

Abstract
The synthesis of biomimetic hydrogel nanoparticles coated with natural cell membrane is described. Compared to existing strategy of wrapping cell membrane onto pre-formed nanoparticle substrates, this new approach forms the cell membrane-derived vesicles first, followed by growing nanoparticle cores in situ. It adds significant controllability over the nanoparticle properties and opens unique opportunities for a broad range of biomedical applications.
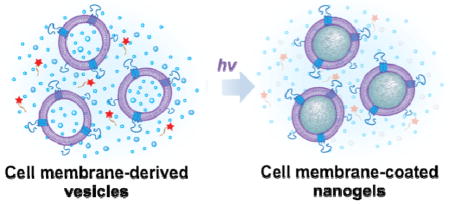
Keywords: nanogel, cell membrane, macromolecular inhibitor, in situ polymerization, red blood cell
Cell membrane-cloaked nanoparticles represent an emerging and intriguing nanodelivery platform that consists of a synthetic nanoparticle core and a natural cell membrane shell.[1] Such core-shell structured biomimetic nanoparticles have garnered increasing interests owing to their unique strength in mimicking natural properties displayed by the source cells while preserving a high degree of synthesis flexibility of the cores.[2,3] As a result, a number of cell membrane-cloaked nanoparticle systems have been developed, which involved the use of membranes from red blood cells, white blood cells, cancer cells, and bacteria while the synthetic cores were made of a variety of materials such as polymers, gold, and silica.[4–7] These biomimetic nanoparticles have enabled novel and promising therapeutic strategies in cancer drug delivery, biodetoxification, and vaccine development.[3,6–8]
From nanoparticle synthesis point of view, however, the existing cell membrane-cloaked nanoparticles were exclusively prepared through a nanoparticle-templated membrane coating route.[9,10] That is, synthetic nanoparticles were pre-made and used as cores to template the subsequent membrane coating. In this process, whether the membranes can be effectively coated onto the cores is determined by the interfacial interactions between the biological membranes and the synthetic cores.[9,10] This indicates that some nanomaterials might not be coatable because of their unfavorable surface properties.[10] In contrast to this nanoparticle-templated membrane coating technology, one would be curious about the possibility of growing nanoparticle cores in situ inside the cell membrane-derived vesicles. Anticipated advantages of such an approach are abundant including ensured complete coating of the cores, facile encapsulation of payloads, and easy control over the size and rigidity of the resulting biomimetic nanoparticles. Inspired by this new nanoparticle synthesis route, herein we demonstrate the first example of using cell membrane-derived vesicles as a template to synthesize polymeric cores via in situ polymerization to prepare cell membrane-coated hydrogel nanoparticles (denoted as “nanogels”).
A critical challenge to synthesize the nanogels is how to selectively and effectively inhibit the polymerization reaction outside of the vesicles while keeping the inside reaction alive.[11,12] To solve this problem, we synthesized a unique macromolecular inhibitor that is impermeable to cell membranes. Figure 1 illustrates the overall concept of this study. Specifically, we use cell membrane-derived vesicles as a nanoreactor to encapsulate the reaction mixture including monomers, crosslinkers, and initiators, which also serve as a template to control the final nanogel size (Figure 1A).[13,14] We choose acrylate polymerization as a model reaction due to its extensive use in hydrogel formulations.[15,16] To prevent possible macrogelation of any unencapsulated precursors outside of the vesicles, we synthesize a membrane-impermeable macromolecular radical scavenger that can selectively inhibit polymerization outside of the vesicles (Figure 1B). After adding the macromolecular inhibitor, gelation is induced by ultraviolet (UV) irradiation, leading to the formation of cell membrane-coated hydrogels, named nanogels (Figure 1C). Instead of wrapping membranes onto pre-formed nanoparticle cores, this nanogel formulation accomplishes membrane coating without the need of knowing the nanoparticle’s coatability and provides unprecedented flexibility to choose materials with desired composition and properties, thereby enabling the cell membrane coating technique for a wider range of applications.
Figure 1.
