

Abstract
Cellular inhibitor of apoptosis proteins (c-IAP) 1 and 2 are widely expressed ubiquitin protein ligases that regulate a variety of cellular functions, including the sensitivity of T cells to co-stimulation. 4-1BB is a TNFR family member that signals via a complex that includes TRAF family members and the c-IAPs to upregulate NF-κB and ERK, and has been implicated in memory T-cell survival. Here we show that effector and memory T cells from mice expressing a dominant negative E3-inactive c-IAP2 (c-IAP2H570A) have impaired signaling downstream of 4-1BB. When infected with LCMV, unlike mice in which c-IAPs were acutely downregulated by c-IAP antagonists, the primary response of c-IAP2H570A mice was normal. However, the number of antigen-specific CD8+ but not CD4+ T cells declined more rapidly and to a greater extent in c-IAP2H570A mice than in wild type (WT) controls. Studies with T-cell adoptive transfer demonstrated that the enhanced decay of memory cells was T-cell-intrinsic. Thus, c-IAP E3 activity is required for 4-1BB co-receptor signaling and maintenance of CD8+ T-cell memory.
Keywords: signal transduction, ubiquitination, T-cell memory, LCMV, 4-1BB, CD8+ memory T cell
Introduction
Cellular inhibitor of apoptosis protein 1 and 2 (c-IAP1 and c-IAP2) are part of a phylogenetically-conserved family of proteins whose molecular signature is the presence of one or more Baculovirus IAP Repeat (BIR) domains mediating protein-protein interactions. c-IAPs have a C-terminal RING domain that confers ubiquitin protein ligase (E3) activity [1], which has been found to be involved in several signaling pathways. One example is their role in Tumor Necrosis Factor α (TNF-α) signaling via TNF receptor (TNFR) 1, in which RIP1 is ubiquitinated by c-IAP1- and c-IAP2 [2–6]. The most widely appreciated activity of c-IAPs is their role in regulating the activation of the Nuclear Factor for κB (NF-κB) family of transcription factors. NF-κB, which exists as a homo/heterodimer composed of components of a five member family, regulates many cellular processes involved in growth and survival. NF-κB can be activated by two mechanisms. The majority of the NF-κB-activating receptors, such as those for cytokines and growth factors, use the canonical pathway in which the NF-κB-binding molecule IκB is phosphorylated and degraded, allowing NF-κB to move from the cytosol to the nucleus [7]. Activation of the non-canonical pathway by a limited number of receptors, on the other hand, results in stabilization of NF-κB-inducing kinase (NIK) and proteolytic processing of the p100 NF-κB family member to the transcriptionally active fragment, p52 [8]. Both of these pathways rely upon ubiquitination by c-IAP, followed by proteasomal degradation (IκB) or processing (p100 to p52). For example, TNF receptor 1 engagement leads to formation of a complex consisting of c-IAP1, c-IAP2, TNFR-associated factor (TRAF) 1, TRAF2, and receptor interacting protein (RIP). The c-IAPs ubiquitinate RIP1, which then acts as a scaffold to recruit the regulatory subunit of the IκB kinase, NEMO, leading to IκB phosphorylation, ubiquitination, and degradation [9, 10]. In the case of non-canonical signaling, a constitutive complex that includes c-IAP1, c-IAP2, TRAF family members, and NIK is disrupted, resulting in relief from constitutive ubiquitination/degradation and upregulation of NIK protein levels, initiating downstream signaling [8, 11].
An effective T-cell-mediated immune response involves robust expansion of antigen-specific T cells followed by a sharp contraction, leaving a small pool of long-lived memory cells that provide protective immunity when the pathogen is reencountered [12]. The generation and maintenance of T-cell memory depends on cell surface costimulatory molecules, especially those belonging to the TNFR superfamily, such as 4-1BB, CD27, OX40, GITR, and TNFR2 [13–15]. CD27 is expressed at high levels on resting T cells, whereas GITR is barely detectable. In contrast, 4-1BB, OX40, and TNFR2 are not expressed at all on naive T cells but are induced upon TCR-mediated stimulation [16, 17]. Although all of these molecules have been shown to enhance the proliferation, maintenance, and quality of antigen-specific T cells, 4-1BB has emerged as key survival factor, particularly for CD8+ T cells [18–20]. 4-1BB engagement results in the association of its cytoplasmic tail with intracellular TRAF 1 and 2, leading to activation of nuclear factor κB (NF-κB) and the mitogen activated protein (MAP) kinase signaling cascade [21]. Activation of these pathways leads to the production of effector cytokines such as IL-2, transcription of the pro-survival factors Bcl-XL and Bfl1, and downregulation of the pro-apoptotic molecule Bim [20–22]. Deficiency of 4-1BB or its ligand, 4-1BBL, has little effect on CD8+ T-cell expansion after infection with strong replicating pathogens such as LCMV, but impairs memory cell survival [16, 23]. Recent findings using RNA interference have suggested that c-IAPs play a positive role in activation of NF-κB downstream 4-1BB [24], leading to the possibility that in the absence of c-IAP memory T-cell survival would be affected.
The role of c-IAPs during viral infection has been recently analyzed by pharmacological depletion with a Smac mimetic, which resulted in a substantial decrease in the acute response to LCMV that was at least in part due to production of TNF-α in response to this “IAP antagonist” [25]. Here we investigate the role of c-IAP E3-activity using a genetic model in which a point mutation in the c-IAP2 RING domain disrupts E3 function (c-IAP2H570A). As a result, c-IAP E3 activity is either absent (c-IAP2) or inhibited due to a dominant negative effect (c-IAP1) [26]. Naïve c-IAP2H570A T cells are costimulation-independent in that they proliferate after engagement of TCR alone [27], and they succumb to non-pathogenic Toxoplasma gondii infection due to very high effector cytokine levels produced during the primary effector response [27]. Using these mice, we have analyzed 4-1BB signaling and both the acute and memory response to LCMV. We find that signaling downstream 4-1BB, and consequently the maintenance of a functional and effective pool of memory T cells, requires c-IAP E3 activity.
