

Abstract
The lung must maintain a proper barrier between airspaces and fluid filled tissues in order to maintain lung fluid balance. Central to maintaining lung fluid balance are epithelial cells which create a barrier to water and solutes. The barrier function of these cells is mainly provided by tight junction proteins known as claudins. Epithelial barrier function varies depending on the different needs within the segments of the respiratory tree. In the lower airways, fluid is required to maintain mucociliary clearance, whereas in the terminal alveolar airspaces a thin layer of surfactant enriched fluid lowers surface tension to prevent airspace collapse and is critical for gas exchange. As the epithelial cells within the segments of the respiratory tree differ, the composition of claudins found in these epithelial cells is also different. Among these differences is claudin-18 which is uniquely expressed by the alveolar epithelial cells. Other claudins, notably claudin-4 and claudin-7, are more ubiquitously expressed throughout the respiratory epithelium. Claudin-5 is expressed by both pulmonary epithelial and endothelial cells. Based on in vitro and in vivo model systems and histologic analysis of lungs from human patients, roles for specific claudins in maintaining barrier function and protecting the lung from the effects of acute injury and disease are being identified. One surprising finding is that claudin-18 and claudin-4 control lung cell phenotype and inflammation beyond simply maintaining a selective paracellular permeability barrier. This suggests claudins have more nuanced roles for the control of airway and alveolar physiology in the healthy and diseased lung.
Keywords: claudin, tight junction, pulmonary edema, cystic fibrosis, alveolarization, airway, alveolus, vascular endothelium
Graphical Abstract
1. Introduction
Epithelia form selective barriers that regulate inner luminal organ spaces, including those found in the respiratory system. Key among the structures which promote epithelial barrier function are tight junctions, structures at cell-cell interfaces composed of transmembrane, cytosolic and cytoskeletal proteins that interact in a coordinated manner to regulate tissue barriers. The core tight junction transmembrane proteins are the claudins. Claudins on adjacent cells associate with each other to form paracellular channels of varying permeability that create a barrier by regulating the paracellular compartment between cells. Importantly, although the cell composition varies throughout the respiratory tree, epithelial tight junctions provide a unified cell-mediated barrier between airspaces and fluid containing tissues.
Several reviews on tight junctions and claudins in the lung have been published over the last few years [1–3]. This review provides an update to current knowledge about the importance of claudin diversity for lung barrier function. This is underscored by recently characterized claudin knockout mouse models demonstrating that multiple claudins play a crucial role in preserving lung barrier function.
2. Multiplicity of epithelial cells lining the respiratory tract
As a conduit for gas exchange, the respiratory tract consists of functionally distinct segments ranging from the conducting airways to the terminal airspaces where gas exchange occurs (Figure 1). The conducting airways are further divided into functionally distinct anatomical segments: the upper, lower, and distal airways. The upper airway consists of the nasal and oral cavities, pharynx, and larynx and is predominantly involved in sensory function, air humidification and gross filtration of airborne particulates. The lower airway begins with the trachea which then branches into two primary bronchi. Primary bronchi extend and branch through secondary and tertiary bronchi followed by 5–20 generations of bronchioles. The bronchial tree provides a semi-rigid cartilaginous framework to support the terminal airspaces that have the flexibility and compliance required for air influx and efflux.
Figure 1. Epithelial diversity along the respiratory tree.

A. Airways are divided into four main segments: the trachea, the branching bronchi, the terminal bronchi, and the alveolar space. Each segment contains a unique mix of cell types that have specialized functions. B. The lower airway, proximal to the bifurcation of the left and right bronchus, consists of mostly ciliated cells whose main function is to sweep mucus secreted by goblet cells (blue) out of the airways. Columnar and other cells (e.g. serous cells) also contribute to the airway barrier. Basal cells (not shown) are localized to the basement membrane but do not contribute to the tight junction barrier. C. Distal to the tracheal bifurcation are bronchiolar cells that consist mainly of ciliated, columnar and club cells. Club cells secrete a specialized form of pulmonary surfactant as opposed to mucus and provide a transition zone between the airway and alveolar space. D. The alveolar space is the location of gas exchange and consists mainly of squamous type I and cuboidal type II cells. Tight junctions between these cells form at apical cell-cell interaction sites. The alveolar sac maintains surface tension through surfactant secreted by type II cells preventing alveolar collapse. E. Gas exchange occurs efficiently through type I cells, which make up the vast majority of alveolar surface area.
The main airspace surface of the trachea, as well as conducting airways distal to the trachea, are lined by a pseudostratified epithelium which includes ciliated cells, mucus secreting goblet cells, columnar cells, serous cells and basal cells which altogether form a permeability barrier. Conducting airways are also peppered with submucosal glands containing goblet, duct, and serous cells which contribute to mucus secretion. A major function of the lower airway is clearance of particulates from the lower airspaces. This is facilitated by the concerted efforts of goblet cells and ciliated cells that serve as a “mucociliary escalator” moving foreign material upward and out of the lung [4]. Regulation of the airway surface liquid (ASL) underneath the mucus layer is critical to proper mucociliary clearance [5]. Airway epithelial cells maintain ASL balance by the concerted action of plasma membrane channels (which regulate transcellular fluid and ion transport) and tight junctions (which regulate the paracellular route). Roles for claudins in regulating paracellular ion and water diffusion are described below.
Conducting lower airways are kept open to the atmosphere by supportive cartilaginous rings which gradually thin as the airway diameter decreases. At the most distal portion of the conducting airways are terminal bronchioles, the smallest branches of the conducting airway with a diameter of 5 mm or less. Terminal bronchioles interconnect the conducting airways with the terminal airspaces, known as alveoli, where gas exchange occurs. In contrast to the conducting airways, the terminal airspaces of the alveolar epithelium are coated with pulmonary surfactant which serves to reduce the surface tension of the air filled alveoli under the pressure of the fluid filled tissue. Thus, bronchioles serve as transition zones from the mucus dominated airspace as reflected by their distinct cellular composition [6, 7]. In contrast to larger bronchioles which are dominated by ciliated and goblet cells, terminal bronchioles lack submucosal glands and are enriched for club cells (formerly known as Clara cells, [8]). Instead of mucus, club cells are secretory cells that produce a form of pulmonary surfactant containing surfactant protein A (SP-A), SP-B and Club Cell specific protein 10 (CC-10) [9]. The surfactant produced by club cells differs from surfactant produced by alveolar epithelial cells, which lacks CC-10 but instead contains SP-C and therefore has different surface active properties [10].
The terminal airspaces are covered by the alveolar epithelium, which consists of type I and type II alveolar epithelial cells (Figure 1). Although type II alveolar epithelial cells constitute 60% of the alveolar cells, they only cover <5% of the alveolar surface because of their size and cuboidal shape [11]. Conversely, type I alveolar epithelial cells are large, squamous and extremely thin to allow the diffusion of gases between the terminal airspaces and capillaries. Given their large surface area, type I cell junctions are mainly responsible for alveolar epithelial barrier function [3]. However, pulmonary microvascular endothelial cell tight junctions also contribute to alveolar barrier function by maintaining a tight seal to prevent leakage from the bloodstream. Failure of the microvascular barrier while the alveolar epithelial barrier is maintained leads to tissue edema [12, 13], whereas failure of the alveolar epithelial barrier causes airspace flooding and increases susceptibility to acute respiratory distress syndrome (ARDS) [14]. Besides the prevention of fluid leak, alveolar epithelial cells maintain lung fluid balance by regulating ion transport to promote fluid resorption, comparable to conducting airway epithelium [15–17].
A key point in considering intercellular junctions throughout lung epithelia is that their nature as intermixed cell populations (such as pseudostratified monolayers) means that they are enriched for heterocellular interfaces between cells of different phenotype. Thus, in order to form functional tight junctions, cells of different phenotype that are in direct contact need to express compatible claudins. Consistent with this, Flynn et al. [18] showed by confocal immunofluorescence microscopy that claudin-1, -3, and -7 localize to tight junctions formed between goblet and ciliated airway epithelial cells. Moreover, type I and type II alveolar epithelial cells were shown to have multiple claudins localized to tight junctions despite having differential claudin expression [19, 20].
3 Claudin structure and composition in tight junctions
Tight junctions are protein complexes that connect neighboring cells with each other to create a barrier that regulates paracellular diffusion of ions and solutes. Morphologically, tight junctions encircle the cell and connect them with their surrounding cells [21]. Freeze fracture electron microscopy showed that tight junctions appear as a meshwork of strands at the apical end of the lateral cell membrane that differ in complexity and continuity, provoking the conclusion that the amount of strands correlate with the degree of sealing. However, studies have shown that the number of horizontally arranged strands does not always correlate with increased barrier function [22, 23] suggesting that the protein composition [24] and dynamics [25] within tight junctions are crucial for regulating barrier function.
3.1 Claudin family proteins
Claudins are a family of tetraspan transmembrane proteins that form the structural basis for tight junction permeability [26–28]. Topographical prediction for all claudin proteins demonstrate the same secondary structure: four transmembrane (TM) domains, N- and C-terminal domains (NT and CT, respectively) oriented towards the cytosol, two extracellular (EC) domains and a one short intracellular loop domain. In humans, there are 27 claudins which range in size from 207 to 305 amino acids (with apparent molecular weights between 21 and 34 kDa). Based on sequence homology, mammalian claudins are sub-divided into so-called classic and non-classic claudins [29]. The classic claudins show a high degree of structural similarity, particularly in the second EC domain, and generally have a short C-terminal cytosolic domain. By contrast, the second EC domain of non-classic claudins has more heterogeneity [29]. Non-classic claudins also tend to have longer C-terminal domains, suggesting the potential for increased interaction with cytoplasmic scaffold proteins. In the lung, the most prevalent classic claudins confirmed to be expressed are claudin-1, -3, -4, -5, and -7 [1, 30]. Claudin-1 expression in the developing and adult human lung is strikingly enriched in the lower airway bronchial epithelium throughout development but not in the small transitional airways, unlike claudin-4 and -7 which are expressed throughout the airway [31, 32]. Other classic claudins likely to be present include claudin-8 and claudin-10 [33–36]. By far, the most prevalent non-classic claudin in lung is claudin-18 which is highly and specifically expressed by the alveolar epithelium yet undetectable in normal airways [1, 37].
In order to form a functional barrier, claudins interact with each other within the same cellular membrane (polymeric cis-interactions) and between two cells via head to head interactions (polytypic trans-interactions). Claudin interactions can be classified as homomeric (polymeric cis-interaction) and homotypic (trans-interaction) when the polymer consists of the same claudin. Heteromeric or heterotypic claudin interactions occur between two different claudins either in the same cell or between two adjacent cells, respectively.
Claudins have also been categorized by whether they have the capacity to form paracellular channels (pore forming) or whether they restrict paracellular permeability (sealing claudins) [29, 38]. Lung epithelial cells tend to favor expression of sealing claudins (e.g. claudin-1, -3,-4, -5, -7 and -18), and do not express high levels of pore forming claudins (claudin-2, -10, -15) [1, 3]. Since the surface liquid that lines the lung epithelium is critical to maintain function, sealing claudins are likely to serve an important role in limiting uncontrolled leak of water, ions, and metabolites between the apical and basolateral compartments. However, there is a report that the pore forming claudin-10 is specifically expressed by club cells, although the functional significance of this is not known at present [34]. Also, whether claudin-7 is an anion channel forming or sealing claudin is somewhat controversial [38] and likely depends upon the context of expression since cells co-expressing compatible claudins have the capacity to form paracellular channels with unique permeability characteristics. For example, claudin-4 and claudin-8 interact to form anion channels [39]. Also, claudin-16 and claudin-19 have an obligate heteromeric interaction required to form cation channels [40]. Roles for heteromeric and/or heterotypic interactions between claudins in maintaining a healthy barrier in the human lung remain to be determined.
