–31. doi: 10.1016/j.neuro.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benskey M, Lee KY, Parikh K, Lookingland KJ, Goudreau JL. Sustained resistance to acute MPTP toxicity by hypothalamic dopamine neurons following chronic neurotoxicant exposure is associated with sustained up-regulation of parkin protein. Neurotoxicology. 2013;37C:144–153. doi: 10.1016/j.neuro.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Braak E. Pathoanatomy of Parkinson's disease. J Neurol. 2000;247:3–10. doi: 10.1007/PL00007758. [DOI] [PubMed] [Google Scholar]
- Chen H, Chan DC. Mitochondrial dynamics--fusion, fission, movement, and mitophagy—in neurodegenerative diseases. Human Mol Genet. 2009;18(R2):R169–76. doi: 10.1093/hmg/ddp326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SW, Gerencser AA, Lee DW, Rajagopalan S, Nicholls DG, Andersen JK, Brand MD. Intrinsic Bioenergetic Properties and Stress Sensitivity of Dopaminergic Synaptosomes. Journal of Neuroscience. 2011;31:4524–4534. doi: 10.1523/JNEUROSCI.5817-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole RW, Jinadasa T, Brown CM. Measuring and interpreting point spread functions to determine confocal microscope resolution and ensure quality control. Nature Protocols. 2011;6(12):1929–41. doi: 10.1038/nprot.2011.407. [DOI] [PubMed] [Google Scholar]
- Connolly BS, Lang AE. Pharmacological treatment of Parkinson disease: a review. JAMA. 2014;311(16):1670–1683. doi: 10.1001/jama.2014.3654. [DOI] [PubMed] [Google Scholar]
- Cook C, Stetler C, Petrucelli L. Disruption of protein quality control in Parkinson's disease. Cold Spring Harb Perspect Med. 2012;2(5):a009423. doi: 10.1101/cshperspect.a009423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cookson J, Hodgson R, Wildgust HJ. Prolactin, hyperprolactinemia and antipsychotic treatment: a review and lessons for treatment of early psychosis. J Psychopharmacol. 2012;26(5):42–51. doi: 10.1177/0269881112442016. [DOI] [PubMed] [Google Scholar]
- Davison EJ, Pennington K, Hung CC, Peng J, Rafiq R, Ostareck-Lederer A, Ostareck DH, Ardley HC, Banks RE, Robinson PA. Proteomic analysis of increased Parkin expression and its interactants provides evidence for a role in modulation of mitochondrial function. Proteomics. 2009;9(18):4284–97. doi: 10.1002/pmic.200900126. [DOI] [PubMed] [Google Scholar]
- Dunkley PR, Jarvie PE, Robinson PJ. A rapid Percoll gradient procedure for preparation of synaptosomes. Nature Protocols. 2008;3:1718–28. doi: 10.1038/nprot.2008.171. [DOI] [PubMed] [Google Scholar]
- Flameng W, Borgers M, Daenen W, Stalpaert G. Ultrastructural and cytochemical correlates of myocardial protection by cardiac hypothermia in man. J Thorac Cardiovasc Surg. 1980;79:413–24. [PubMed] [Google Scholar]
- Ford CP. The role of D2-autoreceptors in regulating dopamine neuronal activity and transmission. Neuroscience. 2014;282:13–22. doi: 10.1016/j.neuroscience.2014.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gohil VM, Sheth SA, Nilsson R, Wojtovich AP, Lee JH, Perocchi F, Chen W, Clish CB, Ayata C, Brookes PS, Mootha VK. Nutrient-sensitized screening for drugs that shift energy metabolism from mitochondrial respiration to glycolysis. Nat Biotech. 2010;28:249–255. doi: 10.1038/nbt.1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman SJ, Taylor R, Zhang Y, Shengkan J. Autophagy and the degradation of mitochondria. Mitochondrion. 2010;10(4):309–15. doi: 10.1016/j.mito.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo M. What have we learned from Drosophila models of Parkinson's disease? Prog Brain Res. 2010;184:3–16. doi: 10.1016/S0079-6123(10)84001-4. [DOI] [PubMed] [Google Scholar]
- Gray E, Whittaker V. Isolation of nerve endings from brain - an electron-microscopic study of cell fragments derived by homogenization and centrifugation. J Anat. 1962;96:79–88. [PMC free article] [PubMed] [Google Scholar]