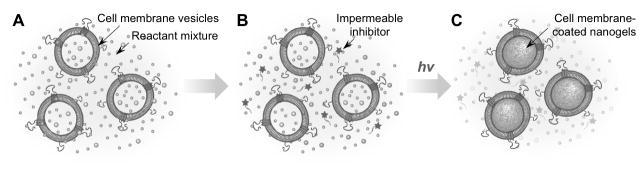
Schematic illustrations showing the preparation procedure of cell membrane-coated nanogels. (A) Cell membrane-derived vesicles are formed from cell membrane ghosts together with desirable monomer, crosslinker, and photo initiator via an extrusion method. (B) The mixture is added with a membrane-impermeable macromolecular inhibitor. (C) The hydrogelation process is then initiated and allowed to proceed under UV light at room temperature.
In the study, we first synthesized the membrane-impermeable macromolecular inhibitor by conjugating (2, 2, 6, 6-Tetramethylpiperidin-1-yl)oxyl (TEMPO), a popular membrane-permeable free radical scavenger, to polyethylene glycol (PEG) (Figure 2A).[17] The use of a macromolecular inhibitor to block the undesirable reaction outside of the cell membrane-derived vesicles simplifies the formulation process and minimizes the risk of protein denaturation and content leakage from the vesicles, particularly when compared to conventional approaches such as dialysis or dilution used in preparation of liposome-containing hydrogels.[18,19] Furthermore, conjugation of PEG is known to effectively prevent small molecules from permeating across cell membranes.[20] Compared to TEMPO conjugated to resins such as silica beads, the use of PEG is also likely to minimize unwanted interactions known to occur between the resin materials and cell membranes.[5,21]
Figure 2.
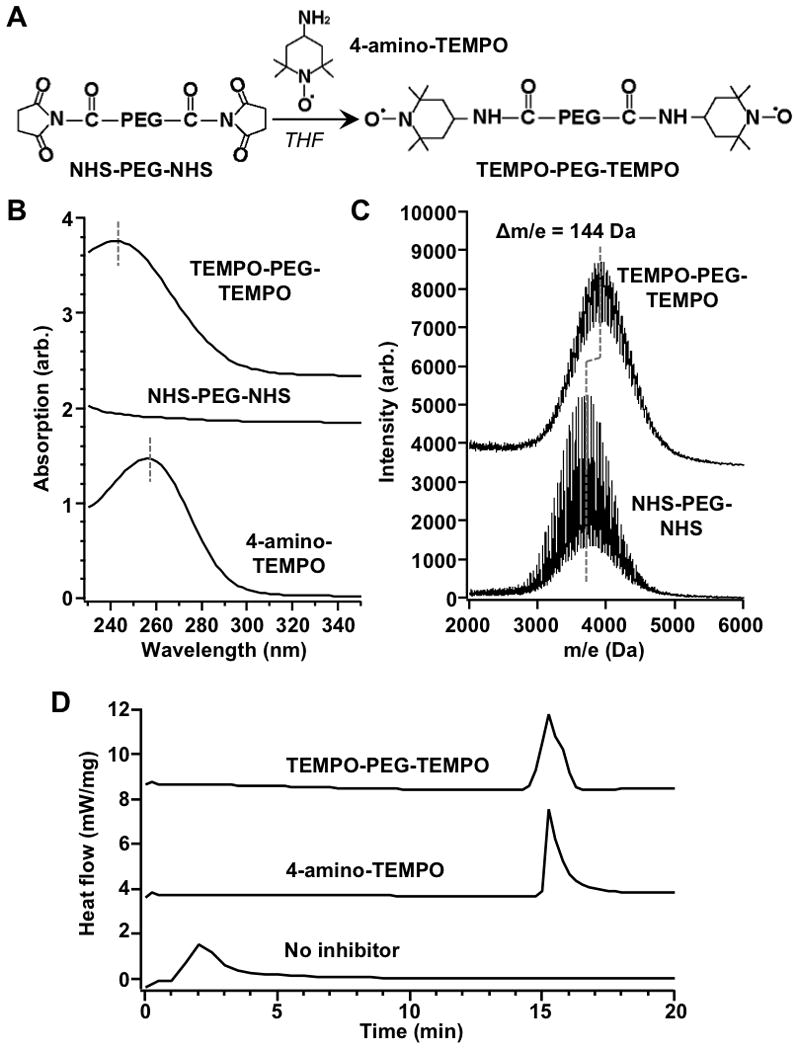
Synthesis and characterization of macromolecular inhibitor, TEMPO-PEG-TEMPO. (A) Schematic synthesis of TEMPO-PEG-TEMPO through EDC/NHS coupling. (B) UV/Vis absorption spectra of TEMPO-PEG-TEMPO, NHS-PEG-NHS and 4-amino-TEMPO. (C) MALDI-TOF MS spectra of TEMPO-PEG-TEMPO and NHS-PEG-NHS. (D) Inhibition capabilities of TEMPO-PEG-TEMPO and 4-amino-TEMPO quantified by DSC.