Results
Impaired 4-1BB-induced signaling in c-IAP2H570A T cells
In vitro studies have shown that engagement of 4-1BB on T cells induces the activation of the canonical NF-κB pathway in a c-IAP-dependent manner [18–20, 24]. We studied the role of c-IAP E3 activity in this process by taking advantage of mice in which endogenous c-IAP2 has been replaced with an E3-inactive point mutant, c-IAP2H570A, that also acts as dominant negative for endogenous c-IAP1[26]. 4-1BB was undetectable on resting WT and c-IAP2H570A splenic CD8+ naïve and memory T cells (data not shown) but present on both to a similar degree after stimulation with anti-CD3/CD28 (Fig. 1A). Activated WT and c-IAP2H570A T cells were cultured with agonistic anti-4-1BB and canonical NF-κB activation was assessed by measuring IκBα degradation (Fig. 1B). As expected, WT T cells exhibited rapid degradation and then re-synthesis of IκBα. In contrast, IκBα, whose levels in IAP2H570A T cells are constitutively higher due to upregulation via non-canonical NF-κB [26], was not degraded in c-IAP2H570A T cells in response to signaling via 4-1BB. Notably, IκBα degradation downstream of two other major TNFR family members that can promote memory T cell survival, OX-40 and CD27, was similar in WT and mutant T cells (Supporting Information Fig. 1A and 1B). Engagement of 4-1BB also induces ERK phosphorylation, which drives transcription of anti-apoptotic genes [21]. In WT T cells, ERK was rapidly phosphorylated after stimulation with 4-1BB, but there was little if any induced phosphorylation in c-IAP2H570A T cells (Fig. 1C). A small percentage of CD8+ T cells in bone marrow (BM) expresses 4-1BB [28], the levels being similar in c-IAP2H570A mice (data not shown). Consistent with the data obtained on pre-activated T cells (Fig. 1A–C), stimulation with either 4-1BBL (Fig. 1D) or agonistic anti-4-1BB (Fig. 1E) induced rapid ERK phosphorylation in WT but not in c-IAP2H570A resting T cells from the BM. To evaluate the impact of loss of c-IAP E3 activity on 4-1BB signaling in vivo, we infected mice with LCMV Armstrong strain. Infection of WT and c-IAP2H570A mice induced expression of 4-1BB on splenic CD8+ T cells specific for the immunodominant MHC I-restricted LCMV epitope GP33 and the subdominant epitope GP276 (Fig. 1F). Notably, 4-1BB engagement induced ERK phosphorylation in WT but not in c-IAP2H570A CD8+ T cells (Fig. 1G, upper panels). As expected, stimulation of CD8+ T cells with PMA/ionomycin induced ERK phosphorylation to a similar degree in WT and c-IAP2H570A cells (Fig. 1G, lower panels), confirming a role for c-IAPs E3 activity in the regulation of signaling downstream 4-1BB. Therefore, although 4-1BB is expressed on c-IAP2 mutant T cells, its signaling is severely impaired.
Figure 1. Impaired 4-1BB-induced IκBα degradation and ERK phosphorylation in c-IAP2H570A T cells.
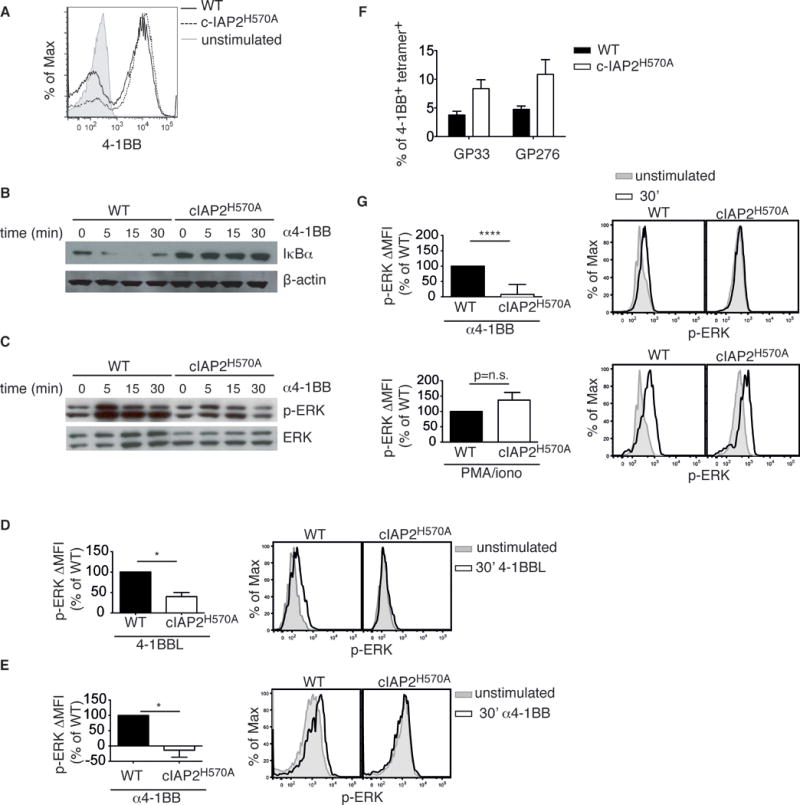
(A) 4-1BB expression on WT and c-IAP2H570A purified T cells stimulated for 48 h with antibodies to CD3 and CD28. Activated WT and c-IAP2H570A purified T cells were stimulated with antibodies to 4-1BB and analyzed by flow cytometry. One representative histogram from 2 independent experiments with 2 mice per genotype is shown. (B) IκBα degradation and (C) ERK phosphorylation were analyzed by immunoblotting at the indicated times. Lanes were rearranged for clarity. ß-actin (B) and total ERK (C) were used as loading controls. The data shown in B, and C are representative of 2 independent experiments each. (D, E) T cells purified from BM of WT or c-IAP2H570A mice were stimulated for 30 min with (D) 4-1BBL or (E) α4-1BB, stained for phospho-ERK, and analyzed by flow cytometry. Bar graphs show the average of p-ERK ΔMFI expressed as percentage of WT ± SD, and a representative histogram is shown. The data shown in D and E are representative of 2 independent experiments, for a total of 3 mice per genotype. (F) WT and c-IAP2H570A mice were infected with LCMV Armstrong and analyzed 7 days later. The bar graph depicts the frequency of CD8+ GP33- and GP276-tetramer-binding 4-1BB-positive cells in c-IAP2H570A mice. (G) CD8+ T cells purified from WT or c-IAP2H570A mice at day 7 of LCMV infection were stimulated with α4-1BB or PMA/ionomycin for 30 minutes and stained for p-ERK. (F, G) Bar graphs show the average of p-ERK ΔMFI expressed as percentage of WT and a representative histogram from one pool of 4 mice per genotype is shown. The data show in F and G represent the average pooled from 2 independent experiments, n=8 mice per genotype. Data are expressed as mean ± SEM unless otherwise specified; *p ≤ 0.05, **** p ≤ 0.0001 with Student’s t-test.