3.2 High resolution structure of claudins
Based on orientation and functional evidence, claudin extracellular domains have been demonstrated to regulate paracellular permeability and trans claudin-claudin interactions between adjacent cells [41, 42]. Understanding the basis for extracellular claudin-claudin interactions was illuminated when the structure of mouse claudin-15 was determined with a crystal diffraction resolution of 2.4 Å [43] (Figure 2). In this structure, it was shown that claudins are formed by four TM domains that form a left-handed four helix bundle. Except for the TM3 domain, the length of the other TM domains matched the diameter of the lipid bilayer underscoring that claudins are firmly embedded into the plasma membrane. Interestingly, the EC domains of claudin-15 were not loops but in fact formed a β-sheet structure that consists of five β-strands. Four of these β-strands are formed by the EC1 domain and the fifth β-strand is provided by the EC2 domain (Figure 2). Cysteine residues within EC1 stabilize the β-sheet structure, as predicted by biochemical analysis [44]. The EC1 domain was suggested to be responsible for the charge-selective permeability of claudins [44, 45]. This hypothesis is supported by the structure of claudin-15 [42]. Homology modeling revealed a similar EC conformation for other ion selective channels such as claudin-10b [43].
Figure 2. Structure of claudin ion selective pores.
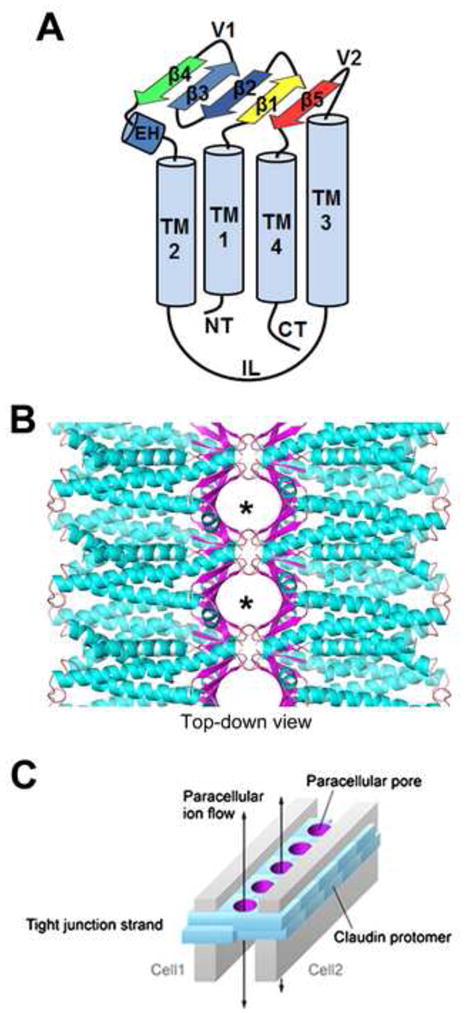
A. Claudin proteins are multi-pass transmembrane proteins that contain intracellular amino terminal (NT) and carboxy terminal (CT) ends, four transmembrane domains (TM1-4), an intracellular loop (IL) and an extracellular (EC) β-sheet domain where interactions between claudins occur. The EC domains consist of a small extracellular α-helix (EH) and five anti-parallel β-strands (β1–5) which form the interacting β-sheet. Based on this structural model, two variable region loops (V1 and V2) are positioned to regulate heterotypic interactions. B. The EC β-sheet (purple) interacts to form paracellular ion or metabolite selective pores (asterisks), where the specific amino acids of the β-sheets comprise the pore lining residues that confer ion/molecule selectivity. C. A simplified schematic of the paracellular pore structures (purple) formed by homo- or heterotypic interactions between claudins. Figure modified from [42] with permission.
3.3 Structural determinants of claudin-claudin interactions
Earlier studies suggested homo- and heterotypic claudin interactions are determined by the EC domains [46–48]. Suzuki et al. [43] found variable regions within the EC domains between the β-strands, variable region 1 (V1, between β-strand 3 and 4) and variable region 2 (V2, between TM3 and β-strand 5), suggesting that V1 and V2 loop regions were involved in hetero- and homotypic interactions of claudin-15 [42] (Figure 2). Cis interactions were suggested to be mediated by interactions between EC1 and TM3. Residue M68 located in the EC1 helix fits into a pocket formed by residues F146, F147 and L158 located in the extracellular part of TM3 and the beginning of the fifth β-strand allowing to form a polymer [42]. In addition, the structure revealed that the claudin-15 monomer contains complementary electrostatic potentials on opposite sides of the molecule which allow claudin-15 to form a linear polymer (cis interactions) within the same membrane. These findings are in apparent contrast to studies of claudin-2 which inferred a role for TM2 in cis-interactions [49], although this could reflect claudin-specific differences in regulating cis interactions. Moreover, posttranslational modifications such as palmitoylation that promote partitioning into cholesterol-enriched membrane microdomains also have the potential to influence claudin cis interactions [50].
3.4 Regulation of claudin assembly by other tight junction proteins
High resolution structural models of claudins do not yet incorporate other components of tight junctions which are critical for tight junction assembly [51]. This includes other classes of transmembrane proteins known to regulate tight junction formation, such as MARVEL proteins (e.g. occludin [52–54]) and Ig superfamily proteins (e.g. Junctional Adhesion Molecule-A (JAM-A) [55]; Coxsackie and Adenovirus Receptor (CAR) [56]).
Occludin, an important regulator of tight junction stability and function, is under the transcriptional control of TTF1/NKX2.1 [57], which is a critical transcription factor required for lung development that also regulates transcription of claudin-1 [57] and claudin-6 [58]. Although this suggests the potential for coordinate regulation of occludin and these claudins, roles for claudin-1 and claudin-6 in lung development are not known at present.
Occludin also biochemically interacts with claudins in tight junction strands [52, 59]. Consistent with a role in regulating paracellular permeability, decreased lung occludin has been associated with several conditions known to promote pulmonary edema, including exposure to environmental toxicants, ventilator induced lung injury (VILI) and chronic exposure to alcohol [60–64]. Also, rats pretreated with a PKCα inhibitor (bisindolylmaleimide) were protected from VILI, which correlated with an increase in total lung occludin protein [65]. Whether diminished occludin expression associated with VILI has an effect on claudins was not determined nor was it determined whether occludin was necessary for protection from VILI.
Dozens of different cytosolic proteins have been found to associate with the tight junction complex [66, 67]. Cytosolic tight junction proteins can be subdivided into those containing PDZ binding domains and those that do not. PDZ binding proteins primarily serve as a structural backbone to crosslink junction-associated proteins, although several also act as transcription factors [68]. Non-PDZ containing proteins are mainly kinases, GTPases, phosphatases, and transcription factors that control tight junction assembly as well as cell proliferation and differentiation [69, 70].
Cytosolic PDZ domain containing scaffold proteins include Zonula Occludens (ZO) -1, -2, and -3 and the PALS/PATJ polarity complex which interacts with crumbs family transmembrane proteins [68, 71]. Interestingly, crumbs3-deficient mice have a significant lung phenotype, where the lungs are filled with proteinaceous material and there is significant epithelial blebbing, however, tight junctions in crumbs3-deficient mice appear morphologically normal [72]. Whether lung epithelial tight junctions are impaired in crumbs3-deficient mice or whether their claudin composition is altered has not yet been determined.
ZO-1 and ZO-2 are the most widely studied scaffold proteins. They are known to directly bind to PDZ motifs on the extreme C-terminus found on nearly all claudins and function to link them to the actin cytoskeleton [73]. Roles for ZO-1 and ZO-2 in promoting lung epithelial barrier function are inferred by decreased ZO-1 and ZO-2 associated with conditions where the alveolar barrier is impaired [61, 62, 64, 74–76]. Nonetheless, ZO-1 and ZO-2 provide more than an inert scaffold for tight junction assembly, since MDCK cells deficient in both ZO-1 and ZO-2 were still able to form tight junctions with a transepithelial resistance comparable to wild type cells [77]. However, it was shown that the permeability to small molecules was significantly increased in ZO-1+ZO-2 deficient cells due to a higher apical contractility of the perijunctional actin-myosin ring [77]. Previously, it was hypothesized that paracellular flux of molecules larger than the actual claudin pore is accomplished by partial strand breaks that are regulated through contraction of the perijunctional actin-myosin ring [78], but elimination of the more active perijunctional actin-myosin ring in the ZO-1+ZO-2 deficient MDCK cells could not diminish the increased paracellular permeability [77]. These findings suggest a likely role for ZO-1 and ZO-2 in regulating claudin dynamics and turnover by controlling interactions with the cytoskeleton as opposed to merely serving as a direct crosslink between claudins and filamentous actin [3, 69].
Scaffold proteins underlie the mechanism behind the observation that JAM-A deficient mice have permeability defects in both the gut [79] and the lung [80]. The effects of JAM-A deficiency on the alveolar epithelium are claudin-specific as revealed by the effects of mild systemic endotoxemia, which specifically disrupted the assembly of claudin-18 into tight junctions, but claudin-4 was unaffected [80]. This was downstream of the effects of endotoxemia on ZO-1 and ZO-2, which were significantly more disrupted in JAM-A deficient mice than wild type controls and consistent with a model where claudin-18 is more tightly associated with these scaffold proteins than claudin-4 [19]. Moreover, these data suggest that, in contrast with occludin, JAM-A does not directly interact with claudins but instead, JAM-A serves to recruit cytoplasmic scaffold proteins where they promote assembly of claudins and other transmembrane proteins into tight junctions [81].
ZO-1 also provides a link between airway barrier function and the Cystic Fibrosis Transmembrane Regulator (CFTR) chloride channel [82, 83], which is important for maintenance of airway surface liquid composition [5, 84, 85], mucociliary clearance [86, 87], and innate immune responses. [4, 88, 89]. There are considerable data demonstrating that airway epithelial cells derived from CF patients have deficits in barrier function as compared to cells expressing wild type CFTR [90–92]. However, measures of tight junction permeability such as transepithelial resistance are sensitive to whether primary cells are being examined or a cell line model is being used. The method of immortalization can also influence barrier function, so results obtained in vitro do need to be interpreted with caution [93]. Biochemical data support a role for CFTR directly interacting with ZO-1 via a PDZ motif [82] as well as indirectly through another scaffold protein, Na+/H+ exchanger regulatory factor 1 (NHERF1) [83]. This parallels the scaffold protein recruitment function of JAM-A and provides a putative mechanism for impaired epithelial paracellular barrier function in CF. The pathologic consequences of a deficit in airway epithelial barrier function remain to be determined, but changes in tight junction permeability are likely to contribute to the overall fluid imbalance that is a hallmark of CF [5].
4. Roles for specific claudins in lung epithelial function
4.1 Claudin-18
There are two main splice variants of claudin-18, one of which is mainly expressed in the lung (Claudin-18.1) and the other is predominantly expressed in the stomach (Claudin-18.2) [37]. The stomach and lung isoforms of claudin-18 differ slightly in their NT, TM1 and EC1 domains. The EC1 domains of the lung and stomach isoforms are 87% identical, however differences in key specific EC1 amino acids might result in subtle changes in specificity of paracellular permeability. Based on MDCK cells overexpressing the stomach isoform, claudin-18.2 has a selective sealing function against sodium and protons but not chloride [94]. Whether claudin-18.1 has a comparable selectivity remains to be determined.