The conjugation was achieved by using 4-amino-TEMPO and NHS-PEG-NHS (Mw = 3400 Da) through EDC/NHS coupling. The conjugation product was purified through dialysis and then lyophilized. To confirm the coupling between PEG and TEMPO, UV/Vis spectroscopy is first used to analyze the product. As shown in Figure 2B, 4-amino-TEMPO shows a maximum absorption at ~245 nm, whereas the NHS-PEG-NHS does not show obvious absorption at 220–350 nm. After the conjugation, an absorption peak centered at ~258 nm was observed, indicating the coupling between PEG polymer chain and the TEMPO moiety. A slight red shift of the maximum absorption of TEMPO-PEG-TEMPO compared to unconjugated TMEPO is attributed to amide bond formation, which known to disturb the photo-excitation of TEMPO moiety.[22] To further confirm the conjugation reaction, matrix-assisted laser desorption and ionization time-of-flight mass spectrometry (MALDI-TOF MS) was used to analyze the product. Compared to NHS-PEG-NHS, the spectrum of the conjugate shifted 144 Da. This molecular weight increase matches the structural change from NHS-PEG-NHS to TEMPO-PEG-TEMPO, confirming the successful synthesis of the macromolecule inhibitor.
Following the synthesis, we examined the efficiency of TEMPO-PEG-TEMPO in inhibiting the free radical polymerization by using differential scanning calorimetry (DSC) to monitor the exothermic heat of the reaction. Under our experiment conditions, without adding inhibitors to the reactant mixture, the exothermic peak was observed approximately 1 min after heating. With the presence of 4-amino-TEMPO, the exothermic peak was not observed until approximately 15 minutes after heating, a significant delay suggesting an effective polymerization inhibition by TEMPO. We next added equivalent amount of TEMPO-PEG-TEMPO to the reactant mixture and also observed the exothermic peak with a similar delay. These results demonstrate that the ability of TEMPO in scavenging free radicals was maintained after PEG conjugation and the TEMPO-PEG-TEMPO conjugates were as effective as the free TEMPO in inhibiting acrylate polymerization.
After having synthesized the macromolecular inhibitor, we proceeded to formulate the cell membrane-coated nanogels. We chose red blood cells (RBC) membrane as a model membrane due to the extensive studies and broad applications of RBC-membrane-coated nanoparticles.[23] In his study, purified mouse RBCs underwent a hypotonic treatment to remove their intracellular contents.[4] The resulting RBC ghosts were then added into the solution containing acrylamide as the monomer, N,N′-methylenebis acrylamide as the crosslinker, and lithium phenyl-2,4,6-trimethylbenzoylphosphinate as the initiator.[24,25] The mixture was extruded through 100 nm porous membranes to generate RBC membrane-derived vesicles containing the reactants within the vesicles. Note that the same concentration of the reactants remains in the solution outside of the vesicles as well. In addition, the extrusion process was performed in PBS with Mg2+ and Ca2+, a condition long established to prevent the formation of in-side-out RBC vesicles.[26,27] Following the vesicle formation, TEMPO-PEG-TEMPO was added into the solution and the hydrogelation process was initiated and proceeded under irradiation with 365 nm UV light at room temperature for 5 min.
The resulting RBC nanogels were stained with uranyl acetate and visualized with transmission electron microscopy (TEM). As shown in Figure 3A, the nanogels showed a closed thin shell structure with a thickness of approximately 10~15 nm. After treatment with Triton X-100 and proteinase K, the thin shell disappeared, likely due to the dissolution of the cell membrane. Meanwhile, dark spherical particles were observed, attributable to the synthetic nanogel cores. These morphological characteristics are consistent with the core-shell structure of the nanogel. Next, we encapsulated a fluorescent dye, calcein (excitation/emission = 495/515 nm) to the precursor solution followed by nanogel preparation. The calcein-loaded nanogels were then treated with Trition X-100, followed by filtration through an Amicon ultra filter with a molecular weight cutoff of 100 kDa to remove the released calcein. The corresponding RBC membrane-derived vesicles were tested in parallel as a control group. It was found that significantly lower amount of calcein was leaked out of the nanogel particles as compared to the vesicles (Figure 3B), indicating the formation of the hydrogel cores and their capability to effectively retain encapsulated cargoes.