Normal acute response in LCMV-infected c-IAP2H570A mice
Although 4-1BBL-deficient mice mount an effective primary response against influenza virus [29], it has been reported that the response to LCMV is inhibited by acute deletion of c-IAPs with IAP-antagonists [25]. To address this, we analyzed the immune response of c-IAP2H570A mice to LCMV. LCMV infection resulted in massive expansion of GP33- and GP276-specific CD8+ T cells, peaking at day 7, to a similar extent in WT and c-IAP2H570A mice (Fig. 2A, 2B and Supporting Information Fig. 2). At the peak of the response, both WT and c-IAP2H570A CD8+ GP33+ T cells had a CD44hiCD62Llo effector phenotype with no differences in expression of IL-7Rα, or the frequency of IL-7RhiKLRG1lo cells, which are thought to be memory precursors (Fig. 2C). Similarly, there were no differences in these activation/differentiation markers between WT and c-IAP2H570A CD8+ cells recognizing the subdominant epitope GP276 (data not shown). No difference was found in expression of the transcription factors T-bet and Eomes (Fig. 2D), which increase during the effector stage and regulate the ratio between effector and central cells in the memory compartment [12]. Examination of the activation/differentiation markers CD122, CD25, CD69, CD27, and CXCR3 also revealed no differences between WT and c-IAP2H570A cells (data not shown). The expansion of CD4+ T cells specific for the MHC-II restricted epitope GP66 was slightly higher in the mutant mice (Supporting Information Fig. 3A), but the activation markers CD11a and CD49d [30] were expressed to the same extent as on their WT counterparts (Supporting Information Fig. 3B), and no difference was found in T-bet expression (Supporting Information Fig. 3C). Although the absolute cell numbers and the expression of surface markers were normal at the peak of the response, day 7, c-IAP2H570A antigen-specific CD8+ (and CD4+) T cells produced more TNF-α, IFN-γ, and IL-2 than WT T cells upon restimulation with viral peptides (Fig. 2E, 2F and Supporting Information Fig. 3D). Therefore, as with CD4+ T cells in T. gondii-infected mice [27], effector cytokine production was increased in c-IAP2H570A virus-specific T cells. Importantly, both strains had cleared the virus by 7 days after infection (Fig. 2G). Therefore, lack of c-IAP E3 activity did not affect the acute response to LCMV.
Figure 2. c-IAP2H570A mice mount a normal acute response to LCMV Armstrong.
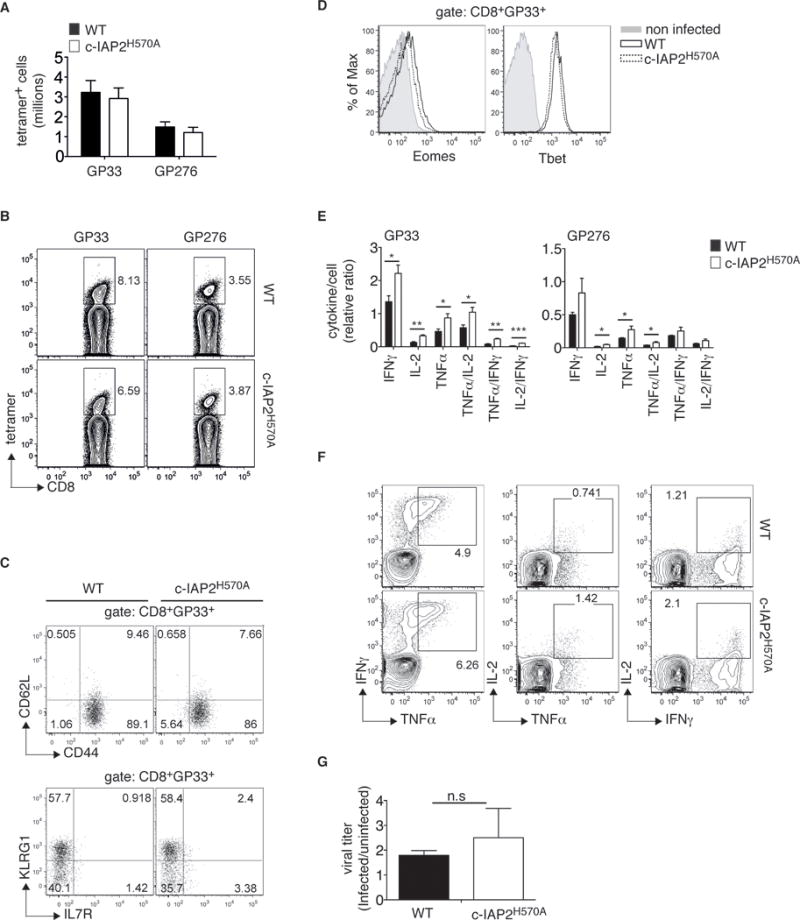
WT and c-IAP2H570A mice were infected with LCMV Armstrong and analyzed after 7 days by flow cytometry. (A) Bar graph depicts the absolute number of splenic CD8+GP33+ T cells and CD8+GP276+ T cells). Bars show the average of 4 pooled experiments (n=14 WT and 13 c-IAP2H570A). (B) One representative dot plot of tetramer frequency is shown from one pool of 3 WT and 4 c-IAP2H570A. (C, D) GP33+CD8+ T cells were analyzed on day 7 after infection. (C) Representative dot plots from one pool of 3 WT and 4 c-IAP2H570A out of 4 independent experiments (n=14 WT and 13 c-IAP2H570A) are shown. Numbers show percentage of the indicated population gated in GP33+CD8+ T cells. (D) Representative histograms from one pool of 3 WT and 4 c-IAP2H570A mice out of 2 independent experiments, (n= 7 WT and 8 c-IAP2H570A mice) are shown. (E) Splenocytes were restimulated in vitro with GP33 or GP276 peptide 7 days after LCMV infection and evaluated for cytokine production by intracellular staining. Bar graphs show the production of cytokines as a function of the number of tetramer positive cell. Data represent the average of 4 WT and 3 c-IAP2H570A mice and are representative of 2 independent experiments for a total of 7 WT and 6 c-IAP2H570A (F) dot plots show a representative staining of cells stimulated with GP33 peptide, gated on CD8, from a pool of 4 WT and 3 c-IAP2H570A mice. (G) Viral RNA from splenocytes of infected WT or c-IAP2H570A mice (3 mice per each genotype) was analyzed by quantitative RT-PCR. One representative out of 2 independent experiments is shown. All bar graphs represent mean ± SEM; *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001; Student’s t-test was used for statistical evaluation.