Claudin-18.1 in the lung is overwhelmingly expressed by the alveolar epithelium, suggesting a key role in regulating pulmonary barrier function [35, 95]. Thus to investigate roles for claudin-18.1 in lung barrier function, two different groups produced claudin-18 deficient mice, one of which was a deletion of exons 2 and 3 [96] and the other was a 17.3 kb deletion spanning exons 1–5 [97]. Surprisingly, in both cases, claudin-18 deficient mice were not only viable but had a mild apparent phenotype with respect to lung barrier function. This is due to a significant increase in alveolar fluid clearance rate in claudin-18 deficient mice associated with increased epithelial sodium channel (ENaC) and Na+/K+-ATPase activity as well as the activation of the CFTR chloride channel [96]. This increase in fluid clearance also enabled the mice to be resistant to VILI [96]. Although increased channel function correlated with loss of claudin-18.1, it is not known how these pathways are mechanistically linked.
Despite the ability of claudin-18 deficient mice to maintain lung fluid balance, a general permeability defect was demonstrated by assays in vivo using labeled macromolecules such as albumin as tracers that revealed a significantly higher rate of paracellular flux between the airspace and circulation [96, 97]. Interestingly, changes in paracellular flux for tracers such as dextran was not observed in the stomach of claudin-18.2 knockout mice [98], indicating a context or isoform dependent aspect of claudin-18 function in lung as opposed to stomach.
Transmission electron microscopy revealed that junctional architecture between alveolar type I cells is disrupted in the absence of claudin-18, where type I-type I junctions appeared ruffled and splayed as compared to the normal overlapping junctions as observed in wild type mice [97]. Consistent with a role for claudin-18 in regulating alveolar tight junction morphology, Li et al. [96] found by immunofluorescence that junctional ZO-1 had areas exhibiting enlarged zones of separation (or gaps) in the absence of claudin-18 as opposed to normal cells which had few areas with disrupted tight junction morphology. These changes in tight junction morphology correlated with changes in the actin cytoskeleton, where claudin-18 deficient alveolar epithelial cells had an increase in formation of perinuclear aggregates with radial fibers that were connected with the plasma membrane [96]. Actin stress fibers have previously been associated with tight junction destabilization and increased paracellular flux [20, 99–101]. These data also indicate that claudin-18 is likely to regulate tight junction turnover which may be a mechanism to directly control paracellular flux, which is underscored by the finding that in claudin-18 deficient mice, claudin-3 and claudin-4 were markedly increased while occludin was significantly decreased [96]. A potential clue to the mechanism for this effect comes from the observation that plasma levels for the pro-inflammatory cytokine IL-1β are significantly increased in claudin-18 deficient mice [98]. IL-1β has been demonstrated to cause pulmonary inflammation and is associated with emphysema and aberrant airway remodeling [102, 103], impaired airway barrier function [104], and decreased claudin-18 incorporation into alveolar epithelial tight junctions [105].
How claudin-18 deficiency leads to changes in lung ion channel activity and function is not known however, there are significant changes in cell architecture and gene expression as a result of loss of claudin-18 [96, 97]. In particular, loss of claudin-18 significantly decreased in mRNA levels of the transcription factor early growth response-1 (Egr-1) [106]. Egr-1 deficient mice also show lower sensitivity to VILI, suggesting that alveolar ion channels may be under the control of Egr-1. LaFemina et al. [97] also showed that claudin-18 is important for postnatal lung development. By gross histology, claudin-18 deficient mice showed impaired alveolarization after 4 weeks post natal as compared to 3 day old claudin-18 knockout mice which were largely normal in appearance. Bronchiolar lavage fluid of claudin-18 deficient mice was enriched for the alveolar type I cell type specific protein podoplanin, indicating significant type I cell damage [97]. Claudin-18 knockout mice had alveolar type II cell hyperplasia as well, suggesting a potential deficiency in alveolar type I cell differentiation. Clearly claudin-18 plays diverse and complex roles in regulating the alveolar epithelium, and perhaps lung epithelial differentiation in general, which is likely to be an active area of investigation over the next decade.
4.2 Claudin-4
Claudin-4 is expressed by epithelial cells throughout the respiratory tree, as detected by immunohistochemistry of fetal and adult lungs [31, 32]. The effects of claudin-4 on paracellular permeability are context dependent. Depending on overall tight junction composition, claudin-4 has been shown to serve either as selective sodium barrier without affecting chloride selectivity [107, 108], as a chloride pore forming claudin [39] or as a barrier forming claudin [19].
Studies using an adenoviral expression system to increase claudin-4 expression by primary rat alveolar epithelial cells within the physiologically relevant range showed a ~30% increase in transepithelial resistance without any changes in either the PNa/PCl permeability ratio or paracellular flux [19]. Conversely, siRNA knockdown of claudin-4 in human distal lung epithelial cells decreased transepithelial resistance [103]. However, alveolar epithelial cells from claudin-4 knockout mice lacked a change in transepithelial resistance or ion permeability whereas paracellular flux to large fluorescent dextran (155 kDa) tracers was increased [109]. The difference between acute, partial claudin-4 depletion in human cells and long term complete claudin-4 deficiency may reflect compensatory remodeling of tight junctions or may be due to cell or species specific differences in tight junction permeability, comparable to what was observed in claudin-18 deficient mice. Also, whether claudin-4 has the same influence on paracellular permeability in airway cells versus alveolar cells has not been directly examined.
Physiologically, claudin-4 has been associated with a protective effect against lung injury [19, 103, 110]. In patients, increased claudin-4 levels were associated with better lung fluid clearance and decreased physiological respiratory impairment [110]. Consistent with this, claudin-4 is upregulated 12–16-fold in response to mechanical trauma (VILI) or hyperoxia in mice [103, 109]. One hypothesis for the beneficial effect of claudin-4 in promoting fluid clearance is that it provides a pathway for paracellular chloride efflux to balance sodium transport through channels such as ENaC [15].
Lungs of claudin-4 deficient mice are morphologically normal and have minor defects in steady state fluid balance, which was largely attributed to decreased Na+/K+-ATPase activity [109]. Nonetheless, claudin-4 deficient mice showed increased sensitivity to VILI and lung injury due to hyperoxia, where lungs became significantly edematous. However, these results were confounded by the mice being on a mixed background of BALB/c, 129S6/SvEvTac and C57BL/6 [109] which is known to have an impact on severity of acute lung injury and oxidative stress [111]. Analysis of mice on a uniform background will enable strain dependent differences in roles for claudin-4 to be elucidated.
A protective role for claudin-4 was also inferred using a blocking peptide derived from Clostridium perfringens enterotoxin (CPE), which exacerbated the injurious effects of high tidal volume mechanical ventilation on alveolar permeability and impaired fluid clearance [103]. These data led to the expectation that claudin-4 deficient mice would have a serious impairment in lung epithelial barrier function. However, as was the case for claudin-18 deficient mice, loss of claudin-4 resulted in a mild lung phenotype even in response to injury [109].
Gene expression analysis of injured claudin-4 deficient mice revealed increased activation of genes downstream of TNF and IL-1β, however, in order to see this effect, the mice needed to be stratified into “high” vs “low” injury groups based on having post injury bronchioalveolar lavage (BAL) fluid protein greater than 1.2 mg/ml [109]. Severely injured claudin-4 deficient mice also showed increased Egr1 [109]. These differences intriguingly parallel the injurious effect of increased IL-1β and protective effect of low Egr1 in claudin-18 deficient mice [96, 98], which further supports roles for lung claudins in regulating cell physiology beyond control of paracellular permeability.
4.3 Claudin-7
Claudin-7 is expressed throughout the respiratory tract during development and in the adult lung [31, 32]. The permeability characteristics of claudin-7 have proven to be difficult to elucidate, indicating that claudin-7 function is largely context sensitive [38]. A role for claudin-7 in restricting paracellular chloride permeability was initially inferred by examining LLC-PK1 cells overexpressing claudin-7 which showed a simultaneous decrease in chloride and increase in sodium permeability [112]. By contrast, claudin-7 depletion of mIMCD-3 cells resulted in a 40% increase in chloride permeability and a 45% increase in sodium permeability, suggesting a more general sealing function for claudin-7 [39]. As a further complication, claudin-7 permeability appears to be sensitive to phosphorylation by WNK4 kinase. WNK4 kinase phosphorylation of claudin-7 correlated with increased chloride permeability [113]. However, it was not established whether this was due to a conformational change in claudin-7 or whether phosphorylation altered the ability of claudin-7 to incorporate into tight junctions.
Claudin-7 deficient mice die within two weeks after birth [114] and so their lung phenotype has not been interrogated. Claudin-7 deficiency leads to an overall defect in water homeostasis and the mice suffer from chronic dehydration [114]. Furthermore, both the small and large intestine of the claudin-7 knockout mouse showed signs of inflammation downstream of increased expression and activity of extracellular matrix remodeling enzymes matrix metalloprotease-3 (MMP-3) and MMP-7, which was recapitulated by silencing claudin-7 by siRNA in a lung epithelial cell line [115]. MMPs in general are associated with increased severity of lung injury [116] suggesting a potential mechanism where loss of claudin-7 could amplify ARDS. Conversely, EGF has a protective effect with is associated with increased claudin-7 and claudin-4 expression and function [117].
In claudin-7 deficient mice, expression of other claudins that are important for kidney function were not affected, although claudin-1 localization in the gut is impaired, primarily due to a defect in formation of a complex with claudin-7 and integrin α2 [115]. However, there were several changes in ion and water channels, including induction of ENaC, aquaporin-2 and Na+/K+-ATPase-α1 which together led to increased sodium absorption. Increased sodium absorption, in concert with decreased chloride secretion, underlies a defect in airway surface liquid (ASL) that is a hallmark of CF [118, 119]. The concordance between the claudin-7 and CFTR-deficient epithelium raises the possibility that claudin-7 may play a role in regulating apical chloride, perhaps by reducing paracellular chloride flux. Consistent with this, in CF there is a 4-fold decrease in passive intestinal chloride absorption that acts as a potential compensatory mechanism [120]. Whether this occurs in the airway and whether the paracellular route plays a role in regulating chloride balance is not known at present. However, this also raises the potential for a therapeutic approach to CF by targeting claudin expression as a means to either reduce chloride efflux from the apical surface and/or augmenting paracellular chloride flux from the basal to apical surface as a means to compensate for loss of chloride secretion by CFTR.
4.4 Claudin-5
Claudin-5 is most prominently expressed by the vascular endothelium and the pulmonary microcirculation is no exception [30, 31]. The phenotype of claudin-5 deficient mice reflects a function for this claudin in sealing the vasculature, since these mice have a selective blood brain barrier permeability defect which allows small molecules (<0.8 kDa) to penetrate into the cerebral spinal fluid resulting in immediate perinatal lethality [121]. Claudin-5 is also clearly expressed by the human airway epithelium [31, 32, 122]. The high levels of claudin-5 expressed by the endothelium tend to obscure the ability to detect claudin-5 expression by alveolar epithelial cells in histologic sections [30, 35], however, claudin-5 mRNA and protein are readily measurable in isolated fetal human alveolar epithelial cells [95]. Rodent alveolar epithelial cells also express claudin-5 [19, 123–125].