Figure 3.

Confirming the formation of RBC membrane-coated nanogels. (A) Representative TEM images RBC membrane-coated nanogels and the nanogel cores after the treatment with Triton X-100 and proteinase K (scale bar = 100 nm). (B) RBC membrane-derived vesicles and RBC membrane-coated nanogels were loaded with calcein, treated with Triton X-100, filtered to collect the released calcein. The released calcein from both samples was then quantify by a fluorecsent spectrophotometer. (C) Size and size distribution of RBC membrane-derived vesicles and the same vesicles treated by Triton X-100 and proteinase K measured by DLS. (D) Size and size distribution of RBC membrane-coated nanogels and the same nanogels treated by Triton X-100 and proteinase K measured by DLS.
To further confirm the nanogel formation, we used dynamic light scattering (DLS) to analyze nanoparticle sizes.[28,29] Prior to the UV irradiation, the RBC membrane-derived vesicles exhibited a hydrodynamic size of 101 ± 0.2 nm in diameter with a unimodal size distribution (Figure 3C). When added with Triton-X and proteinase K, the size of the vesicles dramatically reduced to 10.5 ± 0.3 nm in diameter, as the added surfactants solubilized the phospholipid bilayers to form small lipid micelles.[28,29] On the other hand, upon exposure to UV light the vesicles showed a diameter of 105 ± 0.2 nm with a similar size distribution to that of the un-irradiated vesicles (Figure 3D). However, when Triton-X and proteinase K were added to the nanogel solution, two particle peaks were observed: a small micelle peak at 9.8 ± 0.1 nm and a large particle peak with a diameter of 94 ± 0.3 nm. These results can be explained by the formation of cross-linked hydrogel cores within RBC membrane vesicles: while the cell membrane shell was still soluble with the surfactant, the crosslinked hydrogel cores were not. The insoluble part of the nanogel had an approximate 11 nm decrease in diameter as compared to the entire nanogel particles, which was consistent with the dissolution of the RBC membrane layer.[4] Together, these results confirmed the successful formation of the RBC membrane-coated nanogels via the cell membrane-templated polymerization approach.
Herein, we report a facile approach of synthesizing cell membrane-coated nanogels by using cell membrane-derived vesicles to template inner core gelation. Compared to the existing strategy of wrapping cell membrane onto pre-formed nanoparticle substrates, this new approach uses vesicles to ‘guide’ the core formation, hence overcoming the restriction imposed by ‘coatability’ of nanoparticle cores and adding significant flexibility to the synthesis and use of cell membrane-coated nanoparticle platform. To further simplify the formulation process, we synthesized a membrane-impermeable macromolecular inhibitor that quenches macrogelation of unencapsulated precursors outside of the vesicles without an effect on the polymerization within the vesicles. This selective inhibition approach eliminates potential risks for content leakage and protein denaturation during the nanogel preparation process. While we chose acrylate polymerization as a model system for this study, the principle can be readily extended to many other crosslinking mechanisms and different types of materials. By properly selecting polymerizable building blocks, a range of physicochemical properties and responsiveness can be introduced into the nanogel platform.[30,31] Meanwhile, various other types of cell membranes collected from RBCs,[32,33] leukocytes,[5] cancer cells,[6] and bacteria[7] can be used to prepare the corresponding cell membrane-coated nanogels for more versatile applications. As the nanogel design becomes increasingly sophisticated, the compatibility among gelation precusors, polymerization inhibitors, and various types of payloads needs to be considered. In this regard, novel chemistry for advanced hydrogel synthesis is rapidly emerging, providing gelation methods with superior biocompatibility, high efficiency, precise localization, and versatile controllability.[34, 35] Overall, the reported cell membrane-templated gelation technique is anticipated to result in a new class of biomimetic nanoparticles that will open some unique opportunities for a broad range of biomedical applications.