Reduced LCMV-specific memory CD8+ T-cell number and altered TCM/TEM ratio in c-IAP2H570A mice
4-1BBL-deficient animals have a severe impairment in memory cell maintenance after influenza virus infection [29]. In agreement with this, the numbers of GP33- and GP276-specific c-IAP2H570A CD8+ T cells were reduced by approximately 50% compared to WT as early as 25 days after infection (Fig. 3A and 3B). Notably, memory GP66-specific c-IAP2H570A CD4+ T cells were not reduced (Supporting Information Fig. 4A), consistent with the fact that CD4 T cell survival is relatively independent of 4-1BB [18] but instead relies on OX-40, whose signaling was normal in mutant T cells (Supporting Information Fig. 1A). Memory T cells can be divided into central memory T cells (TCM), characterized by greater proliferative potential, and effector memory T cells (TEM), having a higher degree of effector function such as cytokine and granzyme B production, [12]. Whereas all memory cells are CD44hi, TEM are CD62Llo and TCM are CD62Lhi. In addition, TCM and TEM include subpopulations characterized by markers that define a degree of specialization toward one or the other phenotype. In particular, CD27 and CXCR3 have been associated with a self-renewing central memory-like phenotype [12, 31–33]. Analysis of antigen-specific CD8+ cells ≥60 days after LCMV infection showed no differences between WT and c-IAP2H570A mice in the expression of CD44 and CD62L, but a profound decrease in the CD27hiCXCR3+ population (Fig. 3C, 3D and Supporting Information Fig. 5A and 5B). Moreover, CD8+ c-IAP2H570A T cells specific for both GP33 and GP276 had higher expression of KLRG-1 after the effector phase had peaked (Fig. 3E and 3F). Despite the differences in number and cell surface phenotype, T-bet and Eomes were expressed normally in c-IAP2H570A antigen-specific cells (Fig. 3G). c-IAP2H570A CD8+ T cells produced more effector cytokines 35 days after infection, but no difference ≥60 days after infection (Supporting Information Fig. 5C, Fig. 3H and 3I). CD44 and CD62L expression on CD4+ GP66+ T cells was similar between WT and c-IAP2H570A mice, and no difference was found in cytokine production after restimulation with the specific peptide (Supporting Information Fig. 4B and 4C). Therefore, CD8+ memory T-cell survival requires c-IAP E3 activity, the lack of which alters the balance between TEM and TCM.
Figure 3. Reduced number of CD8+ memory T cells in LCMV-infected c-IAP2H570A mice.
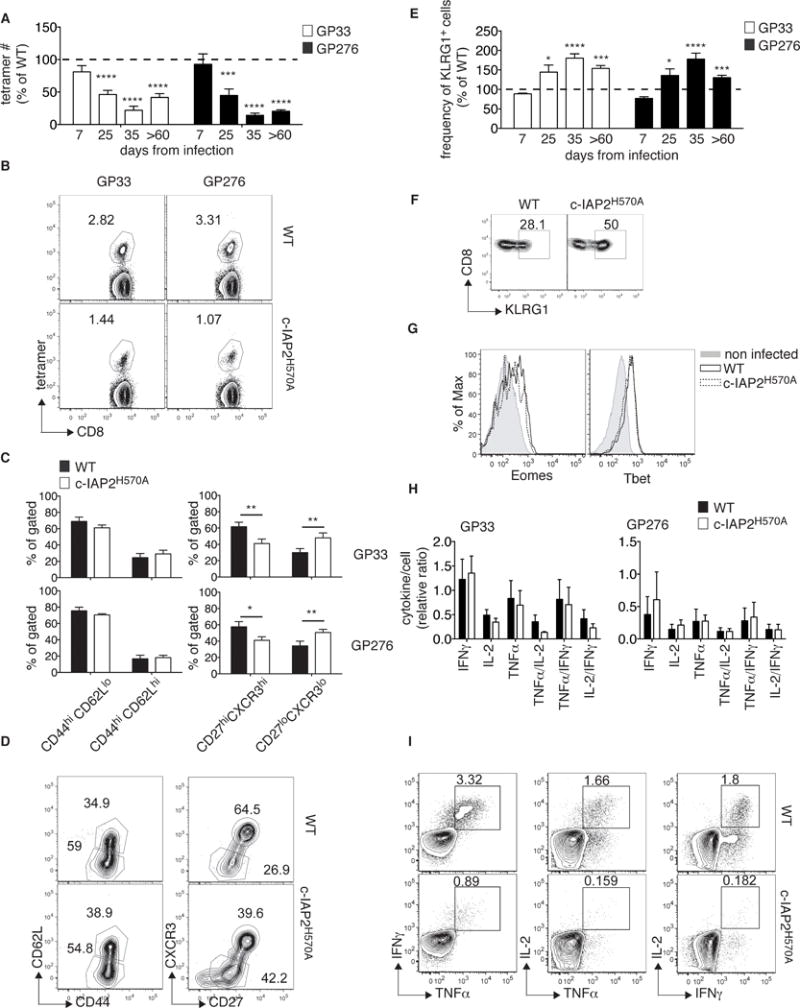
WT and c-IAP2H570A mice were infected with LCMV Armstrong and analyzed after 60 or more days by flow cytometry. (A) GP33+CD8+ and GP276+CD8+ T cells from c-IAP2H570A mice are shown as a percentage of WT at the peak (day 7, n= 14 WT; 13 c-IAP2H570A, pooled from 4 independent experiments), contraction (day 25, average of day 22, 23, and 25, n= 14 per group, pooled from 3 independent experiments), early memory (day 35, n=5 per genotype, pooled from 2 independent experiments), and late memory (day ≥60 n= 10 per genotype, pooled from 3 independent experiments) phases of the LCMV response. (B) A representative dot plot of the staining obtained after ≥60 days is shown from one pool of 3 WT and 4 c-IAP2H570A mice. (C) Bar graphs and (D) representative dot plots show the percentage of the indicated populations analyzed ≥60 days after infection (n=6 per group, pooled from 2 independent experiments with 3 mice per genotype each). (E) The percentage of KLRG1-expressing c-IAP2H570A CD8+GP33+, and CD8+GP276+ T cells is depicted as a percentage of WT during the peak (day 7 n= 8 WT; 7 c-IAP2H570A, pooled from 2 independent experiments), contraction (day 25, n= 8 per group, pooled from 2 independent experiments, average of day 23 and 25), early memory (day 35, n=5 per genotype, pooled from 2 independent experiments), and late memory (day ≥60 n=10 per genotype, pooled from 2 independent experiments) phases of the LCMV response. (F) A representative dot plot of KLRG1 expression, gated in CD8+GP33+ cells at day ≥60 from one pool of 3 WT and 3 c-IAP2H570A mice . (G) Histograms show the expression of Eomes and T-bet on CD8+GP33+ T cells ≥60 days after infection (from one pool of 3 WT and 3 c-IAP2H570A mice out of 2 independent experiments). Splenocytes collected ≥60 days after infection were restimulated in vitro with GP33 or GP276 peptide and evaluated for cytokine production by intracellular staining. (H) The bar graph shows the production of cytokine per tetramer positive cell. These data represent the average of 3 independent experiments (n=9 WT, n=10 c-IAP2H570A). (I) A representative flow cytometric profile of the result obtained (from one pool of 3 WT and 3 c-IAP2H570A mice out of 3 independent experiments). All bar graphs represent mean ± SEM; *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001 ****p ≤ 0.0001; Student’s t-test was used for statistical evaluation.