The alveolar air-liquid barrier reflects the combined function of tight junctions in both the pulmonary microvasculature and the alveolar epithelium [17, 126, 127]. Because of the dual nature of this barrier, the close apposition of alveolar endothelial and epithelial tight junctions and the fact that both tissues express claudin-5 has led to some apparently contradictory data related to roles for claudin-5 in regulating alveolar barrier function. In brief, claudin-5 is likely to have a protective effect by increasing the barrier function of the endothelial part of the barrier [128–133]. However, increased claudin-5 expression decreases alveolar epithelial barrier function [100, 122, 125].
Related to the endothelial barrier, there was no effect on barrier function at baseline in studies using pulmonary artery endothelial cells in vitro where claudin-5 was silenced using siRNA [128]. However, cells treated with claudin-5 siRNA had increased permeability to fluorescein (0.4 kDa) after thrombin treatment as compared to controls expressing normal levels of claudin-5. Simvastatin, which normally bolsters endothelial barrier function, also had diminished efficacy as demonstrated by increased paracellular flux of fluorescein into the alveolar space, although transepithelial resistance and large molecule permeability were not affected [128], consistent with the claudin-5 deficient mouse model [121]. Critically, intratracheal claudin-5 siRNA administered in vivo did not change small molecule permeability at baseline and had only a small protective effect on endotoxin induced lung injury [128]. Nonetheless, total lung claudin-5 tends to inversely correlate with severity of lung injury. For example, studies assessing different mouse strains to acrolein-induced acute lung injury (ALI) showed that claudin-5 expression was increased in more injury-resistant strains as compared to injury-sensitive strains [132]. In HIV-1 infected patients with interstitial pneumonitis, claudin-5 was down-regulated in lung tissue [133]. Further experiments in a mouse model of HIV-infection confirmed this finding which was associated with increased lung infiltration of CD4+ and CD8+ monocytes in HIV-1 infected mice [133].
A potential clue to the dual nature of the alveolar barrier comes from studies using the cancer chemotherapeutic agent imatinib that promotes blood brain barrier function and thus might be expected to have a protective effect in ARDS [134]. However, imatinib treated mice have a complex response to lung injury in that they are protected from endotoxin induced lung injury but have an increased sensitivity to VILI [135]. Although the mechanism underlying this divergence in response is not known at present, we speculate that this is due to differential regulation of the endothelial and epithelial parts of the alveolar barrier.
In support of a deleterious role for claudin-5 on the epithelial barrier, several studies have associated increased claudin-5 expression by lung epithelial cells with decreased barrier function [100, 122, 125]. This is particularly striking in alcoholic lung syndrome, which is associated with increased susceptibility to ARDS [136–139]. In a rat model of chronic alcohol abuse, claudin-5 protein expressed by type II alveolar epithelial cells was significantly increased [124]. This increase in claudin-5 persisted in model type I cells derived from “alcoholic” type II cells which also exhibited impaired barrier function as compared with cells derived from control fed rats. A more direct demonstration of claudin-5 was revealed when airway cells were transfected to overexpress claudin-5, which decreased transepithelial resistance [122]. These data are suggestive; however, further work is needed to identify a molecular basis for the ability of claudin-5 to impair alveolar epithelial barrier function and to assess whether specifically targeting epithelial claudin-5 is a therapeutically viable option for promoting lung barrier function.
4.5 Claudin-3
Claudin-3 is expressed in the developing human lung in both the airway and alveoli. Expression in the developing and adult human lung is ubiquitous upon transition to the canalicular period of branching bronchi biogenesis (weeks 16–28) and beyond, staining most epithelia present in the respiratory tree including type II alveolar epithelial cells [31, 32]. By contrast, claudin-3 expression by type I cells is relatively low compared with other lung epithelia in general and especially in comparison with type II cells [20, 31, 32]
Adenoviral expression of claudin-3 in primary rat model type I alveolar epithelial cell at the level found in type II cells led to decreased transepithelial resistance, increased short circuit currents, and higher paracellular flux of calcein (0.6 kDa) and dextran (10 kDa) [19]. This contrasts with other studies, where increased claudin-3 expression by other types of epithelial cells and NIH/3T3 fibroblasts has an overall sealing function [140, 141]. These apparently conflicting data are likely to reflect a context sensitivity for claudin-3 function that differs for alveolar epithelial cells vs. other cell types. Moreover, claudin-3 is primarily found in type I-type II cell junctions in the alveolus. Since type II cells cover less than 5% of the alveolar surface [11], type II-type I tight junctions provide a relatively small part of the alveolar barrier as compared to type I-type I tight junctions which have vanishingly low levels of claudin-3 [20]. This suggests the hypothesis that claudin-3 enables type I-type II cell junctions to have unique characteristics distinct from type I-type I cell junctions [3]. Interestingly, claudin-3 is unique in that it mediates several heterotypic trans interactions between claudins, including claudin-1, claudin-2 and claudin-5 [142, 143]. By contrast, claudin-3 and claudin-4 do not have a trans interaction, however, they do interact as cis-heteromers [142, 144]. Specific roles for claudin-3, and particularly whether its unique heterotypic/heteromeric compatibility is needed to form fully functional type I–type II cell junctions, remain to be determined.
5. Summary and perspectives
There is a growing literature addressing how individual claudins regulate lung barrier function, ranging from in vitro studies to claudin-deficient mice. Analysis of transgenic mice deficient in two different key lung epithelial claudins, claudin-18 and claudin-4, had a surprisingly mild phenotype with respect to barrier function. Of course altering claudin expression to elucidate function is subject to compensatory changes in other tight junction and channel proteins and thus make it difficult to use these approaches to fully interpret specific roles for specific claudins in control of lung barrier function. Moreover, the fact that the lung can withstand large changes in claudin expression likely reflects the critical nature of the air-liquid barrier for gas exchange and blood oxygenation in that there are several overlapping mechanisms to prevent airspace flooding and clear fluid from the airspace.
Claudin-4 and claudin-18 deficient mice also show phenotypes beyond a simple deficit in barrier function. Alveolar epithelial gene expression is significantly changed in both cases and there is an increase in pro-inflammatory responses, which make the lung more susceptible to injury. It remains to be determined whether increased inflammation is directly downstream to changes in barrier function. Alternatively, inflammation could be independent of changes to tight junction permeability and instead due to changes in the control of lung cell phenotype due to loss of claudin-4 or claudin-18.
There are hints that other claudins also may have non-junctional roles in regulating lung inflammation as well. For instance, claudin-1 was detected in human airway smooth muscle cells, which lack tight junctions, where gene transcription correlated with improved cellular survival and proliferation upon stimulation with pro-inflammatory cytokines IL-1β and TNF-α [145]. What this means for the airway epithelial barrier is unknown at present, but claudin-1 may have a role in the pathogenesis of asthma, based on the location of expression by airway smooth muscle cells.
The morphologic abnormality in claudin-18 deficient mice is profound and suggests that alveolarization may be under the direct control of claudin-18. A role for claudin-18 in alveolar formation also raises the possibility that interfering with claudin-18 expression and/or function may underlie diseases such as pulmonary fibrosis or chronic obstructive pulmonary disorder (COPD), where alveolar morphology is pathologically disrupted.
Roles for claudins in airway barrier function remain relatively less explored than for the alveolus, particularly when considering claudins beyond the most prominently expressed alveolar claudins. For instance, claudin-1 and claudin-8 were specifically down regulated in so-called healthy smokers, which was recapitulated in cigarette smoke extract treated airway cells in vitro, suggesting that changes in claudin expression may presage more severe lung disease or cancer [33]. Expression of the pore forming claudin-10 by club cells [34] may reflect a unique niche for increased paracellular flux in the most distal part of the airway. For instance, increased paracellular flux between club cells could play a role in transmitting chemotactic signals to promote homing of immune cells for transmigration into the terminal airspaces. Whether this is the case remains to be determined.
As mentioned above, there are several indications that tight junctions are diminished in CF, which is related to at least two roles for CFTR, namely, chloride channel activity and structural recruitment of scaffold proteins to the tight junction complex. With respect to decreased chloride secretion, the paracellular route provides the potential to regulate ion homeostasis by either promoting chloride diffusion or limiting apical chloride efflux. Whether this is therapeutically targetable, particularly in the absence of CFTR structurally integrated into tight junctions remains to be determined. Also, the deleterious effects of misfolded mutants of CFTR need to be considered in addition to the loss of CFTR activity, since ER stress and disruption of protein quality control can potentially have a negative impact on claudin assembly into tight junctions. Understanding the mechanistic basis for claudin trafficking and assembly will help elucidate whether this is the case.
Highlights.
Airway and alveolar epithelial barrier function require tight junctions with distinct properties
Claudin-deficient animal models reveal redundancy in mechanisms which maintain air-liquid barriers
Claudins have roles beyond forming tight junctions that regulate lung morphogenesis and inflammation
Acknowledgments
We thank Dr. Wendy Neveu (Emory) for helpful discussions. This work was supported by NIH R01-HL116958, R01- EB018842 (MK), NIH T32-AA013528 (SAM), MCCART13I0 and MCCART13P0 from the (MK), and the German Academic Exchange Service (DAAD) (BS).