Experimental Section
Materials
For synthesis, 4-amino-2,2,6,6-tetramethylpiperidine-1-oxyl (4-aimno-TEMPO), triethylamine (TEA), and tetrahydrofuran (THF) were purchased from Sigma-Aldrich (St. Louis, USA). Succinimidyl succinate-poly(ethylene glycol)-succinimidyl succinate (NHS-PEG-NHS, average molecular weight = 3400 g mol−1) was purchased from Laysan Bio, Inc. (Alabama, USA). For inhibition efficiency study, thermal initiator azodiisobutyronitrile (AIBN) and dimethyl sulfoxide (DMSO) were also purchased from Sigma-Aldrich. For polymerization, acrylamide (monomer) and N,N′-methylenebis(acrylamide) (cross-linker) were purchased from Sigma-Aldrich. Triton X-100 was purchased from Fisher Scientific (Fair Lawn, USA). The photoinitiator lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP) was prepared by following a published protocol.[24,25]
Synthesis and characterization of TEMPO-PEG-TEMPO
The inhibitor TEMPO-PEG-TEMPO was synthesized by conjugating HNS-PEG-NHS and 4-amino-TEMPO in THF with the presence of TEA. Typically, 100 mg of NHS-PEG-NHS (about 0.03 mmol) and 20 mg of 4-amino-TEMPO (about 0.12 mmol) were dissolved in 5 mL of THF. Then 10 μL of TEA was added. The reaction mixture was stirred for 24 h at room temperature under nitrogen condition, followed by solvent removal through evaporation. The remaining brown viscous liquid was repeatedly washed with hexane to remove unreacted 4-amino-TEMPO. The conjugation product was further purified through dialysis and then lyophilized. The final TEMPO-PEG-TEMPO product was collected as a pale brown powder and its molecular weight was confirmed by MALDI-TOF MS.
Inhibition capability study
The inhibition capability of 4-aimno-TEMPO and TEMPO-PEG-TEMPO was compared by monitoring the exothermic heat of acrylate polymerization with DSC (TA-DSC 2920, TA Instrument, New Castle, USA). Specifically, 4-aimno-TEMPO and TEMPO-PEG-TEMPO were added to a mixture solution of acrylamide (200 mg/mL) and AIBN (50 mg/mL) in DMSO, reaching final concentrations of 0.03 and 0.06 mmol/g, respectively. For DSC, 10 ± 0.2 mg of sample was used. The initial temperature was set at 60°C. The heating rate was maintained at 5°C min−1 under nitrogen atmosphere at a flow rate of 80 mL min−1.
Nanogel preparation
RBC vesicles were prepared by following a previously published protocol.[1,3] Briefly, whole blood withdrawn from male ICR mice (Charles River Laboratories, Wilmington, MA) was centrifuged at ×800 g for 5 min at 4°C to remove the plasma and the buffy coat. The resulting packed RBCs were washed and the RBC ghosts were prepared through a hypotonic medium treatment. To prepare RBC vesicles as a polymerization template, the collected RBC ghosts were mixed with acrylamide (0.71 mmol), N,N′-methylenebis acrylamide (0.03 mmol), and LAP (5 μmol) in PBS containing 1 mM Mg2+ and 1 mM Ca2+. Then the mixture was sonicated for 5 min with a bath sonicator (FS30D, Fisher Scientific) at a frequency of 42 kHz and power of 100 W, and then extruded serially through 400 nm and then 100 nm polycarbonate porous membranes with an Avanti mini extruder (Avanti Polar Lipids). Following the extrusion, TEMPO-PEG-TEMPO (0.007 mmol) was added into the solution. After thoroughly deoxygenating with blowing nitrogen gas, the solution was irradiated with UV light for 5 min (Black Ray® Lamp, 365 nm, model UVL-56, Upland, USA).
Nanogel characterization
Transmission electron microscopy (TEM) imaging was carried out by first glow discharging carbon-coated 400 square mesh copper grids (Electron Microscopy Sciences). Particle suspensions (25 μL) were left on the grid for 1 min before being washed off with 10 drops of water. Grids were then negatively stained with 3 drops of 1% uranyl acetate (Sigma Aldrich). For filtration study, the polymerization precursor solution was added with calcein (excitation/emission = 495/515 nm) (5 mg/mL), followed by the same synthesis procedure. Unencapsulated calcein was removed by dialysis using a dialyzer (Harvard Apparatus, Holliston, USA) equipped with a dialysis membrane (0.05 μm Isopore Membrane filters, Merck Millipore Ltd. Billerica, USA). The RBC membrane-derived vesicles and the RBC membrane-coated nanogel samples were then treated with Trition X-100 and filtered through an Amicon ultra filter with a molecular weight cutoff of 100 kDa. The fluorescence intensity of the filtrates was measured with a fluorescent spectrophotometer (Infinite M200, TECAN, Switzerland). The dynamic light scattering (DLS) measurements were carried on a Zetasizer Nano ZS (model ZEN3600 from Malvern Instruments). To dissolve RBC membranes, the solutions were added with 0.25 mL 5 wt% Triton X-100 together with 20 μg Proteinase K (New England Biolabs, Inc., Beverly, USA).