- Green DR, Van Houten B. Mitochondrial quality control. Cell. 2011;147:950–950. doi: 10.1016/j.cell.2011.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregerson KA, Selmanoff M. Rapid release of [3H]dopamine from median eminence and striatal synaptosomes. J Neurochem. 1987;48(4):1222–30. doi: 10.1111/j.1471-4159.1987.tb05650.x. [DOI] [PubMed] [Google Scholar]
- Haugland RP. Handbook of Fluorescent Probes and Research Chemicals. 6th. Eugene, OR: Molecular Probes Inc; 1996. [Google Scholar]
- Heo JM, Rutter J. Ubiquitin-dependent mitochondrial protein degradation. Int J Biochem Cell Biol. 2011;43(10):1422–6. doi: 10.1016/j.biocel.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huotari M, Santha M, Lucas LR, Karaylorgou M, Gogos JA, Mannisto PT. Effect of dopamine uptake inhibition on brain catecholamine levels and locomotion in catechol-O-methyltransferase-disrupted mice. J Pharmacol Exp Ther. 2002;303(3):1309–16. doi: 10.1124/jpet.102.043042. [DOI] [PubMed] [Google Scholar]
- Jellinger Kurt A. Pathology of Parkinson's Disease. Molecular and Chemical Neuropathology. 1991;14(3):153–97. doi: 10.1007/BF03159935. [DOI] [PubMed] [Google Scholar]
- Kaasik A, Safiulina D, Zharkovsky A, Veksler V. Regulation of mitochondrial matrix volume. Am J Physiol Cell Physiol. 2007;292:C157–C163. doi: 10.1152/ajpcell.00272.2006. [DOI] [PubMed] [Google Scholar]
- Koh H, Chung J. PINK1 as a molecular checkpoint in the maintenance of mitochondrial function and integrity. Mol Cells. 2012;34(1):7–13. doi: 10.1007/s10059-012-0100-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langston J William, Forno Lysia S. The Hypothalamus in Parkinson Disease. Annals of Neurology. 1978;3(2):129–33. doi: 10.1002/ana.410030207. [DOI] [PubMed] [Google Scholar]
- Lehéricy S, Sharman MA, Santos CLD, Paquin R, Gallea C. Magnetic resonance imaging of the substantia nigra in Parkinson's disease. Movement Disorders. 2012;27:822–830. doi: 10.1002/mds.25015. [DOI] [PubMed] [Google Scholar]
- Lemasters JJ, Ramshesh VK. Imaging of mitochondrial polarization and depolarization with cationic fluorophores. Methods in Cell Bio. 2007;80:283–95. doi: 10.1016/S0091-679X(06)80014-2. [DOI] [PubMed] [Google Scholar]
- Liang CL, Wang TT, Luby-Phelps K, German DC. Mitochondria mass is low in mouse substantia nigra dopamine neurons: Implications for Parkinson's disease. Experimental Neurology. 2007;203:370–80. doi: 10.1016/j.expneurol.2006.08.015. [DOI] [PubMed] [Google Scholar]
- Lindley SE, Gunnet JW, Lookingland KJ, Moore KE. 3,4-Dihydroxyphenylacetic acid concentrations in the intermediate lobe and neural lobe of the posterior pituitary gland as an index of tuberohypophysial dopaminergic neuronal activity. Brain Research. 1990;506(1):133–8. doi: 10.1016/0006-8993(90)91209-y. [DOI] [PubMed] [Google Scholar]
- Lookingland KJ, Moore KE. Functional neuroanatomy of hypothalamic dopaminergic neuroendocrine systems. Handbook of Chemical Neuroanatomy. 2005;21:435–523. Chapter 8. [Google Scholar]
- Lowry JP, Fillenz M. Evidence for uncoupling of oxygen and glucose utilization during neuronal activation in rat striatum. J Physiol. 1997;498:497–501. doi: 10.1113/jphysiol.1997.sp021875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matzuk Martin M, Saper Clifford B. Preservation of Hypothalamic Dopaminergic Neurons in Parkinson's Disease. Annals of Neurology. 1985;18(5):552–55. doi: 10.1002/ana.410180507. [DOI] [PubMed] [Google Scholar]
- Mizuno Y, Ohta S, Tanaka M, Takamiya S, Suzuki K, Sato T, Oya H, Ozawa T, Kagawa Y. Deficiencies in Complex I subunits of the respiratory chain in Parkinson's disease. Biochemical and Biophysical Research Communications. 1989;163:1450–55. doi: 10.1016/0006-291x(89)91141-8. [DOI] [PubMed] [Google Scholar]
- Moss J, Bolam JP. A dopaminergic axon lattice in the striatum and its relationship with cortical and thalamic terminals. J Neurosci. 2008;28:11221–230. doi: 10.1523/JNEUROSCI.2780-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls DG. Bioenergetics and transmitter release in the isolated nerve terminal. Neurochem Res. 2003;28(10):1433–1441. doi: 10.1023/a:1025653805029. [DOI] [PubMed] [Google Scholar]