Abbreviations
- ARDS
acute respiratory distress syndrome
- ASL
airway surface liquid
- CAR
Coxsackie and Adenovirus Receptor
- CC10
club cell specific protein 10
- CFTR
cystic fibrosis transmembrane conductance regulator
- COPD
chronic obstructive pulmonary disorder
- EC
extracellular
- ENaC
epithelial sodium channel
- JAM-A
Junctional Adhesion Molecule-A
- MMP
matrix metalloprotease
- PALS
proteins associated with Lin-7
- PATJ
Pals1-associated tight junction protein
- PDZ
PSD95, Dlg1, ZO-1
- SP
surfactant protein
- TM
transmembrane
- VILI
ventilator induced lung injury
- ZO
Zonula Occludens
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Soini Y. Claudins in lung diseases. Respir Res. 2011;12:70. doi: 10.1186/1465-9921-12-70. http://dx.doi.org/10.1186/1465-9921-12-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Frank JA. Claudins and alveolar epithelial barrier function in the lung. Ann N Y Acad Sci. 2012;1257:175–183. doi: 10.1111/j.1749-6632.2012.06533.x. http://dx.doi.org/10.1111/j.1749-6632.2012.06533.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Koval M. Claudin heterogeneity and control of lung tight junctions. Annu Rev Physiol. 2013;75:551–567. doi: 10.1146/annurev-physiol-030212-183809. http://dx.doi.org/10.1146/annurev-physiol-030212-183809. [DOI] [PubMed] [Google Scholar]
- 4.Whitsett JA, Alenghat T. Respiratory epithelial cells orchestrate pulmonary innate immunity. Nat Immunol. 2014;16:27–35. doi: 10.1038/ni.3045. http://dx.doi.org/10.1038/ni.3045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tarran R, Button B, Boucher RC. Regulation of normal and cystic fibrosis airway surface liquid volume by phasic shear stress. Annu Rev Physiol. 2006;68:543–561. doi: 10.1146/annurev.physiol.68.072304.112754. http://dx.doi.org/10.1146/annurev.physiol.68.072304.112754. [DOI] [PubMed] [Google Scholar]
- 6.Crapo JD, Barry BE, Gehr P, Bachofen M, Weibel ER. Cell number and cell characteristics of the normal human lung. Am Rev Respir Dis. 1982;126:332–337. doi: 10.1164/arrd.1982.126.2.332. [DOI] [PubMed] [Google Scholar]
- 7.Crystal RG, Randell SH, Engelhardt JF, Voynow J, Sunday ME. Airway epithelial cells: current concepts and challenges. Proc Am Thorac Soc. 2008;5:772–777. doi: 10.1513/pats.200805-041HR. http://dx.doi.org/10.1513/pats.200805-041HR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Winkelmann A, Noack T. The Clara cell: a “Third Reich eponym”? Eur Respir J. 2010;36:722–727. doi: 10.1183/09031936.00146609. http://dx.doi.org/10.1183/09031936.00146609. [DOI] [PubMed] [Google Scholar]
- 9.Boggaram V. Regulation of lung surfactant protein gene expression. Front Biosci. 2003;8:d751–764. doi: 10.2741/1062. [DOI] [PubMed] [Google Scholar]
- 10.Beers MF, Mulugeta S. Surfactant protein C biosynthesis and its emerging role in conformational lung disease. Annu Rev Physiol. 2005;67:663–696. doi: 10.1146/annurev.physiol.67.040403.101937. http://dx.doi.org/10.1146/annurev.physiol.67.040403.101937. [DOI] [PubMed] [Google Scholar]
- 11.Crapo JD, Young SL, Fram EK, Pinkerton KE, Barry BE, Crapo RO. Morphometric characteristics of cells in the alveolar region of mammalian lungs. Am Rev Respir Dis. 1983;128:S42–46. doi: 10.1164/arrd.1983.128.2P2.S42. [DOI] [PubMed] [Google Scholar]
- 12.Mehta D, Ravindran K, Kuebler WM. Novel regulators of endothelial barrier function. Am J Physiol Lung Cell Mol Physiol. 2014;307:L924–935. doi: 10.1152/ajplung.00318.2014. http://dx.doi.org/10.1152/ajplung.00318.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Albertine KH. Histopathology of Pulmonary Edema and the Acute Respiratory Distress Syndrome. In: Matthay MA, Ingbar DH, editors. Pulmonary Edema. New York, NY: Marcel Dekker, Inc; 1998. pp. 37–83. [Google Scholar]
- 14.Ware LB, Matthay MA. Alveolar fluid clearance is impaired in the majority of patients with acute lung injury and the acute respiratory distress syndrome. Am J Respir Crit Care Med. 2001;163:1376–1383. doi: 10.1164/ajrccm.163.6.2004035. [DOI] [PubMed] [Google Scholar]
- 15.Eaton DC, Helms MN, Koval M, Bao HF, Jain L. The contribution of epithelial sodium channels to alveolar function in health and disease. Annu Rev Physiol. 2009;71:403–423. doi: 10.1146/annurev.physiol.010908.163250. [DOI] [PubMed] [Google Scholar]
- 16.Kim KJ, Malik AB. Protein transport across the lung epithelial barrier. Am J Physiol Lung Cell Mol Physiol. 2003;284:L247–259. doi: 10.1152/ajplung.00235.2002. [DOI] [PubMed] [Google Scholar]
- 17.Mehta D, Bhattacharya J, Matthay MA, Malik AB. Integrated control of lung fluid balance. Am J Physiol Lung Cell Mol Physiol. 2004;287:L1081–1090. doi: 10.1152/ajplung.00268.2004. [DOI] [PubMed] [Google Scholar]
- 18.Flynn AN, Itani OA, Moninger TO, Welsh MJ. Acute regulation of tight junction ion selectivity in human airway epithelia. Proc Natl Acad Sci U S A. 2009;106:3591–3596. doi: 10.1073/pnas.0813393106. http://dx.doi.org/10.1073/pnas.0813393106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mitchell LA, Overgaard CE, Ward C, Margulies SS, Koval M. Differential effects of claudin-3 and claudin-4 on alveolar epithelial barrier function. Am J Physiol Lung Cell Mol Physiol. 2011;301:L40–49. doi: 10.1152/ajplung.00299.2010. http://dx.doi.org/10.1152/ajplung.00299.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lafemina MJ, Rokkam D, Chandrasena A, Pan J, Bajaj A, Johnson M, et al. Keratinocyte growth factor enhances barrier function without altering claudin expression in primary alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2010;299:L724–734. doi: 10.1152/ajplung.00233.2010. http://dx.doi.org/10.1152/ajplung.00233.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schneeberger EE, Walters DV, Olver RE. Development of intercellular junctions in the pulmonary epithelium of the foetal lamb. J Cell Sci. 1978;32:307–324. doi: 10.1242/jcs.32.1.307. [DOI] [PubMed] [Google Scholar]
- 22.Martinez-Palomo A, Erlij D. Structure of tight junctions in epithelia with different permeability. Proc Natl Acad Sci U S A. 1975;72:4487–4491. doi: 10.1073/pnas.72.11.4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Colegio OR, Van Itallie C, Rahner C, Anderson JM. Claudin extracellular domains determine paracellular charge selectivity and resistance but not tight junction fibril architecture. Am J Physiol Cell Physiol. 2003;284:C1346–1354. doi: 10.1152/ajpcell.00547.2002. [DOI] [PubMed] [Google Scholar]
- 24.Stevenson BR, Anderson JM, Goodenough DA, Mooseker MS. Tight junction structure and ZO-1 content are identical in two strains of Madin-Darby canine kidney cells which differ in transepithelial resistance. J Cell Biol. 1988;107:2401–2408. doi: 10.1083/jcb.107.6.2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sasaki H, Matsui C, Furuse K, Mimori-Kiyosue Y, Furuse M, Tsukita S. Dynamic behavior of paired claudin strands within apposing plasma membranes. Proc Natl Acad Sci U S A. 2003;100:3971–3976. doi: 10.1073/pnas.0630649100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tsukita S, Furuse M. Claudin-based barrier in simple and stratified cellular sheets. Curr Opin Cell Biol. 2002;14:531–536. doi: 10.1016/s0955-0674(02)00362-9. [DOI] [PubMed] [Google Scholar]
- 27.Lal-Nag M, Morin PJ. The claudins. Genome Biol. 2009;10:235. doi: 10.1186/gb-2009-10-8-235. http://dx.doi.org/10.1186/gb-2009-10-8-235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gunzel D, Fromm M. Claudins and other tight junction proteins. Compr Physiol. 2012;2:1819–1852. doi: 10.1002/cphy.c110045. http://dx.doi.org/10.1002/cphy.c110045. [DOI] [PubMed] [Google Scholar]
- 29.Krause G, Winkler L, Mueller SL, Haseloff RF, Piontek J, Blasig IE. Structure and function of claudins. Biochim Biophys Acta. 2008;1778:631–645. doi: 10.1016/j.bbamem.2007.10.018. http://dx.doi.org/10.1016/j.bbamem.2007.10.018. [DOI] [PubMed] [Google Scholar]
- 30.Lappi-Blanco E, Lehtonen ST, Sormunen R, Merikallio HM, Soini Y, Kaarteenaho RL. Divergence of tight and adherens junction factors in alveolar epithelium in pulmonary fibrosis. Hum Pathol. 2013;44:895–907. doi: 10.1016/j.humpath.2012.08.016. http://dx.doi.org/10.1016/j.humpath.2012.08.016. [DOI] [PubMed] [Google Scholar]
- 31.Kaarteenaho R, Merikallio H, Lehtonen S, Harju T, Soini Y. Divergent expression of claudin -1, -3, -4, -5 and -7 in developing human lung. Respir Res. 2010;11:59. doi: 10.1186/1465-9921-11-59. http://dx.doi.org/10.1186/1465-9921-11-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kaarteenaho-Wiik R, Soini Y. Claudin-1, -2, -3, -4, -5, and -7 in usual interstitial pneumonia and sarcoidosis. J Histochem Cytochem. 2009;57:187–195. doi: 10.1369/jhc.2008.951566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shaykhiev R, Otaki F, Bonsu P, Dang DT, Teater M, Strulovici-Barel Y, et al. Cigarette smoking reprograms apical junctional complex molecular architecture in the human airway epithelium in vivo. Cell Mol Life Sci. 2011;68:877–892. doi: 10.1007/s00018-010-0500-x. http://dx.doi.org/10.1007/s00018-010-0500-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zemke AC, Snyder JC, Brockway BL, Drake JA, Reynolds SD, Kaminski N, et al. Molecular staging of epithelial maturation using secretory cell-specific genes as markers. Am J Respir Cell Mol Biol. 2009;40:340–348. doi: 10.1165/rcmb.2007-0380OC. http://dx.doi.org/10.1165/rcmb.2007-0380OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ohta H, Chiba S, Ebina M, Furuse M, Nukiwa T. Altered expression of tight junction molecules in alveolar septa in lung injury and fibrosis. Am J Physiol Lung Cell Mol Physiol. 2012;302:L193–205. doi: 10.1152/ajplung.00349.2010. http://dx.doi.org/10.1152/ajplung.00349.2010. [DOI] [PubMed] [Google Scholar]