Decreased c-IAP2H570A anti-viral memory cell number is T-cell-intrinsic
Because c-IAP1 and -2 are widely expressed and the c-IAP2 RING mutation is not tissue-specific, we considered the possibility that the loss of CD8+ memory T cells might be due to effects of this mutation in other cells. To test this, splenocytes from CD45.2 WT or CD45.2 cIAP2 mutant mice were collected at day 7 after LCMV infection, when the vast majority of CD8+ T cells are LCMV-specific. Samples were normalized to CD8 T-cell number, mixed at a 1:1 ratio, CFSE labeled, and transferred to naïve WT recipients. After 14 days, c-IAP2H570A donor CD8 cells were approximately half that of WT and expressed higher level of KLRG1 (Fig. 4A, 4B, 4C and Supporting Information Fig. 6). Therefore, T cell c-IAP E3 activity is required for survival of LCMV-specific CD8+ T memory cells and increased death during contraction occurs in a T-cell-intrinsic manner.
Figure 4. The decreased number of c-IAP2H570A memory cell is T-cell-intrinsic.
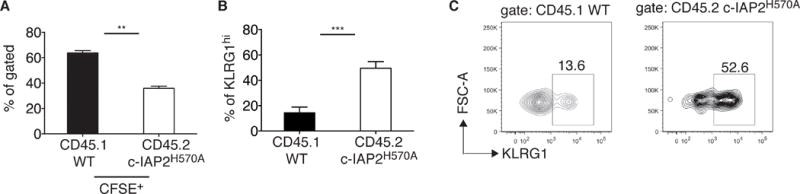
CD45.1 WT and CD45.2 c-IAP2H570A mice were infected with LCMV and splenocytes collected after 7 days. CD8 T cell number was normalized and the two genotypes were mixed at 1:1 ratio, CFSE labeled, and injected into 5 unimmunized CD45.2 WT recipients. (A) The ratio between CFSE+ CD45.1+WT and CD45.2 c-IAP2H570A and (B) the frequency of KLRG1hi CD8+ cells was determined by flow cytometry after 14 days. (C) A representative dot plot is shown. Data are representative of 2 independent experiments, for a total of 8 mice. Bar graphs represent mean ± SEM; **p ≤ 0.01, ***p ≤ 0.001; Student’s t-test was used for statistical evaluation.
Increased apoptotic death of c-IAP2H570A CD8+ cells during acute homeostatic proliferation
It has been reported that stimulation via 4-1BB promotes the proliferation of memory CD8 T cells in lymphopenic hosts [34], and increases their antigen-independent survival [35]. This prompted us to investigate the ability of WT and mutant T cells to reconstitute a lymphopenic host via acute homeostatic proliferation. WT and c-IAP2H570A T cells were mixed at a 1:1 ratio, CFSE-labeled, and adoptively transferred into RAG1-deficient mice. Although WT and mutant CD8+ T cells proliferated to a similar extent, the number of c-IAP2H570A CD8 T cells recovered 20–29 days after transfer was lower than WT T cells (Fig. 5A and B). This was due to enhanced apoptosis of c-IAP2H570A CD8 T cells, which were positive for the activated form of caspase-3 (Fig. 5C). In addition, among CD8 T cells, we observed a decrease in the c-IAP2H570A KLRG1+ population (Fig. 5D), a subset that was viable based on vital dye staining but Annexin V+, indicating that they were in the process of undergoing apoptotic death. These results indicate that lack of c-IAP E3 activity affects CD8 T cells survival during acute homeostatic proliferation.
Figure 5. Apoptotic death of CD8+ c-IAP2H570A cell during acute homeostatic proliferation.
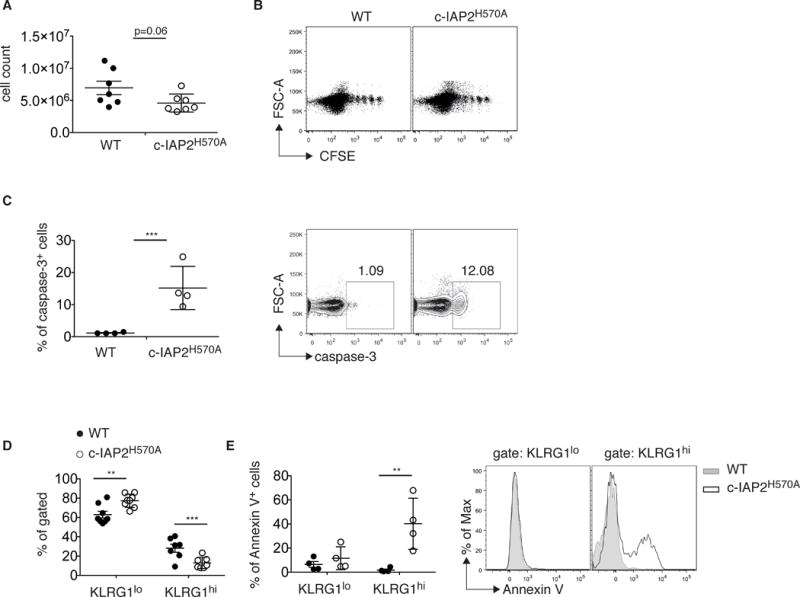
T cells were purified from CD45.1 WT and CD45.2 c-IAP2H570A and adoptively transferred into RAG1-deficient hosts. Recipients were analyzed by flow cytometry after 20 (n=3) and 29 (n=4) days and results were pooled. (A) Graphs depict the absolute numbers of CD45.1 WT and CD45.2 c-IAP2H570A CD8 T cells. (B) A representative dot plot of CFSE dilution on day 29 of reconstitution is shown. Each dot shows an individual mouse and data is from a pool of 4 mice at each time point. (C) Frequency and a representative dot plot of cells positive for activated caspase-3 29 days after transfer. Each dot shows an individual mouse. (D) Frequency of KLRG1-low and -high cells and (E) of Annexin V-positive cells gated in KLRG1-low and -high cells 29 days after transfer are shown. Each dot shows an individual mouse. One representative histogram of four is shown. All bar graphs represent mean ± SEM; **p ≤ 0.01, ***p ≤ 0.001; Student’s t-test was used for statistical evaluation.