- Noble JE, Bailey MJA. Quantitation of protein. In: Burgess RR, Deutscher MP, editors. Guide to Protein Purification. 2nd. Elsevier Academic Press Inc; San Diego: 2009. pp. 73–95. [Google Scholar]
- Pendergrass W, Wolf N, Poot M. Efficacy of MitoTracker Green™ and CMXrosamine to measure changes in mitochondrial membrane potentials in living cells and tissues. Cytometry Part A. 2004;61A:162–169. doi: 10.1002/cyto.a.20033. [DOI] [PubMed] [Google Scholar]
- Pompey SN, Michaely P, Luby-Phelps K. Quantitative Fluorescence Colocalization to Study Protein–Receptor Complexes. In: Williams MA, Daviter T, editors. Protein-Ligand Interactions. Humana Press; 2013. pp. 439–453. [DOI] [PubMed] [Google Scholar]
- Reynolds ES. The use of lead citrate at high pH as an electron-opaque stain in electron microscopy. J Cell Biol. 1963;17:208–212. doi: 10.1083/jcb.17.1.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandyk R, Iacono RP, Bamford CR. The Hypothalamus in Parkinson Disease. The Italian Journal of Neurological Sciences. 1987;8(3):227–234. doi: 10.1007/BF02337479. [DOI] [PubMed] [Google Scholar]
- Schapira AHV, Cooper JM, Dexter D, Jenner P, Clark JB, Marsden CD. Mitochondrial complex I deficiency in Parkinson's disease. The Lancet. 1989;333:1269. doi: 10.1016/s0140-6736(89)92366-0. [DOI] [PubMed] [Google Scholar]
- Smith Y, Bennett BD, Bolam JP, Parent A, Sadikot AF. Synaptic relationships between dopaminergic afferents and cortical or thalamic input in the sensorimotor territory of the striatum in monkey. J Comp Neurol. 1994;344:1–19. doi: 10.1002/cne.903440102. [DOI] [PubMed] [Google Scholar]
- Spurr A. A low-viscosity epoxy resin embedding medium for electron microscopy. J Ultrastructure Res. 1969;26:31–43. doi: 10.1016/s0022-5320(69)90033-1. [DOI] [PubMed] [Google Scholar]
- Sugiura A, McLelland GL, Fon EA, McBride HM. A new pathway for mitochondrial quality control: mitochondrial-derived vesicles. EMBO J. 2014;33(19):2142–56. doi: 10.15252/embj.201488104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulzer D, Surmeier DJ. Neuronal vulnerability, pathogenesis, and Parkinson's disease. Mov Disord. 2013;28(6):715–24. doi: 10.1002/mds.25187. [DOI] [PubMed] [Google Scholar]
- Sun X, Liao NK, Yu JJ. Prognostic Value of a Mitochondrial Functional Score in Prostate Cancer. J Int Med Res. 2012;40:371–76. doi: 10.1177/147323001204000139. [DOI] [PubMed] [Google Scholar]
- Tanaka A. Parkin-mediated selective mitochondrial autophagy, mitophagy: Parkin purges damaged organelles from the vital mitochondrial network. FEBS Lett. 2010;584:1386–92. doi: 10.1016/j.febslet.2010.02.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor EB, Rutter J. Mitochondrial quality control by the ubiquitin-proteasome system. Biochem Soc Trans. 2011;39(5):1509–13. doi: 10.1042/BST0391509. [DOI] [PubMed] [Google Scholar]
- Vaughan RA, Foster JD. Mechanisms of dopamine transporter regulation in normal and disease states. Trends Pharmacol Sci. 2013;34(9):489–96. doi: 10.1016/j.tips.2013.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf ME, Kapatos G. Flow cytometric analysis of rat striatal nerve terminals. J Neurosci. 1989;9(1):94–105. doi: 10.1523/JNEUROSCI.09-01-00094.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yelnik J, Francois C, Percheron G. Spatial relationship between striatal axonal endings and pallidal neurons in macaque monkeys. Adv Neurol. 1997;74:45–56. [PubMed] [Google Scholar]
- Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. 2011;12:9–14. doi: 10.1038/nrm3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle RJ, van der Bliek AM. Mitochondrial fission, fusion and stress. Science. 2012;337(6098):1062–65. doi: 10.1126/science.1219855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamzami N, Metivier D, Kroemer G. Quantitation of mitochondrial transmembrane potential in cells and in isolated mitochondria. Methods in Enzymology. 2000;322:208–213. doi: 10.1016/s0076-6879(00)22021-1. [DOI] [PubMed] [Google Scholar]