- 36.Rezaee F, Georas SN. Breaking barriers. New insights into airway epithelial barrier function in health and disease. Am J Respir Cell Mol Biol. 2014;50:857–869. doi: 10.1165/rcmb.2013-0541RT. http://dx.doi.org/10.1165/rcmb.2013-0541RT. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Niimi T, Nagashima K, Ward JM, Minoo P, Zimonjic DB, Popescu NC, et al. claudin-18, a novel downstream target gene for the T/EBP/NKX2. 1 homeodomain transcription factor, encodes lung- and stomach-specific isoforms through alternative splicing. Mol Cell Biol. 2001;21:7380–7390. doi: 10.1128/MCB.21.21.7380-7390.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gunzel D, Yu AS. Claudins and the modulation of tight junction permeability. Physiol Rev. 2013;93:525–569. doi: 10.1152/physrev.00019.2012. http://dx.doi.org/10.1152/physrev.00019.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hou J, Renigunta A, Yang J, Waldegger S. Claudin-4 forms paracellular chloride channel in the kidney and requires claudin-8 for tight junction localization. Proc Natl Acad Sci U S A. 2010;107:18010–18015. doi: 10.1073/pnas.1009399107. http://dx.doi.org/10.1073/pnas.1009399107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hou J, Renigunta A, Konrad M, Gomes AS, Schneeberger EE, Paul DL, et al. Claudin-16 and claudin-19 interact and form a cation-selective tight junction complex. J Clin Invest. 2008;118:619–628. doi: 10.1172/JCI33970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rossa J, Protze J, Kern C, Piontek A, Gunzel D, Krause G, et al. Molecular and structural transmembrane determinants critical for embedding claudin-5 into tight junctions reveal a distinct four-helix bundle arrangement. Biochem J. 2014;464:49–60. doi: 10.1042/BJ20140431. http://dx.doi.org/10.1042/BJ20140431. [DOI] [PubMed] [Google Scholar]
- 42.Suzuki H, Tani K, Tamura A, Tsukita S, Fujiyoshi Y. Model for the architecture of claudin-based paracellular ion channels through tight junctions. J Mol Biol. 2015;427:291–297. doi: 10.1016/j.jmb.2014.10.020. http://dx.doi.org/10.1016/j.jmb.2014.10.020. [DOI] [PubMed] [Google Scholar]
- 43.Suzuki H, Nishizawa T, Tani K, Yamazaki Y, Tamura A, Ishitani R, et al. Crystal structure of a claudin provides insight into the architecture of tight junctions. Science. 2014;344:304–307. doi: 10.1126/science.1248571. http://dx.doi.org/10.1126/science.1248571. [DOI] [PubMed] [Google Scholar]
- 44.Angelow S, Yu AS. Cysteine mutagenesis to study the structure of claudin-2 paracellular pores. Ann N Y Acad Sci. 2009;1165:143–147. doi: 10.1111/j.1749-6632.2009.04038.x. [DOI] [PubMed] [Google Scholar]
- 45.Colegio OR, Van Itallie CM, McCrea HJ, Rahner C, Anderson JM. Claudins create charge-selective channels in the paracellular pathway between epithelial cells. Am J Physiol Cell Physiol. 2002;283:C142–147. doi: 10.1152/ajpcell.00038.2002. [DOI] [PubMed] [Google Scholar]
- 46.Lim TS, Vedula SR, Hunziker W, Lim CT. Kinetics of adhesion mediated by extracellular loops of claudin-2 as revealed by single-molecule force spectroscopy. J Mol Biol. 2008;381:681–691. doi: 10.1016/j.jmb.2008.06.009. http://dx.doi.org/10.1016/j.jmb.2008.06.009. [DOI] [PubMed] [Google Scholar]
- 47.Lim TS, Vedula SR, Hui S, Kausalya PJ, Hunziker W, Lim CT. Probing effects of pH change on dynamic response of Claudin-2 mediated adhesion using single molecule force spectroscopy. Exp Cell Res. 2008;314:2643–2651. doi: 10.1016/j.yexcr.2008.05.015. http://dx.doi.org/10.1016/j.yexcr.2008.05.015. [DOI] [PubMed] [Google Scholar]
- 48.Piehl C, Piontek J, Cording J, Wolburg H, Blasig IE. Participation of the second extracellular loop of claudin-5 in paracellular tightening against ions, small and large molecules. Cell Mol Life Sci. 2010;67:2131–2140. doi: 10.1007/s00018-010-0332-8. http://dx.doi.org/10.1007/s00018-010-0332-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Van Itallie CM, Mitic LL, Anderson JM. Claudin-2 forms homodimers and is a component of a high molecular weight protein complex. J Biol Chem. 2011;286:3442–3450. doi: 10.1074/jbc.M110.195578. http://dx.doi.org/10.1074/jbc.M110.195578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Van Itallie CM, Gambling TM, Carson JL, Anderson JM. Palmitoylation of claudins is required for efficient tight-junction localization. J Cell Sci. 2005;118:1427–1436. doi: 10.1242/jcs.01735. [DOI] [PubMed] [Google Scholar]
- 51.Van Itallie CM, Anderson JM. Architecture of tight junctions and principles of molecular composition. Semin Cell Dev Biol. 2014;36C:157–165. doi: 10.1016/j.semcdb.2014.08.011. http://dx.doi.org/10.1016/j.semcdb.2014.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cording J, Berg J, Kading N, Bellmann C, Tscheik C, Westphal JK, et al. In tight junctions, claudins regulate the interactions between occludin, tricellulin and marvelD3, which, inversely, modulate claudin oligomerization. J Cell Sci. 2013;126:554–564. doi: 10.1242/jcs.114306. http://dx.doi.org/10.1242/jcs.114306. [DOI] [PubMed] [Google Scholar]
- 53.Raleigh DR, Boe DM, Yu D, Weber CR, Marchiando AM, Bradford EM, et al. Occludin S408 phosphorylation regulates tight junction protein interactions and barrier function. J Cell Biol. 2011;193:565–582. doi: 10.1083/jcb.201010065. http://dx.doi.org/10.1083/jcb.201010065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Van Itallie CM, Fanning AS, Holmes J, Anderson JM. Occludin is required for cytokine-induced regulation of tight junction barriers. J Cell Sci. 2010;123:2844–2852. doi: 10.1242/jcs.065581. http://dx.doi.org/10.1242/jcs.065581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Severson EA, Parkos CA. Structural determinants of Junctional Adhesion Molecule A (JAM-A) function and mechanisms of intracellular signaling. Curr Opin Cell Biol. 2009;21:701–707. doi: 10.1016/j.ceb.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Coyne CB, Bergelson JM. CAR: a virus receptor within the tight junction. Adv Drug Deliv Rev. 2005;57:869–882. doi: 10.1016/j.addr.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 57.Runkle EA, Rice SJ, Qi J, Masser D, Antonetti DA, Winslow MM, et al. Occludin is a direct target of thyroid transcription factor-1 (TTF-1/NKX2-1) J Biol Chem. 2012;287:28790–28801. doi: 10.1074/jbc.M112.367987. http://dx.doi.org/10.1074/jbc.M112.367987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jimenez FR, Lewis JB, Belgique ST, Wood TT, Reynolds PR. Developmental lung expression and transcriptional regulation of claudin-6 by TTF-1, Gata-6, and FoxA2. Respir Res. 2014;15:70. doi: 10.1186/1465-9921-15-70. http://dx.doi.org/10.1186/1465-9921-15-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mrsny RJ, Brown GT, Gerner-Smidt K, Buret AG, Meddings JB, Quan C, et al. A key claudin extracellular loop domain is critical for epithelial barrier integrity. Am J Pathol. 2008;172:905–915. doi: 10.2353/ajpath.2008.070698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhao T, Liu M, Gu C, Wang X, Wang Y. Activation of c-Src tyrosine kinase mediated the degradation of occludin in ventilator-induced lung injury. Respir Res. 2014;15:158. doi: 10.1186/s12931-014-0158-2. http://dx.doi.org/10.1186/s12931-014-0158-2. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 61.You K, Xu X, Fu J, Xu S, Yue X, Yu Z, et al. Hyperoxia disrupts pulmonary epithelial barrier in newborn rats via the deterioration of occludin and ZO-1. Respir Res. 2012;13:36. doi: 10.1186/1465-9921-13-36. http://dx.doi.org/10.1186/1465-9921-13-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Caraballo JC, Yshii C, Westphal W, Moninger T, Comellas AP. Ambient particulate matter affects occludin distribution and increases alveolar transepithelial electrical conductance. Respirology. 2011;16:340–349. doi: 10.1111/j.1440-1843.2010.01910.x. http://dx.doi.org/10.1111/j.1440-1843.2010.01910.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lehmann AD, Blank F, Baum O, Gehr P, Rothen-Rutishauser BM. Diesel exhaust particles modulate the tight junction protein occludin in lung cells in vitro. Part Fibre Toxicol. 2009;6:26. doi: 10.1186/1743-8977-6-26. http://dx.doi.org/10.1186/1743-8977-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fan X, Joshi PC, Koval M, Guidot DM. Chronic alcohol ingestion exacerbates lung epithelial barrier dysfunction in HIV-1 transgenic rats. Alcohol Clin Exp Res. 2011;35:1866–1875. doi: 10.1111/j.1530-0277.2011.01531.x. http://dx.doi.org/10.1111/j.1530-0277.2011.01531.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liu M, Gu C, Wang Y. Upregulation of the tight junction protein occludin: effects on ventilation-induced lung injury and mechanisms of action. BMC Pulm Med. 2014;14:94. doi: 10.1186/1471-2466-14-94. http://dx.doi.org/10.1186/1471-2466-14-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gonzalez-Mariscal L, Tapia R, Chamorro D. Crosstalk of tight junction components with signaling pathways. Biochim Biophys Acta. 2008;1778:729–756. doi: 10.1016/j.bbamem.2007.08.018. http://dx.doi.org/10.1016/j.bbamem.2007.08.018. [DOI] [PubMed] [Google Scholar]
- 67.Steed E, Balda MS, Matter K. Dynamics and functions of tight junctions. Trends Cell Biol. 2010;20:142–149. doi: 10.1016/j.tcb.2009.12.002. http://dx.doi.org/10.1016/j.tcb.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 68.Guillemot L, Paschoud S, Pulimeno P, Foglia A, Citi S. The cytoplasmic plaque of tight junctions: a scaffolding and signalling center. Biochim Biophys Acta. 2008;1778:601–613. doi: 10.1016/j.bbamem.2007.09.032. http://dx.doi.org/10.1016/j.bbamem.2007.09.032. [DOI] [PubMed] [Google Scholar]
- 69.Ivanov AI, Parkos CA, Nusrat A. Cytoskeletal regulation of epithelial barrier function during inflammation. Am J Pathol. 2010;177:512–524. doi: 10.2353/ajpath.2010.100168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shen L, Weber CR, Raleigh DR, Yu D, Turner JR. Tight junction pore and leak pathways: a dynamic duo. Annu Rev Physiol. 2011;73:283–309. doi: 10.1146/annurev-physiol-012110-142150. http://dx.doi.org/10.1146/annurev-physiol-012110-142150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pieczynski J, Margolis B. Protein complexes that control renal epithelial polarity. Am J Physiol Renal Physiol. 2011;300:F589–601. doi: 10.1152/ajprenal.00615.2010. http://dx.doi.org/10.1152/ajprenal.00615.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Whiteman EL, Fan S, Harder JL, Walton KD, Liu CJ, Soofi A, et al. Crumbs3 is essential for proper epithelial development and viability. Mol Cell Biol. 2014;34:43–56. doi: 10.1128/MCB.00999-13. http://dx.doi.org/10.1128/MCB.00999-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Umeda K, Ikenouchi J, Katahira-Tayama S, Furuse K, Sasaki H, Nakayama M, et al. ZO-1 and ZO-2 independently determine where claudins are polymerized in tight-junction strand formation. Cell. 2006;126:741–754. doi: 10.1016/j.cell.2006.06.043. [DOI] [PubMed] [Google Scholar]