Acknowledgments
This work is supported by the National Institute of Diabetes and Digestive and Kidney Diseases of the National Institutes of Health under Award Number R01DK095168 and by the Defense Threat Reduction Agency Joint Science and Technology Office for Chemical and Biological Defense under Grant Number HDTRA1-14-1-0064. J.Z. acknowledges support by National Natural Science Foundation of China (No. 31470925) and China Scholarship Council (No. 201308120084).
Contributor Information
Dr. Jianhua Zhang, Department of Nanoengineering and Moores Cancer Center, University of California, San Diego, La Jolla, CA 92093, USA. Department of Polymer Science and Engineering, School of Chemical Engineering and Technology, Tianjin University, Tianjin, 300072, China
Dr. Weiwei Gao, Department of Nanoengineering and Moores Cancer Center, University of California, San Diego, La Jolla, CA 92093, USA
Dr. Ronnie H. Fang, Department of Nanoengineering and Moores Cancer Center, University of California, San Diego, La Jolla, CA 92093, USA
Prof. Anjie Dong, Department of Polymer Science and Engineering, School of Chemical Engineering and Technology, Tianjin University, Tianjin, 300072, China
Prof. Liangfang Zhang, Email: zhang@ucsd.edu, Department of Nanoengineering and Moores Cancer Center, University of California, San Diego, La Jolla, CA 92093, USA
References
- 1.Hu CMJ, Zhang L, Aryal S, Cheung C, Fang RH, Zhang L. Proc Natl Acad Sci U S A. 2011;108:10980. doi: 10.1073/pnas.1106634108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hu CMJ, Fang RH, Copp J, Luk BT, Zhang L. Nat Nanotechnol. 2013;8:336. doi: 10.1038/nnano.2013.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hu CMJ, Fang RH, Luk BT, Zhang L. Nat Nanotechnol. 2013;8:933. doi: 10.1038/nnano.2013.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gao W, Hu CMJ, Fang RH, Luk BT, Su J, Zhang L. Adv Mater. 2013;25:3549. doi: 10.1002/adma.201300638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Parodi A, Quattrocchi N, van de Ven LA, Chiappini C, Evangelopoulos M, Martinez JO, Brown BS, Khaled SZ, Yazdi IK, Vittoria Enzo M, Isenhart L, Ferrari M, Tasciotti E. Nat Nanotechnol. 2013;8:61. doi: 10.1038/nnano.2012.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fang RH, Hu CMJ, Luk BT, Gao W, Copp JA, Tai Y, O’Connor DE, Zhang L. Nano Lett. 2014;14:2181. doi: 10.1021/nl500618u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gao W, Fang RH, Thamphiwatana S, Luk BT, Li J, Angsantikul P, Zhang Q, Hu CMJ, Zhang L. Nano Lett. 2015;15:1403. doi: 10.1021/nl504798g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Copp JA, Fang RH, Luk BT, Hu CMJ, Gao W, Zhang K, Zhang L. Proc Natl Acad Sci U S A. 2014;111:13481. doi: 10.1073/pnas.1412420111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hu CMJ, Fang RH, Luk BT, Chen KNH, Carpenter C, Gao W, Zhang K, Zhang L. Nanoscale. 2013;5:2664. doi: 10.1039/c3nr00015j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Luk BT, Hu CMJ, Fang RH, Dehaini D, Carpenter C, Gao W, Zhang L. Nanoscale. 2014;6:2730. doi: 10.1039/c3nr06371b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Raemdonck K, Demeester J, De Smedt S. Soft Matter. 2009;5:707. [Google Scholar]