Discussion
Ubiquitination is a versatile and common strategy used to regulate the immune response through post-translational modification of target proteins. The ubiquitin protein ligases c-IAP1 and -2 have been shown to be required for activation of a number of innate-immune signaling pathways, such as those downstream Toll-like receptors (TLRs), nucleotide-binding oligomerization domain (NOD)-1 and NOD-2, Nucleotide-Binding Domain and Leucine-Rich Repeat Containing Family (NLR), and retinoic acid-inducible gene (RIG)-1/ Melanoma Differentiation-Associated protein (MDA)-5 [36]. The role of c-IAP in adaptive immunity has mostly been linked to the activation of NF-κB by tumor necrosis factor receptor (TNFR) superfamily members. For example, c-IAP1/2 E3 activity is required downstream of CD40 and BAFFR occupancy for normal B cell development and function, and in their absence B cells develop a phenotype resembling anomalies found in human GALT lymphoma and multiple myeloma [26, 37]. The role of c-IAP in T cell function is less well known. One such function appears to be to limit T cell activation. Smac mimetics, also called IAP antagonists, are compounds that bind to and induce the activity of c-IAP1 and -2, causing their self-ubiquitination and degradation [10]. T cells so-treated proliferate to TCR-mediated activation more robustly than untreated cells, losing their requirement for co-stimulation via CD28 and increasing the efficacy of an antitumor vaccine [38]. We obtained similar results using the E3-dead and dominant negative c-IAP2H570A mutation used in this present report, and found that these mice mounted an exaggerated primary response to T. gondii, resulting in lethal levels of CD4+ T cell-derived effector cytokines [27]. The acute response of c-IAP2H570A mice to LCMV in the present study was also characterized by high production of effector cytokines, in this case by responding CD8+ T cells, but not so high as to cause mortality. This, and the availability of peptide-MHC I tetramers that allow one to follow antigen-specific T-cell subsets, made it possible to study the consequence of c-IAP E3 loss on anti-viral responses. The major reported consequence of c-IAP E3 loss appears to be the upregulation of NIK and activation of the non-canonical NF-κB pathway [26, 27]. Pharmacologic inhibition [27] and NIK knockdown with siRNA [24] supports the notion that non-canonical NF-κB is responsible for T-cell costimulation-independence. It is noteworthy, then, that a recent report found that NIK-deficient T cells were able to mount relatively normal acute response to LCMV but failed to maintain memory. Indeed, 65 days after LCMV infection both the absolute number of memory T cells and their ability to produce IFN-γ and TNF-α were sharply decreased [39]. These results argue that non-canonical NF-κB promotes memory T-cell maintenance. Therefore, one might have expected that the c-IAP2H570A mice, in which there is constitutive activation of non-canonical NF-κB in T cells, would develop a larger pool of memory cells. The fact that we observed the opposite suggests that the primary mechanism determining the survival of memory cells in c-IAP2 mutant mice is the lack of canonical NF-κB signaling through 4-1BB (and perhaps other TNFR family members), although other, unexplored, signaling abnormalities could also be involved.
Although homeostatic proliferation was unaffected by lack of c-IAP2 E3 activity, expansion was compromised because CD8 T cells underwent apoptotic death. This result is in line with a previous study showing that CD8 T cells proliferate to a similar extent in WT and 4-1BBL-deficient irradiated hosts [40]. In another study, no defects were found in proliferation of OT-I CD8 memory T cells after transfer into 4-1BBL-deficient recipients, but survival was decreased [35]. The diminished survival of cIAP2H570A CD8 T cells during acute homeostatic proliferation fits with these observations, supporting the speculation that defective 4-1BB signaling may account, at least in part, for the observed phenotype.
Lack of c-IAP E3 activity had a marginal effect on the maintenance of CD4+ memory T cells. This can perhaps be explained by the fact that 4-1BB, even if expressed on CD4+ T cells, plays a minor role in their survival [18]. An approach using RNA interference to acutely knock down c-IAP levels has suggested that these proteins are positive regulators of 4-1BB signaling [24]. Notably, 4-1BBL-deficient mice infected with influenza mount a normal primary response but the number of virus specific T cells later decline [29]. Another report found that mice expressing a dominant negative TRAF2 have normal primary responses but defective recall responses to influenza infection [41]. The data in the present report strongly support this interpretation, and demonstrate that c-IAP ubiquitin protein ligase activity is essential for CD8+ T-cell survival.
Another study has recently addressed role of c-IAP in the response to LCMV [25]. Ablation of c-IAP2 by treatment with an IAP antagonist resulted in a reduced acute response, arguing for a positive role of IAPs in the orchestration of the anti-viral effector phase. Notably, the effect of the IAP antagonist was blunted in infected TNFα-/- mice. Because degradation of c-IAP1 and -2 in sensitive cells results in NF-κB-dependent TNFα secretion, which was also observed as an approximately 7-fold increase in the blood of the LCMV-infected IAP antagonist-treated versus untreated mice [25], this argues that it is IAP-antagonist-induced TNFα that was largely responsible for the decreased primary response to LCMV. In contrast, T cells from the genetic model, c-IAP2H570A mice, had only slightly elevated numbers of tetramer+ TNFα-producing cells, with no increase in the amount of TNFα produced per cell. These data suggest that the effect of the IAP antagonist on the acute LCMV response was due to enhanced TNFα production, whether due to acute c-IAP depletion or some other mechanism, and does not reflect the physiologic role of c-IAP in anti-viral immunity. It is also possible that c-IAP play a non-enzymatic role in T-cell responses, and in such a case their ablation with IAP antagonist would reveal a phenotype unrelated to their E3 activity. The genetic loss-of-function model used in this study demonstrates that c-IAP ubiquitin protein ligase activity is dispensable for the acute response to LCMV but required for T-cell survival during the contraction phase.
Materials and Methods
Mice
c-IAP2H570A mice [26] were maintained in a National Cancer Institute pathogen-free animal facility and were subsequently backcrossed onto the B6 background for 9 generations and then interbred for these studies. CD45.1 mice were obtained from Frederick Cancer Research Facility (Frederick, MD). Study protocols were approved by Institutional Animal Care and Use Committee of the National Cancer Institute.
Reagents
Fluorochrome-conjugated antibodies to CD4, CD11c, CD44, CD62L, IL7Rα, MHC II, PD-1, phospho-ERK, TCRβ, B220, IFN-γ, TNF-α, IL-2, 4-1BB, Eomes, AnnexinV, caspase-3, 2.4G2 (Fc receptor blocking), purified antibodies to CD3ɛ (145-2c11), CD28 (37.51), CD27 (LG.3A10) and OX-40 (OX-86) for in vitro stimulation, as well as GolgiStop, BD Annexin V binding buffer, and BD fixation and permeabilization buffers, were obtained from BD Biosciences. Fluorochrome-conjugated antibodies against CD8, CD45.1, CD45.2, CD127, CXCR3, and nuclear fixation and permeabilization buffers were purchased from eBioscience. Fluorochrome-conjugated antibodies against T-bet and KLRG1 were purchased from Santa Cruz Biotechnology and SouthernBiotech, respectively. Anti-IκBα was purchased from Santa Cruz, anti-p-ERK and anti-ERK from Cell Signaling, and anti-β actin from Sigma. GP33–41 H-2Db (GP33), GP276–286 H-2Db (GP276), and GP66–77 I–Ab (GP66) were obtained from the National Institutes of Health Tetramer Core Facility at Emory University, Atlanta, GA. GP33–41, GP276-286, and GP61-80 (GP61) peptides were purchased from Peptide 2.0. Agonistic anti-4-1BB (3H3) was purchased from BioXCell. Phorbol myristate acetate (PMA) and ionomicyn were purchased from Sigma-Aldrich. Blue Fluorescent, LIVE/DEAD Fixable Dead Cell Stain, and CFSE were obtained from Life Technologies. Cells were cultured in RPMI 1640 supplemented with 10% fetal calf serum (FCS), 100 μg/ml gentamicin, 2 mM L-glutamine, and 50 μM β-mercaptoethanol.