- 74.Simet SM, Wyatt TA, DeVasure J, Yanov D, Allen-Gipson D, Sisson JH. Alcohol increases the permeability of airway epithelial tight junctions in Beas-2B and NHBE cells. Alcohol Clin Exp Res. 2012;36:432–442. doi: 10.1111/j.1530-0277.2011.01640.x. http://dx.doi.org/10.1111/j.1530-0277.2011.01640.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cohen TS, Gray Lawrence G, Margulies SS. Cultured alveolar epithelial cells from septic rats mimic in vivo septic lung. PLoS One. 2010;5:e11322. doi: 10.1371/journal.pone.0011322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fan X, Staitieh BS, Jensen JS, Mould KJ, Greenberg JA, Joshi PC, et al. Activating the Nrf2-mediated antioxidant response element restores barrier function in the alveolar epithelium of HIV-1 transgenic rats. Am J Physiol Lung Cell Mol Physiol. 2013;305:L267–277. doi: 10.1152/ajplung.00288.2012. http://dx.doi.org/10.1152/ajplung.00288.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fanning AS, Van Itallie CM, Anderson JM. Zonula occludens-1 and -2 regulate apical cell structure and the zonula adherens cytoskeleton in polarized epithelia. Mol Biol Cell. 2012;23:577–590. doi: 10.1091/mbc.E11-09-0791. http://dx.doi.org/10.1091/mbc.E11-09-0791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Turner JR. Molecular basis of epithelial barrier regulation: from basic mechanisms to clinical application. Am J Pathol. 2006;169:1901–1909. doi: 10.2353/ajpath.2006.060681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Laukoetter MG, Nava P, Lee WY, Severson EA, Capaldo CT, Babbin BA, et al. JAM-A regulates permeability and inflammation in the intestine in vivo. J Exp Med. 2007;204:3067–3076. doi: 10.1084/jem.20071416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mitchell LA, Ward C, Kwon M, Mitchell PO, Quintero DA, Nusrat A, et al. Junctional adhesion molecule a promotes epithelial tight junction assembly to augment lung barrier function. Am J Pathol. 2015;185:372–386. doi: 10.1016/j.ajpath.2014.10.010. http://dx.doi.org/10.1016/j.ajpath.2014.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Monteiro AC, Sumagin R, Rankin CR, Leoni G, Mina MJ, Reiter DM, et al. JAM-A associates with ZO-2, Afadin and PDZ-GEF1 to activate Rap2c and regulate epithelial barrier function. Mol Biol Cell. 2013;24:2849–2860. doi: 10.1091/mbc.E13-06-0298. http://dx.doi.org/10.1091/mbc.E13-06-0298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ruan YC, Wang Y, Da Silva N, Kim B, Diao RY, Hill E, et al. CFTR interacts with ZO-1 to regulate tight junction assembly and epithelial differentiation through the ZONAB pathway. J Cell Sci. 2014;127:4396–4408. doi: 10.1242/jcs.148098. http://dx.doi.org/10.1242/jcs.148098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Castellani S, Guerra L, Favia M, Di Gioia S, Casavola V, Conese M. NHERF1 and CFTR restore tight junction organisation and function in cystic fibrosis airway epithelial cells: role of ezrin and the RhoA/ROCK pathway. Lab Invest. 2012;92:1527–1540. doi: 10.1038/labinvest.2012.123. http://dx.doi.org/10.1038/labinvest.2012.123. [DOI] [PubMed] [Google Scholar]
- 84.Reddy MM, Stutts MJ. Status of fluid and electrolyte absorption in cystic fibrosis. Cold Spring Harb Perspect Med. 2013;3:a009555. doi: 10.1101/cshperspect.a009555. http://dx.doi.org/10.1101/cshperspect.a009555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Stoltz DA, Meyerholz DK, Welsh MJ. Origins of cystic fibrosis lung disease. N Engl J Med. 2015;372:351–362. doi: 10.1056/NEJMra1300109. http://dx.doi.org/10.1056/NEJMra1300109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kurbatova P, Bessonov N, Volpert V, Tiddens HA, Cornu C, Nony P, et al. Model of mucociliary clearance in cystic fibrosis lungs. J Theor Biol. 2015;372:81–88. doi: 10.1016/j.jtbi.2015.02.023. http://dx.doi.org/10.1016/j.jtbi.2015.02.023. [DOI] [PubMed] [Google Scholar]
- 87.Hoegger MJ, Fischer AJ, McMenimen JD, Ostedgaard LS, Tucker AJ, Awadalla MA, et al. Impaired mucus detachment disrupts mucociliary transport in a piglet model of cystic fibrosis. Science. 2014;345:818–822. doi: 10.1126/science.1255825. http://dx.doi.org/10.1126/science.1255825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hartl D, Gaggar A, Bruscia E, Hector A, Marcos V, Jung A, et al. Innate immunity in cystic fibrosis lung disease. J Cyst Fibros. 2012;11:363–382. doi: 10.1016/j.jcf.2012.07.003. http://dx.doi.org/10.1016/j.jcf.2012.07.003. [DOI] [PubMed] [Google Scholar]
- 89.Keiser NW, Birket SE, Evans IA, Tyler SR, Crooke AK, Sun X, et al. Defective Innate Immunity and Hyper-Inflammation in Newborn CFTR-Knockout Ferret Lungs. Am J Respir Cell Mol Biol. 2014 doi: 10.1165/rcmb.2014-0250OC. http://dx.doi.org/10.1165/rcmb.2014-0250OC. [DOI] [PMC free article] [PubMed]
- 90.Coyne CB, Vanhook MK, Gambling TM, Carson JL, Boucher RC, Johnson LG. Regulation of airway tight junctions by proinflammatory cytokines. Mol Biol Cell. 2002;13:3218–3234. doi: 10.1091/mbc.E02-03-0134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.LeSimple P, Liao J, Robert R, Gruenert DC, Hanrahan JW. Cystic fibrosis transmembrane conductance regulator trafficking modulates the barrier function of airway epithelial cell monolayers. J Physiol. 2010;588:1195–1209. doi: 10.1113/jphysiol.2009.182246. http://dx.doi.org/10.1113/jphysiol.2009.182246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Weiser N, Molenda N, Urbanova K, Bahler M, Pieper U, Oberleithner H, et al. Paracellular permeability of bronchial epithelium is controlled by CFTR. Cell Physiol Biochem. 2011;28:289–296. doi: 10.1159/000331742. http://dx.doi.org/10.1159/000331742. [DOI] [PubMed] [Google Scholar]
- 93.Molenda N, Urbanova K, Weiser N, Kusche-Vihrog K, Gunzel D, Schillers H. Paracellular transport through healthy and cystic fibrosis bronchial epithelial cell lines--do we have a proper model? PLoS One. 2014;9:e100621. doi: 10.1371/journal.pone.0100621. http://dx.doi.org/10.1371/journal.pone.0100621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Jovov B, Van Itallie CM, Shaheen NJ, Carson JL, Gambling TM, Anderson JM, et al. Claudin-18: a dominant tight junction protein in Barrett’s esophagus and likely contributor to its acid resistance. Am J Physiol Gastrointest Liver Physiol. 2007;293:G1106–1113. doi: 10.1152/ajpgi.00158.2007. [DOI] [PubMed] [Google Scholar]
- 95.Daugherty BL, Mateescu M, Patel AS, Wade K, Kimura S, Gonzales LW, et al. Developmental regulation of claudin localization by fetal alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2004;287:L1266–1273. doi: 10.1152/ajplung.00423.2003. [DOI] [PubMed] [Google Scholar]
- 96.Li G, Flodby P, Luo J, Kage H, Sipos A, Gao D, et al. Knockout mice reveal key roles for claudin 18 in alveolar barrier properties and fluid homeostasis. Am J Respir Cell Mol Biol. 2014;51:210–222. doi: 10.1165/rcmb.2013-0353OC. http://dx.doi.org/10.1165/rcmb.2013-0353OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.LaFemina MJ, Sutherland KM, Bentley T, Gonzales LW, Allen L, Chapin CJ, et al. Claudin-18 deficiency results in alveolar barrier dysfunction and impaired alveologenesis in mice. Am J Respir Cell Mol Biol. 2014;51:550–558. doi: 10.1165/rcmb.2013-0456OC. http://dx.doi.org/10.1165/rcmb.2013-0456OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hayashi D, Tamura A, Tanaka H, Yamazaki Y, Watanabe S, Suzuki K, et al. Deficiency of Claudin-18 Causes Paracellular H(+) Leakage, Up-regulation of Interleukin-1beta, and Atrophic Gastritis in Mice. Gastroenterology. 2011;142:292–304. doi: 10.1053/j.gastro.2011.10.040. http://dx.doi.org/10.1053/j.gastro.2011.10.040. [DOI] [PubMed] [Google Scholar]
- 99.Koval M. Keratinocyte growth factor improves alveolar barrier function: keeping claudins in line. Am J Physiol Lung Cell Mol Physiol. 2010;299:L721–723. doi: 10.1152/ajplung.00365.2010. http://dx.doi.org/10.1152/ajplung.00365.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ward C, Schlingmann B, Stecenko AA, Guidot DM, Koval M. NF-kB inhibitors impair lung epithelial tight junctions in the absence of inflammation. Tissue Barriers. 2015;3:e982424. doi: 10.4161/21688370.2014.982424. http://dx.doi.org/10.4161/21688370.2014.982424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Overgaard CE, Schlingmann B, Dorsainvil White S, Ward C, Fan X, Swarnakar S, et al. The relative balance of GM-CSF and TGFbeta1 regulates lung epithelial barrier function. Am J Physiol Lung Cell Mol Physiol. 2015 doi: 10.1152/ajplung.00042.2014. ajplung 00042 02014. http://dx.doi.org/10.1152/ajplung.00042.2014. [DOI] [PMC free article] [PubMed]
- 102.Lappalainen U, Whitsett JA, Wert SE, Tichelaar JW, Bry K. Interleukin-1beta causes pulmonary inflammation, emphysema, and airway remodeling in the adult murine lung. Am J Respir Cell Mol Biol. 2005;32:311–318. doi: 10.1165/rcmb.2004-0309OC. http://dx.doi.org/10.1165/rcmb.2004-0309OC. [DOI] [PubMed] [Google Scholar]
- 103.Wray C, Mao Y, Pan J, Chandrasena A, Piasta F, Frank JA. Claudin-4 augments alveolar epithelial barrier function and is induced in acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2009;297:L219–227. doi: 10.1152/ajplung.00043.2009. http://dx.doi.org/10.1152/ajplung.00043.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Saatian B, Rezaee F, Desando S, Emo J, Chapman T, Knowlden S, et al. Interleukin-4 and interleukin-13 cause barrier dysfunction in human airway epithelial cells. Tissue Barriers. 2013;1:e24333. doi: 10.4161/tisb.24333. http://dx.doi.org/10.4161/tisb.24333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Fang X, Neyrinck AP, Matthay MA, Lee JW. Allogeneic human mesenchymal stem cells restore epithelial protein permeability in cultured human alveolar type II cells by secretion of angiopoietin-1. J Biol Chem. 2010;285:26211–26222. doi: 10.1074/jbc.M110.119917. http://dx.doi.org/10.1074/jbc.M110.119917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ngiam N, Peltekova V, Engelberts D, Otulakowski G, Post M, Kavanagh BP. Early growth response-1 worsens ventilator-induced lung injury by up-regulating prostanoid synthesis. Am J Respir Crit Care Med. 2010;181:947–956. doi: 10.1164/rccm.200908-1297OC. http://dx.doi.org/10.1164/rccm.200908-1297OC. [DOI] [PubMed] [Google Scholar]
- 107.Van Itallie CM, Fanning AS, Anderson JM. Reversal of charge selectivity in cation or anion-selective epithelial lines by expression of different claudins. Am J Physiol Renal Physiol. 2003;285:F1078–1084. doi: 10.1152/ajprenal.00116.2003. [DOI] [PubMed] [Google Scholar]