- 12.Sasaki Y, Akiyoshi K. Chem Lett. 2012;41:202. [Google Scholar]
- 13.Sugawara A, Yamane S, Akiyoshi K. Macromol Rapid Commun. 2006;27:441. [Google Scholar]
- 14.Hong JS, Vreeland WN, DePaoli Lacerda SH, Locascio LE, Gaitan M, Raghavan SR. Langmuir. 2008;24:4092. doi: 10.1021/la7031219. [DOI] [PubMed] [Google Scholar]
- 15.Annabi N, Tamayol A, Uquillas JA, Akbari M, Bertassoni LE, Cha C, Camci-Unal G, Dokmeci MR, Peppas NA, Khademhosseini A. Adv Mater. 2014;26:85. doi: 10.1002/adma.201303233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Buwalda SJ, Boere KWM, Dijkstra PJ, Feijen J, Vermonden T, Hennink WE. J Control Release. 2014;190:254. doi: 10.1016/j.jconrel.2014.03.052. [DOI] [PubMed] [Google Scholar]
- 17.Vogler T, Studer A. Synthesis-Stuttgart. 2008:1979. [Google Scholar]
- 18.Park J, Wrzesinski SH, Stern E, Look M, Criscione J, Ragheb R, Jay SM, Demento SL, Agawu A, Limon PL, Ferrandino AF, Gonzalez D, Habermann A, Flavell RA, Fahmy TM. Nat Mater. 2012;11:895. doi: 10.1038/nmat3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liang Y, Kiick KL. Polym Chem. 2014;5:1728. [Google Scholar]
- 20.Gao W, Langer R, Farokhzad OC. Angew Chem Int Ed. 2010;49:6567. doi: 10.1002/anie.201001868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fey T, Fischer H, Bachmann S, Albert K, Bolm C. J Org Chem. 2001;66:8154. doi: 10.1021/jo010535q. [DOI] [PubMed] [Google Scholar]
- 22.Marshall DL, Hansen CS, Trevitt AJ, Bin Oh H, Blanksby SJ. PCCP. 2014;16:4871. doi: 10.1039/c3cp54825b. [DOI] [PubMed] [Google Scholar]
- 23.Gao W, Zhang L. AIChE J. 2015;61:738. [Google Scholar]
- 24.Fairbanks BD, Schwartz MP, Bowman CN, Anseth KS. Biomaterials. 2009;30:6702. doi: 10.1016/j.biomaterials.2009.08.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ma Y, Thiele J, Abdelmohsen L, Xu J, Huck WTS. Chem Commun. 2014;50:112. doi: 10.1039/c3cc46733c. [DOI] [PubMed] [Google Scholar]
- 26.Steck TL, Weinstei Rs, Straus JH, Wallach DFH. Science. 1970;168:255. doi: 10.1126/science.168.3928.255. [DOI] [PubMed] [Google Scholar]
- 27.Lew VL, Hockaday A, Freeman CJ, Bookchin RM. J Cell Biol. 1988;106:1893. doi: 10.1083/jcb.106.6.1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kazakov S, Kaholek M, Teraoka I, Levon K. Macromolecules. 2002;35:1911. [Google Scholar]
- 29.Kazakov S, Kaholek M, Kudasheva D, Teraoka I, Cowman MK, Levon K. Langmuir. 2003;19:8086. [Google Scholar]
- 30.Mitra A, Imae T. Biomacromolecules. 2004;5:69. doi: 10.1021/bm034239u. [DOI] [PubMed] [Google Scholar]
- 31.Toita S, Sawada S-i, Akiyoshi K. J Control Release. 2011;155:54. doi: 10.1016/j.jconrel.2010.12.008. [DOI] [PubMed] [Google Scholar]
- 32.Piao JG, Wang L, Gao F, You YZ, Xiong Y, Yang L. ACS Nano. 2014;8:10414. doi: 10.1021/nn503779d. [DOI] [PubMed] [Google Scholar]
- 33.Fan Z, Zhou H, Li PY, Speer JE, Cheng H. J Mater Chem B. 2014;2:8231. doi: 10.1039/c4tb00980k. [DOI] [PubMed] [Google Scholar]
- 34.Hennink WE, van Nostrum CF. Adv Drug Deliv Rev. 2012;64:223. doi: 10.1016/s0169-409x(01)00240-x. [DOI] [PubMed] [Google Scholar]
- 35.Seliktar D. Science. 2012;336:1124. doi: 10.1126/science.1214804. [DOI] [PubMed] [Google Scholar]