LCMV growth and infection
LCMV Armstrong 53b was grown in baby hamster kidney cells and titer determined as described [42]. B6 and c-IAP2H570A mice were infected i.p. with 2 × 105 PFU of LCMV Armstrong. To determine viral load, total RNA was extracted from splenocytes using RNeasy Mini kits (Qiagen) and reverse transcribed using the Superscript II Reverse Transcriptase kit (Invitrogen) following the manufacturers’ protocols. Viral nucleoproteins were amplified using specific primers as described [43]. Housekeeping ribosomal 18s RNA was amplified to normalize RNA content of the lysate and obtain a ΔCT value.
Immunoblotting
T cells were purified from lymph nodes using Easy Sep T-cell enrichment kits (StemCell Technologies) following the manufacturer’s protocol and the number of live cells was assessed by trypan blue exclusion. Purity was determined by flow cytometry, and for all experiments was >90%. T cells were stimulated for 48 hr with plate-bound anti-CD3ε (1 μg/ml) and anti-CD28 (2 μg/ml), then washed and stimulated with anti-4-1BB, anti-CD27, or anti-OX-40 (all plate-bound at 15 μg/ml). At the indicated times, cells were lysed in RIPA (Pierce). Samples were normalized to protein concentration, resolved by SDS-PAGE, and immunoblotted with the indicated antibodies.
Flow cytometry and cell stimulation
All stainings were performed in PBS plus 0.1% fetal calf serum (FCS) in the presence of 1:500 2.4G2 antibody (Fc block). For detection of intracellular cytokines, 1.5−3 × 106 splenocytes were stimulated with 0.3 μg/ml GP33–41, GP276–268, or GP61-80 for 5 hr or were left untreated, in the presence of BD GolgiStop. Cells were stained with antibodies to surface markers and then fixed. Because restimulation down-regulates TCR levels, the amount of cytokine produced per cell was calculated by dividing the percentage of cytokine-producing cells by the percentage of tetramer-expressing cells prior to stimulation. As specificity controls, each experiment included restimulation of splenocytes from an uninfected mouse and incubation in the presence of GolgiStop alone for each infected mouse-derived sample. Flow cytometry was done with a BD LSRII or BD LSRFortessa cytometer using BD FACSDiva software (BD Biosciences) and data analyzed with FlowJo software (Tree Star).
Phospho-ERK detection by flow cytometry
Phospho-ERK was detected in total T cells purified from bone marrow of uninfected mice or on purified CD8+ T cells (Easy Sep CD8+ T-cell enrichment kit, Stem Cell Techonologies) from LCMV infected mice. In both cases, cells were incubated with plate-bound 4-1BB agonistic antibody (15 μg/ml), 4-1BBL (10 μg/ml), or PMA (20 ng/ml) plus ionomycin (500 ng/ml) for the indicated times and immediately fixed with BD Fixation Buffer I and permeabilized with BD Permeabilization Buffer III before staining.
Adoptive transfer
CD45.1 WT and CD45.2 c-IAP2H570A mice were infected with LCMV Armstrong. After 7 days, spleens were collected from each set of mice and pooled by genotype. Either total splenocytes normalized to CD8+ T cell number or purified CD8+ T cells (Easy Sep CD8 T cell enrichment kit, StemCell Technologies) were mixed at a 1:1 ratio (confirmed by flow cytometry), labeled with CFSE (500 nM), and 107 (splenocytes) or 106 (purified CD8) cells were injected i.v. into unimmunized CD45.1 WT recipients. Fourteen days after transfer, spleens were collected and the ratio between CD45.1 and CD45.2 CFSE+ cells was determined by flow cytometry.
Adoptive transfer
T cells were purified (Easy Sep T cell enrichment kit, StemCell Technologies) from CD45.1 WT and CD45.2 c-IAP2H570A mice, mixed at 1:1 ratio, and CFSE stained. 106 cells were injected i.v. into RAG1-deficient recipients. Spleens were collected and analyzed 20 or 29 days after injection.
Statistical analysis
Statistical analysis was performed using a Student two-tailed unpaired t test with GraphPad Prism software. Error bars indicate standard errors of the mean unless otherwise specified. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p≤ 0.0001.
Supplementary Material
Acknowledgments
We are grateful to the National Institutes of Health Tetramer Core Facility at Emory University for supplying tetramers. This work was supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, NIH.
Abbreviations
- LCMV
lymphocytic choriomeningitis virus
- IAP
inhibitor of apoptosis protein
- ΔMFI
Mεαν Fluorescence Intensity of stimulated sample − Mεαν Fluorescence Intensity of unstimulated sample
Footnotes
Conflict of interest disclosure: The authors declare no commercial or financial conflict of interests.