- 108.Van Itallie C, Rahner C, Anderson JM. Regulated expression of claudin-4 decreases paracellular conductance through a selective decrease in sodium permeability. J Clin Invest. 2001;107:1319–1327. doi: 10.1172/JCI12464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kage H, Flodby P, Gao D, Kim YH, Marconett CN, DeMaio L, et al. Claudin 4 knockout mice: normal physiological phenotype with increased susceptibility to lung injury. Am J Physiol Lung Cell Mol Physiol. 2014;307:L524–536. doi: 10.1152/ajplung.00077.2014. http://dx.doi.org/10.1152/ajplung.00077.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Rokkam D, Lafemina MJ, Lee JW, Matthay MA, Frank JA. Claudin-4 Levels Are Associated with Intact Alveolar Fluid Clearance in Human Lungs. Am J Pathol. 2011;179:1081–1087. doi: 10.1016/j.ajpath.2011.05.017. http://dx.doi.org/10.1016/j.ajpath.2011.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Walkin L, Herrick SE, Summers A, Brenchley PE, Hoff CM, Korstanje R, et al. The role of mouse strain differences in the susceptibility to fibrosis: a systematic review. Fibrogenesis Tissue Repair. 2013;6:18. doi: 10.1186/1755-1536-6-18. http://dx.doi.org/10.1186/1755-1536-6-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Alexandre MD, Lu Q, Chen YH. Overexpression of claudin-7 decreases the paracellular Cl- conductance and increases the paracellular Na+ conductance in LLC-PK1 cells. J Cell Sci. 2005;118:2683–2693. doi: 10.1242/jcs.02406. [DOI] [PubMed] [Google Scholar]
- 113.Tatum R, Zhang Y, Lu Q, Kim K, Jeansonne BG, Chen YH. WNK4 phosphorylates ser(206) of claudin-7 and promotes paracellular Cl(-) permeability. FEBS Lett. 2007;581:3887–3891. doi: 10.1016/j.febslet.2007.07.014. http://dx.doi.org/10.1016/j.febslet.2007.07.014. [DOI] [PubMed] [Google Scholar]
- 114.Tatum R, Zhang Y, Salleng K, Lu Z, Lin JJ, Lu Q, et al. Renal salt wasting and chronic dehydration in claudin-7-deficient mice. Am J Physiol Renal Physiol. 2010;298:F24–34. doi: 10.1152/ajprenal.00450.2009. http://dx.doi.org/10.1152/ajprenal.00450.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ding L, Lu Z, Foreman O, Tatum R, Lu Q, Renegar R, et al. Inflammation and disruption of the mucosal architecture in claudin-7-deficient mice. Gastroenterology. 2012;142:305–315. doi: 10.1053/j.gastro.2011.10.025. http://dx.doi.org/10.1053/j.gastro.2011.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kim JH, Suk MH, Yoon DW, Lee SH, Hur GY, Jung KH, et al. Inhibition of matrix metalloproteinase-9 prevents neutrophilic inflammation in ventilator-induced lung injury. Am J Physiol Lung Cell Mol Physiol. 2006;291:L580–587. doi: 10.1152/ajplung.00270.2005. http://dx.doi.org/10.1152/ajplung.00270.2005. [DOI] [PubMed] [Google Scholar]
- 117.Chen SP, Zhou B, Willis BC, Sandoval AJ, Liebler JM, Kim KJ, et al. Effects of transdifferentiation and EGF on claudin isoform expression in alveolar epithelial cells. J Appl Physiol. 2005;98:322–328. doi: 10.1152/japplphysiol.00681.2004. [DOI] [PubMed] [Google Scholar]
- 118.Hobbs CA, Da Tan C, Tarran R. Does epithelial sodium channel hyperactivity contribute to cystic fibrosis lung disease? J Physiol. 2013;591:4377–4387. doi: 10.1113/jphysiol.2012.240861. http://dx.doi.org/10.1113/jphysiol.2012.240861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Verkman AS, Song Y, Thiagarajah JR. Role of airway surface liquid and submucosal glands in cystic fibrosis lung disease. Am J Physiol Cell Physiol. 2003;284:C2–15. doi: 10.1152/ajpcell.00417.2002. http://dx.doi.org/10.1152/ajpcell.00417.2002. [DOI] [PubMed] [Google Scholar]
- 120.Russo MA, Hogenauer C, Coates SW, Jr, Santa Ana CA, Porter JL, Rosenblatt RL, et al. Abnormal passive chloride absorption in cystic fibrosis jejunum functionally opposes the classic chloride secretory defect. J Clin Invest. 2003;112:118–125. doi: 10.1172/JCI17667. http://dx.doi.org/10.1172/JCI17667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Nitta T, Hata M, Gotoh S, Seo Y, Sasaki H, Hashimoto N, et al. Size-selective loosening of the blood-brain barrier in claudin-5-deficient mice. J Cell Biol. 2003;161:653–660. doi: 10.1083/jcb.200302070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Coyne CB, Gambling TM, Boucher RC, Carson JL, Johnson LG. Role of claudin interactions in airway tight junctional permeability. Am J Physiol Lung Cell Mol Physiol. 2003;285:L1166–1178. doi: 10.1152/ajplung.00182.2003. [DOI] [PubMed] [Google Scholar]
- 123.Koval M, Ward C, Findley MK, Roser-Page S, Helms MN, Roman J. Extracellular Matrix Influences Alveolar Epithelial Claudin Expression and Barrier Function. Am J Respir Cell Mol Biol. 2010;42:172–180. doi: 10.1165/rcmb.2008-0270OC. http://dx.doi.org/10.1165/rcmb.2008-0270OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Fernandez AL, Koval M, Fan X, Guidot DM. Chronic alcohol ingestion alters claudin expression in the alveolar epithelium of rats. Alcohol. 2007;41:371–379. doi: 10.1016/j.alcohol.2007.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Wang F, Daugherty B, Keise LL, Wei Z, Foley JP, Savani RC, et al. Heterogeneity of claudin expression by alveolar epithelial cells. Am J Respir Cell Mol Biol. 2003;29:62–70. doi: 10.1165/rcmb.2002-0180OC. [DOI] [PubMed] [Google Scholar]
- 126.Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev. 2006;86:279–367. doi: 10.1152/physrev.00012.2005. http://dx.doi.org/10.1152/physrev.00012.2005. [DOI] [PubMed] [Google Scholar]
- 127.Overgaard CE, Mitchell LA, Koval M. Roles for claudins in alveolar epithelial barrier function. Ann N Y Acad Sci. 2012;1257:167–174. doi: 10.1111/j.1749-6632.2012.06545.x. http://dx.doi.org/10.1111/j.1749-6632.2012.06545.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Chen W, Sharma R, Rizzo AN, Siegler JH, Garcia JG, Jacobson JR. Role of claudin-5 in the attenuation of murine acute lung injury by simvastatin. Am J Respir Cell Mol Biol. 2014;50:328–336. doi: 10.1165/rcmb.2013-0058OC. http://dx.doi.org/10.1165/rcmb.2013-0058OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wang L, Chiang ET, Simmons JT, Garcia JG, Dudek SM. FTY720-induced human pulmonary endothelial barrier enhancement is mediated by c-Abl. Eur Respir J. 2011;38:78–88. doi: 10.1183/09031936.00047810. http://dx.doi.org/10.1183/09031936.00047810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Wang C, Armstrong SM, Sugiyama MG, Tabuchi A, Krauszman A, Kuebler WM, et al. Influenza Primes Human Lung Microvascular Endothelium to Leak upon Exposure to Staphylococcus aureus. Am J Respir Cell Mol Biol. 2015 doi: 10.1165/rcmb.2014-0373OC. http://dx.doi.org/10.1165/rcmb.2014-0373OC. [DOI] [PubMed]
- 131.Armstrong SM, Wang C, Tigdi J, Si X, Dumpit C, Charles S, et al. Influenza infects lung microvascular endothelium leading to microvascular leak: role of apoptosis and claudin-5. PLoS One. 2012;7:e47323. doi: 10.1371/journal.pone.0047323. http://dx.doi.org/10.1371/journal.pone.0047323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Jang AS, Concel VJ, Bein K, Brant KA, Liu S, Pope-Varsalona H, et al. Endothelial dysfunction and claudin 5 regulation during acrolein-induced lung injury. Am J Respir Cell Mol Biol. 2011;44:483–490. doi: 10.1165/rcmb.2009-0391OC. http://dx.doi.org/10.1165/rcmb.2009-0391OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Li H, Singh S, Potula R, Persidsky Y, Kanmogne GD. Dysregulation of claudin-5 in HIV-induced interstitial pneumonitis and lung vascular injury. Protective role of peroxisome proliferator-activated receptor-gamma. Am J Respir Crit Care Med. 2014;190:85–97. doi: 10.1164/rccm.201106-1151OC. http://dx.doi.org/10.1164/rccm.201106-1151OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Armulik A, Genove G, Mae M, Nisancioglu MH, Wallgard E, Niaudet C, et al. Pericytes regulate the blood-brain barrier. Nature. 2010;468:557–561. doi: 10.1038/nature09522. http://dx.doi.org/10.1038/nature09522. [DOI] [PubMed] [Google Scholar]
- 135.Letsiou E, Rizzo AN, Sammani S, Naureckas P, Jacobson JR, Garcia JG, et al. Differential and opposing effects of imatinib on LPS- and ventilator-induced lung injury. Am J Physiol Lung Cell Mol Physiol. 2015;308:L259–269. doi: 10.1152/ajplung.00323.2014. http://dx.doi.org/10.1152/ajplung.00323.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Overgaard CE, Daugherty BL, Mitchell LA, Koval M. Claudins: control of barrier function and regulation in response to oxidant stress. Antioxid Redox Signal. 2011;15:1179–1193. doi: 10.1089/ars.2011.3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Berkowitz DM, Danai PA, Eaton S, Moss M, Martin GS. Alcohol abuse enhances pulmonary edema in acute respiratory distress syndrome. Alcohol Clin Exp Res. 2009;33:1690–1696. doi: 10.1111/j.1530-0277.2009.01005.x. http://dx.doi.org/10.1111/j.1530-0277.2009.01005.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Moss M, Parsons PE, Steinberg KP, Hudson LD, Guidot DM, Burnham EL, et al. Chronic alcohol abuse is associated with an increased incidence of acute respiratory distress syndrome and severity of multiple organ dysfunction in patients with septic shock. Crit Care Med. 2003;31:869–877. doi: 10.1097/01.CCM.0000055389.64497.11. [DOI] [PubMed] [Google Scholar]
- 139.Moss M, Steinberg KP, Guidot DM, Duhon GF, Treece P, Wolken R, et al. The effect of chronic alcohol abuse on the incidence of ARDS and the severity of the multiple organ dysfunction syndrome in adults with septic shock: an interim and multivariate analysis. Chest. 1999;116:97S–98S. [PubMed] [Google Scholar]
- 140.Furuse M, Furuse K, Sasaki H, Tsukita S. Conversion of zonulae occludentes from tight to leaky strand type by introducing claudin-2 into Madin-Darby canine kidney I cells. J Cell Biol. 2001;153:263–272. doi: 10.1083/jcb.153.2.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Milatz S, Krug SM, Rosenthal R, Gunzel D, Muller D, Schulzke JD, et al. Claudin-3 acts as a sealing component of the tight junction for ions of either charge and uncharged solutes. Biochim Biophys Acta. 2010;1798:2048–2057. doi: 10.1016/j.bbamem.2010.07.014. [DOI] [PubMed] [Google Scholar]
- 142.Daugherty BL, Ward C, Smith T, Ritzenthaler JD, Koval M. Regulation of heterotypic claudin compatibility. J Biol Chem. 2007;282:30005–30013. doi: 10.1074/jbc.M703547200. [DOI] [PubMed] [Google Scholar]
- 143.Furuse M, Sasaki H, Tsukita S. Manner of interaction of heterogeneous claudin species within and between tight junction strands. J Cell Biol. 1999;147:891–903. doi: 10.1083/jcb.147.4.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Piontek J, Fritzsche S, Cording J, Richter S, Hartwig J, Walter M, et al. Elucidating the principles of the molecular organization of heteropolymeric tight junction strands. Cell Mol Life Sci. 2011;68:3903–3918. doi: 10.1007/s00018-011-0680-z. http://dx.doi.org/10.1007/s00018-011-0680-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Fujita H, Chalubinski M, Rhyner C, Indermitte P, Meyer N, Ferstl R, et al. Claudin-1 expression in airway smooth muscle exacerbates airway remodeling in asthmatic subjects. J Allergy Clin Immunol. 2011;127:1612–1621. e1618. doi: 10.1016/j.jaci.2011.03.039. http://dx.doi.org/10.1016/j.jaci.2011.03.039. [DOI] [PubMed] [Google Scholar]