References
- 1.Srinivasula SM, Ashwell JD. Mol Cell. 2008;30:123–135. doi: 10.1016/j.molcel.2008.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mahoney DJ, Cheung HH, Mrad RL, Plenchette S, Simard C, Enwere E, Arora V, et al. Proc Natl Acad Sci U S A. 2008;105:11778–11783. doi: 10.1073/pnas.0711122105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bertrand MJ, Milutinovic S, Dickson KM, Ho WC, Boudreault A, Durkin J, Gillard JW, et al. Mol Cell. 2008;30:689–700. doi: 10.1016/j.molcel.2008.05.014. [DOI] [PubMed] [Google Scholar]
- 4.Varfolomeev E, Goncharov T, Fedorova AV, Dynek JN, Zobel K, Deshayes K, Fairbrother WJ, et al. J Biol Chem. 2008;283:24295–24299. doi: 10.1074/jbc.C800128200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wertz IE, Dixit VM. Cold Spring Harb Perspect Biol. 2010;2:a003350. doi: 10.1101/cshperspect.a003350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moulin M, Anderton H, Voss AK, Thomas T, Wong WW, Bankovacki A, Feltham R, et al. EMBO J. 2012 doi: 10.1038/emboj.2012.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vallabhapurapu S, Karin M. Annu Rev Immunol. 2009;27:693–733. doi: 10.1146/annurev.immunol.021908.132641. [DOI] [PubMed] [Google Scholar]
- 8.Sun SC. Cell Res. 2011;21:71–85. doi: 10.1038/cr.2010.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ben-Neriah Y. Nat Immunol. 2002;3:20–26. doi: 10.1038/ni0102-20. [DOI] [PubMed] [Google Scholar]
- 10.Fulda S, Vucic D. Nat Rev Drug Discov. 2012;11:109–124. doi: 10.1038/nrd3627. [DOI] [PubMed] [Google Scholar]
- 11.Lee S, Challa-Malladi M, Bratton SB, Wright CW. J Biol Chem. 2014;289:30680–30689. doi: 10.1074/jbc.M114.587808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kaech SM, Cui W. Nat Rev Immunol. 2012;12:749–761. doi: 10.1038/nri3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ahlers JD, Belyakov IM. Blood. 2010;115:1678–1689. doi: 10.1182/blood-2009-06-227546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Croft M. Nat Rev Immunol. 2009;9:271–285. doi: 10.1038/nri2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Allam A, Conze DB, Giardino Torchia ML, Munitic I, Yagita H, Sowell RT, Marzo AL, et al. Blood. 2009;114:2121–2130. doi: 10.1182/blood-2009-05-220087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wortzman ME, Clouthier DL, McPherson AJ, Lin GH, Watts TH. Immunol Rev. 2013;255:125–148. doi: 10.1111/imr.12086. [DOI] [PubMed] [Google Scholar]
- 17.Croft M. Nat Rev Immunol. 2003;3:609–620. doi: 10.1038/nri1148. [DOI] [PubMed] [Google Scholar]
- 18.Takahashi C, Mittler RS, Vella AT. J Immunol. 1999;162:5037–5040. [PubMed] [Google Scholar]
- 19.Starck L, Scholz C, Dorken B, Daniel PT. Eur J Immunol. 2005;35:1257–1266. doi: 10.1002/eji.200425686. [DOI] [PubMed] [Google Scholar]
- 20.Lee HW, Park SJ, Choi BK, Kim HH, Nam KO, Kwon BS. J Immunol. 2002;169:4882–4888. doi: 10.4049/jimmunol.169.9.4882. [DOI] [PubMed] [Google Scholar]
- 21.Wang C, Lin GH, McPherson AJ, Watts TH. Immunol Rev. 2009;229:192–215. doi: 10.1111/j.1600-065X.2009.00765.x. [DOI] [PubMed] [Google Scholar]
- 22.Sabbagh L, Srokowski CC, Pulle G, Snell LM, Sedgmen BJ, Liu Y, Tsitsikov EN, et al. Proc Natl Acad Sci U S A. 2006;103:18703–18708. doi: 10.1073/pnas.0602919103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Watts TH. Annu Rev Immunol. 2005;23:23–68. doi: 10.1146/annurev.immunol.23.021704.115839. [DOI] [PubMed] [Google Scholar]
- 24.McPherson AJ, Snell LM, Mak TW, Watts TH. J Biol Chem. 2012;287:23010–23019. doi: 10.1074/jbc.M112.350538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gentle IE, Moelter I, Lechler N, Bambach S, Vucikuja S, Hacker G, Aichele P. Blood. 2014;123:659–668. doi: 10.1182/blood-2013-01-479543. [DOI] [PubMed] [Google Scholar]
- 26.Conze DB, Zhao Y, Ashwell JD. PLoS Biol. 2010;8:e1000518. doi: 10.1371/journal.pbio.1000518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Giardino Torchia ML, Conze DB, Jankovic D, Ashwell JD. J Immunol. 2013;190:549–555. doi: 10.4049/jimmunol.1201697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin GH, Snell LM, Wortzman ME, Clouthier DL, Watts TH. J Immunol. 2013;190:4627–4639. doi: 10.4049/jimmunol.1201854. [DOI] [PubMed] [Google Scholar]
- 29.Bertram EM, Lau P, Watts TH. The Journal of Immunology. 2002;168:3777–3785. doi: 10.4049/jimmunol.168.8.3777. [DOI] [PubMed] [Google Scholar]
- 30.McDermott DS, Varga SM. J Immunol. 2011 doi: 10.4049/jimmunol.1102104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hikono H, Kohlmeier JE, Takamura S, Wittmer ST, Roberts AD, Woodland DL. J Exp Med. 2007;204:1625–1636. doi: 10.1084/jem.20070322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rutishauser RL, Martins GA, Kalachikov S, Chandele A, Parish IA, Meffre E, Jacob J, et al. Immunity. 2009;31:296–308. doi: 10.1016/j.immuni.2009.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Joshi NS, Cui W, Dominguez CX, Chen JH, Hand TW, Kaech SM. J Immunol. 2011;187:4068–4076. doi: 10.4049/jimmunol.1002145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhu Y, Zhu G, Luo L, Flies AS, Chen L. Blood. 2007;109:4882–4889. doi: 10.1182/blood-2006-10-043463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pulle G, Vidric M, Watts TH. J Immunol. 2006;176:2739–2748. doi: 10.4049/jimmunol.176.5.2739. [DOI] [PubMed] [Google Scholar]
- 36.Beug ST, Cheung HH, LaCasse EC, Korneluk RG. Trends Immunol. 2012;33:535–545. doi: 10.1016/j.it.2012.06.004. [DOI] [PubMed] [Google Scholar]
- 37.Gardam S, Turner VM, Anderton H, Limaye S, Basten A, Koentgen F, Vaux DL, et al. Blood. 2011;117:4041–4051. doi: 10.1182/blood-2010-10-312793. [DOI] [PubMed] [Google Scholar]
- 38.Dougan M, Dougan S, Slisz J, Firestone B, Vanneman M, Draganov D, Goyal G, et al. The Journal of Experimental Medicine. 2010;207:2195. doi: 10.1084/jem.20101123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rowe AM, Murray SE, Raue HP, Koguchi Y, Slifka MK, Parker DC. J Immunol. 2013 doi: 10.4049/jimmunol.1301328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Prlic M, Blazar BR, Khoruts A, Zell T, Jameson SC. J Immunol. 2001;167:5664–5668. doi: 10.4049/jimmunol.167.10.5664. [DOI] [PubMed] [Google Scholar]
- 41.Cannons JL, Bertram EM, Watts TH. The Journal of Immunology. 2002;169:2828–2831. doi: 10.4049/jimmunol.169.6.2828. [DOI] [PubMed] [Google Scholar]
- 42.Dutko FJ, Oldstone MB. J Gen Virol. 1983;64:1689–1698. doi: 10.1099/0022-1317-64-8-1689. [DOI] [PubMed] [Google Scholar]
- 43.McCausland MM, Crotty S. J Virol Methods. 2008;147:167–176. doi: 10.1016/j.jviromet.2007.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.